

Abstract
Systemic sclerosis is a complex disease with widespread skin fibrosis and variable visceral organ involvement. Since transforming growth factor-β (TGFβ) has been implicated in driving fibrosis in systemic sclerosis, a mechanism-derived gene expression signature was used to assay TGFβ-responsive gene expression in the skin of patients with systemic sclerosis (SSc). Primary dermal fibroblasts from patients with diffuse SSc (dSSc) and healthy controls were treated with TGFβ, and the genome-wide gene expression was measured on DNA microarrays over a time course of 24 hours. Eight hundred and ninety-four probes representing 674 uniquely annotated genes were identified as TGFβ responsive. Expression of the TGFβ-responsive signature was examined in skin biopsies from 17 dSSc, seven limited SSc (lSSc), three morphea patients, and six healthy controls. The TGFβ-responsive signature was expressed in 10 out of 17 dSSc skin biopsies, but was not found in lSSc, morphea, or healthy control biopsies. Expression of dSSC the TGFβ-responsive signature stratifies patients into two major groups, one of which corresponds to the “diffuse-proliferation” intrinsic subset that showed higher modified Rodnan skin score and a higher likelihood of scleroderma lung disease. The TGFβ-responsive signature is found in only a subset of dSSc patients who could be targeted by specific therapies.
INTRODUCTION
Systemic sclerosis (SSc) is a systemic autoimmune disease characterized by skin fibrosis, internal organ dysfunction, vascular abnormalities, and immune activation. Clinical presentation is heterogeneous and patients with clinically indistinguishable early-stage disease progress to different clinical endpoints. There is a need for objective and quantitative measures of disease severity that can identify relative likelihoods of disease progression.
The most widely recognized classification of SSc groups patients into systemic sclerosis with diffuse scleroderma (dSSc) and systemic sclerosis with limited scleroderma (lSSc), largely by the degree of skin involvement (LeRoy et al., 1988). Efforts have been made to further distinguish the heterogeneity in SSc patients (Barnett et al., 1988a, b; Ferri et al., 2002; Scussel-Lonzetti et al., 2002; Maricq and Valter, 2004), but no further disease classification has been widely accepted.
Genome-wide measurement of gene expression with DNA microarrays can easily distinguish skin biopsies from patients with dSSc from healthy controls (Whitfield et al., 2003; Gardner et al., 2006), and identification of gene expression-based subsets has been reported (Milano et al., 2008). Multiple distinct gene expression signatures were identified among patients with scleroderma (Milano et al., 2008), with some of these groups delineating existing divisions such as those between dSSc and lSSc, and others reflecting more subtle changes in the cellular infiltrates or in the expression of different cellular processes. The signaling pathways underlying these subsets have not been mapped. Here we extend these findings by showing genes responsive to transforming growth factor-β (TGFβ) in vitro are associated with the “diffuse-proliferation” subset of dSSc patients.
Skin fibrosis, generally considered to be driven by TGFβ-activated fibroblasts, is a hallmark of dSSc (Leroy et al., 1989; Smith and LeRoy, 1990; Cotton et al., 1998; Leask and Abraham, 2004; Varga, 2004; Leask, 2006; Verrecchia et al., 2006; Ihn, 2008). Elevated levels of TGFβ have been observed in SSc skin biopsies (Gabrielli et al., 1993; Sfikakis et al., 1993) and TGFβ-activated fibroblasts produce collagen I, III, and V in addition to other matrix proteins such as glycoaminoglycans (Wynn, 2008). Numerous studies have identified TGFβ-regulated genes that show increased expression in SSc fibroblasts, including fibronectin (Xu et al., 1991), collagens α1(I) and α1(III) (Kuroda and Shinkai, 1997), collagen-α2(1) (Jinnin et al., 2006), endoglin (Leask et al., 2002), connective tissue growth factor (Leask, 2004), and cartilage oligomeric matrix protein (Farina et al., 2006). SSc fibroblasts also show an increase in TGFβRI and TGFβRII mRNA levels and surface protein expression when compared with healthy dermal fibroblasts (Kubo et al., 2002), suggesting activation of the TGFβ pathway in these cells. Activation of the platelet-derived growth factor (PDGF) pathway by stimulatory autoantibodies to the PDGF receptor has also been shown and may target many of the same genes (Baroni et al., 2006). A clinical trial using anti-TGFβ therapy in dSSc patients has been reported, but the results were inconclusive (Denton et al., 2007).
We report the use of a mechanism-derived gene expression signature to determine whether a TGFβ-response is found in the gene expression subsets of SSc. We defined a TGFβ-responsive signature over time courses of 24 hours in four independent cultures of dermal fibroblasts (two healthy, two dSSc) and identified 894 probes representing 674 TGFβ-responsive genes. The signature was analyzed in vivo in a microarray data set analyzing skin biopsies from patients with scleroderma and healthy controls (Milano et al., 2008). The TGFβ-responsive signature is found in the “diffuse-proliferation” subset of dSSc skin biopsies. This indicates that TGFβ deregulation, or a related pathway, contributes to pathogenesis in this subset.
RESULTS
We identified the genes responsive to TGFβ on a genome-wide scale with DNA microarrays in adult dermal fibroblasts from patients with dSSc and healthy controls. Four independent primary fibroblast cultures were analyzed, two from dSSc lesional forearm skin biopsie, one from a healthy control forearm skin biopsy, and one commercially available adult dermal fibroblast culture (Table 1). To compare our results to prior studies of TGFβ responses in fibroblasts, we used conditions similar to those previously reported (Chambers et al., 2003; Renzoni et al., 2004). Fibroblasts were cultured for 7–9 passages post explant in 10% serum and then brought to quiescence in 0.1% serum for 24 hours before TGFβ treatment. Fewer than 1% of the cells were in S-phase after 24 hours in 0.1% serum, determined by BrdU incorporation (data not shown).
Table 1.
Primary fibroblast cell cultures
| Cell line | Sex | Age | Disease duration | Biopsy site |
|---|---|---|---|---|
| Control 1 | Male | 47 | — | ND1 |
| Control 2 | Female | 58 | — | Forearm |
| Scleroderma 1 | Female | 62 | 1 year | Forearm |
| Scleroderma 2 | Male | 41 | 1 year | Forearm |
Clinical characteristics of individuals from whose biopsies dermal fibroblasts were obtained.
Adult dermal fibroblast primary cell line obtained from a commercial source. Site not determined (ND).
To determine optimal experimental conditions, a dosage curve was generated with 50–300 pm TGFβ and the cellular response measured by quantitative real-time PCR (qRT-PCR). We found 50 pm TGFβ resulted in an 11-fold induction of plasminogen activator inhibitor 1 (PAI1) mRNA, whereas treatment with 100 pm TGFβ resulted in 16-fold induction (Figure 1). Concentrations of 200–300 pm resulted in decreased relative PAI1 levels, indicating that a concentration higher than 100 pm was saturating. We chose to use 50 pm for all further experiments.
Figure 1. Dosage and time dependence of plasminogen activator inhibitor 1 (PAI1) mRNA expression after TGFβ treatment.
