

Abstract
RNA-protein interaction plays an important role in various cellular processes, such as protein synthesis, gene regulation, post-transcriptional gene regulation, alternative splicing, and infections by RNA viruses. In this study, using Gene Ontology Annotated (GOA) and Structural Classification of Proteins (SCOP) databases an automatic procedure was designed to capture structurally solved RNA-binding protein domains in different subclasses. Subsequently, we applied tuned multi-class SVM (TMCSVM), Random Forest (RF), and multi-class ℓ1/ℓq-regularized logistic regression (MCRLR) for analysis and classifying RNA-binding protein domains based on a comprehensive set of sequence and structural features. In this study, we compared prediction accuracy of three different state-of-the-art predictor methods. From our results, TMCSVM outperforms the other methods and suggests the potential of TMCSVM as a useful tool for facilitating the multi-class prediction of RNA-binding protein domains. On the other hand, MCRLR by elucidating importance of features for their contribution in predictive accuracy of RNA-binding protein domains subclasses, helps us to provide some biological insights into the roles of sequences and structures in protein–RNA interactions.
Keywords: RNA-binding domain, tuned multi-class SVM, Random Forest, Multi-class ℓ1/ℓq -regularized logistic regression, Prediction
1. Introduction
Regulation of biological processes happens through association and dissociation of macromolecules, i.e., protein, RNA and DNA. Furthermore, functional components of cells are frequently complex assemblies of macromolecules. At the molecular level, RNA-protein complexes play an important role in various cellular processes, such as protein synthesis, gene regulation, post-transcriptional gene regulation, alternative splicing, and infections by RNA viruses. Therefore, it is important to understand the principle of RNA-protein interactions and prediction of RNA-binding proteins is essential in identifying the cellular processes in which RNA-protein complexes are involved.
It is commonly accepted that RNA recognition by proteins is mainly mediated by specific kinds of RNA-binding domains (RBDs) (Morozova et al., 2006; Shulman-Peleg et al., 2008). The RBDs can be classified into different subclasses based on their basic binding motifs, e.g., the KH domain, the double-stranded RNA-binding domain (dsRBD), and the zinc finger motif (Chen and Varani, 2005). Although in recent years, new RBDs have been identified (Parker and Barford, 2006), an increasing amount of evidence on non-coding RNAs suggest that new RBDs will be identified (Lingel and Sattler, 2005).
In order to recognize the RNA functional importance in close relationship with protein in its activities, computational studies of RNA-protein complexes have been significantly increased (Ellis et al., 2007; Jones et al., 2001). Recently, a variety of approaches have been proposed to study RNA-protein interactions (Lunde et al., 2007). Although some interesting results have been obtained, the precise details of the RNA-protein interaction are far from being fully understood. For this reason, it is strongly recommended to develop reliable computational methods to accurately predict RNA-binding proteins and analyze important features in RNA-protein interaction.
Homology-based methods are the most common method to identify the class of unknown proteins at sequence or structure level. These methods are limited by the absence of experimentally annotated homologous proteins in protein databases. Hence it is strongly encouraged to develop computational tools to identify RNA-binding proteins (RBPs) using sequence- and structure-derived features. Most of previous investigations, predict RBPs using sequence- derived features (Han et al., 2004; Shao et al., 2009; Yu et al., 2006). In addition to sequence-based methods, up to now, only one investigation by Shazman and Mandel-Gutfreund (2008) developed a structural-based method to predict RBPs. Shazman and Mandel-Gutfreund developed a multiSVM-based method using four subgroups of features including: (i) Largest patch parameters (such as patch size and patch surface accessibility), (ii) Protein parameters (such as molecular weight) (iii) Cleft/patch parameters (such as the overlap between the largest, second largest, and third largest clefts, and largest patch), and (iv) Parameters related to other surface patches (such as number of residues in the lysine out patch and in the negative patch), to describe the global composition of each protein. Using the jackknife test, they reported a 75.61% accuracy of prediction for three subclasses of RBPs; tRNA-, rRNA-, and mRNA-binding proteins. In comparison with our work, it is limit to three classes of RBPs, and they have done a non-accurate manually data collection. Despite the availability of several methods, identification of RBPs using sequence information with high accuracy is still a major challenge.
Here we present a comprehensive performance evaluation of some state of the art predictor methods on an important problem, i.e., classifying RBDs using sequence- and structure-derived information. Combining a diverse set of features, we developed three different methods including; tuned multi-class SVM (TMCSVM), Random Forest (RF), and Multi-class ℓ1/ℓq -regularized logistic regression (MCRLR). By applying these methods, we have shown that we can classify RBDs based on their RNA target (7S, double-stranded, tRNA, rRNA, or mRNA). In all of five different subclasses of RBPs, no exclusive RNA-binding motif is present. However, in such cases in addition to successful classifying RBPs, we discovered dissimilar sequence and structural features.
2. Materials and methods
2.1. Automatic dataset harvesting
Based on the fact that most of similar works on prediction of RNA-binding proteins, manually collected and annotated datasets, in this work, in order to do a more accurate and automated data harvesting we constructed a dataset of non-redundant RNA-binding protein domains using two main datasets including: (i) Gene Ontology Annotated (GOA) database, available at http://www.ebi.ac.uk/GOA/, which cover ~2.5 million reports of associated protein chains with Gene Ontology (GO) terms, and (ii) 40% non-redundant set of Structural Classification of Proteins (SCOP) 1.75 from ASTRAL website. Based on GO classification, RNA binding root involves 28 leaves. Our first step of automatic procedure was one by one search for RNA binding subclasses GO IDs in GOA database to find associated protein chains to each subclass of RNA binding GO IDs. Briefly, GO is a major bioinformatics tool for the unification of biology. More specifically, one of the aims is annotation of genes and gene products. GO contains three ontologies that describe the molecular functions, biological processes, and cellular components of proteins (Ashburner et al., 2000). For more details and comprehensive discussion we refer to the paper (Chou and Shen, 2006), as well as the discussions as elaborated in (Chou and Shen, 2008). The second step was search across SCOP 1.75 to capture non-redundant RNA binding protein domains in different subclasses (Fig. 1). We eliminated protein domains, which associated to more than one RNA binding subclass.
Figure 1.
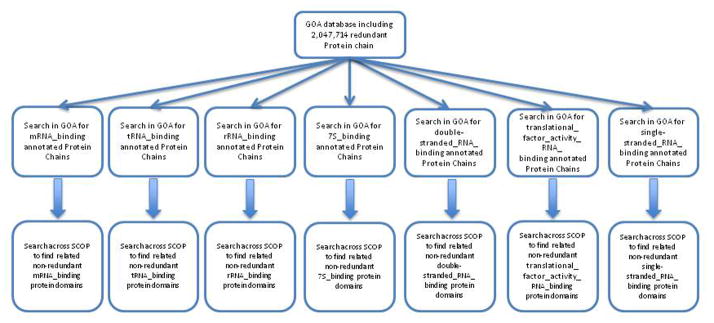
The proposed automatic procedure for dataset harvesting
2.2 Feature generation
In this study a combination of sequence- and structure-derived features were used for prediction of RNA-binding protein domains. Our representation of the protein sequence in this study is a general form of Chou’s pseudo amino acid composition (Chou, 2011). Indeed, to avoid losing many important information hidden in protein sequences, the pseudo amino acid composition (PseAAC) was proposed (Chou, 2001; Chou, 2005) to replace the simple amino acid composition (AAC) for representing the sample of a protein. For a summary about its recent development and applications, see a comprehensive review (Chou, 2009). Ever since the concept of PseAAC was proposes by Chou in 2001, its has rapidly penetrated into almost all the fields of protein attribute prediction (Chen et al., 2009; Ding et al., 2009; Esmaeili et al., 2010; Georgiou et al., 2009; Guo et al., 2011; Hayat and Khan, 2012; Hu et al., 2011; Li et al., 2012; Lin, 2008; Liu et al., 2012; Mei, 2012; Mohabatkar, 2010; Mohabatkar et al., 2011; Nanni et al., 2012; Qin et al., 2012; Qiu et al., 2009; Qiu et al., 2011; Yu et al., 2010; Zhang and Fang, 2008; Zhao et al., 2012; Zou et al., 2011). According to Eq.6 of a recent comprehensive review (Chou, 2011), the form of PseAAC can be generated and formulated as
| (1) |
where T is a transpose operator, while the subscribe Ω is an integer and its value as well as the components ψ1, ψ2, … will be defined by a series of feature extractions as elaborated below.
In addition to sequence-derived features, structure-derived features were generated in this study. Totally, 267 different sequence- and structure-derived features were generated using several information sources, which can be classified into six major subgroups including:
Sequence-derived features including: (i) composition of all 20 amino acids (20 features), (ii) composition of amino acids in 9 different physicochemical groups including tiny, small, aliphatic, aromatic, polar, non-polar, charged, acidic, and basic amino acids groups (9 features), (iii) pI, the isoelectric point (1 feature), (iv) molecular weight (1 feature), and (v) number of residues and number of atoms (2 features). This subgroup of features was generated using seqinr package (version 3.0–3) in R environment.
Secondary structure features including: (i) composition of all 20 amino acids and composition of amino acids in physicochemical groups, within three different secondary structures, i.e., helix, sheet and random coil (87 features), and (ii) composition of 6 different secondary structures, i.e., H (α-helix), G (310 helix), E (extended β-strand), B (isolated β-bridge), T (turn), and S (bend) (6 features). Secondary structure parameters in each protein domain were computed using the output of the program DSSP (Kabsch and Sander, 1983). In order to calculate secondary structures structures in three different secondary structures, the six structures were reduced into three classes (H,G → H, E → E, all other states to C).
