

Abstract
The phospholipid phosphatidylserine (PS) is an early marker exploited for detecting apoptosis (PS externalization in the cell membrane bilayer) and one factor that is associated with increased amyloid plaque deposition in transmissible spongiform encephalopathies (TSEs). PS can therefore be considered as a promising target for diagnosis or treatment of diseases. Aptamers (short nucleic acid sequences) are a particularly attractive class of materials among those currently considered for targeting PS. Here we applied an entropy based seed-and-grow strategy to design a DNA aptamer template to bind specifically to PS. The binding properties of designed aptamers were investigated computationally and experimentally. The studies identify the sequence, 5′-AAAGAC-3′, as the preferred template for further modifications and studies toward its practical implementations.
Introduction
Phospholipids play vital roles in cellular structure and function. In eukaryotic cell membranes, the major structural phospholipid components are the glycerophospholipids phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), phosphatidylinositol (PI) and phosphatidic acid (PA) (van Meer et al., 2008). PC comprises more than 50% of the phospholipids and functions as a neutral type lipid, which is responsible for creating membrane planar bilayers. While PS exists in relatively low concentrations (∼10%) in the cell membrane, it plays crucial roles in various cellular processes. Particularly, two processes, apoptosis and amyloid aggregation, have attracted our interest. The redistribution of PS from inner to outer plasma membranes is referred to as PS externalization and has been shown to be an early marker of apoptosis (Kerr et al., 1972; Martin et al., 1995; ELMORE, 2007; WEINBERG, 2007; Smrz et al., 2008; BLANKENBERG, 2009; Rastogi et al., 2009). Annexin V (annexin A5), a naturally occurring human phosphatidylserine-binding protein, has been most extensively investigated as either a radionuclide-containing or fluorescent probe to detect PS externalization (Martin et al., 1995). However, the annexins have exhibited a variety of disadvantages including high uptake in normal tissues, long half-life in non-target tissues, high radiation burden with radiolabeled tracers, and laborious labeling (Boersma et al., 2005). In addition, the presence of PS is found to alter aggregation pathways and increases amyloid aggregation in TSEs (Robinson and Pinheiro, 2010). The amyloid aggregation is found to trigger apoptosis and leads to neuronal cell death (Chiesa et al., 2000; Thellung et al., 2011). Antibodies and aptamers have been developed as either diagnostic agents or therapeutic inhibitors targeting a pathogenic factor, prion protein (Proske et al., 2002; Rhie et al., 2003; Campana et al., 2009; Quadrio et al., 2011). Yet the effects of the most promising immunotherapy remain controversial (Heppner and Aguzzi, 2004). Therefore, both cases could benefit from finding alternative constructs for either diagnosis or treatments targeting PS.
Aptamers possess many desirable properties; being characterized by strong and specific binding to cellular targets, ease of synthesis, and attractive pharmacokinetics (Nimjee et al., 2005; Keefe et al., 2010). However, the development of aptamers as therapeutic agents has been disappointingly slow to reach the marketplace and there is only one aptamer, Macugen, which has been approved as a commercially available therapeutic agent. Although binding aptamers can be identified by the technique of systematic evolution of ligands by exponential enrichment (SELEX) (Turek and Gold, 1990; James, 2000; Hamula et al., 2006; JAMES, 2007; Lao et al., 2009), this technology is hampered by difficulties in the design of randomized DNA/RNA libraries and the complexity of the selection process (Keefe et al., 2010). Nevertheless, the desirable properties of aptamers remain a compelling motivation to pursue their implementation as both therapeutic and diagnostic tools.
In this work we describe the use of an in silico approach, entropic fragment-based approach (EFBA) (Tseng et al., 2011), to design aptamer templates that selectively bind to PS. To our knowledge, this is the first exploration of in silico techniques directed at this important biological target. In addition, we have prepared the identified binding aptamers as fluorescent analogs and investigated their binding behavior with a validated laboratory assay using liposome technology. These studies have identified a preferred template with the potential for further modifications to improve binding and selectivity. Practical implementation of the aptamer as an imaging tool or a therapeutic agent is discussed.
