

Abstract
Mycobacterium tuberculosis (Mtb), the etiological agent of tuberculosis, is the leading cause of bacterial infectious disease mortality. Biotin protein ligase (BirA) globally regulates lipid metabolism in Mtb through the posttranslational biotinylation of acyl coenzyme A carboxylases (ACCs) involved in lipid biosynthesis and is essential for Mtb survival. We previously developed a rationally designed bisubstrate inhibitor of BirA that displays potent enzyme inhibition and whole-cell activity against multidrug resistant and extensively drug resistant Mtb strains. Here we present the design, synthesis, and evaluation of a focused series of inhibitors, which are resistant to cyclonucleoside formation, a key decomposition pathway of our initial analogue. Improved chemical stability is realized through replacement of the adenosyl N-3 nitrogen and C-5′ oxygen atom with carbon as well as incorporation of a bulky group on the nucleobase to prevent the required syn-conformation necessary for proper alignment of N-3 with C-5′.
Keywords: Adenylate-forming, adenylation, antibiotic, biotin protein ligase, isothermal titration calorimetry, tuberculosis
Tuberculosis (TB), caused by members of the Mycobacterium tuberculosis (Mtb) complex, remains the leading source of bacterial infectious disease mortality.1,2 Current treatment for susceptible Mtb strains requires at least 6 months of drug therapy using a combination of four drugs (i.e., isoniazid, rifampin, ethambutol, and pyrazinamide), which were discovered over 40 years ago.3 The development and dissemenation of multidrug resistant (MDR) and extensively drug resistant (XDR) Mtb strains significantly limits our current treatment options. Therefore, there is a pressing need for new TB drugs, ideally with novel mechanisms of action.4,5
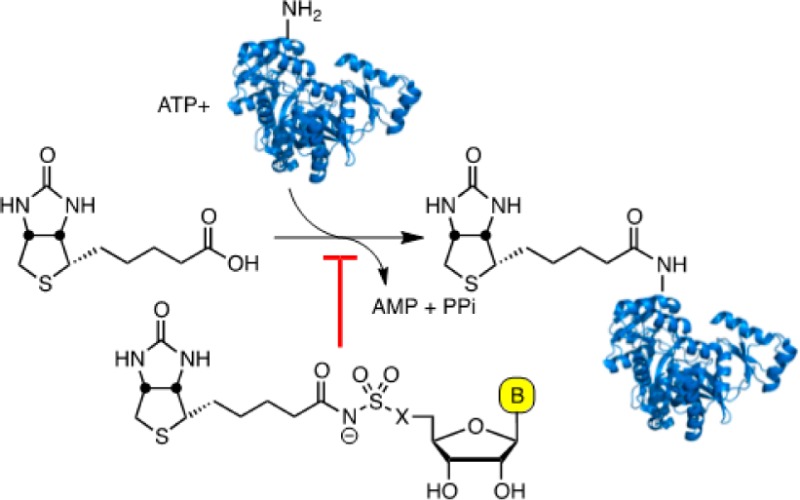
In Mtb, biotin protein ligase encoded by the gene birA globally regulates lipid metabolism through the posttranslational biotinylation of acyl coenzyme A carboxylases (ACCs), which catalyze the first committed step in lipid biosynthesis, and pyruvate coenzyme A carboxylase (PCC) an enzyme in the gluconeogenesis pathway that is necessary for growth on fatty acids.6 Consequently, BirA represents an extremely attractive target for the development of new antitubercular agents. BirA is responsible for the covalent ligation of biotin onto biotin-dependent enzymes and accomplishes this through two partial reactions (Figure 1A) .7 In the first half-reaction, BirA catalyzes the condensation of biotin 1 and ATP to produce biotinyl-5′-AMP (Bio-AMP, 2) and pyrophosphate. In the second half-reaction, BirA binds a biotin-dependent protein 3 and then transfers the biotin group of 2 onto the ϵ-amino group of a conserved lysine residue in 3 to afford the biotinylated protein 4.
Figure 1.