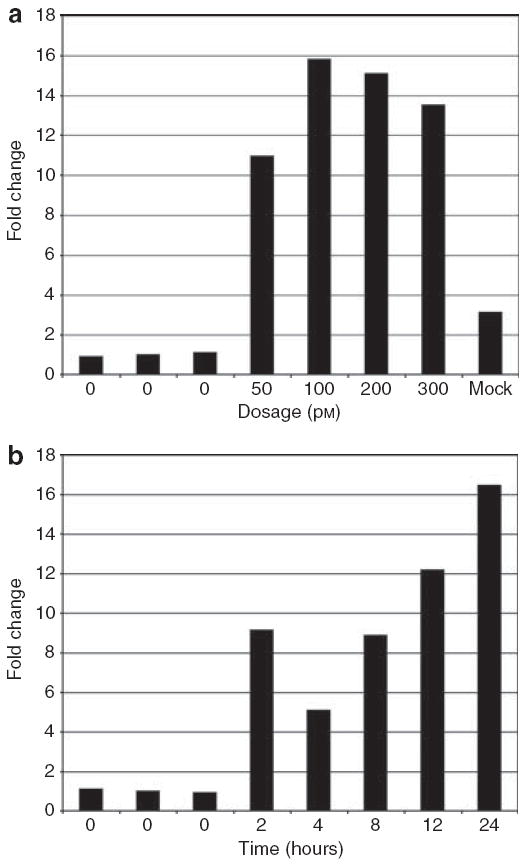
To optimize the concentration of TGFβ and the length of time course to analyze, cells were treated with varying doses of TGFβ, and time points were collected from 2 to 24 hours. (a) Normal dermal fibroblasts (NDFs) in 0.1% serum were treated with 50, 100, 200, or 300 pm TGFβ for 2 hours. Levels of PAI1 mRNA were measured in triplicate by Taqman qRT-PCR, normalized to 18S rRNA; fold change is relative to the average of three independent, untreated samples. (b) NDFs were treated with 50 pm TGFβ and PAI1 mRNA levels were measured in triplicate at 0, 2, 4, 8, 12, and 24 hours post-treatment. Fold change is relative to the average of the zero time points.
Quiescent cells were exposed to 50 pm TGFβ and total RNA was collected at 0, 2, 4, 8 12, and 24 hours after treatment. A mock time course without TGFβ, but in otherwise identical conditions, was performed in a single culture of normal dermal fibroblasts. The mock time course identifies genes that may respond to treatment effects that are independent of TGFβ. Total RNA from each sample was amplified, labeled, and hybridized to whole-genome DNA microarrays in a common reference design. Eight microarrays were hybridized for each time course resulting in a total of 40 microarray hybridizations.
Genome-wide response to TGFβ in adult dermal fibroblasts
To characterize the genome-wide response to TGFβ across the four independent fibroblasts cultures, probes were selected that changed at least 1.74-fold in at least eight of the 32 arrays. Eight hundred and ninety-four TGFβ-responsive probes were selected representing 674 uniquely annotated genes (Figure 2a and Supplementary Data File 1). The gene expression data for eight probes corresponding to seven known TGFβ targets in dermal fibroblasts is shown (Figure 2b). Examination of these probes in the mock, no TGFβ, control shows that the observed changes in gene expression are specific to a TGFβ response (Figure 2a).
Figure 2. Genome-wide response to TGFβ in adult dermal fibroblasts.
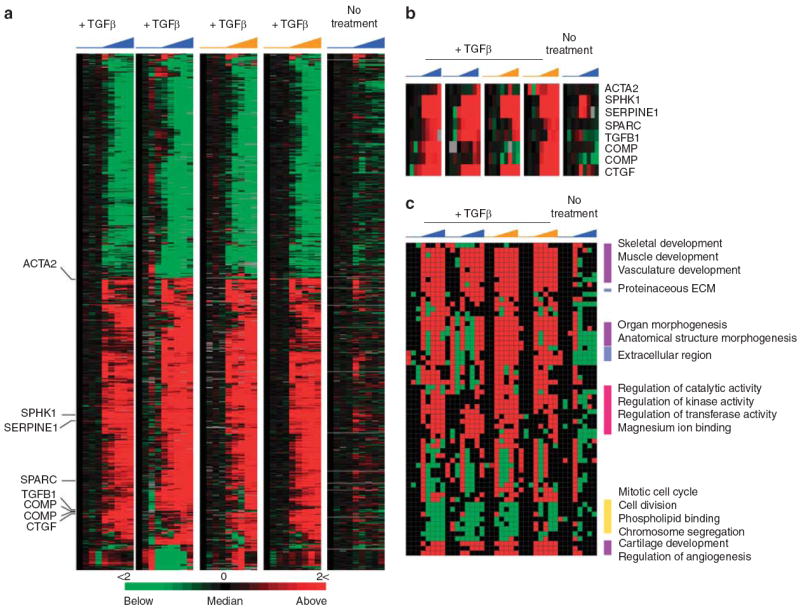
(a) Shown are the 894 probes, representing 674 annotated genes, with a 1.74-fold or greater change in expression over 24 hours following treatment with 50 pm TGFβ. Four independent primary cell cultures were treated with TGFβ, two from healthy control subjects (blue), and two from SSc patients (orange). A mock time course was performed using identical conditions with the omission of TGFβ. Time of treatment, from 0 to 24 hours, is indicated. Each row represents a probe, and each column represents a time point. (b) Expression data for the genes previously reported as being TGFβ responsive (Supplementary Table S2) and found among the 894 probes. (c) Module map of GO terms in the genome-wide response to TGFβ. Each column represents a microarray, and each row represents an enriched GO term. Only modules that were significantly enriched (P < 0.05) in at least 16 microarrays analyzed are shown. Select modules are indicated.
To identify differential TGFβ responses between normal and scleroderma fibroblasts, the data from TGFβ-treated normal fibroblast was compared to that of TGFβ-treated SSc fibroblasts with significance analysis of microarrays (SAM) using slope and area functions, in a two-class unpaired time-course analysis. Only a single gene, Early Growth Response-1 (EGR1), was significantly differentially induced at an false discovery rate (FDR) <5%. Since one would expect this number or more to be called as falsely significantly by chance alone, this is likely a false positive. Another recent study also failed to find significant differences in the TGFβ responses in cultured lung fibroblasts from SSc patients and normal controls (Chambers et al., 2003).
TGFβ module map
To summarize the biological programs represented by genes in the TGFβ-responsive signature, we created a module map that identifies groups of Gene Ontology (GO) terms (biological process, cellular component, or molecular function) coordinately regulated by TGFβ in cultured adult dermal fibroblasts. Each row shows a unique module that is a set of coordinately regulated genes involved in similar processes.
Modules with significantly enriched genes (P<0.05, hypergeometric distribution) in at least 16 of the 20 arrays were selected. A subset of modules is shown (Figure 2c); the full figure with all modules is available as Supplementary Figure S1. Those induced after TGFβ treatment included proteinaceous extracellular matrix, extracellular matrix, and extracellular space, associated with collagen production, extracellular matrix deposition, and remodeling (Figure 2c). TGFβ is a mediator of fibrosis in different organs including skin, kidneys, lungs, and liver, and is necessary for tissue remodeling and wound healing (Varga and Whitfield, 2009; Wynn, 2008).
Modules associated with development programs included angiogenesis, skeletal development, muscular development, vasculature development, multicellular organismal development, organ morphogenesis, and anatomical structure morphogenesis. The role of TGFβ and its family members in embryogenesis, tissue specification, and organ development through to tissue maintenance and repair have been extensively characterized (Wall and Hogan, 1994; Padgett et al., 1997; Mummery, 2001).
Among the enriched, downregulated GO terms were mitotic cell cycle, cell division, and chromosomal segregation, indicating that TGFβ downregulates the proliferation signature (Whitfield et al., 2006) under low-serum conditions.
TGFβ-responsive gene expression underlies the “diffuse-proliferation” intrinsic subset
To determine whether the TGFβ-responsive signature was expressed in the intrinsic subsets of scleroderma, we analyzed the expression of the 894 TGFβ-responsive signature probes (674 genes) in the data set of Milano et al. (2008), which included dSSc, lSSc, morphea, and healthy controls. Expression data for each of the 894 probes was extracted from the set of 75 microarrays and organized by hierarchical clustering (Figure 3). Organization of the samples was compared to the sample groupings reported using an intrinsic gene set (Milano et al., 2008). The group expressing the TGFβ-responsive signature is similar to the diffuse-proliferation subset (Supplementary Table S1), suggesting that deregulated TGFβ signaling, or a related pathway, may underlie this subset. Several related pathways and modulators have been implicated in SSc, including endoglin, which has increased expression on endothelial cells (Dharmapatni et al., 2001), and fibroblasts (Leask et al., 2002), endothelin-1, which is a downstream mediator of TGFβ responses, (Shi-Wen et al., 2007), and the related PDGF pathway, implicated by the presence of stimulatory autoantibodies to the PDGF receptor (Baroni et al., 2006). Activation of the PDGF pathway in SSc has proven controversial (Classen et al., 2009). Consistent with deregulation of the TGFβ and PDGF pathways, several studies have demonstrated that imatinib mesylate prevents fibrosis in murine models of fibrosis (Daniels et al., 2004; Distler et al., 2007; Akhmet-shina et al., 2008). Additionally, a case report showed a gene expression response associated with disease improvement in two patients after treatment with imatinib mesylate (Chung et al., 2009).