Solvent accessibility features including: composition of all 20 amino acids and composition of amino acids in physiochemical groups, within three different solvent accessibility states, i.e., buried, intermediate, and exposed (87 features). Based on the standard ranges of solvent accessibility values (SAV), three kinds of solvent accessibility states are defined. Buried state, B, is endowed to residues having 0≤SAV≤0.16, intermediate state, I, to residues having 0.16<SAV≤0.36, and exposed state, E, to residues having 0.36<SAV≤1. Solvent accessibility values of residues were computed using ASAView program (Ahmad et al., 2004).
Hydrogen bonds features: The hydrogen bond from the backbone CO (i) to the backbone NH (i+N), is expressed by the symbol H-bond (i, i+N). In this study we computed frequencies of H-bond (i, i+N) for N=-5,-4,-3, …, 3, 4, 5. Furthermore, total hydrogen bonds, parallel- and antiparallel hydrogen bonds were computed. The values of these features were divided by length of protein domains. The output of the program DSSP (Kabsch and Sander, 1993) was used to generate these features (13 features).
Electrostatic properties features: eight electrostatic properties features including net molecular charge, net molecular charge per atom, overall molecular dipole moment in debyes, net molecular dipole moment per atom, number of positively charged residues, and number of negatively charged residues were calculated using the Protein Dipole Moments Server (http://bip.weizmann.ac.il/dipol/).
Patch features: main, second and third patch sizes, main patch’s molecular weight, composition of all 20 amino acids and composition of amino acids in physico-chemical groups, within the main patch were calculated (33 features). In order to extract all continuous positive patches on the proteins surface the PatchFinder algorithm (Stawiski et al., 2003) was used. The patches were sorted based on the number of grid points contained within the patch, and the largest three patches were selected.
2.3. Predictor methods
In this study, we used three different predictor methods including tuned multi-class SVM (TMCSVM), Random Forest (RF), and multi-class regularized logistic regression (MCRLR) to classify RBDs to three and five subclasses. The jackknife test was used to training and testing on databases. Through the jackknife test, one case is removed from the database and training is done using the remaining cases; then testing is done using the removed case. This procedure is repeated until all cases are tested. Although this method is time-consuming, it is more useful for the small databases such as ours. In addition to jackknife we also used self-consistency test to evaluate the prediction results. Both of jackknife and self-consistency are thought to be the most rigorous and objective methods for evaluation of prediction.
Among the independent dataset test, sub-sampling (e.g., 5 or 10-fold cross-validation) test, and jackknife test, which are often used for examining the accuracy of a statistical prediction method (Chou and Zhang, 1995), the jackknife test was deemed the least arbitrary that can always yield a unique result for a given benchmark dataset, as elucidated in (Chou and Shen, 2008) and demonstrated by Eqs.28–32 of (Chou, 2011). Therefore, the jackknife test has been widely recognized and increasingly used by investigators to test the power of various prediction methods (see, e.g., (Chen et al., 2009; Chou et al., 2011; Chou et al., 2012; Ding et al., 2009; Esmaeili et al., 2010; Georgiou et al., 2009; Gu et al., 2010; Jiang et al., 2008; Lin, 2008; Li and Li, 2008; Lin et al., 2008; Lin and Wang, 2011; Li et al., 2012; Mei, 2012; Mohabatkar, 2010; Mohabatkar et al., 2011; Qiu et al., 2010; Wu et al., 2011; Wu et al., 2012; Xiao et al., 2011a; Xiao et al., 2011b; Xiao et al., 2012; Yu et al., 2010; Zeng et al., 2009; Zhang and Fang, 2008; Zhang et al., 2008; Zhou et al., 2007).
2.3.1. Tuned multi-class support vector machine
Basically, support vector machine (SVM) is a kind of learning machines based on statistical learning theory. They have three remarkable characteristics including: the absence of minima, the sparseness of the solution, and implementation using the kernel Adatron algorithm. The kernel Adatron maps inputs to a high-dimensional feature space, and then optimally separates data into their respective classes by isolating those inputs which fall close to the data boundaries. Therefore, the kernel Adatron is especially effective in separating sets of data which share complex boundaries. Because of seeking a global optimized solution and avoiding over-fitting in the SVM training process, dealing with a large number of features is possible. SVMs can only be used for classification, not for function approximation. The theory and algorithms of SVMs can be found in Vapnik (1995, 1998).
In this study, we applied the tune function using e1071-package of R environment (version 2.11-1) to develop our multi-class SVM based method. Multi-class SVM in e1071 uses the “one-against-one” strategy, i.e., binary classification between all pairs, followed by voting. On the other hand, the tune function uses Grid Search to find the best functions. Using the tune function through jackknife procedure, it provides as many simulations as the number of cases in databases to select optimum structure each time.
2.3.2. Random Forest
Random forest (RF) was developed by Breiman in 2001 (Vapnik, 1998). The RF classification extends the concept of decision trees and has been successfully used in various biological problems (Dudoit et al., 2002; Statnikov et al., 2008; Jia and Hu, 2011; Kandaswamy et al., 2011; Lin et al., 2011; Pugalenthi et al., 2012; Qiu and Wang, 2011; Shameer et al., 2011). RF is a collection of decision trees instead of one tree, where each tree is trained using a bootstrap sample from the training dataset. The trees are then grown using a randomly selected subset of predictors at each node. After constructing all trees, a new object can then be classified based on the class label with the most votes, where every vote is decided by every tree in the forest. Finally, predictive performance is estimated using the observations left out of the bootstrap sample, termed the out-of-bag (OOB) observations. An appeal of RF is that the forest of trees contains a large amount of information about the relationship between the variables and observations. This information can be used for prediction, clustering, imputing missing data, and detecting outliers. The RF algorithm was implemented by the randomForest (version 4.6-2) R package (Liaw, 2002). We used tune randomForest (tuneRF) function. The number of trees and stepFactor were set to 1000 and 2, respectively. However, there are default values for different features, which are provided by the program and we used in this work.
2.3.3. Multi-class ℓ1/ℓq-regularized logistic regression
A multi-class ℓ1/ℓq-regularized logistic regression model that we used in this study is a generalization of the ℓ1-regularization logistic regression. Development of such strong theoretical guarantees, and great empirical success method is from recent studies in areas such as machine learning, statistics, and applied mathematics (Bach, 2008; Duchi and Singer, 2009; Kowalski, 2009; Negahban et al., 2009; Yuan and Lin, 2006).
The multi-class ℓ1/ℓq-regularized logistic regression is an expression of the form
| (2) |
where indicates vector of size 1 × n, n is number of features for i-th protein domain of the ℓ-th RBDs subclass, wiℓ is the weight for , yiℓ is the response of aiℓ, and cℓ is the intercept for the ℓ-th RBDs subclass. To construct multi-class ℓ1/ℓq-regularized logistic regression we used mcLogisticR function of SLEP package (version 4.0) which is written in Matlab. In this function, the elements in y are required to be m × k matrix including elements of 1 or −1 (m is the number of protein domains and k is the number of RBDs subclasses).
3. Results
3.1. Construction of dataset
Constructed dataset cover 7 out of 28 RNA binding subclasses with at least one protein domain member, including 7S RNA binding (10 protein domains), double-stranded RNA binding (16 protein domains), mRNA binding (11 protein domains), rRNA binding (29 protein domains), tRNA binding (16 protein domains), translational factor activity RNA binding (2 protein domains), and single-stranded RNA binding (1 protein domain). The RBDs of our dataset are summarized in Table 1. In construction of our methods we eliminated subclasses with less than 10 protein domain. In addition we constructed methods for prediction of five subclasses (i.e. tRNA-, rRNA-, mRNA-, 7S-, and double-stranded binding domain subclasses) and three subclasses (i.e. tRNA-, rRNA-, and mRNA-binding domain subclasses).
Table 1.
Summarized RNA binding domains in our dataset.
