

Abstract
Double-stranded(ds) RNA has diverse roles in gene expression and regulation, host defense, and genome surveillance in bacterial and eukaryotic cells. A central aspect of dsRNA function is its selective recognition and cleavage by members of the ribonuclease III (RNase III) family of divalent-metal-ion-dependent phosphodiesterases. The processing of dsRNA by RNase III family members is an essential step in the maturation and decay of coding and noncoding RNAs, including miRNAs and siRNAs. RNase III, as first purified from Escherichia coli, has served as a biochemically well-characterized prototype, and other bacterial orthologs provided the first structural information. RNase III family members share a unique fold (RNase III domain) that can dimerize to form a structure that binds dsRNA and cleaves phosphodiesters on each strand, providing the characteristic 2 nt, 3′-overhang product ends. Ongoing studies are uncovering the functions of additional domains, including, inter alia, the dsRNA-binding and PAZ domains that cooperate with the RNase III domain to select target sites, regulate activity, confer processivity, and support the recognition of structurally diverse substrates. RNase III enzymes function in multicomponent assemblies that are regulated by diverse inputs, and at least one RNase III-related polypeptide can function as a noncatalytic, dsRNA-binding protein. This review summarizes the current knowledge of the mechanisms of catalysis and target site selection of RNase III family members, and also addresses less well understood aspects of these enzymes and their interactions with dsRNA. WIREs RNA 2014, 5:31–48. doi: 10.1002/wrna.1195
Double-stranded(ds) RNA is created through the pairing of complementary sequences. A ubiquitous motif in biological systems, dsRNA exhibits a limited conformational plasticity, but exerts a multitude of biological effects. dsRNA can possess a transient, dynamic nature, as seen in antisense RNA binding to target sites. Intramolecular base-pairing provides local double-helical structures that can function as architectural elements or as protein recognition sites. In the latter case, protein binding to dsRNA enables processes such as RNA transport and localization. dsRNA invokes potent cellular responses, as a single molecule of dsRNA can trigger interferon production in vertebrate cells. The cellular response underscores the important aspect of dsRNA as an indicator of pathogenic conditions. In fact, dsRNA is the chromosomal material of many viruses, and cell membrane receptors specifically recognize dsRNA as an initial event in the response pathway.1–5
The enzymatic cleavage of dsRNA is a conserved reaction of fundamental importance in cellular and viral gene expression and regulation, host defense, and genome surveillance. A conserved enzymatic mechanism has been characterized that catalyzes the coordinate cleavage of both RNA strands at selected target sites. Specifically, dsRNA processing is accomplished by members of a ribonuclease family, including the prototypical ribonuclease III (RNase III) of bacterial cells. Ongoing studies on RNase III, and in particular the enzyme from Escherichia coli, have revealed a global role for dsRNA processing in bacterial gene expression and regulation.6,7 The subsequent characterization of eukaryotic family members has extended the essential involvement of dsRNA processing in many additional pathways.8–10 In particular, analysis of the components of RNA interference (RNAi) and related pathways identified two RNase III family members, Dicer and Drosha, as essential functional partners in RNAi.10 Understanding how dsRNA is processed by RNase III family members, and characterizing the many consequences of this reaction remain as key objectives.
This review addresses current knowledge of dsRNA processing by RNase III family enzymes. Emphasis will be placed on the mechanisms of catalysis and target site selection. The salient physicochemical properties of dsRNA also will be summarized. Owing to page limitations this review is unable to include the important contributions from many laboratories that are studying dsRNA processing and gene regulation. However, the reviews cited above may serve as alternative sources.
PHYSICAL PROPERTIES OF dsRNA
The RNase III mechanism of dsRNA recognition, phosphodiester hydrolysis, and product release necessarily reflects the unique attributes of dsRNA. In contrast to the DNA double helix, dsRNA is structurally conservative, existing essentially as an A-form helix with an 11-fold pitch. The preferential adoption of the A-helix reflects the uniform presence of ribose 2′-hydroxyl groups that stabilize the C3′-endo sugar conformation.11 The sugar pucker in turn provides a shallow, solvent-exposed minor groove and a narrow, deep major groove. The 2′-hydroxyl groups that line each side of the minor groove create a polar, extensively hydrated surface, and many dsRNA–protein interactions, including those seen with RNase III family members, rely on the tracks of 2′-hydroxyl groups to provide binding energy and to distinguish dsRNA from DNA, or DNA•RNA hybrids. Alterations in solvent conditions yield the A′-helix that exhibits a 12 base-pair (bp) pitch and a widened major groove.12 The A′-helix has been implicated in specific protein-dependent RNA transport processes involving protein–major groove interactions.13
A prominent feature of dsRNA is its thermodynamic stability that is conferred in part by metal cation interactions. Two modes of binding have been described, in which a metal (e.g., Mg2+) either can be sequestered within the major groove or occupy sites that span the major groove entrance.14 These interactions effectively reduce interstrand repulsion by providing significant charge screening,15 and also contribute to the ability of dsRNA to resist condensation caused by double helix denaturation, as seen with DNA.16 The metal ion interactions allow close packing of helices to provide stable structures, as seen in ribozymes and riboswitches. Another prominent feature of dsRNA is its relative rigidity, as evidenced by a persistence length greater than that for DNA.17,18 While dsRNA is not responsive to sequences than can permanently kink dsDNA,19,20 specific secondary structural elements such as bulges, loops, and mismatches can provide flexible joints.21,22 As discussed below, these elements can function as key substrate reactivity determinants.
THE RIBONUCLEASE III FAMILY
Catalysis of dsRNA cleavage is achieved by a unique fold, termed the RNase III domain. The domain has the capacity to dimerize, providing a stable homodimeric structure with a catalytic site in each subunit. The RNase III domain (˜150 aa) exhibits a characteristic consensus sequence of ˜10 aa, containing conserved residues that bind divalent metal ions important for catalytic activity, or stabilize the dimeric structure.9,10,23 RNase III enzymes exhibit additional domains (Figure 1) that function in concert with the RNase III domain. The additional domains contribute to substrate binding, broaden substrate range, participate in cleavage site selection, confer regulation, and enable protein–protein interactions (see below).9,10,23 RNase III enzymes are highly conserved in the Bacteria and Eukarya, but are only sporadically observed in the Archaea. From an evolutionary perspective, an RNase III enzyme may first have appeared in an early bacterium, then entered the eukaryotic lineage via an endosymbiont. Subsequent elaboration of the structure by acquisition of additional domains created the diversity of eukaryotic family members.
Figure 1.
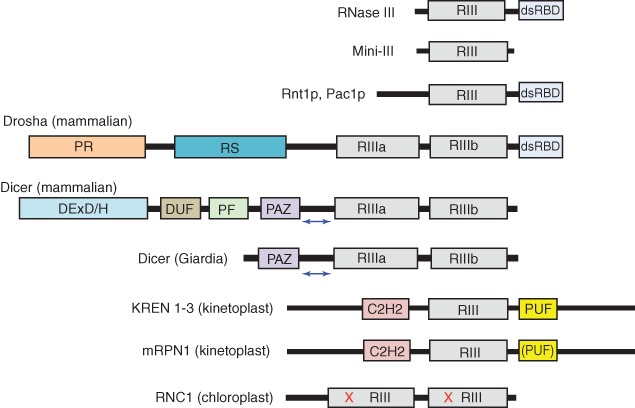
Domain structures of ribonuclease III (RNase III) family polypeptides. Domain key: RIII (gray), RNase III domain; dsRBD (light blue), dsRNA-binding domain; PR (orange), proline-rich domain; RS (Blue), arginine/serine-rich domain; DExD/H (blue), helicase domain; DUF (brown), DUF283 domain; PF (green), platform domain; PAZ (purple), PAZ domain; C2H2 (pink): C2H2 Zinc finger domain; PUF (yellow), Pumilio/FBF homology domain. The blue double-headed arrow between the PAZ and RIIIa domains of human and giardia Dicer indicates the dsRNA ‘measuring’ segment. The parentheses in the PUF domain of mRPN1 indicate that the identity of the domain has not been confirmed. For RNC1, the red letters ‘X’ refer to inactivating point mutations of catalytic site residues. Diagrams are not to scale.
Ribonuclease III
Bacterial RNase III polypeptides (∼220 aa) consist of an N-terminal RNase III domain (˜150 aa) and a C-terminal dsRNA-binding domain (dsRBD), connected by a short (∼7 aa) linker (Figure 1). The dsRBD (∼65 aa) incorporates a single copy of the dsRNA-binding motif (dsRBM), a conserved αβββα fold that occurs in one or more copies in other dsRNA-binding proteins.24 A shorter form of RNase III that lacks the dsRBD, named Mini-III, also occurs in specific bacterial lineages.25 Mini-III is catalytically active, so the dsRBD is not a conserved requirement for activity (see below). In this regard, a truncated form of E. coli RNase III that lacks the dsRBD retains catalytic activity under specific conditions in vitro.26
The purification of native RNase III to homogeneity required large-scale cultures of E. coli, reflecting the low abundance of the enzyme (see below). Essentially pure enzyme was obtained by a multistep protocol involving differential centrifugation, ammonium sulfate precipitation, ion exchange chromatography, and dsRNA affinity chromatography as the final step.27 A polypeptide molecular weight (MW) of ∼25,000 and a holoenzyme MW of ∼50,000 indicated a homodimeric active form.27 Other studies established a catalytic requirement for divalent metal ion, preferably Mg2+, and that RNase III can cleave cellular substrates in vitro at the same sites observed in vivo, without the need for additional factors (reviewed by Court).6
The first reported structure of the RNase III domain involved the successful crystallization of the enzyme from Aquifex aeolicus (Aa) that had the dsRBD deleted.28 The Aa-RNase III domain exhibits (1) a novel, essentially all-α-helical fold; (2) a dimeric structure with an extensive subunit interface; and (3) a symmetrically positioned divalent metal ion (Mn2+ or Mg2+) in each subunit, coordinated to conserved carboxylic acid side chains important for catalytic activity.28 The subunit interface is stabilized by symmetric, hydrophobic ‘ball-and-socket’ motifs, with the dimensions of the shallow valley, defined by the subunit interface, appropriate for accommodation of a dsRNA. Subsequent studies described structures of full-length Aa-RNase III bound to 11 or 12 bp dsRNAs, where in several cases the catalytic site was inactivated by point mutation.29–31 Together with a 2.0 Å structure of full-length enzyme from Thermotoga maritima, crystallized in the absence of dsRNA or divalent metal ion (unpublished; PDB entry 1O0W), these structures revealed the positional flexibility of the dsRBD, and suggested how RNase III can engage dsRNA in a noncatalytic manner. In this regard, a 2.1 Å structure of full-length RNase III of Mycobacterium tuberculosis (PDB entry 2A11) revealed only the RNase III domain, as the flexibility of the linker conferred significant positional variability to the dsRBDs.32 Subsequent studies attempted to define the structure of a precatalytic complex. To this end, the D44N mutation was incorporated into Aa-RNase III, with the expectation that it would suppress phosphodiester cleavage, thus allowing formation of an enzyme-substrate assemblage that approximates the precatalytic complex.33 However, the mutation did not fully inactivate the enzyme, and dsRNA cleavage occurred during crystallization. This reaction was apparently followed by rebinding of product, providing a structure with features of a post-catalytic complex (PDB entry 2EZ6)33 (see below).