Figure 3. TGFβ-responsive genes are deregulated in the “diffuse-proliferation” subset of scleroderma.
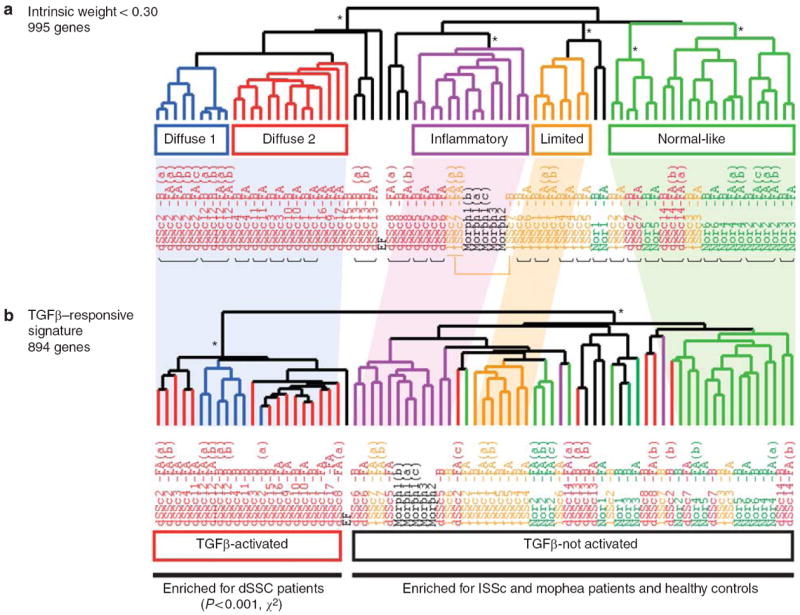
(a) Intrinsic subsets of scleroderma as described by Milano et al. (2008). (b) Organization of 75 arrays from Milano et al. by hierarchical clustering of 894 TGFβ-responsive signature genes. Shading is indicative of positioning of subsets identified by intrinsic genes relative to that identified by the TGFβ-responsive signature. *P < 0.001.
Samples with high expression of the TGFβ-responsive signature include only dSSc skin biopsies, and this enrichment is statistically significant (P<1 × 10−4, χ2-test). One patient classified as diffuse-proliferation and one patient classified as inflammatory did not show consistent expression of the TGFβ-responsive signature (Supplementary Table S1). Therefore, there is not a precise overlap of the TGFβ-responsive group and the proliferative group identified by Milano et al. The group without the TGFβ-responsive signature contains a subset of the dSSc biopsies, all lSSc biopsies, morphea samples, and healthy controls. Therefore, the TGFβ-responsive signature, a set of genes independent from the 995 intrinsic genes, is expressed in a subset of dSSc patients, but is not expressed in lSSc, morphea, or healthy controls.
TGFβ-responsive gene expression in dSSc patients
To determine whether the TGFβ-responsive gene signature was consistently found in both lesional and non-lesional skin, or associated with specific clinical phenotypes, we analyzed the signature in the diffuse patients alone. The expression values for each of the 894 genes in the TGFβ-responsive signature were extracted from the skin biopsy data set and the 53 microarrays representing only patients with dSSc and normal controls. Samples were ordered by hierarchical clustering; the resulting sample dendrogram shows clear bifurcation of the skin biopsies (Figure 4a). The left branch, highlighted in red, is comprised solely of samples from dSSc patients, while the right branch includes the remaining dSSc samples and all healthy controls. Samples from two patients, dSSc2 and dSSc8, split between the two groups and, therefore, could not be assigned to either group. Of the patients who were consistently classified, each showed deregulation of the TGFβ-responsive gene signature in both lesion forearm and non-lesion back biopsies. Therefore, the systemic nature of the disease extends to specific pathways.
Figure 4. The TGFβ-responsive signature distinguishes a subset of dSSc patients.
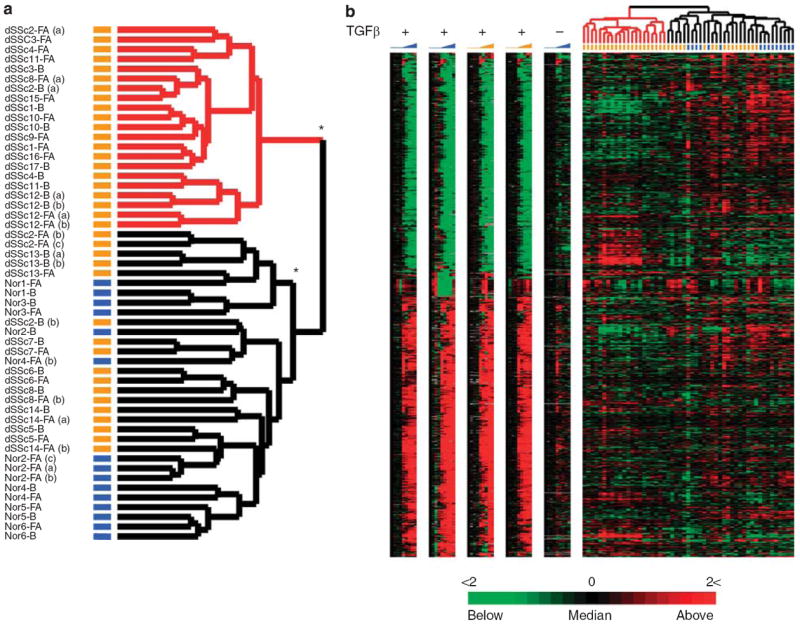
(a) Patient sample dendrogram resulting from hierarchical clustering of 53 arrays probing gene expression in skin biopsies of patients with dSSc (orange bars) and healthy control (blue bars). The samples were clustered using the 894 TGFβ-responsive probes that comprise the signature. Two major groups of samples are evident: TGFβ-activated (red) and TGFβ-not-activated (black). Technical replicates are designated by a letter (a, b or c) following patient and biopsy site identification. Statistically significant clusters as determined by SigClust are marked by *(P < 0.001). (b) Individual TGFβ time courses are aligned with the gene expression data from dSSc and healthy control biopsies, and illustrate the heterogeneity of the in vitro–derived TGFβ-responsive signature in skin biopsies.
The significance of the classification was determined with Statistical Significance of Clustering (SigClust) (Liu et al., 2008), which tests the robustness of the sample bifurcation and found that the clustering is highly significant (P<0.001). We find the TGFβ-activated group to be a single statistically significant cluster. The TGFβ-not-activated group forms two statistically significant clusters. We did not further investigate this additional sub-grouping given the relatively small size of the groups.
Alignment of the skin biopsy gene expression data with that from the in vitro TGFβ time courses reveals that expression of the signature is heterogeneous (Figure 4b). We identified the subset of the 894 probes driving the sample bifurcation. A two-class unpaired SAM analysis identified 474 probes to be significantly differentially expressed between the two groups (FDR < 1.15%; Figure 5). The centroid for the 474 differentially expressed probes is shown (Figure 5 and Supplementary Data File 2). The activation of the TGFβ-responsive signature in each patient sample was determined by calculating the Pearson correlation coefficients between the centroid and the gene expression for each skin biopsy (Figure 5). For simplicity, we have termed the group on the left (red dendrogram) TGFβ-activated because of high positive correlation coefficients associated with these samples, and the group on the right (black dendogram) TGFβ-not activated owing to the predominantly negative correlation coefficients.