Materials and Methods
Rationale of aptamer-designed theory
The foundation of the EFBA lies in the answer to the question:
“Given the structural information of the target, what is the preferred probability distribution of having an aptamer that is most likely to interact with the target?” (Tseng et al., 2011). EFBA integrates the methods of information processing and seed-and-grow strategy to answer this question. Specifically, three methods regarding information processing are utilized: (1) a method of assigning probability distributions based on a limited amount of information denoted by MaxEnt (Jaynes, 1957a, 1957b); (2) a method of updating probability prior distributions when new information becomes available denoted by ME (Shore and Johnson, 1980; Shore and Johnson, 1981; Caticha, 2004; Caticha and Giffin, 2007); and (3) a selection criterion for different probability distributions associated with various types of nucleotides (TSENG, 2006; Chen et al., 2007; Tseng and Caticha, 2007).
There are three steps in EFBA. First, it determines the probability distribution of the preferred first nucleotide (seed) that interacts with the target based on information such as total or interaction energy of the nucleotide–target complex using MaxEnt. Second, given the preferred nucleotide from the previous step, it determines the probability distribution of preferred dimer using ME. By repeating this same procedure, one can obtain the joint probability distribution of an L-mer nucleotide sequence (PL-mer) that is most likely to interact with the target. Third, it applies the entropic criterion defined by the relative entropy, 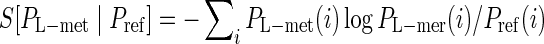 , to determine the preferred sequence and its length L based on the following argument. According to the works of TSENG, 2006 and Chen et al., 2007, the reference probability (Pref) is set to be a uniform distribution, which represents our complete ignorance regarding the interaction, and i labels conformations of the sequence in the previous two steps. Based on the information theory, this relative entropy, which is not a thermodynamic entropy, measures differences between probability distribution PL-mer and the uniform reference. When the relative entropy in the growth process is saturated at a minimum value, it indicates that the effect of the corresponding nucleotide on the interactions of the complex is at a global minimum; namely, the portion grown in the aptamer after the saturation step plays an insignificant role in the interactions, and the preferred length L is the number of nucleotides grown before the saturation step. In summary, EFBA generates preferred sequences by ruling out the ones that do not satisfy entropic criterion. In practice, the design theory is carried out by an algorithm prototype, which is described below.
, to determine the preferred sequence and its length L based on the following argument. According to the works of TSENG, 2006 and Chen et al., 2007, the reference probability (Pref) is set to be a uniform distribution, which represents our complete ignorance regarding the interaction, and i labels conformations of the sequence in the previous two steps. Based on the information theory, this relative entropy, which is not a thermodynamic entropy, measures differences between probability distribution PL-mer and the uniform reference. When the relative entropy in the growth process is saturated at a minimum value, it indicates that the effect of the corresponding nucleotide on the interactions of the complex is at a global minimum; namely, the portion grown in the aptamer after the saturation step plays an insignificant role in the interactions, and the preferred length L is the number of nucleotides grown before the saturation step. In summary, EFBA generates preferred sequences by ruling out the ones that do not satisfy entropic criterion. In practice, the design theory is carried out by an algorithm prototype, which is described below.
Design of PS binding DNA aptamer templates
The tertiary structures of a single PS and PC were obtained in Tseng et al. (Tseng et al., 2011). PC was used for selectivity studies of designed aptamers. The force field parameters to characterize PC and PS were generated using Antechamber (Wang et al., 2004; Wang et al., 2006). To remove atoms with close contact in the structure of PS, PC and the building blocks (four nucleotides), they were energy minimized using the steepest descent method in Amber 10 (Case et al., 2008) for the first 10 cycles and then followed by conjugate gradient for another 1,000 cycles at 300K before the design. The potential cutoff distance was set to 12 Å.
Note that there are no theories yet to indicate which type of information is likely to allow EFBA to generate the sequences preferred over all other combinations. Besides, it will require a universal criterion to define which random sequence is least favored and is suitable for negative control in affinity studies. Based on the nature of EFBA described above, we are uncertain that the sequences ruled out by EFBA to be least favored and qualified for negative control. However, according to information theory, we should expect to see the sequences bind strongly with PS when the criterion is mostly relevant to the binding. Namely, the preference of binding depends on the degree of relevance in information. Therefore, we considered two designs using two different common criteria, the total energy and the interaction energy of the aptamer-PS complex, respectively. That will result in two sets of sequences with different binding affinity quantified by relative entropy. In this way, we can expect one set of sequences to be preferred over the other by exploring more possible compositions and structures of sequences. Therefore, it provides a suitable control to identify the most preferred candidate(s).