(A) BirA catalyzed biotinylation of ACC, a biotin dependent enzyme. (B) The prototypical bisubstrate inhibitor 5′-O-[N-(biotinyl)sulfamoyl]adenosine is in equibibrium between the anticonformation (5) around the glycosidic linkage and the syn-conformation (6), which can react intramolecularly to liberate N-biotinylsulfamic acid 7 and cycloadenosine 8.
We reported a rationally designed inhibitor of BirA, termed Bio-AMS (9, Figure 2), with potent enzyme inhibition and antitubercular activity against MDR and XDR Mtb strains.6 Polyak, Abell, Wilce, and co-workers have also disclosed some novel 1,2,3-triazole-biotin nucleosides and biotin analogues that are selective inhibitors of the biotin protein ligase from Staphylococcus aureus.8−10 Bio-AMS 9 is a bisubstrate inhibitor, which mimics the intermediate 2 by replacing the labile acyl-phosphate linkage with an acyl-sulfamide moiety. The related analogue 5(11,12) with an acyl-sulfamate linkage, another typical acyl-phosphate bioisostere, decomposes through cyclonucleoside formation to afford N-biotinylsulfamic acid 7 and 3,5′-cyclo-5′-deoxyadenosine 8 (Figure 1B).6 The greater stability of 9 is due to the decreased nucleofugality of the sulfamide. Herein, we report the design, synthesis, and evaluation of a systematic series of Bio-AMS analogues that are completely resistant to cyclonucleoside formation (Figure 2). In analogue 10, substitution of the 5′-oxygen atom of 5 with a methylene group converts the labile acyl-sulfamate moiety to a chemically stable acyl-sulfonamide. In analogue 11, the N-3 nitrogen atom of adenine is replaced by a methine group thereby eliminating the internal nucleophile. We also conceived analogues 12 and 13 that prevent cyclonucleoside formation through the incorporation of bulky substituents on the adenine ring that hinder the required syn-conformation around the glycosidic linkage, necessary for proper alignment of N-3 with C-5′.
Figure 2.
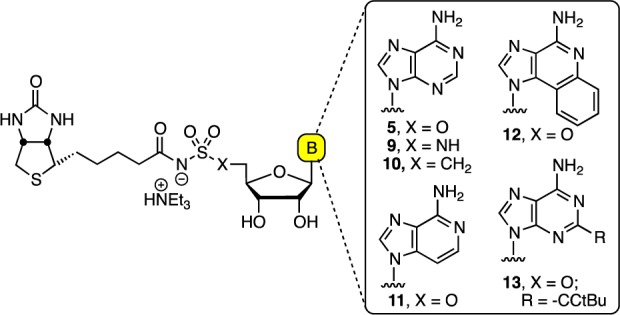
Structure of 5, Bio-AMS 9, and analogues 10–13.
Synthesis of the noncleavable sulfonamide 10 was accomplished by Dess–Martin oxidation of protected adenosine 14(13) to aldehyde 15 that is in equilibrium with hemiaminal 16 (Scheme 1). The N6-bis-Boc groups in 15 attenuate the nucleophilicity at N-3 suppressing hemiaminal formation and decomposition via depurination. After an aqueous workup the unstable crude aldehyde 15 was subjected to a modified Horner–Wittig olefination employing the dilithium salt of N-Boc-diphenylphosphorylmethanesulfonamide to afford exclusively the (E)-vinylsulfonamide 17.14,15 Catalytic hydrogenation of the alkene followed by chemoselective deprotection of the three Boc groups by treatment with trifluoroacetic acid (TFA) in dichloromethane furnished sulfonamide 18. Biotinylation of 18 with (+)-biotin N-hydroxysuccinimide ester (biotin-NHS) mediated by Cs2CO3 and subsequent acetonide deprotection with 50% aqueous TFA afforded 10 (Scheme 1). Sulfonamide 10 exhibits excellent chemical stability with no degradation upon long-term storage as determined by HPLC analysis.
Scheme 1. Synthesis of 10.

Reagents and conditions: (a) Dess–Martin periodinane, CH2Cl2, 25 °C; (b) Ph2P(O)CH2SO2NHBoc, LiHMDS, THF–DMF, −78 → 25 °C, 45% over 2 steps; (c) Pd/C, H2, MeOH; (f) TFA, CH2Cl2, 0 °C, 87% over 2 steps; (e) biotin–NHS, Cs2CO3, DMF, 0 → 25 °C; (f) 50% aqueous TFA, 0 °C, 60% over 2 steps.