In summary, the homodimeric structure of RNase III, and the relative positions of the catalytic sites with respect to the dsRNA binding surface form the structural basis for the characteristic action of RNase III and its family members: the scissile phosphodiesters of the bound dsRNA are on the same face of the double helix and on opposite sides of the minor groove; and each can be simultaneously accommodated in a catalytic site. As such, hydrolysis of both linkages would create product ends exhibiting 2-nucleotide(nt), 3′-overhangs. Additional details on the catalytic mechanism are discussed below.
RNase III Catalytic Mechanism
RNase III activates water as a nucleophile to hydrolyze target site phosphodiesters, creating 3′-hydroxyl, 5′-phosphomonoester product termini. An oxygen isotope incorporation study of Ec-RNase III action on a model substrate revealed the essential irreversibility of the hydrolytic step.34 RNase III requires a divalent metal ion for catalysis, and it was shown for Ec-RNase III that in addition to Mg2+, the transition metal ions Mn2+, Ni2+, and Co2+ also support catalysis, with differing efficiencies.34 Given the intracellular abundance of Mg2+, and its ability to support optimal catalytic activity, this metal most likely is the biologically relevant species. A direct involvement of the metal ion in water activation is indicated by the dependence of the rate of the hydrolytic step on the metal ion pK.34 Since RNase III can bind dsRNA in the absence of Mg2+, metal occupancy of the catalytic sites is not a prerequisite for substrate binding.35
A kinetic study of Ec-RNase III measured the rate of cleavage of a model substrate containing a single scissile bond as a function of Mg2+ concentration. Single-turnover conditions were applied, such that the rate reflected the hydrolytic step. The concentration dependence of the rate indicated that two Mg2+ ions participate in phosphodiester hydrolysis.36 This is consistent with crystal structures of wild-type Aa-RNase III bound to dsRNA cleavage products, exhibiting two Mg2+ ions in each catalytic site that are coordinated to a set of highly conserved carboxylic acid side chains (PDB entries 2NUF, 2NUG).37 The adjacency of the two metal ions and their interaction with the scissile phosphodiester linkage fit the well-studied two-metal-ion catalytic mechanism, wherein one metal (MgA) binds and activates the water nucleophile, and the second metal (MgB) facilitates departure of the 3′-oxygen atom (Figure 2a). Both metal ions are jointly coordinated to the side chain of a highly conserved, functionally essential glutamic acid (E110 in Aa-RNase III, and E117 in Ec-RNase III).37 Additional carboxylic acid side chains and associated water molecules contribute to the proper positioning of MgA and MgB (Figure 2a). Since there is no evidence for the involvement of additional functional groups in the catalytic site, catalysis appears to be largely driven by the two metals. Ca2+ ion does not support catalysis, perhaps reflecting its larger radius and differing ligand coordination properties.38 However, since Ca2+ can stabilize RNase III-substrate complexes35 it is possible that Ca2+ binding to the catalytic sites reduces local negative charge density, thereby promoting stable binding of substrate. While phosphodiester hydrolysis depends upon two closely positioned metal ions, there is structural evidence for a third metal binding site, near the A and B sites, whose occupancy either by a third metal ion—or transiently by one of the two catalytic metals—may contribute to the reaction pathway.37 Since product release is rate-limiting in the steady-state,34 and may also be subject to positive control by an additional process (see below), the third metal site may be important for this step.
Figure 2.
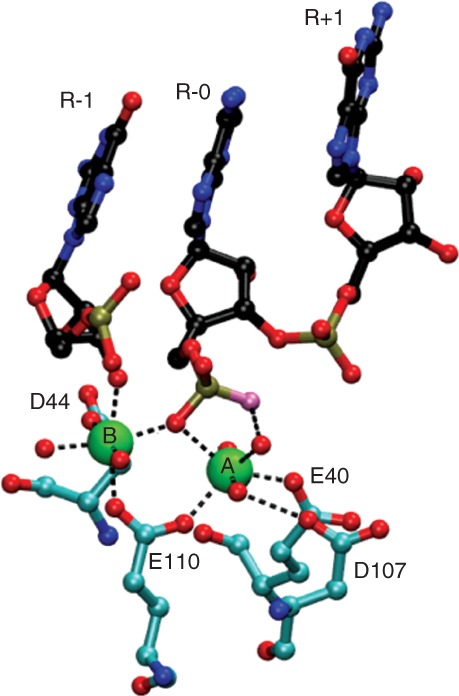
Ribonuclease III (RNase III) catalytic site structure. Shown are the interactions, presumably subsequent to phosphodiester hydrolysis, in the catalytic site of the 1.7 Å structure of wild-type Aa-RNase III bound to cleaved dsRNA (PDB entry 2NUG).37 The two Mg2+ ions (A and B) are shown in green and are coordinated to the E40, D44, D107, and E110 side chains. Metal-oxygen coordinate bonds are shown as dotted black lines. The oxygen atom of the water nucleophile that is bonded to phosphorus (yellow-gold) is colored in lavender. Water molecules are shown as red spheres. The nucleotides (R-1, R-0, and R+1) are colored in blue (nitrogen), black (carbon) and red (oxygen), and the numbering refers to their position with respect to the scissile phosphodiester (between R-0 and R-1).
The structures of Aa-RNase III-substrate complexes and the RNA-free structure of T. maritima RNase III (Figure 3), along with biochemical studies, led to a proposed catalytic pathway for dsRNA cleavage.37,39 (Figure 4). Initial engagement of substrate by a dsRBD is followed by engagement by the dimeric RNase III domain and the second dsRBD, forming the precatalytic complex. Phosphodiester cleavage is followed by disengagement of the products from the RNase III domain, then their release from each dsRBD. The catalytic pathway does not appear to involve an unwinding or other major distortion of the double helix, as none of the reported crystal structures reveal any unusual RNA conformations. Thus, there may only be a modest distortion of the double helix during phosphodiester cleavage. Other studies have shown that the catalytic sites are functionally independent, as mutational inactivation of one site does not affect the function of the other site.39,40 Thus, cleavage of both phosphodiesters would occur following dsRNA binding, but would not be coupled in an obligatory manner.
Figure 3.
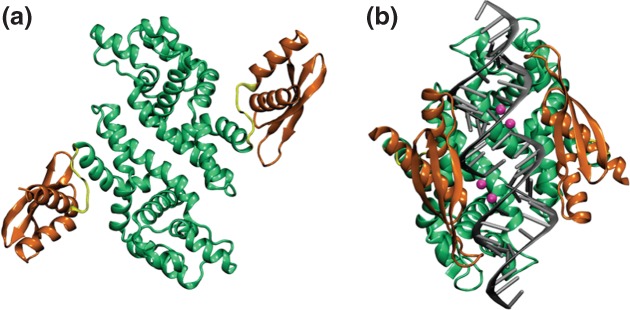
Ribonuclease III (RNase III) structures. (a) 2.0 Å structure of Thermotoga maritima RNase III, determined by the Joint Center for Structural Genomics, University of California at San Diego (PDB entry 1O0w). The structure displays the two dsRBDs (dark orange) in symmetric, extended positions with respect to the RNase III domain (green). The flexible linkers are shown in yellow. (b) A 1.7 Å structure of Aquifex aeolicus (Aa) RNase III bound to dsRNA (PDB entry 2NUG).37 The color scheme in panel (a) also is used here, and the two Mg2+ ions in each catalytic site are colored in cyan. RNA is shown in gray. The phosphodiester bonds at each catalytic site have been hydrolyzed, so the complex contains two dsRNA segments.
Figure 4.
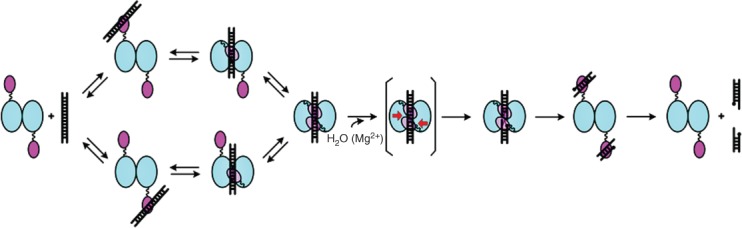
Proposed catalytic pathway for ribonuclease III (RNase III). The diagram is a modification of a proposed scheme.31,39 Initial recognition of dsRNA (black) by a dsRBD (pink) is followed by engagement of the RNase III domain (light blue) and the second dsRBD to form a precatalytic complex. Phosphodiester hydrolysis (red arrows) provides a post-catalytic complex with products still bound by the RNase III domain. Release of the products from the RNase III domain is followed by release from the dsRBDs. The rate-limiting step may be release of products from the RNase III domain (see text). (Reprinted with permission from Ref 7. Copyright 2011 Springer Science+Business Media).
RNase III Substrate Recognition and Target Site Selection
Understanding how dsRNA target sites are selected is central to understanding RNase III function. A long-known feature of the bacterial family members, in particular Ec-RNase III, is the ability to degrade long dsRNAs of broad sequence content to ∼11–15 bp products.6,9 The questions arise as to whether this mode of action can be mechanistically reconciled with its site-specific action on cellular substrates, and also whether it has functional importance. Crystallographic33,37 and biochemical41–43 studies show that target site recognition by RNase III involves protein contacts with at least two specific segments in the substrate, termed the proximal box (pb; 4 bp) and distal box (db; 2 bp) (with the two terms referring to the positions of each box relative to the target site) (Figure 5a). The involvement of the pb and db in target site selection was shown by the ability of a duplicated pb–db pair in a model substrate to direct cleavage at the predicted additional site.42 A third region, termed the middle box (mb), is positioned between the pb and db, and also contacts protein.33,37
Figure 5.
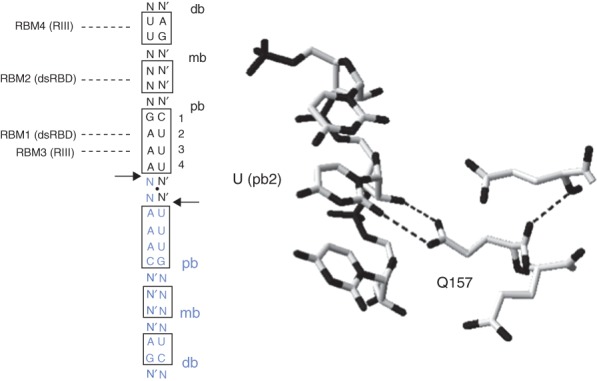
Reactivity determinants of ribonuclease III (RNase III) substrates. (a) Diagrammatic structure of an idealized dsRNA substrate of Ec-RNase III. The length of the helix from the end of each db is 22 bp (two A-helical turns). The boxed bp indicate the distal box (db), middle box (mb) and proximal box (pb). The twofold symmetry of the substrate is indicated by the black dot centered between the two cleavage sites (black arrows), and blue color of the lower portion of the substrate. Note that the symmetry does not include specific bp sequence. N-N′ indicate a standard bp of unspecified sequence. The sites of interaction of RBM1, RBM2, RBM3, and RBM4 with the db, mb, and pb are indicated by dashed lines. (b) Sequence-dependent interaction of the conserved glutamine side chain of the dsRBD α1 helix (RBM1) with the O2 uracil atom. Taken from the Aa-RNase III(D44N)-dsRNA crystal structure (PDB entry 2EZ6).32 The dotted lines indicate probable hydrogen bonds of the Q157 carboxamide group with the O2 atom and the adjacent 2′-hydroxyl group.