Figure 5. A subset of the TGFβ-responsive signature ideally differentiates the two groups.
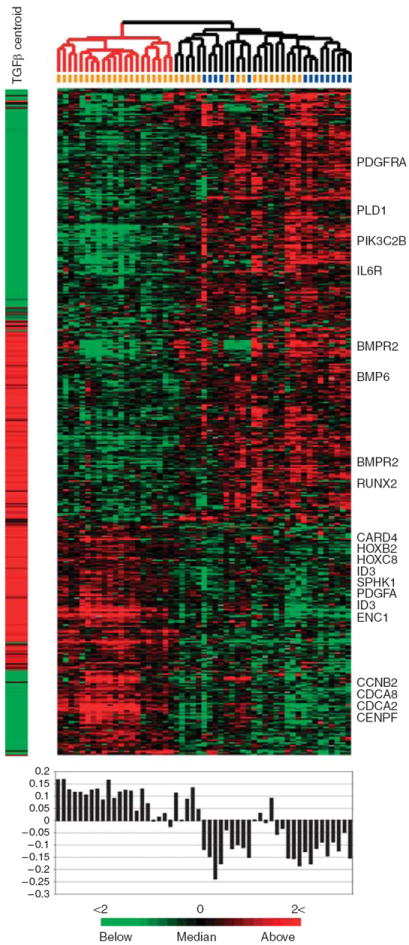
SAM was used to identify 474 probes from the initial set of 894 probes that showed consistent, statistically significant differential expression between the TGFβ-activated and TGFβ-not-activated groups. The centroid representing the average of the TGFβ-responsive gene signature at maximal induction (12 and 24 hours) is shown to the left of the heat map. Pearson correlations between the centroid and each array were calculated and are plotted directly beneath each array.
The genes in Figure 5 can be classified into two groups based on their expression patterns in the fibroblast time courses and skin biopsies. Those concordantly and those discordantly expressed between fibroblasts and skin, and further characterized as up- or downregulated in fibroblasts upon TGFβ treatment. Genes found concordantly upregulated in TGFβ-stimulated fibroblasts and skin included canonical targets ID3, PDGFA, SPHK1 (Supplementary Table S2), genes involved in organ and tissue development (HOXB2 and HOXC8), and genes associated with apoptosis (ENC1 and CARD4). Genes concordantly downregulated included those involved in cell signaling (PDGFRA and IL6R) and phospholipids binding (PLD1 and PIK3C2B).
Genes induced in the TGFβ-treated fibroblasts, but with low expression in the TGFβ-activated skin biopsies, were enriched for developmental processes and signaling (RUNX2, BMPR2, and BMP6). Genes showing decreased expression in TGFβ-treated fibroblasts, but increased expression in the skin biopsies, were those associated with cell cycle (the proliferation signature; Whitfield et al., 2006; CDCA2, CDCA8, CCNB2, and CENPF). High expression of these genes was observed previously in the diffuse-proliferation subset and we found increased KI67 staining in the epidermis of the skin biopsies (Milano et al., 2008).
Validation of TGFβ signature genes in patient samples
We validated the expression of three genes from the TGFβ-responsive signature, which were also the most highly differentially expressed between the groups with and without the TGFβ-responsive gene signature (Figure 6). We analyzed the expression of E2F7, which is also a cell-cycle-regulated gene (Whitfield et al., 2002), growth differentiation factor-6, which is a member of the TGFβ superfamily, and actin-α2. Each gene was measured in representative patient samples by qRT-PCR (Figure 6a–c) and compared to the expression levels from the DNA microarray (Figure 6d–f), in all cases the gene expression follows the same trends between the TGFβ-activated versus TGFβ-not-activated groups.
Figure 6. Validation of TGFβ-responsive genes in dSSc skin.
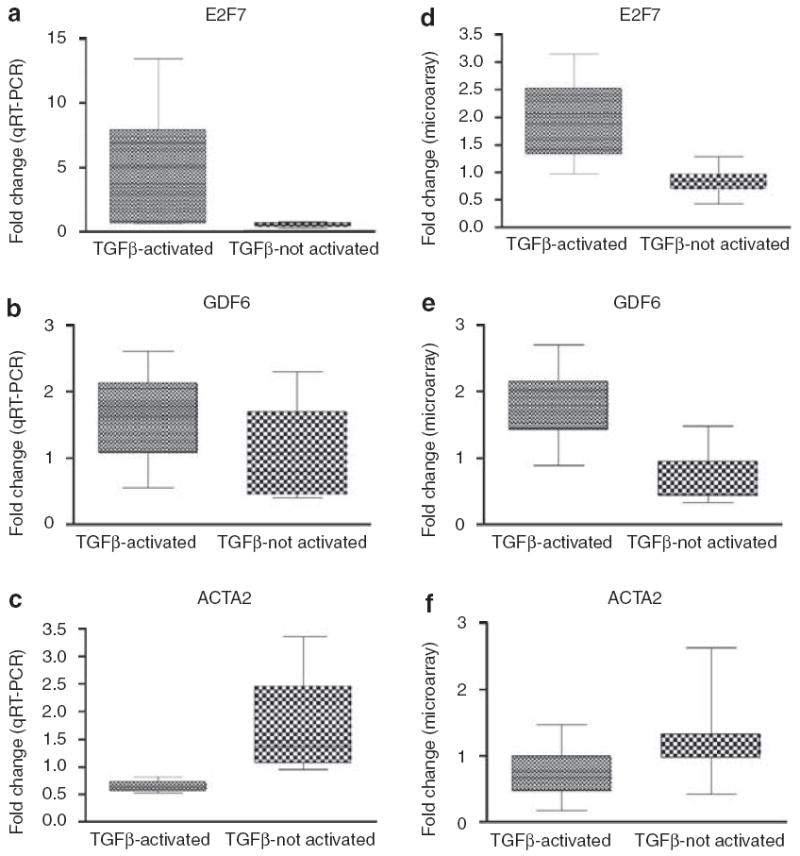
Relative mRNA levels of three genes, E2F7 (a, d), growth differentiation factor-6 (b, e), and actin-α2 (c, f) were determined using Fast Taqman qRT-PCR on select patient samples from the TGFβ-activated and TGFβ-not-activated patient groups. Trends of mRNA levels determined by qRT-PCR were reflective of those measured on DNA microarrays (d–f). All data were normalized to the mean relative expression ratio.
The TGFβ-responsive signature is associated with more severe skin and lung disease
We examined the severity and incidence of available clinical parameters, calculated both by patient and by biopsy, to determine whether the dSSc patients in the TGFβ-activated group showed phenotypic differences from those in the TGFβ-not-activated group. Since patients dSSc2 and dSSc8 could not be accurately assigned, they were excluded from the analysis, leaving 10 patients in the TGFβ-activated group and five in the TGFβ-not-activated group. Each group was analyzed for differences in clinical covariates (Table 2). Student’s t-tests were used to analyze differences in patient age, disease duration as defined by onset of first non-Raynaud symptoms, mean modified Rodnan skin score (MRSS) (0–51), Raynaud phenomenon (0–10), and digital ulcers (0–3). All other parameters for which we had clinical data, including incidence of interstitial lung disease (ILD), impaired renal function, and gastrointestinal involvement were scored as either present or absent and a χ2-test used to assess differences between the groups (Table 2). When individual biopsies were considered, the TGFβ-activated group showed significantly higher skin scores (mean = 26.9 ± 2.04) than the TGFβ-not-activated group (mean = 17.8 ± 1.95; P = 0.0061). When performed on a per patient basis, the difference in the means is weakly significant (P = 0.11). Examination of the dot plots for MRSS shows consistent difference in the means, with no significant outliers (Figure 7). We found that ILD was significantly more prevalent among the TGFβ-activated samples (7 of 16) as compared with TGFβ-not-activated group (0 of 10; P = 0.014; Table 2), with an odds ratio of ≈ 16.58. When patients rather than biopsies are considered, then five out of 10 patients in the TGFβ-activated group had ILD, whereas none of the five patients in the TGFβ-not-activated group had ILD (P = 0.053). Since this type of exploratory analysis has not been performed previously, we are more concerned about type-II errors (false negatives) than type-I errors (false positives). As such, we consider the P-value of 0.053 to be strongly suggestive, although it does not meet a strict 0.05 significance level, and should be considered a hypothesis that should be tested in a larger cohort of patients. The analysis by biopsy is important because it shows the TGFβ signature is reproducible from different skin samples (lesion forearm and non-lesion back) from the same patient. We found only a weak association with disease duration (biopsies, P = 0.086; patients, P = 0.14). Examination of the dot plots of disease duration shows that the TGFβ-activated group is skewed by two outliers from patients with disease durations of 20 years (Figure 7). We found no significant associations with any of the other clinical variables, including potential immune modulating therapies or with anti-topoisomerase-I status (Table 2).