| No. | Protein Domain | GO term | Class | Fold | Superfamily | Family | Domain | Species |
|---|---|---|---|---|---|---|---|---|
| 1 | d1914a1 | 7S_RNA_binding | α+β | SRP9/14 | SRP9/14 | SRP9/14 | SRP9 | Mouse |
| 2 | d1914a2 | 7S_RNA_binding | α+β | SRP9/14 | SRP9/14 | SRP9/14 | SRP14 | Mouse |
| 3 | d1hq1a_ | 7S_RNA_binding | α | SPBD | SPBD | SPBD | SSBP Ffh | EC |
| 4 | d1jida_ | 7S_RNA_binding | α+β | SRP19 | SRP19 | SRP19 | SRP19 | Human |
| 5 | d1kvva_ | 7S_RNA_binding | α+β | SRP19 | SRP19 | SRP19 | SRP19 | AAF |
| 6 | d1lnga_ | 7S_RNA_binding | α+β | SRP19 | SRP19 | SRP19 | SRP19 | AMJ |
| 7 | d1ls1a1 | 7S_RNA_binding | α | FHUDB | D-SRP/SRP receptor G-proteins | D-SRP/SRP receptor G-proteins | SSBP Ffh | TA |
| 8 | d1ls1a2 | 7S_RNA_binding | α/β | P-loop NTP hydrolases | P-loop NTP hydrolases | Nitrogenase iron protein-like | GTPase domain of SSBP Ffh | TA |
| 9 | d1qb2a_ | 7S_RNA_binding | α | SPBD | SPBD | SPBD | SRP54M | Human |
| 10 | d1qzxa2 | 7S_RNA_binding | α | SPBD | SPBD | SPBD | SSBP Ffh | ASS |
| 11 | d1di2a_ | DS_RNA_binding | α+β | dsRBD-like | dsRBD-like | dsRBD | dsRBD A | XL |
| 12 | d1ekza_ | DS_RNA_binding | α+β | dsRBD-like | dsRBD-like | dsRBD | Staufen, domain III | DM |
| 13 | d1o0wa1 | DS_RNA_binding | α | RNase III domain-like | RNase III domain-like | RNase III catalytic domain-like | RNase III ECD | TM |
| 14 | d1o0wa2 | DS_RNA_binding | α+β | dsRBD-like | dsRBD-like | dsRBD | RNase III, C-terminal domain | TM |
| 15 | d1t4oa_ | DS_RNA_binding | α+β | dsRBD-like | dsRBD-like | dsRBD | RNase III, C-terminal domain | SC |
| 16 | d1uhza_ | DS_RNA_binding | α+β | dsRBD-like | dsRBD-like | dsRBD | staufen homolog 2 | Mouse |
| 17 | d1uila_ | DS_RNA_binding | α+β | dsRBD-like | dsRBD-like | dsRBD | ATP-dep RNA helicase A, Dhx9 | Mouse |
| 18 | d1whna_ | DS_RNA_binding | α+β | dsRBD-like | dsRBD-like | dsRBD | Dus2l | Mouse |
| 19 | d1whqa_ | DS_RNA_binding | α+β | dsRBD-like | dsRBD-like | dsRBD | ATP-dependent RNA helicase A, Dhx9 | Mouse |
| 20 | d1x47a1 | DS_RNA_binding | α+β | dsRBD-like | dsRBD-like | dsRBD | Dgcr8 protein | Human |
| 21 | d1x48a1 | DS_RNA_binding | α+β | dsRBD-like | dsRBD-like | dsRBD | dsRNA-dependent protein kinase pkr | Mouse |
| 22 | d1x49a1 | DS_RNA_binding | α+β | dsRBD-like | dsRBD-like | dsRBD | dsRNA-dependent protein kinase pkr | Mouse |
| 23 | d2dixa1 | DS_RNA_binding | α+β | dsRBD-like | dsRBD-like | dsRBD | Interferon -ids RNA DPK activator A | Human |
| 24 | d2dmya1 | DS_RNA_binding | α+β | dsRBD-like | dsRBD-like | dsRBD | Spermatid perinuclear RBP | Human |
| 25 | d2nuga1 | DS_RNA_binding | α | RNase III domain-like | RNase III domain-like | RNase III catalytic domain-like | RNase III ECD | AA |
| 26 | d2nuga2 | DS_RNA_binding | α+β | dsRBD-like | dsRNA-binding domain-like | dsRBD | RNase III, C-terminal domain | AA |
| 27 | d1afwa1 | mRNA_binding | α/β | Thiolase-like | Thiolase-like | Thiolase-related | Thiolase | SC |
| 28 | d1j1ja_ | mRNA_binding | α | α-α superhelix | Translin | Translin | Translin | Human |
| 29 | d1kvka1 | mRNA_binding | α+β | RP S5 domain 2-like | RP S5 domain 2-like | GHMP Kinase, N-terminal domain | Mevalonate kinase | RN |
| 30 | d1kvka2 | mRNA_binding | α+β | Ferredoxin-like | GHMP Kinase, C-terminal domain | Mevalonate kinase | Mevalonate kinase | RN |
| 31 | d1l5ja1 | mRNA_binding | α | α-α superhelix | Aconitase B, N-terminal domain | Aconitase B, N-terminal domain | Aconitase B, N-terminal domain | EC |
| 32 | d1l5ja2 | mRNA_binding | α/β | The “swivelling” β/β/α domain | LeuD/IlvD-like | LeuD-like | Aconitase B, second N-terminal domain | EC |
| 33 | d1l5ja3 | mRNA_binding | α/β | Aconitase iron-sulfur domain | Aconitase iron-sulfur domain | Aconitase iron-sulfur domain | Aconitase B, C-terminal domain | EC |
| 34 | d1p5fa_ | mRNA_binding | α/β | Flavodoxin-like | Class I glutamine AM-like | DJ-1/PfpI | DJ-1 | Human |
| 35 | d1q67a_ | mRNA_binding | β | PH domain-like barrel | PH domain-like | Dcp1 | Dcp1 | SC |
| 36 | d1xlya_ | mRNA_binding | α | RNA-binding protein She2p | RNA-binding protein She2p | RNA-binding protein She2p | RNA-binding protein She2p | SC |
| 37 | d3gcba_ | mRNA_binding | α/β | Cysteine proteinases | Cysteine proteinases | Papain-like | Bleomycin hydrolase | SC |
| 38 | d1a32a_ | rRNA_binding | α | S15/NS1 RNA-binding domain | S15/NS1 RNA-binding domain | RP S15 | RP S15 | BST |
| 39 | d1diva1 | rRNA_binding | α+β | RP L9 C-domain | RP L9 C-domain | RP L9 C-domain | RP L9 C-domain | BST |
| 40 | d1dmga_ | rRNA_binding | α/β | RP L4 | RP L4 | RP L4 | RP L4 | TM |
| 41 | d1egaa2 | rRNA_binding | α+β | α-lytic protease prodomain-like | Prokaryotic type KH domain | Prokaryotic type KH domain | GTPase Era C-terminal domain | EC |
| 42 | d1feua_ | rRNA_binding | β | RP L25-like | RP L25-like | RPL25-like | RP TL5 (general stress protein CTC) | TT |
| 43 | d1i4ja_ | rRNA_binding | α+β | RP L22 | RP L22 | RP L22 | RP L22 | TA |
| 44 | d1i6ua_ | rRNA_binding | α+β | RP S8 | RP S8 | RP S8 | RP S8 | AMJ |
| 45 | d1iqva_ | rRNA_binding | α | Ribosomal protein S7 | RP S7 | RP S7 | RP S7 | APH |
| 46 | d1loua_ | rRNA_binding | α+β | Ferredoxin-like | RP S6 | RP S6 | RP S6 | TT |
| 47 | d1n0ua1 | rRNA_binding | β | R/I/E factor common domain | Translation proteins | EF | eEF-2, domain II | SC |
| 48 | d1n0ua2 | rRNA_binding | α/β | P-loop NTP hydrolases | P-loop NTP hydrolases | G proteins | eEF-2, N-terminal (G) domain | SC |
| 49 | d1n0ua3 | rRNA_binding | α+β | RP S5 domain 2-like | RP S5 domain 2-like | TMC | eEF-2, domain IV | SC |
| 50 | d1n0ua4 | rRNA_binding | α+β | Ferredoxin-like | EF-G C-terminal domain-like | EF-G/eEF-2 domains III and V | eEF-2 | SC |
| 51 | d1n0ua5 | rRNA_binding | α+β | Ferredoxin-like | EF-G C-terminal domain-like | EF-G/eEF-2 domains III and V | eEF-2 | SC |
| 52 | d1pkpa1 | rRNA_binding | α+β | RP S5 domain 2-like | RP S5 domain 2-like | TMC | RP S5, N-terminal domain | BST |
| 53 | d1pkpa2 | rRNA_binding | α+β | dsRBD-like | dsRBD-like | RP S5, N-terminal domain | RP S5, N-terminal domain | BST |
| 54 | d1rl6a1 | rRNA_binding | α+β | RP L6 | RP L6 | RP L6 | RP L6 | BST |
| 55 | d1rl6a2 | rRNA_binding | α+β | RP L6 | RP L6 | RP L6 | RP L6 | BST |
| 56 | d1seia_ | rRNA_binding | α+β | RP S8 | RP S8 | RP S8 | RP S8 | BST |
| 57 | d1vmba_ | rRNA_binding | α+β | Ferredoxin-like | RP S6 | RP S6 | RP S6 | TM |
| 58 | d1vqoa1 | rRNA_binding | β | SH3-like barrel | TP SH3-like domain | C-terminal domain of RP L2 | C-terminal domain of RP L2 | AHM |
| 59 | d1vqoa2 | rRNA_binding | β | OB-fold | Nucleic acid-binding proteins | Cold shock DNA-binding domain-like | N-terminal domain of RP L2 | AHM |
| 60 | d1wf3a1 | rRNA_binding | α/β | P-loop NTP hydrolases | P-loop NTP hydrolases | G proteins | GTPase Era, N-terminal domain | TT |
| 61 | d1wf3a2 | rRNA_binding | α+β | α-lytic protease prodomain-like | Prokaryotic type KH domain | Prokaryotic type KH domain | GTPase Era C-terminal domain | TT |
| 62 | d1whia_ | rRNA_binding | β | RP L14 | RP L14 | RP L14 | RP L14 | BS |
| 63 | d2cqla1 | rRNA_binding | α+β | RP L6 | RP L6 | RP L6 | RP L6 | Human |
| 64 | d2j5aa1 | rRNA_binding | α+β | Ferredoxin-like | RP S6 | RP S6 | RP S6 | AA |
| 65 | d2v3ka1 | rRNA_binding | α/β | α/β knot | α/β knot | EMG1/NEP1-like | EMG1 | SC |
| 66 | d3bbda1 | rRNA_binding | α/β | α/β knot | α/β knot | EMG1/NEP1-like | RBP NEP1 | MJ |
| 67 | d1dm9a_ | SS_ RNA_binding | α+β | α-L RNA-binding motif | α-L RNA-binding motif | Heat shock protein 15 kD | HSP 15 Kd | EC |
| 68 | d1d7qa_ | TFA_ RNA_binding | β | OB-fold | Nucleic acid-binding proteins | Cold shock DNA-binding domain-like | eIF1a | Human |
| 69 | d2if1a_ | TFA_ RNA_binding | α+β | eIF1-like | eIF1-like | eIF1-like | eIF- 1 (SUI1) | Human |
| 70 | d1a6fa_ | tRNA_binding | α+β | RP S5 domain 2-like | RP S5 domain 2-like | RNase P protein | RNase P protein | BSU |