The distal box is important for recognition, as its deletion significantly weakens enzyme binding.41 The positioning of the db ∼11 bp from the target site serves to establish a minimum substrate length (∼11 bp) necessary for efficient reaction. The db does not exhibit a conserved sequence, either among substrates of a given RNase III, or across species. However, bp substitutions in the db reduce Ec-RNase III substrate reactivity,41,42 while having minimal effects on Aa-RNase III substrate reactivity.43 A short, nonconserved region in the RNase III domain, termed RBM4, interacts with the db.33,37 Thus, an emerging picture is an idiosyncratic yet energetically important db–RNase III interaction.7 Structural studies of RNase III-substrate complexes with anticipated precatalytic features can be expected to provide further insight on this interaction.
Structural and biochemical studies on the RNase III–pb interaction have shown how bp sequence controls reactivity and participates in target site selection. Side chains in the N-terminal portion of the dsRBD N-terminal α helix (termed RBM1) form an array of hydrogen bonds with functional groups within the pb, including ribose 2′-hydroxyl groups on both strands.33,37 One interaction of interest occurs in the minor groove, and involves a hydrogen bond between the carboxamide side chain of a highly conserved glutamine (Q157 in Aa-RNase III) and the O2 pyrimidine atom of the AU bp at pb position 2 (Figure 5b).33,37 The carboxamide group also forms a second hydrogen bond with an adjacent ribose 2′-OH group. These are functionally important interactions, as mutation of the glutamine to an alanine strongly reduces RNase III catalytic activity due to a significantly weakened affinity for substrate.43 Interestingly, pb position 2 also is the site of inhibition by bp substitution. Thus, while the AU and UA bp confer equivalent reactivity, a CG or GC bp blocks binding of Ec-RNase III.41–43 The purine 2-amino group is responsible for inhibition, as incorporation of an amino group on the adenine C2 atom of the AU pair (forming a 2,6-diaminopurine•U bp) also confers inhibition, and that removal of the 2-amino group from G (forming an inosine-cytosine pair) restores processing activity.42 On the basis of crystallographic data, the 2-amino group may prevent dsRBD binding by a steric clash with the glutamine side chain, prohibiting formation of the hydrogen bonds.42,43 The apparent invariance of the glutamine in the RBM1 site of bacterial RNase III sequences, and the preference for a UA or AU bp at pb position 2 underscores the important contribution of this interaction to binding of enzyme in the correct register. The absence of a conserved glutamine in the corresponding position of the dsRBDs of eukaryotic RNase III enzymes (AWN, unpublished) is consistent with different mechanisms for target site selection (see below).
The inhibitory action of GC/CG bp substitution at pb position 2 may have a role in minimizing ‘off-target’ effects of RNase III. Antideterminants were originally described as tRNA sequence or structural features that block inappropriate recognition by noncognate aminoacyl-tRNA synthetases or other proteins.44 For dsRNA structures whose functions would be destroyed by RNase III action, the presence of antideterminant bp at key positions would prevent enzyme recognition, and without disruption of the regular double-helical structure. A qualitatively different type of bp-dependent inhibition is seen at pb position 4, where a GC or CG substitution prevents substrate cleavage, but does not inhibit binding of Ec-RNase III.42 This type of substitution may afford dsRNA structures that function as RNase III binding sites. In this regard, an in vitro selection (SELEX) study identified an RNA that can bind RNase III but is resistant to cleavage, due in part to a GC/CG bp adjacent to the cleavage site.45
In summary, RNase III recognition of substrate involves at least one sequence-specific, energetically important interaction of the dsRBD with the pb, and an idiosyncratic interaction of the RNase III domain with the db. Interestingly, a truncated form of Ec-RNase III that lacks the dsRBD also cleaves substrate at the canonical site, under conditions of reduced salt concentration and using Mn2+ in place of Mg2+.26 Thus, the dsRBD and RNase III domain engage substrate in a complementary register. This coincident specificity may provide an important reinforcement of target site selectivity, with the dsRBD performing a key role in initial discrimination of substrate. The mechanism by which the RNase III domain identifies the target site is not known, and the db interaction may be insufficient to achieve this. The natural RNase III variant, Mini-III lacks the dsRBD, yet site-specifically cleaves the immediate precursor to the mature 23S rRNA.25 While optimal activity of Mini-III requires ribosomal protein L3,46 the latter protein is not required for specificity. Finally, the proximal boxes of cellular substrates exhibit significant degeneracy. The generally constrained lengths (∼<20 bp) of the double-helical structures of many cellular substrates may compensate for the degeneracy to provide a single target site. Regarding the apparent paradox of RNase III action (see above), the degeneracy of recognition sites also would allow RNase III to degrade much longer dsRNAs that are created, for example, by association of genomic transcripts.47,48 Thus, the long-noted ability of RNase III to degrade long dsRNAs in vitro may have important functions in vivo.
While RNase III target sites are established by double-helical sequence elements, an important additional aspect of substrate reactivity is whether one or both target site strands are cleaved. This is an important distinction as it can determine product function and stability. For example, single-strand cleavage can create hairpin structures at RNA 3′-ends that act as stability determinants.49 Secondary structural elements such as loops, bulges, or mismatches at target sites create deviations from a regular double-helical structure, and would allow only one of the two strands to productively engage a catalytic site. Cleavage of the phosphodiester would be followed by product release. While internal loops can be redesigned to change the pattern of cleavage,50 a full understanding of the role of RNA secondary structure in modulating the pattern of reactivity awaits further study.
Regulation of RNase III
As with metabolic pathways in general, RNA processing and decay reactions and the associated enzymes are subject to regulation. This not only controls ribonucleotide flux, but as (or more) importantly, controls gene expression. Regulation of ribonuclease levels and activity can confer global control of the production of the processed products. Several lines of evidence demonstrate that dsRNA processing is regulated at multiple levels through control of RNase III. It has been estimated that there are several hundred copies of RNase III in the E. coli cell, the levels of which increase in proportion to growth rate.6 The limited amount of RNase III indicates a stringent regulation of expression. While there is no apparent transcriptional control, regulation of Ec-RNase III expression is imposed at the post-transcriptional level by the action of the enzyme on its own mRNA. Specifically, cleavage of an irregular hairpin in the mRNA 5′-UTR creates a new, unstructured 5′-end with a phosphomonoester group. This structure is recognized in turn by RNase E, leading to rapid decay of the mRNA.51 The negative autoregulation not only limits production of enzyme but also allows for upregulation of enzyme level with increased growth rate. Here, the increased number of substrates—in particular the abundantly produced rRNA precursors that are processed by RNase III—engage a larger proportion of the available enzyme, thereby reducing the level of autoregulation.
RNase III catalytic activity is subject to positive regulation. The T7 bacteriophage expresses a protein kinase that phosphorylates RNase III and enhances catalytic activity ∼fourfold, as measured in vitro.52 Phosphorylation occurs on a serine in the RNase III domain,53 and biochemical analyses reveal that the covalent modification facilitates product release, which is the rate-limiting step in the catalytic pathway (S. Gone, M. Prieto and AWN, manuscript in preparation). The T7-induced catalytic enhancement of RNase III may serve to meet the demands placed on limited amounts of enzyme to process the abundant phage transcripts, most of which contain at least one RNase III target site, and whose maturation is required for optimal production of phage protein.54
RNase III is negatively regulated by the macrodomain protein, YmdB. Cohen and coworkers showed that ectopic overexpression of YmdB reduces RNase III activity, and that RNase III activity also is downregulated when cells undergo cold shock, which increases YmdB levels.55 YmdB-dependent regulation of RNase III appears to be a post-transcriptional feature of the global shift in physiological state in response to lower temperatures and perhaps other stress conditions. The mechanism of YmdB-dependent inhibition of RNase III is unclear, but it is thought to be dependent upon the direct interaction of the two proteins.55 YmdB also binds ADP-ribose, and hydrolyzes O-acetyl ADP-ribose.56 The latter compound is a product of sirtuin-catalyzed protein deacetylation, which uses NAD as cosubstrate.56 Whether these activities are related to YmdB regulation of RNase III is unclear, but brings up the possibility of functional crosstalk between dsRNA-dependent gene regulation, the protein acetylome, and cellular redox state.
RNase III activity in vivo also is responsive to osmotic stress. The E. coli proU operon encodes an uptake system for the osmoprotectants proline betaine and glycine betaine.57 The proU operon transcript is relatively stable (half-life ∼65 second) under conditions of high osmolarity, but under conditions of reduced osmolarity undergoes rapid degradation (half-life <4 second) through an initiating cleavage by RNase III.57 The rapid degradation ensures efficient inhibition of proU expression and further uptake of osmoprotectants. Other substrates are anticipated whose reactivity towards RNase III is responsive to changes in growth conditions. In summary, RNase III levels and catalytic activity are regulated at multiple levels, including mRNA stability, covalent modification, protein binding, and changes in ionic conditions, and reflect transitions between different physiological states. Additional regulatory mechanisms are predicted for other family members.
Yeast RNase III
RNase III family members within the fungi include Rnt1p of Saccharomyces cerevisiae and Pac1p of Schizosaccharomyces pombe. Rnt1p and Pac1p cleave hairpin structures in pre-rRNAs, pre-mRNAs, and transcripts containing noncoding RNAs such as snoRNAs, as part of the respective maturation pathways. The enzymes also are involved in RNA quality control.8,9 The Rnt1p polypeptide possesses a single RNase III domain, a C-terminal dsRBD, and an additional N-terminal domain that functions in nuclear localization and interaction with additional factors58 (Figure 1). Many Rnt1p substrates contain a consensus [A/u]GNN tetraloop that functions as a recognition determinant.59,60 Rnt1p recognizes the tetraloop in a shape-dependent (rather than sequence-dependent) manner, and cleaves the stem ∼14–16 bp from the structure. Tetraloop recognition involves the dsRBD α1 helix, which possesses an extended structure relative to the α1 helix of other RNases III.61 The elongated helix provides an expanded hydrophobic core, conferring a conformational plasticity to the dsRBD that is important for forming a functionally competent dsRBD-hairpin complex.61,62 The conformational changes that occur in the dsRBD and tetraloop are consistent with an induced-fit mechanism that can distinguish substrates from other hairpin structures.61,62
Additional features of the Rnt1p-substrate interaction contribute to processing reactivity. Specific bp sequence elements can modulate substrate reactivity, and a network of hydrogen bonds provides an energetically important contribution to Rnt1p binding.63,64 A phylogenetic-based substrate alignment analysis revealed a statistically significant exclusion of the UA bp from the position adjacent to the tetraloop.65 Substitution of the UA bp at that position inhibited substrate cleavage but did not perturb enzyme binding.65 The mechanism of inhibition is unclear but is dependent on the purine 6-amino group immediately 3′ to the fourth nucleotide of the tetraloop. The inhibition does not appear to disrupt the tetraloop–dsRBD interaction, but instead perturbs a downstream event, perhaps engagement of the RNase III domain.65 The UA bp may afford an antideterminant that is used in other stem-loop structures that are not Rnt1p substrates.