In practice, the design algorithm first generated a spatial grid space, which covers possible locations for nucleotides in the vicinity of the head group of PS. Given this grid space, the probability distribution of the first nucleotide interacting with PS then was determined using MaxEnt based on the input information such as corresponding total energy, which were calculated using Amber 10 with consideration of latest force field ff03 (Case et al., 2008). Entropic selection was applied to determine which nucleotide highly likely interacts with the head group of PS. After the first nucleotide was determined, the same algorithm switched to utilize ME repeatedly to determine the updated joint probability distribution of the rest of nucleotides. Note that an orientation grid space, which consists of possible orientations of nucleotides to be connected to the previous one, was created. Entropic selection was then applied to determine the preferred nucleotide sequence and the length. The technical details are reported in Tseng et al. (Tseng et al., 2011).
In silico studies of aptamer-PS binding
To provide theoretical insights into the binding affinity and selectivity of designed aptamers at the molecular level, molecular dynamic (MD) simulations using Amber 11 with consideration of latest force field ff03 were utilized (Case et al., 2010). Following the concept of Monte Carlo method, we considered five different relative locations and orientations randomly generated in each aptamer-phospholipid complex as initial structures. Each location and orientation specific complex was energy minimized using the steepest descent method for the first ten cycles and then followed by a conjugate gradient for another 1,000 cycles. During the equilibration of the complex using the explicit water TIP3P model, we first applied Langevin dynamics during the process of heating up the system for 200 ps, with targets being restrained using a harmonic potential having a force constant k=100 N/m to homogeneously distribute water molecules in the system. We then introduced pressure regulation in the simulation to equilibrate solvent density for another 200 ps in addition to temperature regulation. Then, we performed a 10-nanosecond (ns) explicit water MD simulation at 300K and solution at pH 7 for each complex as a production run. Note that the targets were gently restrained with a harmonic potential with a force constant k=10 N/m applied to the phosphorus in PS or PC. To further elucidate the binding selectivity of the best candidate suggested from binding assays, 20-ns explicit water MD simulations were conducted. Three quantities, the separation distance of centers of mass of aptamers and phospholipid (dapt-lipid), electrostatic (ES) and van der Waals (vdW) energy, and solvent accessible area (SA) were utilized to analyze simulations.
In vitro studies of aptamer-liposome binding
Binding targets required to assay the designed aptamers were prepared using liposome technology for its simplicity (Hope et al., 1986). The liposomes were prepared with 1,2-dipalmitoyl-sn-glycero-3-phosphatidylcholine (DPPC), 1,2-dipalmitoyl-sn-glycero-3-phospho-L-serine (sodium salt) (DPPS), and cholesterol (all in powder form) obtained from Avanti Polar Lipids. They were used without further purification. DNA designed aptamer sequences containing the 6-carboxyfluorescein (6-FAM) fluorescent tag attached at the 3′ end of the DNA sequence were obtained from Integrated DNA Technologies. Ninety-six-well assay plates for fluorescence assays using FLUOstar OPTIMA (BMG LABTECH GmbH) were from Corning Inc. There were eight replicates of samples prepared for each aptamer concentration in the studies. A 2:1 ratio of PC to cholesterol was prepared by dissolving 100 mg of DPPC and 24.45 mg of cholesterol in 10 mL of chloroform to prepare control liposomes that do not contain PS. PS-containing liposomes contained a combination of DPPC and DPPS in a 10 to 1 molar ratio.
The protocol of binding assays
The detailed protocol published previously (Tseng et al., 2011) is summarized as follows. Ten milliliters of a chloroform solution of the phospholipids and cholesterol was evaporated to dryness by gently blowing nitrogen gas into the solution in a test tube. The dried lipid film created on the walls of the test tube was kept overnight to remove remnants of chloroform; 10 mL of HEPES buffer was then added to the dried lipid film and the mixture was vortexed at room temperature (∼25°C) to create lipid vesicles (primarily multilamellar) in buffer (e.g., see Hope et al., 1986). One milliliter of the liposome formulation in HEPES buffer was transferred to each Eppendorf tube, followed by addition of appropriate volume of the test fluorescent aptamer as a solution in Tris/EDTA buffer (1 mM stock or stock with further dilution) to ensure the desired experimental aptamer concentration in solution.