| 71 | d1dj0a_ | tRNA_binding | α+β | Pseudouridine synthase | Pseudouridine synthase | Pseudouridine synthase I TruA | Pseudouridine synthase I TruA | EC |
| 72 | d1fl0a_ | tRNA_binding | β | OB-fold | Nucleic acid-binding proteins | Myf domain | EMAP II | Human |
| 73 | d1gd7a_ | tRNA_binding | β | OB-fold | Nucleic acid-binding proteins | Myf domain | TRBP111 homolog CsaA | TT |
| 74 | d1jjca_ | tRNA_binding | α+β | Class II aaRS and biotin synthetases | Class II aaRS and biotin synthetases | Class I A-tRNA S- like, catalytic domain | PheRS alpha subunit | TT |
| 75 | d1nz0a_ | tRNA_binding | α+β | RP S5 domain 2-like | RP S5 domain 2-like | RNase P protein | RNase P protein | TM |
| 76 | d1ou5a1 | tRNA_binding | α | Poly A PCT region-like | Poly A PCT region-like | Poly A PCT region-like | tRNA CCA-adding E, C-terminal domains | HM |
| 77 | d1ou5a2 | tRNA_binding | α+β | Nucleotidyltransferase | Nucleotidyltransferase | Poly A polymerase head domain-like | tRNA CCA-adding E, head domain | HM |
| 78 | d1pyba_ | tRNA_binding | β | OB-fold | Nucleic acid-binding proteins | Myf domain | TRBP111 | AA |
| 79 | d1r6la1 | tRNA_binding | α+β | RP S5 domain 2-like | RP S5 domain 2-like | Ribonuclease PH domain 1-like | Ribonuclease PH, domain 1 | PseA |
| 80 | d1r6la2 | tRNA_binding | α+β | RPH domain 2-like | RPH domain 2-like | Ribonuclease PH domain 2-like | Ribonuclease PH, domain 2 | PseA |
| 81 | d1rqga1 | tRNA_binding | α | ABD of a subclass of class I AA-tRNA-S | ABD of a subclass of class I AA-tRNA-S | ABD of a subclass of class I AA-tRNA-S | MetRS | PA |
| 82 | d1rqga2 | tRNA_binding | α/β | AN-α hydrolase-like | Nucleotidylyl transferase | Class I A-tRNA S, catalytic domain | MetRS | PA |
| 83 | d2c5sa1 | tRNA_binding | α/β | AN-α hydrolase-like | AN-α hydrolase-like | ThiI-like | TBP ThiI, N-ter D | BA |
| 84 | d2c5sa2 | tRNA_binding | α+β | THUMP domain | THUMP domain-like | THUMP domain | TBP ThiI, N-ter D | BA |
| 85 | d2iy5a1 | tRNA_binding | α | Long alpha-hairpin | tRNA-binding arm | PheRS | PheRS | TT |
DS_RNA_binding: double-stranded_RNA_binding, SS_RNA_binding: single-stranded_RNA_binding, TFA_RNA_binding: translational_factor_activity_RNA_binding, SRP: Signal recognition particle alu RNA binding heterodimer, SPBD: Signal peptide-binding domain, FHUDB: Four-helical up-and-down bundle, P-loop NTP hydrolases: P-loop containing nucleoside triphosphate hydrolases, R/I/E factor common domain: Reductase/isomerase/elongation factor common domain, RP: Ribosomal protein, Class II aaRS and BS: Class II aaRS and biotin synthetases, Poly A PCT region-like: Poly A polymerase C-terminal region-like, RPH domain 2-like: Ribonuclease PH domain 2-like, ABD of a subclass of class I AA-tRNA-S: Anticodon-binding domain of a subclass of class I aminoacyl- tRNA synthetases, AN-α hydrolase-like: Adenine nucleotide alpha hydrolase-like, TP SH3-like domain: Translation proteins SH3-like domain, D-SRP/SRP receptor G-proteins: Domain of the SRP/SRP receptor G-proteins, Class I glutamine AM-like: Class I glutamine amidotransferase-like, AAF: Archaeon Archaeoglobus fulgidus, TT: Thermus thermophiles, BA: Bacillus anthracis, PA: Pyrococcus abyssi, PseA: Pseudomonas aeruginosa, AA: Aquifex aeolicus, TM: Thermotoga maritime, AMJ: Archaeon Methanococcus jannaschii, MJ: Methanococcus jannaschii, SC: Saccharomyces cerevisiae, BST: Bacillus stearothermophilus, EC: Escherichia coli, BSU: Bacillus subtilis, AHM: Archaeon Haloarcula marismortui, APH: Archaeon Pyrococcus horikoshii, TA:Thermus aquaticus, TM: Thermotoga maritime, HM: Human mitochondrial, RN: Rattus norvegicus, DM: Drosophila melanogaster, XL: Xenopus laevis, ASS: Archaeon Sulfolobus solfataricus; SSBP: Signal sequence binding protein Ffh, RNase III ECD: RNase III endonuclease catalytic domain, PheRS: Phenylalanyl-tRNA synthetase, TBP ThiI, N-ter D: Thiamine biosynthesis protein ThiI, N-terminal domain, MetRS: Methionyl-tRNA synthetase, TRBP: tRNA-binding protein, tRNA CCA-adding E: tRNA CCA-adding enzyme, PheRS: Phenyl-tRNA synthetase, HSP: Heat shock protein, RBP: Ribosome biogenesis protein, EMG: Essential for mitotic growth, RNase III ECD: RNase III endonuclease catalytic domain, Interferon-ids RNA DPK activator A: Interferon-inducible double stranded RNA-dependent protein kinase activator A, Dus: dihydrouridine synthase, TMC: Translational machinery components.
3.2. ANOVA analysis for feature selection
In order to consider the effect of number of features on performance of methods, ANOVA was used to select significantly different features between three and five RNA-binding protein domain subclasses. Table 2 and Table 3 have shown 10 top features with the lowest p-values. From ANOVA results, RNA binding subclasses show an obvious difference in sequence- and structure-based features. Figure 2 shows difference of shape, size of RBDs, size of main patch and frequency of two important charged amino acids, i.e., Arg and Lys, in five different RBD subclasses. In addition reduced models were constructed using selected features with significant level of <.05, which were 45 and 102 features in three and five subclasses, respectively.
Table 2.
Indicating ten top selected features between three RNA-binding domain subclasses by using ANOVA analysis.
| Number | Feature | P-value |
|---|---|---|
| 1 | Number of Arg in intermediate regions | 2.0E-04 |
| 2 | Molecular weight of RBDs | 5.0E-04 |
| 3 | Number of Ser in buried regions | 7.0E-04 |
| 4 | Number of Cys in main patch | 0.001 |
| 5 | Number of basic amino acids in sequence | 0.0012 |
| 6 | Number of Glu in sheet | 0.0026 |
| 7 | Number of charged amino acids in sequence | 0.003 |
| 8 | Number of Arg in exposed regions | 0.0034 |
| 9 | Number of charged amino acids in sheet | 0.0039 |
| 10 | Isoelectric point | 0.0041 |
Table 3.
Indicating ten top selected features between five RNA-binding domain subclasses by using ANOVA analysis.
| Number | Feature | P-value |
|---|---|---|
| 1 | Dipole | 4.00E-10 |
| 2 | Total number of residues | 8.00E-10 |
| 3 | Total number of atoms | 1.40E-09 |
| 4 | Total number of negative residues | 7.40E-09 |
| 5 | Total number of positive residues | 4.93E-08 |
| 6 | Number of Lys in main patch | 8.92E-08 |
| 7 | Molecular weight of RBDs | 8.55E-07 |
| 8 | RM | 2.18E-06 |
| 9 | Number of small amino acids in main patch | 1.50E-05 |
| 10 | Number of Ala in buried regions | 4.62E-05 |
Figure 2.
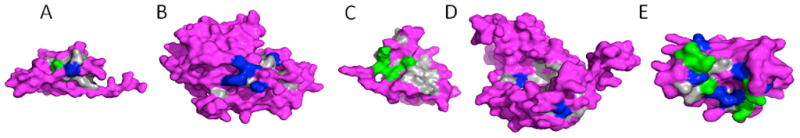
Diversity of features between five different RBDs. (A) sample of 7S RBDs (d1914a1), (B) sample of rRNA RBDs (d2v3ka1), (C) sample of double_stranded RBPs (d1ekza_), (D) sample of mRNA RBDs (d1afwa1), and (E) sample of tRNA RBDs (d1a6fa_). The gray region represents the main patch, blue represents Arg amino acids, and green represents Lys amino acids.
3.3. Tuned multi-class support vector machine analysis
We used a tune function to select optimized structure of TMCSVM through jackknife and self-consistency tests. The most important parameter of TMCSVM topology is kernel function which was searched for the best one among four different kernel functions, i.e., linear, polynomial, radial, and sigmoid. Table 8 and 9 show the highest performance obtained by TMCSVM in overall. TMCSVM and reduced-TMCSVM show the highest rate of 79.31% in prediction of rRNA BDs subclass in comparison with the other methods in five subclasses prediction and also SVM shows the highest rate of 50% for prediction of tRNA BD subclass in three subclasses prediction. Our results confirm that although TMCSVM is a machine learning method, dealing with a large number of features is possible because of seeking a global optimized solution and avoiding over-fitting in the SVM training process. However, obtained results in three subclasses prediction, emphasize that this ability is diminished to limited range of features/samples ratio.
Table 8.