Table 2.
Clinical parameters associated with the TGFβ-responsive signature
| Clinical parameter | Activated (16 biopsies) | Not activated (10 biopsies) | P-value | Activated (10 patients) | Not activated (5 patients) | P-value |
|---|---|---|---|---|---|---|
| Patient age (years) | 46.3±2.55 | 50.6±2.33 | 0.26 | 48.0±3.43 | 50.6±3.50 | 0.64 |
| Disease duration (years) | 7.63±1.41 | 4.00±1.19 | 0.086 | 9.00±2.04 | 4.40±4.07 | 0.14 |
| MRSS | 26.9±2.04 | 17.8±1.95 | 0.00611 | 25.2±2.63 | 17.8±2.92 | 0.11 |
| Lung disease | 7/16 | 0/10 | 0.0142 | 5/10 | 0/5 | 0.053 |
| GI involvement | 14/16 | 6/10 | 0.11 | 9/10 | 3/5 | 0.17 |
| Renal disease | 2/16 | 0/10 | 0.24 | 2/10 | 0/5 | 0.28 |
| Raynaud severity | 5.93±0.57 | 7.00±0.99 | 0.33 | 5.33±0.78 | 7.00±1.48 | 0.29 |
| Digital ulcers | 0.50±0.18 | 0.80±0.38 | 0.44 | 0.50±0.22 | 0.80±0.58 | 0.57 |
| Immunosuppressive therapy | 9/16 | 2/10 | 0.069 | 5/10 | 1/5 | 0.26 |
| Anti-topoisomerase-I3 | 6/12 | 2/6 | 0.502 | 3/6 | 1/3 | 0.64 |
MRSS, modified Rodnan skin score; TGFβ, transforming growth factor-β.
Statistical associations of clinical parameters with the TGFβ-activated and TGFβ-not-activated groups of patients. Associations with MRSS, disease duration, patient age, Raynaud severity, and digital ulcers were calculated using Student’s t-tests (mean ± SEM). Associations with ILD, GI involvement, renal disease, and presence of anti-topoisomerase antibodies were determined by χ2-test
(P<0.01,
P<0.02).
Calculations were performed by biopsy (columns 2–4) and by patient (columns 6–7).
Data on the anti-topoisomerase-I status were only available for six patients (12 biopsies) in the activated group and three patients (six biopsies) patients in the not activated group.
Figure 7. Distributions of MRSS and disease duration of dSSc patients.
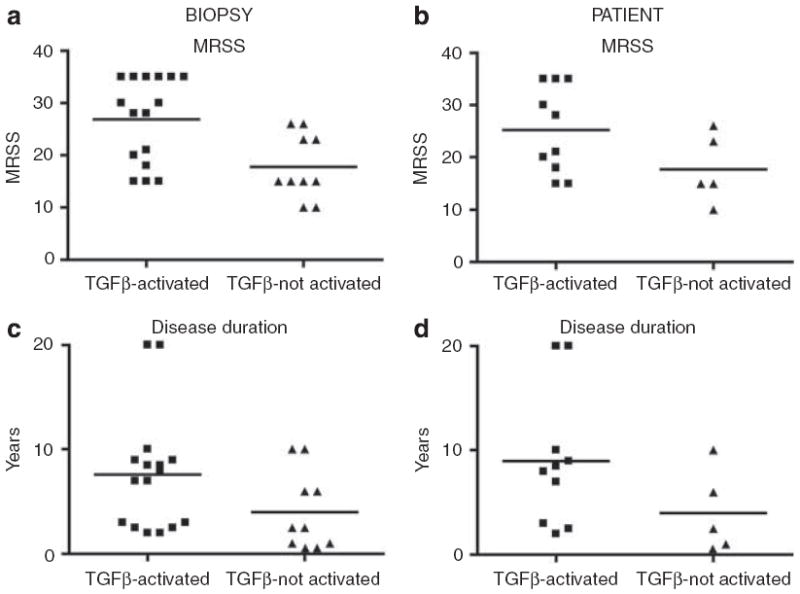
Distributions of MRSS at the time of biopsy were plotted by biopsy (a) and by patient (b) for the TGFβ-activated and TGFβ-not-activated groups. Distributions of disease duration were also plotted by biopsy (c) and by patient (d) for the two groups. P-values for all comparisons are given in Table 2.
DISCUSSION
Experimental determination of a mechanism-derived TGFβ signature in fibroblasts has allowed us to analyze the genes responsive to this pathway in skin biopsies from patients with scleroderma and healthy controls. This has shown that the TGFβ-responsive gene signature is expressed heterogeneously across the spectrum of scleroderma, but shows high expression in a subset of dSSc patients with higher MRSS and increased incidence of lung involvement. The TGFβ-responsive signature is not found in patients with lSSc, morphea, or in healthy controls. Given the established role for TGFβ in mediating fibrosis, it is not surprising that such a signature shows increased expression in dSSc skin, but the heterogeneity of expression among the SSc patients was not expected. This provides further evidence for subsets of patients that can be distinguished by gene expression and demonstrates that this heterogeneity extends to the pathway level. Most importantly, it suggests that TGFβ activation, or activation of a related pathway such as PDGF, may underlie the “diffuse-proliferation” subset of scleroderma.
The finding that a gene signature expressed in skin is associated with the occurrence of lung disease is surprising and to our knowledge is previously unreported. ILD is the leading cause of death among patients with dSSc and approximately 70% of patients show evidence of significant lung pathology at autopsy (Ostojic et al., 2007; Steen and Medsger, 2007). Recent work has developed tools and methods for diagnosis, staging, and characterization of ILD in dSSc patients (Goh et al., 2008); however, biomarkers that reliably predict who will develop lung complications before they become symptomatic would be beneficial. Surprisingly, only a weak association was found between the TGFβ-responsive gene signature and disease duration, raising the possibility that the signature is stable over the disease course in this subset of patients.
Concordance between the TGFβ-responsive gene signature defined in vitro and that found in vivo is variable. Gene expression observed in skin is likely influenced by multiple soluble factors and signals from the extracellular matrix. Thus, the heterogeneity of the signature in skin likely results from differences between single-cell-type cultures stimulated with a single cytokine versus the complexity of whole skin that contains multiple cell types in distinct tissue-specific niches. In addition, many TGFβ-responsive genes are also responsive to other profibrotic cytokines and signaling molecules such as IL13, IL4, spingosine-1 phosphate (JL Sargent and ML Whitfield, unpublished data), endoglin (Dharmapatni et al., 2001; Leask et al., 2002; Fujimoto et al., 2006; Wipff et al., 2007), endothelin-1 (Shi-Wen et al., 2007), but also PDGF and fibroblast growth factor (Gu and Iyer, 2006). Both endothelin-1 (Shi-Wen et al., 2007) and PDGF have been implicated in SSc pathogenesis (Baroni et al., 2006). Therefore, multiple profibrotic signaling pathways converge on this set of genes, which almost certainly contributes to the signature variability in the TGFβ-activated group.