The maturation of a number of snoRNAs, including U18 and snR38, involves Rnt1p-dependent excision from the respective host introns. The structures of the intronic precursors deviate from the canonical [A/u]GNN stem-loop substrates, and their processing requires participation of the nucleolar protein Nop1p, which physically interacts with Rnt1p.66 Rnt1p also interacts with Gar1p, a protein involved in pseudouridylation reactions. The C-terminal portion of Rnt1p, adjacent to the dsRBD, interacts with Gar1p.67 As these interactions imply an association of rRNA processing and RNA modification reactions, Rnt1p may be regarded as a core nucleolytic component of one or more dynamic nuclear/nucleolar protein complexes. The full characterization of these complexes remains an important goal.
Drosha and Microprocessor Function
Over 1000 micro(mi) RNAs are estimated to be encoded in mammalian genomes, and their expression and utilization is regulated by diverse inputs in a cell- and development-specific manner. Mammalian miRNA maturation involves, inter alia, the sequential action of two RNase III family enzymes, Drosha and Dicer. Drosha is localized in the nuclear compartment and acts on primary transcripts synthesized by RNA polymerase II, and that typically contain several miRNAs. Site-specific cleavage within irregular, extended hairpin structures (pri-miRNAs) creates the pre-miRNAs that then are delivered by Exportin5 to the cytoplasm for final maturation by Dicer. Drosha functions within a complex termed the microprocessor that contains a protein, DGCR8, that is required for Drosha action.68–70 There is scant structural information currently available on Drosha, which in part reflects a general challenge in studying large proteins that function in larger complexes. Thus, most current information derives from biochemical studies, including sequence and structural analyses of the pri-miRNAs (see below). The Drosha polypeptide (Figure 1) possesses tandem RNase III domains and a C-terminal dsRBD. The RNase III domains form an intramolecular pseudodimer with two catalytic sites.69 A solution structure determination of the Drosha dsRBD reveals an α1–α1 loop element with a dynamic, extended structure.71 The inability of the Drosha dsRBD to form a stable complex on its own with dsRNA may reflect the atypical mobility and negative charge of the loop, and raises questions on its function in pri-miRNA processing.71,72 The Drosha N-terminal region also contains proline-rich and arginine/serine-rich domains that are implicated in protein–protein and protein–nucleic acid interactions.73
The DGCR8 polypeptide exhibits tandem dsRBDs and an N-terminal WW domain.74 The structure of DGCR8 is not known, but a crystallographic study of the C-terminal region reveals the specific, stable positioning of the two dsRBDs which are packed on the C-terminal helix, and a linker connecting the dsRBDs that exhibits an ordered structure (Figure 6a).76 Molecular dynamic simulations indicate a correlated motion of the two dsRBDs.78 This can be contrasted with the flexible linkers and independent action of the RNase III dsRBDs. DGCR8 also undergoes dimerization in a heme-dependent manner, and is important in providing optimal activity.79,80 The dimerization region is a component of the heme-binding domain, and involves the WW motif.80 The role of heme-dependent dimerization of DGCR8 in microprocessor function awaits further analysis.
Figure 6.
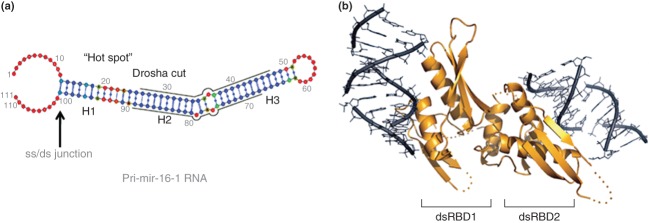
Reactivity determinants of a pri-miRNA substrate for Drosha, and a proposed DGCR8-dsRNA interaction. (a) Features of mammalian pri-miR-16-1 RNA important for reactivity.75 The grey lines indicate the positions of the miRNA sequences, and the Drosha cleavage sites are at the left hand termini of the lines, that upon hydrolysis create 2-nt, 3′-overhang product ends (see text). ‘ss/ds junction’ indicates the region recognized by DGCR8, and ‘Hot Spot’ refers to a deformable region proximal to the Drosha cut site. H1, H2, H3 denote regular helical regions that are punctuated by the deformable regions. The color scheme for individual nucleotides (solid circles) indicates the relative probability of single-strandedness, determined as described75: blue, low probability; green/yellow, medium probability; red, high probability. (Reprinted with permission from Ref 75. Copyright 2013 American Chemical Society). (b) Proposed model of a complex of dsRNA bound to the DGCR8 core, consisting of dsRBD1, dsRBD2, the connecting linker, and the C-terminal helix.76 The fixed relative positions of the two dsRBDs and the corresponding placement of the respective dsRNA-binding surfaces suggest that engagement of a pri-miRNA by the DGCR8 polypeptide requires distortion of the pri-miRNA structure, which would be facilitated by the deformable regions (see panel (a)).77,75 (Reprinted with permission from Ref 76. Copyright 2004 Macmillan Publishers Ltd)
Structural features of pri-miRNAs important for reactivity have been identified through extensive genomic sequence analyses, RNA structure probing, and biochemical assays. Pri-miRNAs exhibit nonuniform structures, and contain double-helical segments punctuated at each full helical turn by bulges, loops, or mismatches (Figure 6b). DGCR8 recognizes the ss/ds junction at the base of the extended stem-loop hairpins and allows engagement of Drosha.68 It was originally proposed that Drosha cleavage sites are identified by their distance (one helical turn) from the ss/ds junction.68 The recognition model was further elaborated by incorporating noncanonical secondary structural elements near the target site as reactivity determinants.77 Structure-probing protocols, coupled with computational modeling75 showed that the elements confer structural plasticity to pri-miRNAs. The deformability may be a prerequisite for engagement by the DGCR8 dsRBDs, whose fixed orientations require a conformational change in the pri-miRNA in order to achieve a stable complex and allow engagement of Drosha.77,75 Moreover, the conformational plasticity also would identify substrates by an induced-fit pathway. Structural studies involving new approaches (e.g., see analysis of Dicer below) are expected to shed light on other Drosha domains and the structural basis of microprocessor function. Recent studies have uncovered a cytoplasmic version of the microprocessor that acts on a viral-encoded pri-miRNA, ultimately providing the mature species (termed ‘virtron’) in a Dicer-dependent manner. The cytoplasmic microprocessor involves Drosha, which is shuttled from the nucleus in a virus-infection-dependent manner, but is not strictly dependent upon DGCR8.81–83 Whether Drosha associates with other factors, forming the cytoplasmic microprocessor, remains to be determined. In a similar light, studies are revealing a broader functional role of DGCR8 in RNA processing pathways that may involve nucleases other than Drosha.84
Dicer
Eukaryotic genomes encode single or multiple forms of Dicer polypeptides. While a key function of Dicer is the conversion of pre-miRNAs to miRNAs, other roles include host defense and genome surveillance mediated by the processing of alternative, dsRNA-based substrates. Dicer polypeptides share a core structure consisting of tandem RNase III domains and a PAZ domain (Figure 1). One or two dsRBDs may be present in the C-terminal region, and the polypeptide also may carry an N-terminal helicase-like domain (Figure 1). The questions addressed here are how Dicer (1) provides precisely-sized products; (2) can act in a processive manner; and (3), achieves flexibility in processing regular dsRNAs as well as hairpin substrates that include pre-miRNAs.
Biochemical analysis of human(h) Dicer first indicated that the tandem RNase III domains form an intramolecular pseudodimer with two catalytic sites.85 A crystallographic analysis of the ‘minimal’ Dicer of Giardia intestinalis (gDicer), containing the PAZ and tandem RNase III domains, confirmed the pseudodimeric RNase III domain, and revealed how product length derives from the physical spacing and functional cooperation of the PAZ and RNase III domains.86 The PAZ domain specifically recognizes the 2-nt, 3′-overhangs of a processed dsRNA terminus and is positioned ∼65 Å from the RNase III catalytic sites (Figure 7a). Engagement of substrate by the two domains allows production of the characteristic ∼25 bp product of gDicer action86 (Figure 7b). Although a comparable crystallographic analysis of hDicer has been sought, the enzyme has been largely refractory to such an approach. Instead, domain-specific labeling, combined with single-molecule electron microscopy provided essential insight on the relative positions and functions of the additional domains, and afforded a structural model for hDicer action.87 The PAZ domain of the L-shaped protein is positioned at the top of the ‘L’, with the tandem RNase III domains occupying the bend (Figure 8a). The physical separation of the two domains establishes the ∼21 bp dsRNA products. Here, PAZ domain recognition of the 2-nt, 3′-overhang and the 5′-phosphomonoester group provides the “3'-counting”86 and ‘5′-counting’88 mechanisms, respectively, for target site selection. The shorter size of the hDicer products compared to the ∼25 bp products of gDicer reflects a translational rotation of the RNase III domain, relative to the PAZ domain.87
Figure 7.
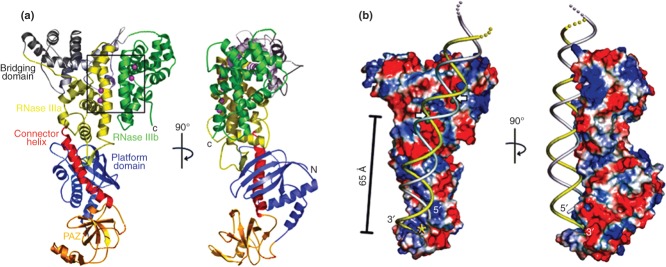
Structural features of a ‘minimal’ Dicer. (a) Two views of the 3.3 Å crystal structure of Dicer from Giardia intestinalis86 (PDB entry 2ffl). The PAZ domain (gold) is linked to the RNase IIIa domain (yellow) by the connector helix, or ‘ruler’ (red) (see also Figure 1), which determines the dsRNA product length. The RNase IIIb domain is shown in green, and the two catalytic sites (within the rectangle) are identified by the two metal (Er3+) ions (purple) in each site (see also Figure 2). (b) Two views of a modeled complex of gDicer bound to dsRNA.86 Blue and red coloration indicate basic and acidic regions, respectively. dsRNA is shown in yellow and gray strands, and the white arrows indicate the sites of cleavage. The yellow star indicates the site in the PAZ domain that binds the dsRNA 3′-overhang. (Reprinted with permission from Ref 86. Copyright 2006 AAAS)
Figure 8.