Results of self-consistency and jackknife tests in prediction of five subclasses
| Test | Method | Rate of correct prediction for each RBD subclasses
|
Overall rate of accuracy | ||||
|---|---|---|---|---|---|---|---|
| 7s (%) | Double-stranded (%) | mRNA (%) | rRNA (%) | tRNA (%) | |||
| Self-consistency | RF | 100 | 100 | 100 | 100 | 100 | 100 |
| Reduced-RF | 100 | 100 | 100 | 100 | 100 | 100 | |
| SVM | 100 | 100 | 100 | 100 | 100 | 100 | |
| Reduced-SVM | 100 | 100 | 100 | 100 | 87.50 | 97.56 | |
| MCRLR | 100 | 100 | 100 | 100 | 100 | 100 | |
| Reduced-MCRLR | 100 | 100 | 100 | 100 | 100 | 100 | |
| Jackknife | RF | 60.00 | 81.25 | 63.64 | 44.83 | 37.50 | 54.88 |
| Reduced-RF | 70.00 | 81.25 | 81.82 | 44.83 | 37.50 | 58.54 | |
| SVM | 40.00 | 87.50 | 54.55 | 79.31 | 50.00 | 67.07 | |
| Reduced-SVM | 50.00 | 87.50 | 45.45 | 79.31 | 43.75 | 65.84 | |
| MCRLR | 60.00 | 100 | 54.55 | 65.52 | 31.25 | 63.41 | |
| Reduced-MCRLR | 50.00 | 87.50 | 45.45 | 65.52 | 43.75 | 60.98 | |
Table 9.
Results of self-consistency and jackknife tests in prediction of three subclasses
| Test | Method | Rate of correct prediction for each RBD subclasses
|
Overall rate of accuracy | ||
|---|---|---|---|---|---|
| mRNA (%) | rRNA (%) | tRNA (%) | |||
| Self-consistency | RF | 100 | 100 | 100 | 100 |
| Reduced-RF | 100 | 100 | 100 | 100 | |
| SVM | 100 | 100 | 100 | 100 | |
| Reduced-SVM | 100 | 100 | 100 | 100 | |
| MCRLR | 100 | 100 | 100 | 100 | |
| Reduced-MCRLR | 100 | 100 | 100 | 100 | |
| Jackknife | RF | 81.82 | 62.07 | 43.75 | 60.71 |
| Reduced-RF | 81.82 | 62.07 | 62.50 | 66.07 | |
| SVM | 63.64 | 82.76 | 37.50 | 66.07 | |
| Reduced-SVM | 63.64 | 79.31 | 62.50 | 71.43 | |
| MCRLR | 54.55 | 82.76 | 62.50 | 71.43 | |
| Reduced-MCRLR | 18.18 | 79.31 | 37.50 | 55.36 | |
3.4. Random Forest analysis
R randomForest package was used to construct RF for prediction of RBD subclasses. In order to optimize performance of RF, we defined cutoffs based on distribution of RBDs in subclasses, i.e., number of RBDs in each subclass divided by total number of RBDs. Obtained results reveal that although RF can predict all of RBDs correctly through self-consistency, performance of jackknife test drastically reduced (Table 8 and 9). However, reduced-RF shows the highest rate in prediction of 7S RBDs subclass (70%), and mRNA RBDs subclass (81.82%) in comparison with the other methods in five subclasses prediction. In addition, RF and reduced-RF show the highest rate of 81.82% for prediction of mRNA BD subclass in three subclasses prediction. Furthermore, from obtained results, it is obvious that number of features in RF training is an important issue and it is independent of number of subclasses. Indeed, RF is over-fitting prone when we train it using large number of features.
3.5. Multi-class ℓ1/ℓq-regularized logistic regression
We ran a MCRLR method on the dataset in five and three subclasses using jackknife and self-consistency. MCRLR provides useful information about preferred and avoided features in each one of RNA binding subclasses. tRNA BD subclasses shows some preferred and avoided with higher average values in comparison with the other subclasses in three- and five subclasses through jackknife and self-consistency procedures (Tables 4–7). Our results confirm previous reported unique properties of tRNA BPs by Shazman and Mandel-Gutfreund (2008).
Table 4.
Indicating average weights of ten top important features computed by MCRLR in the jackknife procedure using all of features for prediction of five RNA-binding domain subclasses. Positive values show preference of the features in related subclasses and negative values show avoidance of the features in related subclasses.
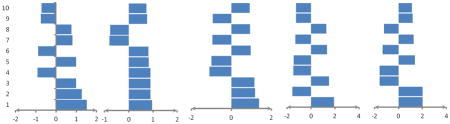
| |||||
|---|---|---|---|---|---|
| No. | Feature/Average Value
|
||||
| 7s RBD subclass | Double -stranded RBD subclass | mRNA RBD subclass | rRNA RBD subclass | tRNA RBD subclass | |
| 1 | Number of Met in MP/1.517 | Number of basic AAs in MP/0.967 | Molecular weight/1.373 | Number of His in MP/1.913 | Number of Arg in IR/2.004 |
| 2 | Number of Met in RC/1.274 | Number of Glu in MP/0.900 | Number of Ser in BR/1.176 | Number of Arg in IR/-1.555 | Number of His in sheet/2.003 |
| 3 | Number of Cys in sheet/0.998 | Dipole/0.900 | Number of Cys in MP/1.142 | Number of Ile in MP/1.499 | Number of Pro in MP/-1.583 |
| 4 | Number of His in seq./-0.920 | Number of Glu in IR/0.891 | Number of Ser in IR/-1.094 | Number of Leu in helix/-1.481 | Number of Ile in MP/-1.576 |
| 5 | Number of Met in ER/0.990 | Number of Tyr in MP/0.831 | Isoelectric point/-1.007 | Number of Met in RC/-1.458 | Number of Asp in BR/1.371 |
| 6 | Number of Glu in MP/-0.886 | Number of Ala in BR/0.811 | Number of Cys in RC/0.969 | Number of Ser in RC/1.330 | Number of Ile in IR/-1.335 |
| 7 | Number of Tyr in helix/0.811 | Number of Gln in RC/-0.799 | Number of charged AAs in seq./-0.949 | Number of Tyr in MP/-1.304 | Number of Met in sheet/1.255 |
| 8 | Number of Glu in IR/0.759 | Number of Arg in MP/-0.779 | Number of Asp in ER/0.934 | Number of Val in helix/1.280 | Number of Phe in ER/-1.226 |
| 9 | Frequency of antiparallel HB/-0.753 | Number of Lys in MP/0.752 | Number of Arg in ER/-0.931 | Number of His in ER/-1.269 | Number of Phe in IR/1.166 |
| 10 | Second patch size/-0.729 | Number of Asp in MP/0.728 | Number of Asp in IR/0.920 | Number of Cys in RC/-1.258 | Number of Gln in IR/1.114 |
MP: main patch, RC: randomcoil, ER: exposed regions, IR: intermediate regions, HB: hydrogen bond, AAs: amino acids, BR: buried regions, Seq.: Sequence.
Table 7.
Indicating average weights of ten top important features computed by MCRLR in the jackknife procedure using selected features by ANOVA analysis for prediction of three RNA-binding domain subclasses. Positive values show preference of the features in related subclasses and negative values show avoidance of the features in related subclasses.
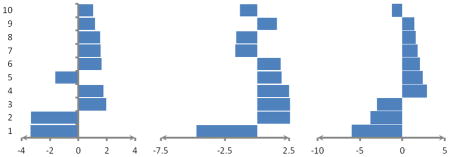
| |||
|---|---|---|---|
| No. | Feature/Average Value
|
||
| mRNA RBD subclass | rRNA RBD subclass | tRNA RBD subclass | |
| 1 | Number of Glu in seq./-3.365 | Number of Ala in helix/-4.742 | Number of Asn in helix/-5.94 |
| 2 | Number of Cys in helix/-3.347 | Number of Glu in seq./3.999 | Number of Leu in seq./-3.765 |
| 3 | Number of Asp in seq./1.950 | Number of Leu in seq./2.571 | Number of Trp in seq./-2.978 |
| 4 | Number of Gln in helix/1.759 | Number of Asn in helix/2.481 | Number of Ala in helix/2.913 |
| 5 | Number of Glu in helix/-1.621 | Number of Cys in helix/1.900 | Number of Gly in helix/2.386 |
| 6 | Number of Val in seq./1.620 | Number of Trp in seq/1.837 | Number of aliphatic AAs in seq./2.048 |
| 7 | Number of Leu in seq./1.570 | Number of Lys in seq./-1.712 | Number of Lys in seq./1.773 |
| 8 | Number of Ala in helix/1.525 | Number of Ile in seq./-1.618 | Number of Ile in seq./1.567 |
| 9 | Number of nonpolar AAs in seq./1.187 | Number of Pro in seq./1.551 | Number of Pro in helix/1.402 |
| 10 | Number of Trp in seq./1.051 | Number of nonpolar AAs in seq./-1.355 | Number of Met in helix/-1.241 |
The results of jackknife and self-consistency tests, which shown in Table 8 and 9, are obtained according to the output of the model. High performance measures of MCRLR model through self-consistency confirm usefulness of defined features in prediction of RBPs subclasses. Results of jackknife tests show that performance of reduced-MCRLR drastically decreased especially in three subclasses prediction using selected features. Rationale for decrease of MCRLR performance is restriction of shrinkage ability using limited number of features (N=45 for prediction of three subclasses). Indeed, ℓ1/ ℓq-regularized constrains the total weight allocated to a set of features, with the end result that some features received zero weight. Additionally, MCRLR shows the highest rate in prediction of double-stranded RBDs in comparison with the other methods.
4. Discussion
Knowledge regarding how bio-macromolecules interact with each other is essential in the understanding of cellular processes. In this study, we investigated interaction of protein and RNA as an important interaction in various cellular processes.
According to a recent comprehensive review (Chou, 2011), to establish a really useful predictor for a protein system, we need to consider: construct a valid benchmark dataset, formulate the protein samples with an effective mathematical expression, develop a powerful algorithm to operate the prediction, evaluate the anticipated accuracy of the predictor, and establish a user-friendly web-server, respectively.