The 674 genes of the TGFβ-responsive signature defined here were compared to two previously published studies of TGFβ-responsive genes in human fibroblasts. Chambers et al. (2003) analyzed human fetal lung fibroblast and identified 122 TGFβ-responsive genes, of which 58 (48%) were identified as TGFβ-responsive in our study. Renzoni et al. (2004) analyzed adult lung fibroblasts and identified 128 TGFβ-responsive genes, of which 33 (26%) were identified in our study. Only 11 genes were identified by both Renzoni and Chambers, all of which are found to be TGFβ-responsive here. The limited overlap found among the different signatures is not surprising given the differences in fibroblasts from different anatomical sites (Chang et al., 2002; Rinn et al., 2006), the use of different microarray platforms, and different analysis techniques.
We have previously described subsets of scleroderma patients distinguished by unique gene expression signatures found in both lesional (forearm) and non-lesional (lower back) skin (Milano et al., 2008). The association of the TGFβ-responsive signature with both the lesional and non-lesional skin is an important validation of the systemic gene expression in skin.
MATERIALS AND METHODS
The gene expression data from lesional forearm and non-lesional back skin of scleroderma, and of healthy skin biopsies, has been previously described (Milano et al., 2008). All work with human subjects or materials was performed with adherence to the Declaration of Helsinki Principles. All subjects signed consent forms approved by the Committee on Human Research at the University of California, San Francisco. Patients met the ACR criteria for systemic sclerosis and were further defined as the diffuse subset. The Committee on the Protection of Human Subjects approved all protocols at Dartmouth Medical School.
All clinical measurements were taken at time of biopsy and have been described in detail previously (Milano et al., 2008). Briefly, as part of the patients’ routine scleroderma standard of care, patients were assessed for MRSS on a 51-point scale, disease duration since first onset of non-Raynaud’s symptoms, a self-reported Raynaud severity score on a 10-point scale, and presence or absence of digital ulcers on a three-point scale. Also recorded were presence (+) or absence (−) of gastrointestinal involvement, scleroderma lung disease, which included patients defined as having pulmonary fibrosis or patients with ILD as determined by ground glass appearance, fibrosis or honeycombing on high-resolution computerized tomography, renal disease, and anti-topoisomerase antibodies. Data from patients on immunosuppressive therapy (mycophenolate mofetil, methotrexate, azathioprine, or prednisone >10 mg daily) were also recorded.
Cells and cell culture
Primary adult human dermal fibroblasts were purchased from Cambrex Bioscience Inc. (East Rutherford, NJ) Additional adult dermal fibroblasts were isolated from explanted healthy or dSSc lesional forearm skin biopsies (Table 1) cultured for at least three passages in DMEM, 10% (v/v) fetal bovine serum, penicillin-streptomycin (100 IU/ml) at 37 °C in a humidified atmosphere with 5% CO2. Cells were passaged every 7 days for 7–10 passages prior to treatment.
BrdU staining
Cell proliferation was assessed with BrdU Labeling and Detection kit I (Roche Applied Sciences, Penzberg, Germany). BrdU incorporation was detected according to the manufacturer’s instructions, counter-stained with 4′-6-diamidino-2-phenylindole, and visualized with an Olympus BX51 microscope.
RNA preparation
For analysis of the TGFβ response, 4 × 105 cells were grown in 100-mm dishes and cultured in DMEM with 10% fetal bovine serum for 48 hours. Cells were brought to quiescence by culturing in low-serum media, DMEM with 0.1% fetal bovine serum, for 24 hours. TGFβ derived from human platelets (R&D Systems, Minneapolis, MN) was diluted into low-serum media and used to treat cells for 0, 2, 4, 8, 12, and 24 hours. Following treatment with TGFβ, cells were lysed in RLT buffer with β-mercaptoethanol and total RNA was isolated with RNeasy minikits (Qiagen, Valencia, CA).
Microarray procedures
Microarray protocols have been described in detail (Milano et al., 2008). Total RNA (300–500 ng) was amplified and labeled according to Agilent Low RNA Input Fluorescent Linear Amplification protocols. Each experimental RNA sample was labeled with Cy3-CTP and competitively hybridized against Universal Human Reference RNA (Stratagene, La Jolla, CA) labeled with Cy5-CTP on Agilent 44,000-element Human microarrays.
Microarrays were scanned with a GenePix 4000B scanner and acquired images quantified with the GenePix Pro 5.1 software (Axon Instruments, Foster City, CA). Technical artifacts and poor-quality spots were flagged and excluded from further analysis. Data were loaded to the UNC Microarray Database.
Data analysis
Data were analyzed as log2 of the lowess-normalized Cy5/Cy3 ratio. A total of 18,696 probes with fluorescent signal at least 1.5 greater than local background and of good quality in at least 80% of arrays were selected for analysis. Each data table was multiplied by negative one to convert the log2 ratios to Cy3/Cy5 ratios and then T0-transformed using the average of triplicate 0-hour samples. TGFβ-responsive probes were selected by fold-change cutoff and genes were hierarchically clustered using the Cluster 3.0 software (Eisen et al., 1998). Centroid values for each gene were calculated by averaging the T0-transformed data for the 12- and 24-hour time points across all TGFβ treatment time courses. The optimal fold-change threshold was determined by comparing genes induced or repressed by the addition of TGFβ over a range of threshold values to a list of 15 known TGFβ targets in human fibroblasts complied from the literature (Supplementary Table S2). At a 1.74 fold-change threshold we identified seven of the 15 (47%) known TGFβ targets in the microarray time-course data. Analyzing this same list of genes in two published TGFβ signatures from pulmonary fibroblasts identified three (20%) (Renzoni et al., 2004) and six (40%) (Chambers et al., 2003) of the fibroblasts targets. A less stringent fold-change threshold did not significantly increase the fraction of known targets identified. Therefore, we elected to use the 1.74 fold-change threshold to define TGFβ-responsive probes.
Module maps were created with Genomica (Segal et al., 2003). Expression data for probes that mapped to the same gene were averaged. Probes lacking entrez gene identifiers were excluded, yielding 10,664 unique genes for analysis. Gene sets of GO biological processes for the module map analysis were obtained from http://genomica.weizmann.ac.il.
Microarray data analyzing scleroderma skin are available from GEO (accession no. GSE9285) (Milano et al., 2008). The patient data set consisting of biopsies from 17 dSSc patients and six healthy controls (53 arrays in total) was downloaded from UNC Microarray Database. A total of 40,818 probes with fluorescent signal at least 1.2-fold greater than local background and of good quality in at least 80% of arrays were selected for analysis.
Statistical analysis of clustering of the patient samples was performed with SigClust (Liu et al., 2008). Heat maps were generated and visualized using TreeView version 1.0.13 (Eisen et al., 1998).
Quantitative real-time PCR
Total RNA (100–200 ng) was reverse-transcribed into single-stranded complementary DNA using SuperScript II reverse transcriptase (Invitrogen, San Diego, CA). Complementary DNA (1.25 mg) was used for each qRT-PCR reaction. Primer probes sets for PAI1 (NM_00602.2), 18S (X03205.1), E2F7 (NM_203394.2), growth differentiation factor-6 (NM_001001557.1), actin-α2 (NM_001141945.1), and glyceraldehyde-3-phosphate dehydrogenase (NM_002046.3) were obtained from Applied Biosystems (Foster City, CA) and analyzed using either the 7300 Real-Time PCR System or the 7,500 Fast Real-Time PCR system. The number of cycles required to generate a detectable fluorescence above background (Ct) was measured for each sample. For dose–response and time-course analyses, fold changes were calculated relative to the average of triplicate untreated samples by the comparative Ct formula 2−ΔΔCt (Livak and Schmittgen, 2001), where ΔCt is the difference between the target gene (PAI1) and the 18S rRNA control, and ΔΔCt is the difference between the ΔCt value of the target gene and the average of the ΔCt values of untreated triplicate samples.
Statistical methods
Statistical calculations were performed using Prism for Macintosh V4.0a (GraphPad Software Inc., La Jolla, CA) or Microsoft Excel with the add-in Winstat. Where indicated, SAM was used to identify significantly differentially expressed genes (Tusher et al., 2001).