The 3-deaza analogue 11 was prepared from 5-amino-imidazole-5-carbonitrile riboside 19(16,17) by an adaptation of the route described by Minnakawa and co-workers for the synthesis of 3-deazaadenosine (Scheme 2).18−20 Sequential acetonide and TBS protection of 19 followed by Sandmeyer iodination provided 20. The copper-free Sonogashira coupling of 20 with TMS-acetylene employing catalytic bis(benzonitrile)palladium chloride gave alkyne 21. Treatment of 21 with 7 N methanolic ammonia at 110 °C in a sealed tube induced a formal aza-Bergmann cyclization to afford 3-deazaadenosine 22. Removal of the TBS group from 22 was optimally accomplished under anhydrous conditions with KOtBu in DMF.21 Sulfamoylation22 with NH2SO2Cl and NaH in DMF furnished 24, which was biotinylated and deprotected using our optimized conditions to afford 11.
Scheme 2. Synthesis of 11.

Reagents and conditions: (a) 2,2-dimethoxypropane, PTSA, acetone, 25 °C; (b) TBSCl, imidazole, DMF, 25 °C; (c) CH2I2, isoamylnitrite, CHCl3, reflux, 55% over 3 steps; (d) Pd(PhCN)2Cl2, NEt3, HC≡CTMS, CH3CN, sealed tube, 100 °C, 70%; (e) 7N NH3/MeOH, 110 °C sealed tube, 77%; (f) KOtBu, DMF, 25 °C, 78%; (g) NH2SO2Cl, NaH, DME, 25 °C, 80%; (h) biotin–NHS, Cs2CO3, DMF, 0 → 25 °C; (i) 50% aqueous TFA, 0 °C, 65% over 2 steps.
The benzannulated analogue 12 was prepared from 5-iodoimidazole-4-carbonitrile riboside 20 in 5 steps as shown in Scheme 3. Initial Suzuki coupling of 20 with 2-aminophenylboronic acid furnished heterobiaryl 25 as an approximately 1:2 mixture of atropisomers.23 Cyclization, likely via an intermediate imidate,24 and attendant desilylation to 26 was effected by refluxing 25 with NaOMe–MeOH. Sulfamoylation was performed under base-free conditions25 with NH2SO2Cl in dimethylacetamide (DMA) to provide 27 in excellent yield. Biotinylation of 27 and subsequent TFA mediated deprotection of the acetonide gave 12.
Scheme 3. Synthesis of 12.

Reagents and conditions: (a) Pd(OAc)2, PPh3, 2-aminophenylboronic acid hydrochloride, Na2CO3, DME–H2O, 90 °C, 75%; (b) NaOMe, MeOH, reflux, 92%; (c) NH2SO2Cl, DMA, 88%. (d) biotin–NHS, Cs2CO3, DMF, 0 → 25 °C; (e) 50% aqueous TFA, 0 °C, 62% over 2 steps.
The 2-alkynyl analogue 13 was synthesized from 2-iodoadenosine 28, prepared in 5 steps from guanosine (Scheme 4).26 Sonogashira coupling was then performed to introduce the 3,3-dimethylbut-1-ynyl group directly onto unprotected 28 as attempts using the acetonide protected nucleoside were sluggish and low yielding.26 The alkynylated intermediate 29 was converted to acetonide 30, sulfamoylated to 31, and biotinylated and deprotected using our optimized conditions to afford 13.
Scheme 4. Synthesis of 13.

Reagents and conditions: (a) Pd(PPh3)2Cl2, CuI, NEt3, 3,3-dimethyl-1-butyne, DMF, 88 °C, 77%; (b) 2,2-dimethoxypropane, PTSA, acetone, 25 °C, 85%; (c) NH2SO2Cl, Et3N, DMF, 0 → 25 °C, 80%. (d) biotin–NHS, Cs2CO3, DMF, 0 → 25 °C; (e) 50% aqueous TFA, 0 °C, 65% over 2 steps.