From previous reports, it is mentioned that the aminoacyl tRNA synthetases, and bacterial factors, which mimic tRNA BPs have highly negatively charged surface (Tworowski et al., 2005; Nakamura and Ito, 2003). But there is no more information about variation in feature distribution in different RBPs. In this study, in addition to multi-class classification of RBDs we tried to do feature selection. In addition to comparable prediction accuracy with TMCSVM, a clear variety of feature distributions was elucidated by using MCRLR. For example, our results demonstrate exciting diversity in distribution of Lys and Arg, two important charged amino acids in interaction and catalytic reaction, in different RBDs subclasses. From our data in tRNA BD subclass, Lys is preferred in sequence and Arg is preferred in intermediate regions with high scores (Tables 4–7). In mRNA BDs subclass, Arg is preferred in sequence, and in double-stranded RBD subclass, Lys is preferred in main domain while Arg is avoided in main domain. In addition, in rRNA BD subclass, Lys is avoided in sequence and it seems that Arg is preferred to be on surface as it has been determined with negative value of being in intermediate regions. In 7S RBD subclass, Arg and Lys have not been selected among top preferred or avoided residues. From our results we can understand that Lys and Arg in tRNA BDs, Arg in mRNA BDs, Lys in double-stranded RBDs, and exposed Arg in rRNA BDs are possibly important in RNA-protein interaction and catalytic reaction of RBDs. Figure 2 illustrates distribution of Arg and Lys in main patches of different RBDs subclasses.
These results emphasize that the tRNA BDs have unique local and global properties that can be utilized for identifying novel proteins possibly involved in tRNA processing. Moreover, it is worth to mention that the size of secondary patch show positive average value in tRNA BDs subclasses and it means secondary patch may have specific properties as mentioned by Shazman and Mandel-Gutfreund (2008). Growth of 3D solved protein databases will be helpful to discover more details about RBDs.
In this study we developed a first of its kind in silico approach for analysis and prediction of RBDs subclasses in three and five subclasses using RF, TMCSVM and MCRLR. In overall, TMCSVM outperforms the other methods, although tuning of SVM is time consuming. On the other hand, MCRLR show some advantages including fast training, report of more important features for RBD prediction, and detection of avoided and preferred features in each subclass.
In addition, RF shows the worst accuracy among three predictor methods which means RF is prone to over-fitting especially when large numbers of features are fed into it. In conclusion, we used two types of predictor methods including: (1) MCRLR as a statistical method and (2) RF and TMCSVM as machine learning methods. Statistical methods are commonly accepted and popularity of these models may be attributed to the interpretability of model parameters and ease of use, although they suffer from their specific limitations. For example, statistical methods use linear combinations of independent variables and, therefore, are not the best adept at modeling grossly nonlinear complex interactions as has been demonstrated in biological systems. On the other hand, machine learning methods are rich and flexible nonlinear systems that show robust performance in dealing with noisy or incomplete data and have the ability to generalize from the input data. They may be better suited than other modeling systems to predict outcomes when the relationships between the variables are complex, multidimensional, and nonlinear as found in complex biological systems. Although machine learning methods can give high prediction accuracy, some problem may be raised in their training. For example in this study we showed that RF as a well-known machine learning method is not well suited for our problem and is prone to over-fitting. “black box” nature, and the empirical nature of model development are other disadvantages of machine learning methods (Tu, 1996).
5. Conclusion
A great challenge in classifying ligand binding proteins (such as RBDs) is to be able to identify to which ligand it will bind. For this purpose, we applied three different predictor methods to classify RNA-binding domains using a large number of sequence and structural features, which was trained on three and five different subclasses of known RBDs classified according to their RNA target. From our results TMCSVM shows the highest prediction accuracy in compare with other methods. Overall, the results we obtained are encouraging, reinforcing the idea that combination of sequence and structural properties of protein domains can give clues to the protein’s interacting partner.
It is important to note that subclassification of the RBDs to three and five subclasses using our multiclass approach is only possible –given the prior knowledge that the protein domain binds RNA. Indeed we have to mention that requiring known protein domains as RNA binding is a limitation of such predictor models.
Finally, our results showed that, in addition to multi class prediction, biological diversity of RBD’s subclasses would be interpretable using state-of-the-art methods like ℓ1/ℓq-regularized logistic regression.
Since user friendly and publicly accessible web-servers represent the future direction for developing practically more useful predictors (Chou and Shen, 2009), we shall make efforts in our future work to provide a web-server for the method presented in this paper.
Table 5.
Indicating average weights of ten top important features computed by MCRLR in the jackknife procedure using selected features by ANOVA analysis for prediction of five RNA-binding domain subclasses. Positive values show preference of the features in related subclasses and negative values show avoidance of the features in related subclasses.
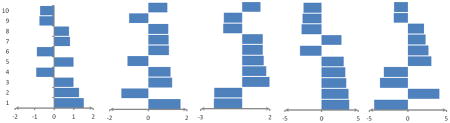
| |||||
|---|---|---|---|---|---|
| No. | Feature/Average Value
|
||||
| 7sRBD subclass | Double-stranded RBD subclass | mRNA RBD subclass | rRNA RBD subclass | tRNA RBD subclass | |
| 1 | Number of Cys in sheet/2.029 | Number of Glu in IR/1.675 | Number of Ser in IR/-2.051 | Number of small AAs in helix/3.773 | Number of Ile in MP/-4.351 |
| 2 | Number of Met in MP/2.011 | Number of Arg in MP/-1.394 | Number of Arg in ER/-2.034 | Number of Ser in RC/3.652 | Number of Arg in IR/4.083 |
| 3 | Number of Met in RC/1.653 | Number of Tyr in MP/1.230 | Number of Cys in MP/1.976 | Number of Ile in MP/3.378 | Number of Small AAs in helix/-3.625 |
| 4 | Frequency of antiparallel HB/-1.54 | Dipole/1.160 | Number of Ser in BR/1.733 | Number of acidic AAs in ER/3.219 | Number of Small AAs in RC/-3.118 |
| 5 | Number of His in seq./-1.370 | Number of Gln in RC/-1.070 | Molecular weight/1.554 | Number of Ser in IR/3.030 | Number of His in seq./3.027 |
| 6 | Number of Met in ER/1.300 | Number of Phe in MP/1.062 | Number of small AAs in BR/1.486 | Number of Tyr in MP/-2.954 | Number of Phe in IR/2.643 |
| 7 | Number of Glu in IR/1.292 | Number of Aromatic AAs in MP/1.047 | Number of Arg in IR/1.482 | Number of Pro in sheet/2.7428 | Number of tiny AAs in sheet/2.288 |
| 8 | Number of Leu in sheet/1.235 | Number of Ser in IE/1.036 | Number of Glu in IR/-1.363 | Number of Met in RC/-2.742 | Frequency of i+3 HB/2.091 |
| 9 | Number of Gln in RC/1.231 | Number of basic AAs in BR/-0.989 | Isoelectric point/-1.319 | Number of Arg in IR/-2.588 | Number of Small AAs in MP/-1.816 |
| 10 | Second patch size/-1.1781 | Number of Met in RC/0.981 | Number of Ile in MP/1.317 | Number of Glu in IR/-2.458 | Number of Phe inRC/-1.790 |
MP: main patch, RC: randomcoil, ER: exposed regions, IR: intermediate regions, HB: hydrogen bond, AAs: amino acids, BR: buried regions, Seq.: Sequence.
Table 6.
Indicating average weights of ten top important features computed by MCRLR in the jackknife procedure using all of features for prediction of three RNA-binding domain Subclasses. Positive values show preference of the features in related subclasses and negative values show avoidance of the features in related subclasses.
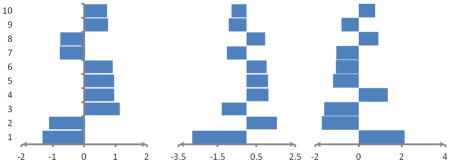
| |||
|---|---|---|---|
| No. | Feature/Average Value
|
||
| mRNA RBD subclass | rRNA RBD subclass | tRNA RBD subclass | |
| 1 | Number of charged AAs in seq./-1.337 | Number of Arg in IR/-2.794 | Number of Arg in IR/2.129 |
| 2 | Number of Ser in IR/-1.132 | Number of Glu in sheet/1.556 | Number of Pro in MP/-1.699 |
| 3 | Number of Arg in seq./1.131 | Number of Phe in helix/-1.278 | Number of Pro in IR/-1.581 |
| 4 | Number of Pro in seq./0.953 | Number of charged AAs in seq./1.109 | Number of Phe in IR/1.339 |
| 5 | Number of Glu in sheet/0.949 | Number of Phe in MP/1.091 | Number of Asn in IR/-1.183 |
| 6 | Number of Ser in BR/0.898 | Number of Ser in IR/1.021 | Number of Val in helix/-1.055 |
| 7 | Number of Phe in IR/-0.782 | Second patch size/-1.015 | Number of Ile in IR/-1.033 |
| 8 | Number of Arg in ER/-0.769 | Number of Cys in helix/0.954 | Number of Phe in helix/0.921 |
| 9 | Number of Arg in IR/0.753 | Number of His in sheet/-0.930 | Number of Cys in helix/-0.790 |
| 10 | Number of Cys in RC/0.712 | Number of Cys in RC/-0.789 | Second patch size/0.766 |
MP: main patch, RC: randomcoil, ER: exposed regions, IR: intermediate regions, AAs: amino acids, BR: buried regions, Seq.: Sequence.
Highlights.
Multi-class prediction of RNA-binding protein domains.
We compared prediction accuracy of three different state-of-the-art predictor methods.
In addition to successful classifying RBPs, we discovered dissimilar sequence and structural features using ℓ1/ℓq-regularized logistic regression.
Our method could be applied to identify novel RNA-binding proteins with unique folds.