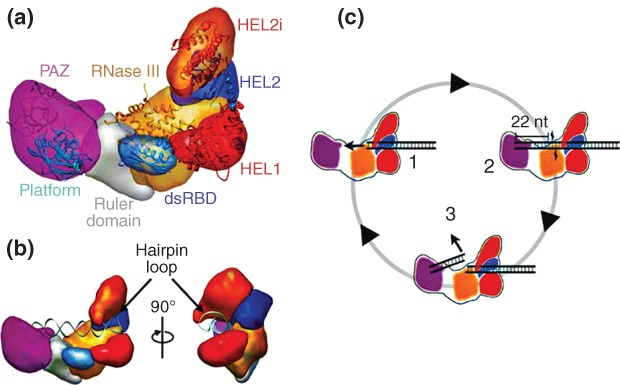
Functional interplay of human(h) Dicer domains in dsRNA processing.87 (a) Domain structure of hDicer as determined by domain-specific tagging and single molecule electron microscopy (EM). Shown are the PAZ (purple), platform (blue), ruler (gray), dsRBD (blue), ribonuclease III (RNase III) (a+b) (orange) and helicase domains, with the latter domain displayed as three subdomains HEL1 (red), HEL2 (blue), and HEL2i (orange). (b) Two views of hDicer bound to a pre-miRNA hairpin, showing the engagement of the loop by the helicase domain, and the opposing 3′-overhang end by the PAZ domain. (c) Proposed processing cycle for hDicer cleavage of dsRNA. In complex 1, the dsRNA is engaged by the helicase domain. In complex 2, cooperation of the PAZ and RNase III domains provide a 22 bp product, while the remainder of the dsRNA remains engaged with the helicase domain. In complex 3, release of the 22 bp product from the PAZ and RNase III domains allows subsequent engagement of the contiguous dsRNA segment by the RNase III and PAZ domains. This cycle explains the processivity of hDicer action, supported by the helicase domain. (Reprinted with permission from Ref 87. Copyright 2012 Macmillan Publishers Ltd)
The hDicer helicase domain is positioned adjacent to the RNase III domain, at the opposite end of the L from the PAZ domain. The position of the helicase domain and its ability to engage dsRNA, confers processivity, wherein engagement of dsRNA allows successive rounds of cleavage prior to release of substrate87 (Figure 8b). The positioning of the helicase domain explains its ability, under specific conditions, to inhibit substrate processing, most likely by the occluding the catalytic sites. This facet of helicase domain function may ensure stringent control of the ribonucleolytic action of Dicer. Additional insight on helicase domain function has emerged from a high-throughput sequencing study of the products of Dicer action on small hairpin(sh) RNAs in vivo. It was determined that placement of the cleavage site 2 nt from the hairpin loop confers maximal accuracy of processing. In addition, an insertion mutation of the helicase domain caused a reduction in processing accuracy for these substrates.89 A ‘loop-counting’ mechanism was proposed for target site selection, in which the helicase domain establishes an additional interaction with the RNA loop (either as a terminal or internal loop), thereby conferring an additional level of cleavage precision not afforded by the PAZ domain alone.89
Biochemical and structural studies of hDicer have been challenged by the size of the polypeptide and the attendant issues of solubility and activity. As an alternative approach, individual domains or domain combinations were purified in recombinant form, and assays of substrate binding and cleavage performed that included single domain omission.90 It was shown that the helicase domain directly interacts with pre-miRNA hairpins, thus rationalizing how Dicer can process substrates structurally distinct from dsRNA, using an alternative substrate recognition pathway.90 The functional role of the dsRBD has been enigmatic, as it is not essential for Dicer action in standard assays. However, it was shown that this domain, in isolated form, binds dsRNA,91 and enables substrate processing by Dicer in the absence of the PAZ domain.90 These findings suggest that the dsRBD may establish a pathway for recognition of alternative substrates, including those not recognized by the PAZ domain.90 The occurrence of Dicer family members with multiple dsRBDs underscores the relevance of alternative recognition pathways connected with substrate diversification. On the basis of these findings it is anticipated that additional substrates will be identified that do not conform to the pre-miRNA or regular dsRNA structures. Dicer interacts with other proteins, including TRBP, PACT, and Ago2.92,93 While the structural details of these interactions are just beginning to be elucidated, their influence on Dicer activity is evident. For example, TRBP alters the manner in which hDicer processes pre-miRNAs, allowing formation of alternative miRNAs (‘isomiRs’) that are one nt longer than the canonical form, and possess different target specificities.94,95
RNase III Enzymes in Trypanosome Kinetoplasts
RNase III family enzymes participate in kinetoplast RNA editing, wherein multiple uridine nucleotides are inserted or deleted at multiple sites in the mRNA precursors, ultimately creating the mature, translationally competent species. The selection of editing sites; the determination whether U insertion or deletion occurs at a given site; and how many U residues are inserted or deleted at each site are determined by trans-acting, noncoding guide(g) RNAs.96 A segment of each gRNA is complementary to clusters of editing sites, allowing formation of an intermolecular duplex that directs endonucleolytic cleavage of the pre-mRNA at the ss/ds junction. The gRNA sequence also determines whether U residues are removed from the cleaved pre-mRNA via exonucleolytic action, or instead are inserted by a uridylyltransferase. A final ligation reaction establishes the mature sequence at the editing site.
Three distinct, multisubunit RNA editing complexes have been characterized that have differing editing site specificities, and that contain an RNase III family polypeptide termed KREN1, KREN2, or KREN3.97–100 These polypeptides are required for the endonucleolytic cleavage step, and they may specifically function in the form of heterodimers that involve partner proteins KREPB4 or KREPB5.101 The latter proteins also have RNase III domains, but sequence analysis and mutational studies reveal that the catalytic sites are nonfunctional.101 If such a heterodimeric complex exists, it would therefore have only a single functional catalytic site in the KREN1, KREN2, or KREN3 subunit, and therefore would cleave only one strand in a single binding event (here, the pre-mRNA at the editing site) while preserving the other strand (the gRNA). In this regard, the ability of RNase III heterodimers to ‘nick’ dsRNA has been shown elsewhere.39,40 Alternatively, the single-strand cleavage pattern may reflect local RNA structure at or near the target site (see also above). Further analyses are needed to determine whether the KREN1-3 polypeptides associate with KREPB4 and KREPB5 to form heterodimeric structures. The KREN1-3 polypeptides exhibit a distinctive modular structure that includes a C2H2 Zinc finger (ZnF) motif (Figure 1). A sequence alignment analysis101 showed that the five RNase III family polypeptides contain a PUF domain, rather than a dsRBD, as had been previously suggested. Since PUF domains of other proteins can recognize RNA in a sequence-dependent manner,102 it is anticipated that the domain functions in cleavage site selection. The presence of the ZnF and PUF domains reflects the unique requirements for the highly specialized and complex process of RNA editing.
The maturation of the gRNAs from polycistronic precursors also involves an additional RNase III family enzyme, mRPN1.103 A knockdown of mRPN1 levels causes a drop in gRNA levels and concomitant accumulation of the precursor forms.103 mRPN1 possesses a homodimeric structure, and mutational analysis identified a glutamic acid essential for activity, and which corresponds to a catalytic site glutamic acid in bacterial RNase III.103 The polypeptide contains a C2H2 ZnF domain and a dsRBD (Figure 1) (although the identity of the latter domain remains to be confirmed—see above). mRPN1 associates with several proteins, one of which (TbRGG2) may mediate gRNA access to the RNA editing complexes.103 The structures of the gRNA precursors and the associated mRPN1 processing sites have not yet been described. However, the maturation of polycistronic gRNA precursors by mRPN1 is reminiscent of the action of E. coli RNase III on the five processing signals within the ∼7000 nt polycistronic mRNA precursor of bacteriophage T7.27,54
Noncatalytic Action of RNase III-like Proteins
Point mutations that selectively disable the catalytic site have been used to show that RNase III can regulate gene expression as a dsRNA-binding protein.104 Structural studies reveal how RNase III can engage dsRNA in a noncatalytic manner.29–31 Also, variants of naturally occurring RNase III substrates, as isolated by in vitro selection, can be bound by RNase III but are resistant to cleavage.45 The question is whether in fact RNase III family members have noncatalytic cellular roles. An example is provided in the chloroplasts of land plants. Here, a polypeptide (RNC1) that plays a role in supporting the splicing of group II introns, possesses tandem RNase III domains that are catalytically silent due to mutation of active site residues105 (Figure 1). The tandem RNase III domains are anticipated to self-associate, forming a dsRNA-binding fold that stabilizes structures important for intron splicing.106 The RNC1 polypeptide associates with at least one additional protein, WTF1, as part of a larger complex that supports splicing.107
BOX 1 Other ways of processing dsRNA, and a question about Archaeal dsRNA.
The processing of dsRNA is not exclusively performed by RNase III family members. Specific members of the RNase A family of vertebrate-secreted proteins are capable of degrading dsRNA, using an alternative catalytic mechanism. These enzymes, in dimeric form, can bind dsRNA and stabilize localized ssRNA structures created through natural ‘breathing’ of the dsRNA. Each strand is cleaved via a phosphotransferase reaction, involving vicinal 2′-OH groups.108 The functions of these enzymes are diverse, and include host defense, response to stress, and angiogenesis.109,110 The Lassa fever virus expresses a 3′→5′ exonuclease that degrades dsRNA (as well as the RNA strand of RNA-DNA hybrids).111,112 The degradation of the RNA strand apparently occurs without major disruption of the double-helical structure.112 Thus, at least one function of this protein is to suppress the host cell innate immune mechanism that recognizes dsRNA.111–113
RNase III orthologs appear to be only accidental to the Archaea. Thus, a question is how dsRNA is recognized and processed in this major branch of life. The Bulge-Helix-Bulge (BHB) splicing endoribonuclease functions in place of RNase III to process the pre-rRNAs, but it is not a dsRNA-specific nuclease.114 If dsRNA processing is a feature of Archaeal RNA metabolism (which is reasonable to assume), either an alternative, dsRNA-specific nuclease exists (e.g., see above), or dsRNA processing is accomplished by the cooperative action of several enzymes, such as a specific exo- or endoribonuclease working in conjunction with, for example, an RNA helicase. In any event, the pathways remain to be understood.
CONCLUSION
The broadening understanding of the role of dsRNA processing in gene expression and regulation continues to spur studies on the structures and mechanisms of RNase III family members, and their functional and physical interactions with other factors. How specificity is achieved in dsRNA cleavage continues to draw attention, especially in the maturation of miRNAs and siRNAs, for which accuracy is essential for correct function, but for which there is evidence for modulation of cleavage site selection as a means to diversify target sites. The demonstration that RNase III enzymes are regulated at multiple levels, as especially shown by bacterial family members, indicates the importance for such regulation in cellular processes. RNase III enzymes function in complexes that exhibit dynamic features and are subject to regulation. Studies are now increasingly focused on determining the components and structural features of the complexes. The regulated activity of these complexes in turn can confer global regulation of dsRNA-dependent processes, of which much remains to be understood. Definition of these networks is needed for a full understanding of the involvement of dsRNA processing on cell physiology in response to diverse inputs.
The question remains as to whether RNase III family members can act in a noncatalytic manner. There is now an example of this in chloroplasts, involving a noncatalytic RNase III polypeptide, and it is anticipated that cellular substrates will be uncovered that bind RNase III in this manner. Specific substrates of RNase III family members are being used as functional modules in synthetic gene networks,115,116 and research on dsRNA processing can be envisioned to extend to the nanotechnology arena.117 RNase III can function in dense nanopatches of dsRNA, and can provide a permanent topographic ‘imprint’ of dsRNA recognition by either a protein or inhibition by an intercalating agent.118 Such approaches can provide the basis for detecting dsRNA and related molecules as disease biomarkers at the single-cell level.