As a result of the tight-binding nature of Bio-AMS 9 and analogues 10–13 and our inability to discriminate their potency by a coupled kinetic assay,6 we turned to isothermal titration calorimetry (ITC) to evaluate the binding affinities of 9–13 to BirA (Table 1). ITC is an extremely useful technique to determine the dissociation constant (KD) of ligand–protein interactions and additionally provides the thermodynamic binding parameters.27 For ligands 9–12 that bound with subnanomolar affinity, we employed competitive displacement experiments with 250–500 μM biotin (KD for BirA–biotin is 0.94 μM)7 to determine ΔG, KD, and n (the number of binding sites per monomer, see Supporting Information for details).28 The binding enthalpy ΔH was obtained by stoichiometric titration of BirA in which the ratio of ligand/KD was greater than 1000, and the entropy term TΔS was then calculated from the Gibbs free energy equation. All compounds bind tightly with KD values ranging from ≤0.04 to 11 nM and binding affinity is solely driven by a large favorable enthalpy. 3-Deaza analogue 11 is the most potent analogue with a KD of 40 pM and an astonishing ΔH of −21.2 kcal/mol due to the large number of favorable electrostatic H-bond interactions between this bisubstrate inhibitor and both substrate binding pockets within the BirA active site.6 The high potency demonstrates deletion of the polar N-3 atom is well tolerated. The increase in the enthalpy term relative to Bio-AMS 9 is likely due to a lower desolvation penalty. Replacement of the 5′-nitrogen atom of 9 with a CH2 moiety in sulfonamide 10 results in a slight attenuation in potency providing a KD of 0.10 nM. Benzannulated analogue 12 is remarkably potent with a KD of 0.23 nM with the loss of enthalpy (relative to 9) being compensating by a more favorable entropy term, presumably due to a greater entropy of desolvation. Alkyne analogue 13 is the least potent of the four compounds studied and displays a KD of only 11 nM with an enthalpy term of −13.5 kcal/mol. This result is readily reconciled based on the cocrystal structure of 9.12 However, the findings that both analogues 12 and 13 retain such good binding affinities indicate that the enzyme is sufficiently flexible to accommodate these bulky substituents as has been observed previously for MbtA, a functionally related adenylating enzyme in M. tuberculosis.29,30
Table 1. Binding Affinities to BirA and Antitubercular Activity of Analogues 9–13a,b.
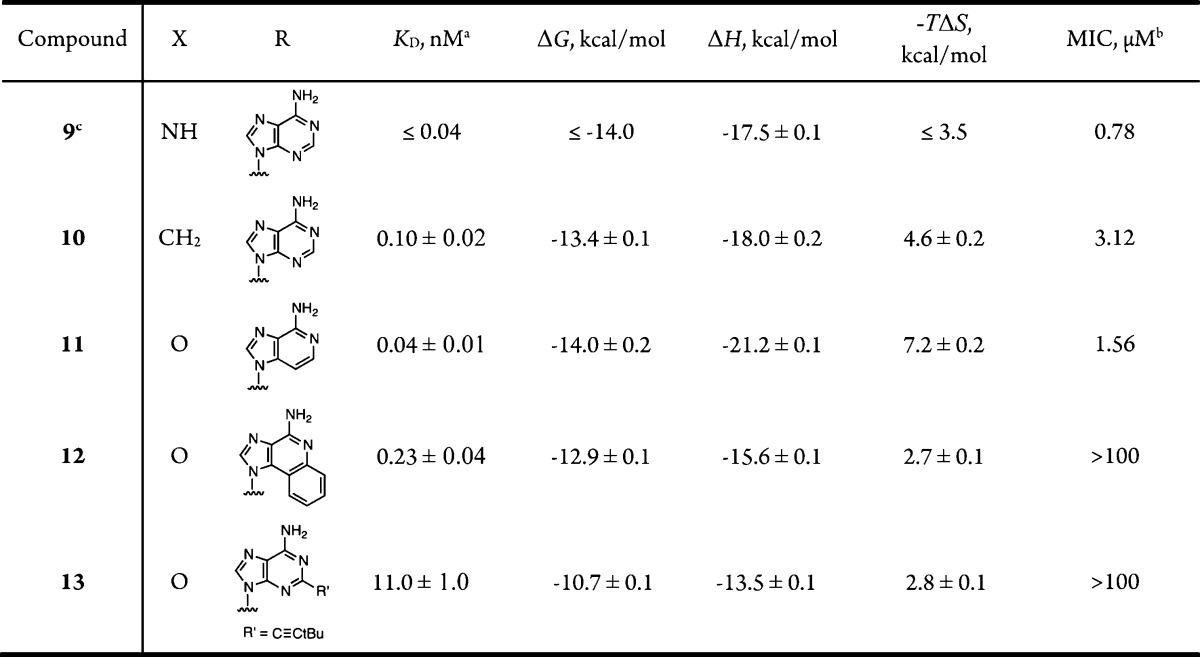
Competitive ITC experiments to determine KD and ΔG were performed, and direct titration experiments were done in duplicate. All analogues showed n values of 1 ± 0.03.