Supplementary Material
Acknowledgments
This work was supported by research grants from the Scleroderma Research Foundation to MKC, HYC, and MLW. Additional support for MLW came from HHMI Biomedical Research Support award no. 76200-560801 to Dartmouth College. HYC received additional support from the Congressionally Directed Medical Research Program. AM also received support from the NIH Autoimmunity and Connective Tissue Biology Training grant (AR007576) from the National Institute of Arthritis, Musculoskeletal and Skin diseases (NIAMS).
Abbreviations
- dSSc
diffuse systemic sclerosis
- COMP
cartilage oligomeriz matrix protein
- GO
Gene Ontology
- ILD
interstitial lung disease
- lSSc
limited systemic sclerosis
- MRSS
modified Rodnan skin score
- PDGF
platelet-derived growth factor
- qRT-PCR
quantitative real-time PCR
- SAM
significance analysis of microarrays
- SSc
systemic sclerosis
- TGFβ
transforming growth factor-β
Footnotes
Data access
Microarray data have been deposited at GEO (http://www.ncbi.nlm.nih.gov/geo/; accession no. GSE12493). The complete data set and searchable versions of the figures are available at http://whitfieldlab.dartmouth.edu/TGFB/.
SUPPLEMENTARY MATERIAL
Supplementary material is linked to the online version of the paper at http://www.nature.com/jid
CONFLICT OF INTEREST
ML Whitfield has filed a patent application for gene expression biomarkers in scleroderma. This intellectual property has been licensed to a startup company, Celdara Medical, LLC, that is aiming to translate the discovery into clinical use. Dr Whitfield holds an interest in this company. The remaining authors state no conflict of interest.
References
- Akhmet-shina A, Dees C, Pileckyte M, Maurer B, Axmann R, Jungel A, et al. Dual inhibition of c-abl and PDGF receptor signaling by dasatinib and nilotinib for the treatment of dermal fibrosis. FASEB J. 2008;22:2214–22. doi: 10.1096/fj.07-105627. [DOI] [PubMed] [Google Scholar]
- Barnett AJ, Miller M, Littlejohn GO. The diagnosis and classification of scleroderma (systemic sclerosis) Postgrad Med J. 1988a;64:121–5. doi: 10.1136/pgmj.64.748.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett AJ, Miller MH, Littlejohn GO. A survival study of patients with scleroderma diagnosed over 30 years (1953–1983): the value of a simple cutaneous classification in the early stages of the disease. J Rheumatol. 1988b;15:276–83. [PubMed] [Google Scholar]
- Baroni SS, Santillo M, Bevilacqua F, Luchetti M, Spadoni T, Mancini M, et al. Stimulatory autoantibodies to the PDGF receptor in systemic sclerosis. N Engl J Med. 2006;354:2667–76. doi: 10.1056/NEJMoa052955. [DOI] [PubMed] [Google Scholar]
- Chambers RC, Leoni P, Kaminski N, Laurent GJ, Heller RA. Global expression profiling of fibroblast responses to transforming growth factor-beta1 reveals the induction of inhibitor of differentiation-1 and provides evidence of smooth muscle cell phenotypic switching. Am J Pathol. 2003;162:533–46. doi: 10.1016/s0002-9440(10)63847-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HY, Chi JT, Dudoit S, Bondre C, van de RM, Botstein D, et al. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci USA. 2002;99:12877–82. doi: 10.1073/pnas.162488599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung L, Fiorentino DF, Benbarak MJ, Adler AS, Mariano MM, Paniagua RT, et al. Molecular framework for response to imatinib mesylate in systemic sclerosis. Arthritis Rheum. 2009;60:584–91. doi: 10.1002/art.24221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Classen JF, Henrohn D, Rorsman F, Lennartsson J, Lauwerys BR, Wikstrom G, et al. Lack of evidence of stimulatory autoantibodies to platelet-derived growth factor receptor in patients with systemic sclerosis. Arthritis Rheum. 2009;60:1137–44. doi: 10.1002/art.24381. [DOI] [PubMed] [Google Scholar]
- Cotton SA, Herrick AL, Jayson MI, Freemont AJ. TGF beta – a role in systemic sclerosis? J Pathol. 1998;184:4–6. doi: 10.1002/(SICI)1096-9896(199801)184:1<4::AID-PATH968>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Daniels CE, Wilkes MC, Edens M, Kottom TJ, Murphy SJ, Limper AH, et al. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J Clin Invest. 2004;114:1308–16. doi: 10.1172/JCI19603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton CP, Merkel PA, Furst DE, Khanna D, Emery P, Hsu VM, et al. Recombinant human anti-transforming growth factor beta1 antibody therapy in systemic sclerosis: a multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum. 2007;56:323–33. doi: 10.1002/art.22289. [DOI] [PubMed] [Google Scholar]
- Dharmapatni AA, Smith MD, Ahern MJ, Simpson A, Li C, Kumar S, et al. The TGF beta receptor endoglin in systemic sclerosis. Asian Pac J Allergy Immunol. 2001;19:275–82. [PubMed] [Google Scholar]
- Distler JH, Jungel A, Huber LC, Schulze-Horsel U, Zwerina J, Gay RE, et al. Imatinib mesylate reduces production of extracellular matrix and prevents development of experimental dermal fibrosis. Arthritis Rheum. 2007;56:311–22. doi: 10.1002/art.22314. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–8. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina G, Lemaire R, Korn JH, Widom RL. Cartilage oligomeric matrix protein is overexpressed by scleroderma dermal fibroblasts. Matrix Biol. 2006;25:213–22. doi: 10.1016/j.matbio.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Ferri C, Valentini G, Cozzi F, Sebastiani M, Michelassi C, La Montagna G, et al. Systemic sclerosis: demographic, clinical, and serologic features and survival in 1,012 Italian patients. Medicine (Baltimore) 2002;81:139–53. doi: 10.1097/00005792-200203000-00004. [DOI] [PubMed] [Google Scholar]
- Fujimoto M, Hasegawa M, Hamaguchi Y, Komura K, Matsushita T, Yanaba K, et al. A clue for telangiectasis in systemic sclerosis: elevated serum soluble endoglin levels in patients with the limited cutaneous form of the disease. Dermatology. 2006;213:88–92. doi: 10.1159/000093846. [DOI] [PubMed] [Google Scholar]
- Gabrielli A, Di Loreto C, Taborro R, Candela M, Sambo P, Nitti C, et al. Immunohistochemical localization of intracellular and extracellular associated TGF beta in the skin of patients with systemic sclerosis (scleroderma) and primary Raynaud’s phenomenon. Clin Immunol Immunopathol. 1993;68:340–9. doi: 10.1006/clin.1993.1136. [DOI] [PubMed] [Google Scholar]
- Gardner H, Shearstone JR, Bandaru R, Crowell T, Lynes M, Trojanowska M, et al. Gene profiling of scleroderma skin reveals robust signatures of disease that are imperfectly reflected in the transcript profiles of explanted fibroblasts. Arthritis Rheum. 2006;54:1961–73. doi: 10.1002/art.21894. [DOI] [PubMed] [Google Scholar]
- Goh NS, Desai SR, Veeraraghavan S, Hansell DM, Copley SJ, Maher TM, et al. Interstitial lung disease in systemic sclerosis: a simple staging system. Am J Respir Crit Care Med. 2008;177:1248–54. doi: 10.1164/rccm.200706-877OC. [DOI] [PubMed] [Google Scholar]
- Gu J, Iyer VR. PI3K signaling and miRNA expression during the response of quiescent human fibroblasts to distinct proliferative stimuli. Genome Biol. 2006;7:R42. doi: 10.1186/gb-2006-7-5-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihn H. Autocrine TGF-beta signaling in the pathogenesis of systemic sclerosis. J Dermatol Sci. 2008;49:103–13. doi: 10.1016/j.jdermsci.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Jinnin M, Ihn H, Mimura Y, Asano Y, Tamaki K. Potential regulatory elements of the constitutive upregulated alpha2(I) collagen gene in scleroderma dermal fibroblasts. Biochem Biophys Res Commun. 2006;343:904–9. doi: 10.1016/j.bbrc.2006.03.037. [DOI] [PubMed] [Google Scholar]