Acknowledgments
The author thanks Ms. Samridhdi Paudyal, Dr. Mercedes Prieto and Dr. Wenzhao Meng for help with the figures, and Dr. Rhonda Nicholson for comments on the manuscript. Research in the author's laboratory is supported in part by grants from the NIH.
REFERENCES
- Haines DS, Strauss KI, Gillespie DH. Cellular response to double-stranded RNA. J Cell Biochem. 1991;46:9–20. doi: 10.1002/jcb.240460104. [DOI] [PubMed] [Google Scholar]
- Nicholson AW. Structure, reactivity and biology of double-stranded RNA. Prog Nucleic Acid Res Mol Biol. 1996;52:1–65. doi: 10.1016/s0079-6603(08)60963-0. [DOI] [PubMed] [Google Scholar]
- Wang Q, Carmichael GG. Effects of length and location on the cellular response to double-stranded RNA. Microbiol Mol Biol Rev. 2004;68:432–452. doi: 10.1128/MMBR.68.3.432-452.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantier MP, Williams BR. The response of mammalian cells to double-stranded RNA. Cytokine Growth Factor Rev. 2007;18:363–371. doi: 10.1016/j.cytogfr.2007.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deFaria IJ, Olmo RP, Silva EG, Marques JT. dsRNA sensing during viral infection: lessons from plants, worms, insects, and mammals. J. Interferon Cytokine Res. 2013;33:239–253. doi: 10.1089/jir.2013.0026. [DOI] [PubMed] [Google Scholar]
- Court D. RNA processing and degradation by RNase III. In: Belasco J, Brawerman G, editors. Control of Messenger RNA Stability. New York: Academic Press, Inc; 1993. pp. 71–116. [Google Scholar]
- Nicholson AW. Ribonuclease III and the role of double-stranded RNA processing in bacterial systems. In: Nicholson AW, editor. Ribonucleases. Nucleic Acids and Molecular Biology 26. Berlin-Heidelberg: Springer-Verlag; 2011. pp. 269–297. http://10.1007/978-3-642-21078-5_11. [Google Scholar]
- Lamontagne B, LaRose S, Boulanger J, Elela SA. The RNase III family: a conserved structure and expanding functions in eukaryotic dsRNA metabolism. Curr Issues Mol Biol. 2001;3:71–78. [PubMed] [Google Scholar]
- Nicholson AW. The ribonuclease III superfamily: forms and functions in RNA maturation, decay, and gene silencing. In: Hannon GJ, editor. RNAi: A Guide to Gene Silencing. Cold Spring Harbor, NY: Cold Spring Harbor Press; 2003. pp. 149–174. [Google Scholar]
- MacRae IJ, Doudna JA. Ribonuclease revisited: structural insights into ribonuclease III family enzymes. Curr Opin Struct Biol. 2007;17:1380145. doi: 10.1016/j.sbi.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Saenger W. Principles of Nucleic Acid Structure. New York, Berlin: Springer-Verlag; 1983. [Google Scholar]
- Tanaka Y, Fujii S, Hiroaki H, Sakata T, Tanaka T, Uesugi S, Tomita K, Kyogoku Y. A′-form RNA double helix in the single crystal structure of r(UGAGCUUCGGCUC) Nucleic Acids Res. 1999;27:949–955. doi: 10.1093/nar/27.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock SL, Ringel I, Ish-Horowicz D, Lukavsky PJ. A′-form RNA helices are required for cytoplasmic mRNA transport in Drosophila. Nat Struct Mol Biol. 2010;17:703–710. doi: 10.1038/nsmb.1813. 10.1038/nsmb.1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson H, Gao Y-G, Sanishvili R, Joachimiak A, Wang AH-J. Hexahydrated magnesium ions bind in the deep major groove and at the outer mouth of A-form nucleic acid duplexes. Nucleic Acids Res. 2000;28:1760–1766. doi: 10.1093/nar/28.8.1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabit SA, Qiu X, Lamb JA, Li L, Meisburger SP, Pollack L. Both helix topology and counterion distribution contribute to the more effective charge screening in dsRNA compared with dsDNA. Nucleic Acids Res. 2009;37:3887–3896. doi: 10.1093/nar/gkp257. 10.1093/nar/gkp257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Pabit SA, Meisburger SP, Pollack L. Double-stranded RNA resists condensation. Phys Rev Lett. 2011;106:108101. doi: 10.1103/PhysRevLett.106.108101. 10.1103/PhysRevLett.106.108101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abels JA, Moreno-Herrero F, van der Heijden T, Dekker C, Dekker NH. Single-molecule measurements of the persistence length of double-stranded RNA. Biophys J. 2005;88:2737–2744. doi: 10.1529/biophysj.104.052811. 10.1529/biophysj.104.052811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faustino I, Pérez A, Orozco M. Toward a consensus view of duplex RNA flexibility. Biophys J. 2010;99:1876–1885. doi: 10.1016/j.bpj.2010.06.061. 10.1016/j.bpj.2010.06.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y-H, Howard MT, Griffith JD. Phased adenine tracts in double-stranded RNA do not induce sequence-directed bending. Biochemistry. 1991;30:5443–5449. doi: 10.1021/bi00236a017. [DOI] [PubMed] [Google Scholar]
- Gast F-U, Hagerman PJ. Electrophoretic and hydrodynamic properties of duplex ribonucleic acid molecules transcribed in vitro: evidence that A-tracts do not generate curvature in RNA. Biochemistry. 1991;30:4268–4277. doi: 10.1021/bi00231a024. [DOI] [PubMed] [Google Scholar]
- Zacharias M, Hagerman PJ. The influence of symmetric internal loops on the flexibility of RNA. J Mol Biol. 1996;257:276–289. doi: 10.1006/jmbi.1996.0162. [DOI] [PubMed] [Google Scholar]
- Zacharias M, Hagerman PJ. Bulge-induced bends in RNA: quantification by transient electric birefringence. J Mol Biol. 1995;247:486–500. doi: 10.1006/jmbi.1995.0155. [DOI] [PubMed] [Google Scholar]
- Ji X. The mechanism of RNase III action: how dicer dices. Curr Top Microbiol Immunol. 2008;320:99–116. doi: 10.1007/978-3-540-75157-1_5. [DOI] [PubMed] [Google Scholar]
- Masliah G, Barraud P, Allain FHT. RNA recognition by double-stranded RNA binding domains: a matter of shape and sequence. Cell Mol Life Sci. 2013;70:1875–1895. doi: 10.1007/s00018-012-1119-x. 10.1007/s00018-012-1119-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redko Y, Bechhofer DH, Condon C. Mini-III, an unusual member of the RNase III family of enzymes, catalyses 23S ribosomal RNA maturation in B. subtilis. Mol Microbiol. 2008;68:1096–1106. doi: 10.1111/j.1365-2958.2008.06207.x. 10.1111/j.1365-2958.2008.06207.x. [DOI] [PubMed] [Google Scholar]
- Sun W, Jun E-J, Nicholson AW. Intrinsic double-stranded-RNA processing activity of Escherichia coli ribonuclease III lacking the dsRNA-binding domain. Biochemistry. 2001;40:14976–14984. doi: 10.1021/bi011570u. [DOI] [PubMed] [Google Scholar]
- Dunn JJ. RNase III cleavage of single-stranded RNA. Effect of ionic strength on the fidelity of cleavage. J Biol Chem. 1976;251:3807–3814. [PubMed] [Google Scholar]
- Blaszczyk J, Tropea JE, Bubunenko M, Routzahn KM, Waugh DS, Court DL, Ji X. Crystallographic and modeling studies of RNase III suggest a mechanism for double-stranded RNA cleavage. Structure. 2001;9:1225–1236. doi: 10.1016/s0969-2126(01)00685-2. [DOI] [PubMed] [Google Scholar]
- Blaszczyk J, Gan J, Tropea JE, Court DL, Waugh DS, Ji X. Noncatalytic assembly of ribonuclease III with double-stranded RNA. Structure. 2004;12:457–466. doi: 10.1016/j.str.2004.02.004. 10.1016/j.str.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Gan J, Tropea JE, Austin BP, Court DL, Waugh DS, Ji X. Intermediate states of ribonuclease III in complex with double-stranded RNA. Structure. 2005;13:1435–1442. doi: 10.1016/j.str.2005.06.014. 10.1016/j.str.2005.06.014. [DOI] [PubMed] [Google Scholar]
- Ji X. Structural basis for non-catalytic and catalytic activities of ribonuclease III. Acta Crystallogr D Biol Crystallogr. 2006;D62:933–940. doi: 10.1107/S090744490601153X. 10.1107/S090744490601153X. [DOI] [PubMed] [Google Scholar]
- Akey DL, Berger JM. Structure of the nuclease domain of ribonuclease III from M. tuberculosis at 2.1 Å. Protein Sci. 2005;14:2744–2750. doi: 10.1110/ps.051665905. 10.1110/ps.051665905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan J, Tropea JE, Austin BP, Court DL, Waugh DS, Ji X. Structural insight into the mechanism of double-stranded RNA processing by ribonuclease III. Cell. 2006;1245:355–366. doi: 10.1016/j.cell.2005.11.034. 10.1016/j.cell.2005.11.034. [DOI] [PubMed] [Google Scholar]
- Campbell FE, Jr, Cassano AG, Anderson VE, Harris ME. Pre-steady-state and stopped-flow fluorescence analysis of Escherichia coli ribonuclease III: insights into mechanism and conformational changes associated with binding and catalysis. J Mol Biol. 2002;317:21–40. doi: 10.1006/jmbi.2002.5413. [DOI] [PubMed] [Google Scholar]
- Li H, Nicholson AW. Defining the enzyme binding domain of a ribonuclease III processing signal. Ethylation interference and hydroxyl radical footprinting using catalytically inactive RNase III mutants. EMBO J. 1996;15:1421–1433. [PMC free article] [PubMed] [Google Scholar]
- Sun W, Pertzev A, Nicholson AW. Catalytic mechanism of Escherichia coli ribonuclease III: kinetic and inhibitor evidence for the involvement of two magnesium ions in RNA phosphodiester hydrolysis. Nucleic Acids Res. 2005;33:807–815. doi: 10.1093/nar/gki197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan J, Shaw G, Tropea JE, Waugh DS, Court DL, Ji X. A stepwise model for double-stranded RNA processing by ribonuclease III. Mol Microbiol. 2008;67:143–154. doi: 10.1111/j.1365-2958.2007.06032.x. 10.1111/j.1365-2958.2007.06032.x. [DOI] [PubMed] [Google Scholar]
- Mordasini T, Curioni A, Andreoni W. Why do divalent metal ions either promote or inhibit enzymatic reactions? J Biol Chem. 2003;278:4381–4384. doi: 10.1074/jbc.C200664200. 10.1074/jbc.C200664200. [DOI] [PubMed] [Google Scholar]
- Meng W, Nicholson AW. Heterodimer-based analysis of subunit and domain contributions to double-stranded RNA processing by Escherichia coli RNase III in vitro. Biochem J. 2008;410:39–48. doi: 10.1042/BJ20071047. 10.1042/BJ20071047. [DOI] [PubMed] [Google Scholar]
- Conrad C, Schmitt JG, Evguenieva-Hackenberg E, Klug G. One functional subunit is sufficient for catalytic activity and substrate specificity for Escherichia coli endoribonuclease III artificial heterodimers. FEBS Lett. 2002;518:93–96. doi: 10.1016/s0014-5793(02)02653-4. [DOI] [PubMed] [Google Scholar]