Minimum inhibitory concentrations (MIC) that resulted in >90% growth inhibition of M. tuberculosis H37Rv were determined by a broth microdilution assay in GAS medium. Experiments were performed twice independently in triplicate. The MIC is defined as the lowest concentration of inhibitors that prevented growth, as determined by measuring the end point OD600 values (see Supporting Information for details).
The earlier reported6KD of this compound is incorrect since competitive ITC was not performed, and thus, the potency was vastly underestimated. The previous ΔH value is also incorrect as the active enzyme concentration was not determined. In all experiments described above, the active enzyme concentration was determined by active site titration as described in the Supporting Information.
The antitubercular activity of 9–13 was next evaluated against Mycobacterium tuberculosis H37Rv (Table 1). The minimum concentration that inhibited greater than 90% of cell growth (MIC) was determined in defined glycerol-alanine salts (GAS) medium lacking biotin. Consistent with the ITC results, 3-deaza 11 possesses the most potent antitubercular activity with an MIC of 1.56 μM followed by sulfonamide 10 with a MIC of 3.12 μM. Analogues 12 and 13 that contain significant modifications to the nucleobase are totally inactive (MIC >100 μM) despite their excellent to modest biochemical potency. We hypothesize that one of the ATP-binding cassette (ABC) transporters present in Mtb, which are responsible for the assimilation of nucleosides and cofactors, facillitates the uptake of these polar nucleoside analogues.31 The observation that analogues 12 and 13 are devoid of whole-cell activity could be due to their inability to be recognized by these ABC transporters.
In conclusion, we have designed and synthesized a focused series of Bio-AMS analogues that are resistant to cyclonucleoside formation. All analogues 10–13 showed remarkable chemical stability and demonstrated high affinities to BirA as measured by isothermal titration calorimetry. Analogues 10 and 11 also displayed potent activity against M. tuberculosis H37Rv, comparable to that of Bio-AMS 9. The lack of antitubercular activity of 12 despite its potent enzyme affinity suggests modifications to the nucleobase may hinder cellular uptake and/or accumulation.
Acknowledgments
We thank Dr. Krzysztof W. Pankiewicz (Center for Drug Design, University of Minnesota) for providing 5-amino-imidazole-5-carbonitrile riboside.
Glossary
ABBREVIATIONS
- ACC
acyl coenzyme A carboxylase
- biotin-NHS
(+)-biotin N-hydroxysuccinimide ester
- GAS
glycerol-alanine salts medium
- ITC
isothermal titration calorimetry
- MIC
minimum inhibitory concentration
- MDR
multidrug resistant
- Mtb
Mycobacterium tuberculosis
- TB
tuberculosis
- XDR
extensively drug resistant
Supporting Information Available
All synthetic procedures, ITC experimental methods, and Mtb whole-cell assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Financial support was provided by the National Institutes of Health Grant AI091790 (to D.S.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- World Health Organization. Global tuberculosis report 2012. http://www.who.int/tb/publications/global_report/en/ (last accessed 02/22/13).