Acknowledgments
We thank Abbas Mahdavi for his assistance in this investigation. This work is partially funded by NIH Grant R00RR024163.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmad S, Gromiha M, Fawareh H, Sarai A. ASAView: database and tool for solvent accessibility representation in proteins. BMC Bioinformatics. 2004;5:51. doi: 10.1186/1471-2105-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. Nature Genetics. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach F. Consistency of the group lasso and multiple kernel learning. Journal of Machine Learning Research. 2008;9:1179–1225. [Google Scholar]
- Breiman L. Random Forests. Machine Learning. 2001;45:5–32. [Google Scholar]
- Chen C, Chen L, Zou X, Cai P. Prediction of protein secondary structure content by using the concept of Chou’s pseudo amino acid composition and support vector machine. Protein & Peptide Letters. 2009;16:27–31. doi: 10.2174/092986609787049420. [DOI] [PubMed] [Google Scholar]
- Chen Y, Varani G. Protein families and RNA recognition. FEBS J. 2005;272:2088–2097. doi: 10.1111/j.1742-4658.2005.04650.x. [DOI] [PubMed] [Google Scholar]
- Chou KC. Prediction of protein cellular attributes using pseudo amino acid composition. PROTEINS: Structure, Function, and Genetics. 2001;43:246–255. doi: 10.1002/prot.1035. (Erratum: ibid, 2001, Vol44, 60) [DOI] [PubMed] [Google Scholar]
- Chou KC. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics. 2005;21:10–19. doi: 10.1093/bioinformatics/bth466. [DOI] [PubMed] [Google Scholar]
- Chou KC. Pseudo amino acid composition and its applications in bioinformatics, proteomics and system biology. Current Proteomics. 2009;6:262–274. [Google Scholar]
- Chou KC. Some remarks on protein attribute prediction and pseudo amino acid composition (50th Anniversary Year Review) J Theor Biol. 2011;273:236–247. doi: 10.1016/j.jtbi.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou KC, Shen HB. Hum-PLoc: A novel ensemble classifier for predicting human protein subcellular localization. Biochem Biophys Res Commun. 2006;347:150–157. doi: 10.1016/j.bbrc.2006.06.059. [DOI] [PubMed] [Google Scholar]
- Chou KC, Shen HB. Cell-PLoc: A package of Web servers for predicting subcellular localization of proteins in various organisms (updated version: Cell-PLoc 2.0: An improved package of web-servers for predicting subcellular localization of proteins in various organisms, Natural Science, 2010, 2, 1090–1103) Nature Protocols. 2008;3:153–162. doi: 10.1038/nprot.2007.494. [DOI] [PubMed] [Google Scholar]
- Chou KC, Shen HB. Review: recent advances in developing web-servers for predicting protein attributes. Natural Science. 2009;2:63–92. (openly accessible at http://www.scirp.org/journal/NS/) [Google Scholar]
- Chou KC, Wu ZC, Xiao X. iLoc-Euk: A Multi-Label Classifier for Predicting the Subcellular Localization of Singleplex and Multiplex Eukaryotic Proteins. PLoS ONE. 2011;6:e18258. doi: 10.1371/journal.pone.0018258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou KC, Wu ZC, Xiao X. iLoc-Hum: Using accumulation-label scale to predict subcellular locations of human proteins with both single and multiple sites. Molecular Biosystems. 2012;8:629–641. doi: 10.1039/c1mb05420a. [DOI] [PubMed] [Google Scholar]
- Chou KC, Zhang CT. Review: Prediction of protein structural classes. Critical Reviews in Biochemistry and Molecular Biology. 1995;30:275–349. doi: 10.3109/10409239509083488. [DOI] [PubMed] [Google Scholar]
- Ding H, Luo L, Lin H. Prediction of cell wall lytic enzymes using Chou’s amphiphilic pseudo amino acid composition. Protein & Peptide Letters. 2009;16:351–355. doi: 10.2174/092986609787848045. [DOI] [PubMed] [Google Scholar]
- Duchi J, Singer Y. Online and batch learning using forward backward splitting. Journal of Machine Learning Research. 2009;10:2899–2934. [Google Scholar]
- Dudoit S, Fridlyan J, Fridlyan TP. Comparison of discrimination methods for the classification of tumors using gene expression data. J Am Stat Assoc. 2002;97:77–87. [Google Scholar]
- Ellis JJ, Broom M, Jones S. Protein RNA interactions: structural analysis and functional classes. Proteins. 2007;66:903–911. doi: 10.1002/prot.21211. [DOI] [PubMed] [Google Scholar]
- Esmaeili M, Mohabatkar H, Mohsenzadeh S. Using the concept of Chou’s pseudo amino acid composition for risk type prediction of human papillomaviruses. J Theor Biol. 2010;263:203–209. doi: 10.1016/j.jtbi.2009.11.016. [DOI] [PubMed] [Google Scholar]
- Georgiou DN, Karakasidis TE, Nieto JJ, Torres A. Use of fuzzy clustering technique and matrices to classify amino acids and its impact to Chou’s pseudo amino acid composition. J Theor Biol. 2009;257:17–26. doi: 10.1016/j.jtbi.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Gu Q, Ding YS, Zhang TL. Prediction of G-Protein- Coupled Receptor Classes in Low Homology Using Chou’s Pseudo Amino Acid Composition with Approximate Entropy and Hydrophobicity Patterns. Protein & Peptide Letters. 2010;17:559–567. doi: 10.2174/092986610791112693. [DOI] [PubMed] [Google Scholar]
- Guo J, Rao N, Liu G, Yang Y, Wang G. Predicting protein folding rates using the concept of Chou’s pseudo amino acid composition. Journal of Computational Chemistry. 2011;32:1612–1617. doi: 10.1002/jcc.21740. [DOI] [PubMed] [Google Scholar]
- Han LY, Cai CZ, Lo SL, Chung MC, Chen YZ. Prediction of RNA-binding proteins from primary sequence by a support vector machine approach. RNA. 2004;10:355–368. doi: 10.1261/rna.5890304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayat M, Khan A. Discriminating Outer Membrane Proteins with Fuzzy KNearest Neighbor Algorithms Based on the General Form of Chou’s PseAAC. Protein & Peptide Letters. 2012;19:411–421. doi: 10.2174/092986612799789387. [DOI] [PubMed] [Google Scholar]
- Hu L, Zheng L, Wang Z, Li B, Liu L. Using pseudo amino Acid composition to predict protease families by incorporating a series of protein biological features. Protein and Peptide Letters. 2011;18:552–558. doi: 10.2174/092986611795222795. [DOI] [PubMed] [Google Scholar]
- Jia SC, Hu XZ. Using Random Forest Algorithm to Predict beta-Hairpin Motifs. Protein and Peptide Letters. 2011;18:609–617. doi: 10.2174/092986611795222777. [DOI] [PubMed] [Google Scholar]
- Jiang X, Wei R, Zhang TL, Gu Q. Using the concept of Chou’s pseudo amino acid composition to predict apoptosis proteins subcellular location: an approach by approximate entropy. Protein & Peptide Letters. 2008;15:392–396. doi: 10.2174/092986608784246443. [DOI] [PubMed] [Google Scholar]
- Jones S, Daley DT, Luscombe NM, Berman HM, Thornton JM. Protein RNA interactions: a structural analysis. Nucleic Acids Res. 2001;29:943–954. doi: 10.1093/nar/29.4.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W, Sander C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1993;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Kandaswamy KK, Chou KC, Martinetz T, Moller S, Suganthan PN, Sridharan S, Pugalenthi G. AFP-Pred: A random forest approach for predicting antifreeze proteins from sequence-derived properties. J Theor Biol. 2011;270:56–62. doi: 10.1016/j.jtbi.2010.10.037. [DOI] [PubMed] [Google Scholar]
- Kowalski M. Sparse regression using mixed norms. Applied and Computational Harmonic Analysis. 2009;27:303–324. [Google Scholar]
- Li BQ, Huang T, Liu L, Cai YD, Chou KC. Identification of Colorectal Cancer Related Genes with mRMR and Shortest Path in Protein-Protein Interaction Network. PLoS ONE. 2012;7:e33393. doi: 10.1371/journal.pone.0033393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FM, Li QZ. Predicting protein subcellular location using Chou’s pseudo amino acid composition and improved hybrid approach. Protein & Peptide Letters. 2008;15:612–616. doi: 10.2174/092986608784966930. [DOI] [PubMed] [Google Scholar]
- Li LQ, Zhang Y, Zou LY, Zhou Y, Zheng XQ. Prediction of Protein Subcellular Multi-Localization Based on the General form of Chou’s Pseudo Amino Acid Composition. Protein & Peptide Letters. 2012;19:375–387. doi: 10.2174/092986612799789369. [DOI] [PubMed] [Google Scholar]
- Liaw A. Wiener M. Classification and regression by randomForest. R News. 2002;2:18–22. [Google Scholar]
- Lin H. The modified Mahalanobis discriminant for predicting outer membrane proteins by using Chou’s pseudo amino acid composition. J Theor Biol. 2008;252:350–356. doi: 10.1016/j.jtbi.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Lin H, Ding H, Feng-Biao Guo FB, Zhang AY, Huang J. Predicting subcellular localization of mycobacterial proteins by using Chou’s pseudo amino acid composition. Protein & Peptide Letters. 2008;15:739–744. doi: 10.2174/092986608785133681. [DOI] [PubMed] [Google Scholar]
- Lin J, Wang Y. Using a novel AdaBoost algorithm and Chou’s pseudo amino acid composition for predicting protein subcellular localization. Protein & Peptide Letters. 2011;18:1219–1225. doi: 10.2174/092986611797642797. [DOI] [PubMed] [Google Scholar]