- Zhang K, Nicholson AW. Regulation of ribonuclease III processing by double-helical sequence antideterminants. Proc Natl Acad Sci U S A. 1997;94:13437–13441. doi: 10.1073/pnas.94.25.13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertzev A, Nicholson AW. Characterization of RNA sequence determinants and antideterminants of processing reactivity for a minimal substrate of Escherichia coli ribonuclease III. Nucleic Acids Res. 2006;34:3708–3721. doi: 10.1093/nar/gkl459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, Nicholson RH, Jaggi R, Nicholson AW. Characterization of Aquifex aeolicus ribonuclease III and the reactivity epitopes of its pre-ribosomal RNA substrates. Nucleic Acids Res. 2011;39:2756–2768. doi: 10.1093/nar/gkq1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudinger J, Hillenbrandt R, Sprinzl M, Giegé R. Antideterminants present in minihelix(Sec) hinder its recognition by prokaryotic elongation factor Tu. EMBO J. 1996;15:650–657. [PMC free article] [PubMed] [Google Scholar]
- Calin-Jageman I, Nicholson AW. RNA structure-dependent uncoupling of substrate recognition and cleavage by Escherichia coli ribonuclease III. Nucleic Acids Res. 2003;31:2381–2392. doi: 10.1093/nar/gkg329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redko Y, Condon C. Ribosomal protein L3 bound to 23S precursor rRNA stimulates its maturation by Min-III ribonuclease. Mol Microbiol. 2009;71:1145–1154. doi: 10.1111/j.1365-2958.2008.06591.x. 10.1111/j.1365-2958.2008.06591.x. [DOI] [PubMed] [Google Scholar]
- Lasa I, Toledo-Arana A, Dobin A, Lasa I, Villanueva M, de los Mozos IR, Vergara-Irigaray M, Segura V, Fagegaltier D, Penadés JR, et al. Genome-wide antisense transcription drives mRNA processing in bacteria. Proc Natl Acad Sci USA. 2011;108:20172–20177. doi: 10.1073/pnas.1113521108. 10.1073/pnas.1113521108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioliou E, Sharma CM, Caldelari I, Helfer A-C, Fechter P, Vandenesch F, Vogel J, Romby P. Global regulatory functions of the Staphylococcus aureus endoribonuclease III in gene expression. PLoS Genet. 2012;8:e1002782. doi: 10.1371/journal.pgen.1002782. 10.1371/journal.pgen.1002782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panayotatos N, Truong K. Cleavage within an RNase III site can control mRNA stability and protein synthesis in vivo. Nucleic Acids Res. 1985;13:2227–2240. doi: 10.1093/nar/13.7.2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin-Jageman I, Nicholson AW. Mutational analysis of an RNA internal loop as a reactivity epitope for Escherichia coli ribonuclease III substrates. Biochemistry. 2003;42:5025–5034. doi: 10.1021/bi030004r. [DOI] [PubMed] [Google Scholar]
- Matsunaga J, Simons EL, Simons RW. RNase III autoregulation: structure and function of rncO, the posttranscriptional “operator”. RNA. 1996;2:1228–1240. [PMC free article] [PubMed] [Google Scholar]
- Mayer JE, Schweiger M. RNase III is positively regulated by T7 protein kinase. J Biol Chem. 1983;258:5340–5343. [PubMed] [Google Scholar]
- Gone S, Nicholson AW. Bacteriophage T7 protein kinase: site of inhibitory autophosphorylation, and use of dephosphorylated enzyme for efficient modification of protein in vitro. Protein Expr Purif. 2012;85:218–223. doi: 10.1016/j.pep.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JJ, Studier FW. Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J Mol Biol. 1983;166:477–535. doi: 10.1016/s0022-2836(83)80282-4. [DOI] [PubMed] [Google Scholar]
- Kim K, Manasherob R, Cohen SN. YmdB: a stress-responsive ribonuclease-binding regulator of E. coli RNase III activity. Genes Dev. 2008;22:3497–3508. doi: 10.1101/gad.1729508. 10.1101/gad.1729508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Vollmar M, Ross MN, Phillips C, Kraehenbuehl R, Slade D, Mehrotra PV, von Delft F, Crosthwaite SK, Gileadi O, et al. Identification of macrodomain proteins as novel O-acetyl-ADP-ribose deacetylases. J Biol Chem. 2011;286:13261–13271. doi: 10.1074/jbc.M110.206771. 10.1074/jbc.M110.206771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalchuk K, Srinivasan M, Schnetz K. RNase III initiates rapid degradation of proU mRNA upon hypo-osmotic stress in Escherichia coli. RNA Biol. 2012;9:1–12. doi: 10.4161/rna.9.1.18228. 10.4161/rna.9.1.18228. [DOI] [PubMed] [Google Scholar]
- Lamontagne B, Tremblay A, Abou ES. The N-terminal domain that distinguishes yeast from bacterial RNase III contains a dimerization signal required for efficient double-stranded RNA cleavage. Mol Cell Biol. 2000;20:1104–1115. doi: 10.1128/mcb.20.4.1104-1115.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebars I, Lamontagne B, Yoshizawa S, Abou Elela S, Fourmy D. Solution structure of conserved AGNN tetraloops: insights into Rnt1p processing. EMBO J. 2001;20:7250–7258. doi: 10.1093/emboj/20.24.7250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Yang PK, Butcher SE, Kang S, Chanfreau G, Feigon J. A novel family of RNA tetraloop structure forms the recognition site for accharomyces cerevisiae RNase III. EMBO J. 2001;20:7240–7249. doi: 10.1093/emboj/20.24.7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman E, Wang Z, Zhang Q, Roy K, Chanfreau G, Feigon J. Intrinsic dynamics of an extended hydrophobic core in the S. cerevisiae RNase III dsRBD contributes to recognition of specific RNA binding sites. J Mol Biol. 2013;425:546–562. doi: 10.1016/j.jmb.2012.11.025. 10.1016/j.jmb.2012.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Hartman E, Roy K, Chanfreau G, Feigon J. Structure of a yeast RNase III dsRBD complex with a noncanonical RNA substrate provides new insights into binding specificity of dsRBDs. Structure. 2011;19:999–1010. doi: 10.1016/j.str.2011.03.022. 10.1016/j.str.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamontagne B, Ghazal G, Lebars I, Yoshizawa S, Fourmy D, Abou ES. Sequence dependence of substrate recognition and cleavage by yeast RNase III. J Mol Biol. 2003;327:985–1000. doi: 10.1016/s0022-2836(03)00231-6. 10.1016/S002202836(03)00231-6. [DOI] [PubMed] [Google Scholar]
- Lavoie M, Abou ES. Yeast ribonuclease III uses a network of multiple hydrogen bonds for RNA binding and cleavage. Biochemistry. 2008;47:8514–8526. doi: 10.1021/bi800238u. 10.1021/bi800238u. [DOI] [PubMed] [Google Scholar]
- Sam M, Henras AK, Chanfreau G. A conserved major groove antideterminant for Saccharomyces cerevisiae RNase III recognition. Biochemistry. 2005;44:4181–4187. doi: 10.1021/bi047483u. 10.1021/bi047483u. [DOI] [PubMed] [Google Scholar]
- Giorgi C, Fatica A, Nagel R, Bozzoni I. Release of U18 snoRNA from its host intron requires interaction of Nop1p with the Rnt1p endonuclease. EMBO J. 2001;20:6856–6865. doi: 10.1093/emboj/20.23.6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamontagne B, Catala M, Yam Y, Larose S, God L, Abou ES. A physical interaction between Gar1p and Rnt1p is required for the nuclear import of H/ACA small nucleolar RNA-associated proteins. Mol Cell Biol. 2002;22:4792–4802. doi: 10.1128/MCB.22.13.4792-4802.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Lee Y, Yeom K-H, Nam J-W, Heo I, Rhee J-K, Sohn SY, Cho Y, Zhang B-T, Kim VN. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell. 2006;125:887–901. doi: 10.1016/j.cell.2006.03.043. 10.1016/j.cell.2006.03.043. [DOI] [PubMed] [Google Scholar]
- Han J, Lee Y, Yeom K-H, Kim Y-K, Jin H, Kim VN. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18:3016–3027. doi: 10.1101/gad.1262504. 10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias S, Cordiner RA, Cáceres JF. Cellular functions of the microprocessor. Biochem Soc Trans. 2013;41:838–843. doi: 10.1042/BST20130011. [DOI] [PubMed] [Google Scholar]
- Mueller GA, Miller MT, DeRose EF, Ghosh M, London RE, Tanaka-Hall TM. Solution structure of the Drosha double-stranded RNA-binding domain. Silence. 2010;1:2. doi: 10.1186/1758-907X-1-2. 10.1186/1758-907X-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wostenberg C, Quarles KA, Showalter SA. Dynamic origins of differential RNA binding function in two dsRBDs from the miRNA “Microprocessor” complex. Biochemistry. 2010;49:10728–10736. doi: 10.1021/bi1015716. 10.1021/bi1015716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin KR, Nicholson RH, Nicholson AW. Mouse ribonuclease III. cDNA structure, expression analysis, and chromosomal location. BMC Genomics. 2002;3:26. doi: 10.1186/1471-2164-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeom K-H, Lee Y, Han J, Suh MR, Kim VN. Characterization of DGCR8/Pasha, the essential cofactor for Drosha in primary miRNA processing. Nucleic Acids Res. 2006;34:4622–4629. doi: 10.1093/nar/gkl458. 10.1093/nar/gkl458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarles KA, Sahu D, Havens MA, Forsyth ER, Wostenberg C, Hastings ML, Showalter SA. Ensemble analysis of primary microRNA structure reveals an extensive capacity to deform near the Drosha cleavage site. Biochemistry. 2013;52:795–807. doi: 10.1021/bi301452a. 10.1021/bi301452a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn SY, Bae WJ, Kim JJ, Yeom K-H, Kim VN, Cho Y. Crystal structure of human DGCR8 core. Nat Struct Mol Biol. 2007;14:847–853. doi: 10.1038/nsmb1294. 10.1038/nsmb1294. [DOI] [PubMed] [Google Scholar]
- Warf MB, Johnson WE, Bass BL. Improved annotation of C. elegans microRNAs by deep sequencing reveals structures associated with processing by Drosha and Dicer. RNA. 2011;17:563–577. doi: 10.1261/rna.2432311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wostenberg C, Noid WG, Showalter SA. MD simulations of the dsRBP DGCR8 reveal correlated motions that may aid pri-miRNA binding. Biophys J. 2010;99:248–256. doi: 10.1016/j.bpj.2010.04.010. 10.1016/j.bpj.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faller M, Matsuanga M, Yin S, Loo JA, Guo F. Heme is involved in microRNA processing. Nat Struct Mol Biol. 2007;14:23–29. doi: 10.1038/nsmb1182. 10.1038/nsmb1182. [DOI] [PubMed] [Google Scholar]