- Koul A.; Arnoult E.; Lounis N.; Guillemont J.; Andries K. The challenge of new drug discovery for tuberculosis. Nature 2011, 469, 483–490. [DOI] [PubMed] [Google Scholar]
- Mitchison D.; Davies G. The chemotherapy of tuberculosis: past, present and future. Int. J. Tuberc. Lung Dis. 2012, 16, 724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry C. E. III; Blanchard J. S. The chemical biology of new drugs in the development for tuberculosis. Curr. Opin. Chem. Biol. 2010, 14, 456–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich C. C.; Boshoff H. I.; Remmel R. P.. Antitubercular Agents. In Burgers Medicinal Chemistry, 7th ed.; Rotella D., Abraham D. J., Eds; Wiley: New York, 2010; pp 713–812. [Google Scholar]
- Duckworth B. P.; Geders T. W.; Tiwari D.; Boshoff H. I.; Sibbald P. A.; Barry C. E. III; Schnappinger D.; Finzel B. C.; Aldrich C. C. Bisubstrate adenylation inhibitors of biotin protein ligase from Mycobacterium tuberculosis. Chem. Biol. 2011, 18, 1432–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushothaman S.; Gupta G.; Srivastava R.; Ramu V. G.; Surolia A. Ligand specificity of group I biotin protein ligase of Mycobacterium tuberculosis . PLoS One 2008, 3, e2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares da Costa T. P.; Tieu W.; Yap M. Y.; Pendini N. R.; Polyak S. W.; Pedersen D. S.; Morona R.; Turnidge J. D.; Wallace J. C.; Wilce M. C. J.; Booker G. W.; Abell A. D. Selective inhibition of biotin protein ligase from Staphylococcus aureus. J. Biol. Chem. 2012, 287, 17823–17832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares da Costa T. P.; Tieu W.; Yap M. Y.; Zvarec O.; Bell J. M.; Turnidge J. D.; Wallace J. C.; Booker G. W.; Wilce M. C. J.; Abell A. D.; Polyak S. W. Biotin analogues with antibacterial activity are potent inhibitors of biotin protein ligase. ACS Med. Chem. Lett. 2012, 3, 509–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieu W.; Soares da Costa T. P.; Yap M. Y.; Keeling K. L.; Wilce M. C.; Wallace J. C.; Booker G. W.; Polyak S. W.; Abell A. D. Optimising in situ click chemistry: the screening and identification of biotin protein ligase inhibitors. Chem. Sci. 2013, 4, 3533–3537. [Google Scholar]
- Brown P. H.; Beckett D. Use of binding enthalpy to drive an allosteric transition. Biochemistry 2005, 44, 3112–3121. [DOI] [PubMed] [Google Scholar]
- Brown P. H.; Cronan J. E.; Grøtli M.; Beckett D. The biotin repressor: modulation of allostery by corepressor analogs. J. Mol. Biol. 2004, 337, 857–869. [DOI] [PubMed] [Google Scholar]
- Ikeuchi H.; Meyer M. E.; Ding Y.; Hiratake J.; Richards N. G. J. A critical electrostatic interaction mediates inhibitor recognition by human asparagine synthetase. Bioorg. Med. Chem. 2009, 17, 6641–6650. [DOI] [PubMed] [Google Scholar]
- Reuter D. C.; McIntosh J. E.; Guinn A. C.; Madera A. M. Synthesis of vinylsulfonamides using the Horner reaction. Synthesis 2003, 2321–2324. [Google Scholar]
- Sundlov J. A.; Shi C.; Wilson D. J.; Aldrich C. C.; Gulick A. M. Structural and functional investigation of the intermolecular interaction between NRPS adenylation and carrier protein domains. Chem. Biol. 2012, 19, 188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw E. A convenient preparation of 5-amino-4-imidazolecarboxamide riboside. Ring opening of N1-p-toluenesulfonyl-inosine. J. Am. Chem. Sci. 1959, 81, 6021–6022. [Google Scholar]
- Minakawa N.; Takeda T.; Sasaki T.; Matsuda A.; Ueda T. Nucleosides and nucleotides. 96. Synthesis and antitumor activity of 5-ethynyl-1-.beta.-d-rebofuranosylimida-zole-4-carboxamide (EICAR) and its derivatives. J. Med. Chem. 1991, 34, 778–786. [DOI] [PubMed] [Google Scholar]