- Lin WZ, Fang JA, Xiao X, Chou KC. iDNA-Prot: Identification of DNA Binding Proteins Using Random Forest with Grey Model. PLoS ONE. 2011;6:e24756. doi: 10.1371/journal.pone.0024756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingel A, Sattler M. Novel modes of protein-RNA recognition in the RNAi pathway. Curr Opin Struct Biol. 2005;15:107–115. doi: 10.1016/j.sbi.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Liu L, Hu XZ, Liu XX, Wang Y, Li SB. Predicting Protein Fold Types by the General Form of Chou’s Pseudo Amino Acid Composition: Approached from Optimal Feature Extractions. Protein & Peptide Letters. 2012;19:439–449. doi: 10.2174/092986612799789378. [DOI] [PubMed] [Google Scholar]
- Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol. 2007;8:479–490. doi: 10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei S. Multi-kernel transfer learning based on Chou’s PseAAC formulation for protein submitochondria localization. J Theor Biol. 2012;293:121–130. doi: 10.1016/j.jtbi.2011.10.015. [DOI] [PubMed] [Google Scholar]
- Mohabatkar H. Prediction of cyclin proteins using Chou’s pseudo amino acid composition. Protein & Peptide Letters. 2010;17:1207–1214. doi: 10.2174/092986610792231564. [DOI] [PubMed] [Google Scholar]
- Mohabatkar H, Mohammad Beigi M, Esmaeili A. Prediction of GABA(A) receptor proteins using the concept of Chou’s pseudo-amino acid composition and support vector machine. J Theor Biol. 2011;281:18–23. doi: 10.1016/j.jtbi.2011.04.017. [DOI] [PubMed] [Google Scholar]
- Morozova N, Allers J, Myers J, Shamoo Y. Protein RNA interactions: exploring binding patterns with a three-dimensional superposition analysis of high resolution structures. Bioinformatics. 2006;22:2746–2752. doi: 10.1093/bioinformatics/btl470. [DOI] [PubMed] [Google Scholar]
- Nanni L, Lumini A, Gupta D, Garg A. Identifying Bacterial Virulent Proteins by Fusing a Set of Classifiers Based on Variants of Chou’s Pseudo Amino Acid Composition and on Evolutionary Information. IEEE/ACM Trans Comput Biol Bioinform. 2012;9:467–475. doi: 10.1109/TCBB.2011.117. [DOI] [PubMed] [Google Scholar]
- Negahban S, Ravikumar P, Wainwright M, Yu B. A unified framework for high dimensional analysis of m-estimators with decomposable regularizers. Advances in Neural Information Processing Systems. 2009:1348–1356. [Google Scholar]
- Parker JS, Barford D. Argonaute: a scaffold for the function of short regulatory RNAs. Trends Biochem Sci. 2006;31:622–630. doi: 10.1016/j.tibs.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Pugalenthi G, Kandaswamy KK, Chou KC, Vivekanandan S, Kolatkar P. RSARF: Prediction of Residue Solvent Accessibility from Protein Sequence Using Random Forest Method. Protein & Peptide Letters. 2012;19:50–56. doi: 10.2174/092986612798472875. [DOI] [PubMed] [Google Scholar]
- Qin YF, Wang CH, Yu XQ, Zhu J, Liu TG, et al. Predicting Protein Structural Class by Incorporating Patterns of Over- Represented k-mers into the General form of Chou’s PseAAC. Protein & Peptide Letters. 2012;19:388–397. doi: 10.2174/092986612799789350. [DOI] [PubMed] [Google Scholar]
- Qiu JD, Huang JH, Liang RP, Lu XQ. Prediction of G-protein-coupled receptor classes based on the concept of Chou’s pseudo amino acid composition: an approach from discrete wavelet transform. Analytical Biochemistry. 2009;390:68–73. doi: 10.1016/j.ab.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Qiu JD, Huang JH, Shi SP, Liang RP. Using the concept of Chou’s pseudo amino acid composition to predict enzyme family classes: an approach with support vector machine based on discrete wavelet transform. Protein & Peptide Letters. 2010;17:715–722. doi: 10.2174/092986610791190372. [DOI] [PubMed] [Google Scholar]
- Qiu JD, Suo SB, Sun XY, Shi SP, Liang RP. OligoPred: A web-server for predicting homo-oligomeric proteins by incorporating discrete wavelet transform into Chou’s pseudo amino acid composition. Journal of Molecular Graphics & Modelling. 2011;30:129–134. doi: 10.1016/j.jmgm.2011.06.014. [DOI] [PubMed] [Google Scholar]
- Qiu Z, Wang X. Improved Prediction of Protein Ligand-Binding Sites Using Random Forests. Protein & Peptide Letters. 2011;18:1212–1218. doi: 10.2174/092986611797642788. [DOI] [PubMed] [Google Scholar]
- Shameer K, Pugalenthi G, Kandaswamy KK, Sowdhamini R. 3dswap-pred: Prediction of 3D Domain Swapping from Protein Sequence Using Random Forest Approach. Protein & Peptide Letters. 2011;18:1010–1020. doi: 10.2174/092986611796378729. [DOI] [PubMed] [Google Scholar]
- Shao X, Tian Y, Wu L, Wang Y, Jing L, Deng N. Predicting DNA- and RNA-binding proteins from sequences with kernel methods. J Theor Biol. 2009;258:289–293. doi: 10.1016/j.jtbi.2009.01.024. [DOI] [PubMed] [Google Scholar]
- Shazman S, Mandel-Gutfreund Y. Classifying RNA-binding proteins based on electrostatic properties. PLoS Comput Biol. 2008;4:e1000146. doi: 10.1371/journal.pcbi.1000146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman-Peleg A, et al. Prediction of interacting single-stranded RNA bases by protein-binding patterns. J Mol Biol. 2008;379:299–316. doi: 10.1016/j.jmb.2008.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Statnikov A, Wang L, Aliferis CF. A comprehensive comparison of random forests and support vector machines for microarray-based cancer classification. BMC Bioinformatics. 2008;9:319. doi: 10.1186/1471-2105-9-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stawiski EW, Gregoret LM, Mandel-Gutfreund Y. Annotating nucleic acid-binding function based on protein structure. J Mol Biol. 2003;326:1065–1079. doi: 10.1016/s0022-2836(03)00031-7. [DOI] [PubMed] [Google Scholar]
- Tu JV. Advantages and disadvantages of using artificial neural networks versus logistic regression for predicting medical outcomes. J Clin Epidemiol. 1996;49:1225–1231. doi: 10.1016/s0895-4356(96)00002-9. [DOI] [PubMed] [Google Scholar]
- Vapnik V. Statistical Learning Theory. Wiley-Interscience; New York: 1998. [Google Scholar]
- Vapnik VN. The Nature of Statistical Learning Theory. Springer; Berlin: 1995. [Google Scholar]
- Wu ZC, Xiao X, Chou KC. iLoc-Plant: a multi-label classifier for predicting the subcellular localization of plant proteins with both single and multiple sites. Molecular BioSystems. 2011;7:3287–3297. doi: 10.1039/c1mb05232b. [DOI] [PubMed] [Google Scholar]
- Xiao X, Wang P, Chou KC. iNR-PhysChem: A Sequence-Based Predictor for Identifying Nuclear Receptors and Their Subfamilies via Physical-Chemical Property Matrix. PLoS ONE. 2012;7:e30869. doi: 10.1371/journal.pone.0030869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Wu ZC, Chou KC. iLoc-Virus: A multi-label learning classifier for identifying the subcellular localization of virus proteins with both single and multiple sites. J Theor Biol. 2011a;284:42–51. doi: 10.1016/j.jtbi.2011.06.005. [DOI] [PubMed] [Google Scholar]
- Xiao X, Wu ZC, Chou KC. A multi-label classifier for predicting the subcellular localization of gram-negative bacterial proteins with both single and multiple sites. PLoS ONE. 2011b;6:e20592. doi: 10.1371/journal.pone.0020592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Guo Y, Li Y, Li G, Li M, et al. SecretP: Identifying bacterial secreted proteins by fusing new features into Chou’s pseudo-amino acid composition. J Theor Biol. 2010;267:1–6. doi: 10.1016/j.jtbi.2010.08.001. [DOI] [PubMed] [Google Scholar]
- Yu X, Cao J, Cai Y, Shi T, Li Y. Predicting rRNA-, RNA-, and DNA-binding proteins from primary structure with support vector machines. J Theor Biol. 2006;240:175–184. doi: 10.1016/j.jtbi.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Yuan M, Lin Y. Model selection and estimation in regression with grouped variables. Journal Of The Royal Statistical Society Series B. 2006;68(1):49–67. [Google Scholar]
- Zeng YH, Guo YZ, Xiao RQ, Yang L, Yu LZ, Li ML. Using the augmented Chou’s pseudo amino acid composition for predicting protein submitochondria locations based on auto covariance approach. J Theor Biol. 2009;259:366–372. doi: 10.1016/j.jtbi.2009.03.028. [DOI] [PubMed] [Google Scholar]
- Zhang GY, Fang BS. Predicting the cofactors of oxidoreductases based on amino acid composition distribution and Chou’s amphiphilic pseudo amino acid composition. J Theor Biol. 2008;253:310–315. doi: 10.1016/j.jtbi.2008.03.015. [DOI] [PubMed] [Google Scholar]
- Zhang GY, Li HC, Gao JQ, Fang BS. Predicting lipase types by improved Chou’s pseudo-amino acid composition. Protein & Peptide Letters. 2008;15:1132–1137. doi: 10.2174/092986608786071184. [DOI] [PubMed] [Google Scholar]
- Zhao XW, Li XT, Ma ZQ, Yin MH. Identify DNA-Binding Proteins with Optimal Chou’s Amino Acid Composition. Protein & Peptide Letters. 2012;19:398–405. doi: 10.2174/092986612799789404. [DOI] [PubMed] [Google Scholar]
- Zhou XB, Chen C, Li ZC, Zou XY. Using Chou’s amphiphilic pseudo-amino acid composition and support vector machine for prediction of enzyme subfamily classes. J Theor Biol. 2007;248:546–551. doi: 10.1016/j.jtbi.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Zou D, He Z, He J, Xia Y. Supersecondary structure prediction using Chou’s pseudo amino acid composition. Journal of Computational Chemistry. 2011;32:271–278. doi: 10.1002/jcc.21616. [DOI] [PubMed] [Google Scholar]