- Senturia R, Faller M, Yin SM, Loo JA, Cascio D, Sawaya MR, Hwang D, Clubb RT, Guo F. Structure of the dimerization domain of DiGeorge Critical Region 8. Protein Sci. 2010;19:1354–1365. doi: 10.1002/pro.414. 10.1002/pro.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro JS, Varble A, Pham AM, Tenoever BR. Noncanonical cytoplasmic processing of viral microRNAs. RNA. 2010;16:2068–2074. doi: 10.1261/rna.2303610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro JS, Langlois RA, Pham AM, Tenoever BR. Evidence for a cytoplasmic microprocessor of pri-miRNAs. RNA. 2012;18:1338–1346. doi: 10.1261/rna.032268.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro JS. Processing of virus-derived cytoplasmic primary-microRNAs. Wiley Interdiscip Rev RNA. 2013;4:463–471. doi: 10.1002/wrna.1169. [DOI] [PubMed] [Google Scholar]
- Macias S, Plass M, Stajuda A, Michlewski G, Eyras E, Cáceres JF. DGCR8 HITS-CLIP reveals novel functions for the microprocessor. Nat Struct Mol Biol. 2012;19:760–766. doi: 10.1038/nsmb.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Kolb FA, Jaskiewicz L, Westhof E, Filipowicz W. Single processing center models for human dicer and bacterial RNase III. Cell. 2004;118:57–68. doi: 10.1016/j.cell.2004.06.017. [DOI] [PubMed] [Google Scholar]
- MacRae IJ, Zhou K, Li F, Repic A, Brooks AN, Cande WZ, Adams PD, Doudna JA. Structural basis for double-stranded RNA processing by Dicer. Science. 2006;311:195–198. doi: 10.1126/science.1121638. 10.1126/science.1121638. [DOI] [PubMed] [Google Scholar]
- Lau PW, Guiley KZ, De N, Potter CS, Carragher B, MacRae IJ. The molecular architecture of human dicer. Nat Struct Mol Biol. 2012;19:436–441. doi: 10.1038/nsmb.2268. 10.1038/nsmb.2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JE, Heo I, Tian Y, Simanshu DK, Chang H, Jee D, Patel DJ, Kim VN. Dicer recognizes the 5′ end of RNA for efficient and accurate processing. Nature. 2011;475:201–205. doi: 10.1038/nature10198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S, Jin L, Zhang Y, Huang Y, Zhang F, Valdmanis PN, Kay MA. The loop position of shRNAs and pre-miRNAs is critical for the accuracy of Dicer processing in vivo. Cell. 2012;151:900–911. doi: 10.1016/j.cell.2012.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma E, Zhou K, Kidwell MA, Doudna JA. Coordinated activities of human dicer domains in regulatory RNA processing. J Mol Biol. 2012;422:466–476. doi: 10.1016/j.jmb.2012.06.009. 10.1016/j.jmb.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wostenberg C, Lary JW, Sahu D, Acevedo R, Quarles KA, Cole JL, Showalter SA. The role of human dicer-dsRBD in processing small regulatory RNAs. PLoS One. 2012;7:e51829. doi: 10.1371/journal.pone.0051829. 10.1371/journal.pone.0051829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau PW, Potter CS, Carragher B, MacRae IJ. Structure of the human dicer-TRBP complex by electron microscopy. Structure. 2009;17:1326–1332. doi: 10.1016/j.str.2009.08.013. 10.1016/j.str.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noland CL, Ma E, Doudna JA. siRNA repositioning for guide strand selection by human dicer complexes. Mol Cell. 2011;43:110–121. doi: 10.1016/j.molcel.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HY, Doudna JA. TRBP alters human precursor microRNA processing in vitro. RNA. 2012;18:2012–2019. doi: 10.1261/rna.035501.112. 10.1261/rna.035501.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga R, Han BW, Hung JH, Xu J, Weng Z, Zamore PD. Dicer partner proteins tune the length of mature miRNAs in flies and mammals. Cell. 2012;151:533–546. doi: 10.1016/j.cell.2012.09.027. 10.1016/j.cell.2012.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson L, Sbicego S, Aphasizhev R. Uridine insertion/deletion RNA editing in trypanosome mitochondria: a complex business. RNA. 2003;9:265–276. doi: 10.1261/rna.2178403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnes J, Trotter JR, Ernst NL, Steinberg A, Stuart K. An essential RNase III insertion editing endonuclease in Trypanosoma brucei. Proc Natl Acad Sci USA. 2005;102:16614–16619. doi: 10.1073/pnas.0506133102. 10.1073/pnas.0506133102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnes J, Trotter JR, Peltan A, Fleck M, Stuart K. RNA editing in Trypanosoma brucei requires three different editosomes. Mol Cell Biol. 2008;28:122–130. doi: 10.1128/MCB.01374-07. 10.1128/MCB.01374-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez A, Panigrahi A, Cifuentes-Rojas C, Sacharidou A, Stuart K, Cruz-Reyes J. Determinants for association and guide RNA-directed endonuclease cleavage by purified RNA editing complexes from Trypanosoma brucei. J Mol Biol. 2008;381:35–48. doi: 10.1016/j.jmb.2008.05.003. 10.1016/j.jmb.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnes J, Soares CZ, Wickham C, Stuart K. Endonuclease associations with three distinct editosomes in Trypanosoma brucei. J Biol Chem. 2011;286:19320–19330. doi: 10.1074/jbc.M111.228965. 10.1074/jbc.M111.228965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnes J, Schnaufer A, Mcdermott SM, Domingo G, Proff R, Steinberg AG, Kurtz I, Stuart K. Mutational analysis of Trypanosoma brucei editosome proteins KREPB4 and KREPB5 reveal domains critical for function. RNA. 2012;18:1897–1909. doi: 10.1261/rna.035048.112. 10.1261/rna.035048.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flipovska A, Rackham O. Modular recognition of nucleic acids by PUF, TALE and PPR proteins. Mol Biosyst. 2012;8:699–708. doi: 10.1039/c2mb05392f. 10.1039/c2mb05392f. [DOI] [PubMed] [Google Scholar]
- Madina BK, Kuppan G, Vashisht AA, Liang Y-H, Downey KM, Wohlschlegel JA, Ji X, Sze S-H, Sacchettini JC, Read LK, et al. Guide RNA biogenesis involves a novel RNase III family endoribonuclease in Trypanosoma brucei. RNA. 2011;17:1821–1830. doi: 10.1261/rna.2815911. 10.1261/rna.2815911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta S, Fernandez L, Kameyama L, Inada T, Nakamura Y, Pappas A, Court DL. Genetic uncoupling of the dsRNA-binding and RNA cleavage activities of the Escherichia coli endoribonuclease RNase III—the effect of dsRNA binding on gene expression. Mol Microbiol. 1998;28:629–640. doi: 10.1046/j.1365-2958.1998.00828.x. [DOI] [PubMed] [Google Scholar]
- Watkins KP, Kroeger TS, Cooke AM, Williams-Carrier RE, Friso G, Belcher SE, van Wijk KJ, Barkan A. A ribonuclease III domain protein functions in group II intron splicing in maize chloroplasts. Plant Cell. 2007;19:2606–2623. doi: 10.1105/tpc.107.053736. 10.1105/tpc.107.053736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeger TYS, Watkins KP, Friso G, van Wijk KJ, Barkan A. A plant-specific RNA-binding domain revealed through analysis of chloroplast group II intron splicing. Proc Natl Acad Sci U S A. 2009;106:4537–4542. doi: 10.1073/pnas.0812503106. 10.1073/pnas.0812503106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olinares PD, Ponnala L, van Wijk KJ. Megadalton complexes in the chloroplast stroma of Arabidopsis thaliana characterized by size exclusion chromatography, mass spectrometry, and hierarchical clustering. Mol Cell Proteomics. 2010;9:1594–1615. doi: 10.1074/mcp.M000038-MCP201. 10.1074/mcp.M000038-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libonati M, Sorrentino M. Degradation of double-stranded RNA by mammalian pancreatic-type ribonucleases. Methods Enzymol. 2001;341:234–248. doi: 10.1016/s0076-6879(01)41155-4. [DOI] [PubMed] [Google Scholar]
- D'Alessio G. The superfamily of vertebrate-secreted ribonucleases. In: Nicholson AW, editor. Ribonucleases. Nucleic Acids and Molecular Biology 26. Berlin-Heidelberg: Springer-Verlag; 2011. pp. 1–34. 10.1007/978-3-642-21078-5_1. [Google Scholar]
- Rosenberg HF. Vertebrate secretory (RNase A) ribonucleases and host defense. In: Nicholson AW, editor. Ribonucleases. Nucleic Acids and Molecular Biology 26. Berlin-Heidelberg: Springer-Verlag; 2011. pp. 36–53. 10.1007/978-3-642-21078-5_2. [Google Scholar]
- Hastie KM, Kimberlin CR, Zandonatti MA, MacRae IJ, Saphire EO. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3′ to 5′ exonuclease activity essential for immune suppression. Proc Natl Acad Sci U S A. 2011;108:2396–2401. doi: 10.1073/pnas.1016404108. 10.1073/pnas.1016404108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie KM, King LB, Zandonatti MA, Saphire EO. Structural basis for the dsRNA specificity of the Lassa virus NP exonuclease. PLoS One. 2012;7:e44211. doi: 10.1371/journal.pone.0044211. 10.1371/journal.pone.0044211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie KM, Bale S, Kimberlin CR, Saphire EO. Hiding the evidence: two strategies for innate immune evasion by hemorrhagic fever viruses. Curr Opin Virol. 2:151–156. doi: 10.1016/j.coviro.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TH, Rozhdestvensky TS, d'Orval BC, Bortolin ML, Huber H, Charpentier B, Branlant C, Bachellerie JP, Brosius J, Hüttenhofer A. RNomics in Archaea reveals a further link between splicing of archaeal introns and rRNA processing. Nucleic Acids Res. 2012;30:921–930. doi: 10.1093/nar/30.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babiskin AH, Smolke CD. Engineering ligand-responsive RNA controllers in yeast through the assembly of RNase III tuning modules. Nucleic Acids Res. 2011;39:5299–5311. doi: 10.1093/nar/gkr090. 10.1093/nar/gkr090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babiskin AH, Smolke CD. Synthetic RNA modules for fine-tuning gene expression levels in yeast by modulating RNase III activity. Nucleic Acids Res. 2011;39:8651–8664. doi: 10.1093/nar/gkr445. 10.1093/nar/gkr445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castronovo M, Stopar A, Coral L, Redhu SK, Vidonis M, Kumar V, Del Ben F, Grassi M, Nicholson AW. Effects of nanoscale confinement on the functionality of nucleic acids: implications for nanomedicine. Curr Med Chem. 2013;20:3539–3557. doi: 10.2174/09298673113209990173. [DOI] [PubMed] [Google Scholar]
- Redhu SK, Castronovo M, Nicholson AW. Digital imprinting of RNA recognition and processing on a self-assembled nucleic acid matrix. Sci Rep. 2013;3:2550. doi: 10.1038/srep02550. 10.1038/srep02550. [DOI] [PMC free article] [PubMed] [Google Scholar]