- Kubo M, Ihn H, Yamane K, Tamaki K. Upregulated expression of transforming growth factor-beta receptors in dermal fibroblasts of skin sections from patients with systemic sclerosis. J Rheumatol. 2002;29:2558–64. [PubMed] [Google Scholar]
- Kuroda K, Shinkai H. Gene expression of types I and III collagen, decorin, matrix metalloproteinases and tissue inhibitors of metalloproteinases in skin fibroblasts from patients with systemic sclerosis. Arch Dermatol Res. 1997;289:567–72. doi: 10.1007/s004030050241. [DOI] [PubMed] [Google Scholar]
- Leask A. Transcriptional profiling of the scleroderma fibroblast reveals a potential role for connective tissue growth factor (CTGF) in pathological fibrosis. Keio J Med. 2004;53:74–7. doi: 10.2302/kjm.53.74. [DOI] [PubMed] [Google Scholar]
- Leask A. Scar wars: is TGFbeta the phantom menace in scleroderma? Arthritis Res Ther. 2006;8:213. doi: 10.1186/ar1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816–27. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- Leask A, Abraham DJ, Finlay DR, Holmes A, Pennington D, Shi-Wen X, et al. Dysregulation of transforming growth factor beta signaling in scleroderma: overexpression of endoglin in cutaneous scleroderma fibroblasts. Arthritis Rheum. 2002;46:1857–65. doi: 10.1002/art.10333. [DOI] [PubMed] [Google Scholar]
- LeRoy EC, Black C, Fleischmajer R, Jablonska S, Krieg T, Medsger TA, Jr, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988;15:202–5. [PubMed] [Google Scholar]
- Leroy EC, Smith EA, Kahaleh MB, Trojanowska M, Silver RM. A strategy for determining the pathogenesis of systemic sclerosis. Is transforming growth factor beta the answer? Arthritis Rheum. 1989;32:817–25. [PubMed] [Google Scholar]
- Liu Y, Hayes DN, Nobel A, Marron JS. Statistical significance of clustering for high-dimension, low-sample size data. J Am Stat Assoc. 2008;103:1281–93. [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(–delta delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Maricq HR, Valter I. A working classification of scleroderma spectrum disorders: a proposal and the results of testing on a sample of patients. Clin Exp Rheumatol. 2004;22:S5–13. [PubMed] [Google Scholar]
- Milano A, Pendergrass SA, Sargent JL, George LK, McCalmont TH, Connolly MK, et al. Molecular subsets in the gene expression signatures of scleroderma skin. PLoS ONE. 2008;3:e2696. doi: 10.1371/journal.pone.0002696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mummery CL. Transforming growth factor beta and mouse development. Microsc Res Tech. 2001;52:374–86. doi: 10.1002/1097-0029(20010215)52:4<374::AID-JEMT1022>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Ostojic P, Cerinic MM, Silver R, Highland K, Damjanov N. Interstitial lung disease in systemic sclerosis. Lung. 2007;185:211–20. doi: 10.1007/s00408-007-9012-3. [DOI] [PubMed] [Google Scholar]
- Padgett RW, Savage C, Das P. Genetic and biochemical analysis of TGF beta signal transduction. Cytokine Growth Factor Rev. 1997;8:1–9. doi: 10.1016/s1359-6101(96)00050-0. [DOI] [PubMed] [Google Scholar]
- Renzoni EA, Abraham DJ, Howat S, Shi-Wen X, Sestini P, Bou-Gharios G, et al. Gene expression profiling reveals novel TGFbeta targets in adult lung fibroblasts. Respir Res. 2004;5:24. doi: 10.1186/1465-9921-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Bondre C, Gladstone HB, Brown PO, Chang HY. Anatomic demarcation by positional variation in fibroblast gene expression programs. PLoS Genet. 2006;2:e119. doi: 10.1371/journal.pgen.0020119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scussel-Lonzetti L, Joyal F, Raynauld JP, Roussin A, Rich E, Goulet JR, et al. Predicting mortality in systemic sclerosis: analysis of a cohort of 309 French Canadian patients with emphasis on features at diagnosis as predictive factors for survival. Medicine (Baltimore) 2002;81:154–67. doi: 10.1097/00005792-200203000-00005. [DOI] [PubMed] [Google Scholar]
- Segal E, Shapira M, Regev A, Pe’er D, Botstein D, Koller D, et al. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nat Genet. 2003;34:166–76. doi: 10.1038/ng1165. [DOI] [PubMed] [Google Scholar]
- Sfikakis PP, McCune BK, Tsokos M, Aroni K, Vayiopoulos G, Tsokos GC. Immunohistological demonstration of transforming growth factor-beta isoforms in the skin of patients with systemic sclerosis. Clin Immunol Immunopathol. 1993;69:199–204. doi: 10.1006/clin.1993.1170. [DOI] [PubMed] [Google Scholar]
- Shi-Wen X, Renzoni EA, Kennedy L, Howat S, Chen Y, Pearson JD, et al. Endogenous endothelin-1 signaling contributes to type I collagen and CCN2 overexpression in fibrotic fibroblasts. Matrix Biol. 2007;26:625–32. doi: 10.1016/j.matbio.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Smith EA, LeRoy EC. A possible role for transforming growth factor-beta in systemic sclerosis. J Invest Dermatol. 1990;95:125S–7S. doi: 10.1111/1523-1747.ep12874998. [DOI] [PubMed] [Google Scholar]
- Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis. 2007;66:940–4. doi: 10.1136/ard.2006.066068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga J. Antifibrotic therapy in scleroderma: extracellular or intracellular targeting of activated fibroblasts? Curr Rheumatol Rep. 2004;6:164–70. doi: 10.1007/s11926-004-0062-8. [DOI] [PubMed] [Google Scholar]
- Varga J, Whitfield ML. Transforming growth factor-beta in systemic sclerosis (scleroderma) Front Biosci. 2009;1:226–35. doi: 10.2741/s22. [DOI] [PubMed] [Google Scholar]
- Verrecchia F, Mauviel A, Farge D. Transforming growth factor-beta signaling through the Smad proteins: role in systemic sclerosis. Autoimmun Rev. 2006;5:563–9. doi: 10.1016/j.autrev.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Wall NA, Hogan BL. TGF-beta related genes in development. Curr Opin Genet Dev. 1994;4:517–22. doi: 10.1016/0959-437x(94)90066-c. [DOI] [PubMed] [Google Scholar]
- Whitfield ML, Finlay DR, Murray JI, Troyanskaya OG, Chi JT, Pergamenschikov A, et al. Systemic and cell type-specific gene expression patterns in scleroderma skin. Proc Natl Acad Sci USA. 2003;100:12319–24. doi: 10.1073/pnas.1635114100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield ML, George LK, Grant GD, Perou CM. Common markers of proliferation. Nat Rev Cancer. 2006;6:99–106. doi: 10.1038/nrc1802. [DOI] [PubMed] [Google Scholar]
- Whitfield ML, Sherlock G, Saldanha AJ, Murray JI, Ball CA, Alexander KE, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell. 2002;13:1977–2000. doi: 10.1091/mbc.02-02-0030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wipff J, Kahan A, Hachulla E, Sibilia J, Cabane J, Meyer O, et al. Association between an endoglin gene polymorphism and systemic sclerosis-related pulmonary arterial hypertension. Rheumatology (Oxford) 2007;46:622–5. doi: 10.1093/rheumatology/kel378. [DOI] [PubMed] [Google Scholar]
- Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu WD, Leroy EC, Smith EA. Fibronectin release by systemic sclerosis and normal dermal fibroblasts in response to TGF-beta. J Rheumatol. 1991;18:241–6. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.