- Minakawa N.; Matsuda A. Nucleosides and nucleotides. 116. Convenient synthesis of 3-deazaadenosine, 3-deaza-guanosine, and 3-deazainosine via ring closure of 5-ethynyl-1-β-d-ribofuranosylimidazole-4-carboxamide or -carboni-trile. Tetrahedron 1993, 49, 557–570. [Google Scholar]
- Minakawa N.; Matsuda A.; Ueda T. Nucleosides and nucleotides. 114. A convenient method for the synthesis of 3-deazapurine nucleosides from AICA-riboside.. Tetrahedron Lett. 1993, 34, 661–664. [Google Scholar]
- Minakawa N.; Sasabuchi Y.; Kiyosue A.; Kojima N.; Matsuda A. Nucleosides and nucleotides. 143. Synthesis of 5-amino-4-imidazolecarboxamide (AICA) deoxyribosides from deoxyinosines and their conversion into 3-deazapurine derivatives. Chem. Pharm. Bull. 1996, 44, 288–295. [Google Scholar]
- Negrón G.; Vásquezy F.; Calderón G.; Cruz R.; Gaviño R.; Islas G. Efficient desilylation of some nucleoside derivatives with potassium tert-butoxide in dimethylformamide. Synth. Commun. 1998, 28, 3021–3027. [Google Scholar]
- Shuman D. A.; Robins M. J.; Robins R. K. The synthesis of nucleoside sulfamates related to nucleocidin. J. Am. Chem. Soc. 1970, 92, 3434–3440. [DOI] [PubMed] [Google Scholar]
- Shi C.; Xiong Z.; Chittepu P.; Aldrich C. C.; Ohlfest J. R.; Ferguson D. M. Discovery of imidazoquinolines with toll-like receptor 7/8 independent cytokine induction. ACS Med. Chem. Lett. 2012, 3, 501–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer F. C.; Peters G. A. Base-catalyzed reaction of nitriles with alcohols. A convenient route to imidates and amidine salts. J. Org. Chem. 1960, 26, 412–418. [Google Scholar]
- Okada M.; Iwashita S.; Koizumi N. A general method for sulfamoylation of a hydroxyl group. Tetrahedron Lett. 2000, 41, 7047–7051. [Google Scholar]
- Matsuda A.; Shinozaki M.; Yamaguchi T.; Homma H.; Nomoto R.; Miyasaka T.; Watanabe Y.; Abiru T. Nucleosides and nucleotides. 103. 2-Alkynyladenosines: a novel class of selective adenosine A2 receptor agonists with potent antihypertensive effects. J. Med. Chem. 1992, 35, 241–252. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y.; Freire E. Finding a better path to drug selectivity. Drug Discovery Today 2011, 16, 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazquez-Campoy A.; Freire E. Isothermal titration calorimetry to determine association constants for high-affinity ligands. Nat. Protoc. 2006, 1, 186–191. [DOI] [PubMed] [Google Scholar]
- Neres J.; Labello N. P.; Somu R. V.; Boshoff H. I.; Wilson D. J.; Vannada J.; Chen L.; Barry C. E. III; Bennett E. M.; Aldrich C. C. Inhibition of siderophore biosynthesis in Mycobacterium tuberculosis with nucleoside bisubstrate analogues: structure–activity relationships of the nucleobase domain of 5′-O-[N-(salicyl)sulfamoyl]adenosine. J. Med. Chem. 2008, 51, 5349–5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupte A.; Boshoff H. I.; Wilson D. J.; Neres J.; Labello N. P.; Somu R. V.; Xing C.; Barry C. E. III; Aldrich C. C. Inhibition of siderophore biosynthesis by 2-triazole substituted analogues of 5′-O-[N-(salicyl)sulfamoyl]adenosine: antibacterial nucleosides effective against Mycobacterium tuberculosis. J. Med. Chem. 2008, 51, 7495–7507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braibant M.; Gilot P.; Content J. The ATP binding cassette (ABC) transport systems of Mycobacterium tuberculosis. FEMS Microbiol. Rev. 2000, 24, 449–467. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.