The aptamer/phospholipid mixture was incubated for 40 minutes in the dark. We then centrifuged the Eppendorf tubes to pellet lipid and the bound aptamers at 25°C using Hettich Rotina 35R (Het-tich America LLP) at 19,520 g for 1 minute. Supernatants (buffer and unbound aptamers) were then removed. HEPES buffer (1 mL) was added to the Eppendorf tubes to resuspend the phospholipid followed by centrifugation and the supernatants were again removed. This process was repeated three times to effectively remove unbound aptamers. The required amount of HEPES buffer was then added to each Eppendorf tube containing bound aptamer/phospholipids to make the final volume 1 mL. One hundred milliliters from this final suspension was loaded onto a 96-well plate, and the fluorescence intensity was measured for each sample (with excitation filter at 485 nm and emission filter at 520 nm). PC software version V1.30 R4 (BMG LABTECH GmbH) was used for data collection.
Results and Discussion
Two sets of DNA aptamer templates
Two designs were obtained with consideration of two criteria: total energy and interaction energy of the aptamer-PS complex. Since the lipid portion of many phospholipids is similar, we have used the head group of PS as shown in top left of Fig. 1 for the design. The top right panel shows its two-dimensional structure. According to our aptamer design theory as summarized above (Tseng et al., 2011), we calculated the relative entropy of the complex of PS and a nucleotide fragment at each step of the growth process to determine an appropriate length that allows the resulting nucleotide fragment to have the highest probability to interact with PS. The bottom left panel of Fig. 1 shows the relative entropy value at each step of the growth process calculated based on total energy of complexes. The preferred nucleotide type is listed above the symbol and the odd numbers beside it (except for the first step), indicate that the nucleotide is connected to the 3′ end of the previous nucleotide in the series and the even numbers indicate attachment at the 5′ end of the sequence. The curve shows that relative entropy first drops dramatically at the second step and climbs back. It then fluctuates around −400 after the fifth step. Although it seems that the relative entropy is minimum at the second step, there is insufficient information to confirm whether this two-aptamer (2-mer) nucleotide fragment will strongly interact with PS. However, according to the changes of relative entropy along the growing step, it suggests that the nucleotides added after either the fifth or sixth step do not further enhance the interactions between the nucleotide fragment and PS. This result indicates that the nucleotide fragment that is most likely to steadily interact with PS needs to have a minimum length around a 6-mer. Therefore, the preferred length is selected to be six nucleotides. The bottom right panel of Fig. 1 then shows that the relative entropy fluctuates around −16 after the 10th step when interaction energy was used for the design and this suggests that the preferred length is 11. The top four sequences result from using the total energy defined by SIApn, and the top two sequences result from using the interaction energy defined by SIIApn. They are listed in Table 1 for further studies.
FIG. 1.

The upper panels show the tertiary structure of the SIAp1 aptamer–phosphatidylserine (PS) complex generated using visual molecular dynamics (VMD) (Humphrey et al., 2006) and the two-dimensional structure of PS. Adenine is colored by blue and guanine is red. The lower panels plot the relative entropy at each step of the designing process using total (lower left) and interaction (lower right) energy as input information are plotted. The preferred nucleotide type is labeled within the boxes; the numbering beside each nucleotide denotes the connection end. The odd numbers, except for the first step, refer to the nucleotide attachment at the 3′ end of the apatmer and the even numbers are for attachment at the 5′ end.
Table 1.
The Two Sets of DNA Aptamer Templates
| Namea | Sequence (5′→3′) | PSb | PCc |
|---|---|---|---|
| SIAp1 |
AAAAGA |
0.23±0.11 |
0.06±0.02 |
| SIAp2 |
AAAGAG |
N/A |
N/A |
| SIAp3 |
TAAAGA |
0.57±0.15 |
0.31±0.15 |
| SIAp4 |
AAAGAC |
0.54±0.19 (0.46±0.13) |
0.41±0.08 (0.30±0.09) |
| SIIAp1 |
CAGAAAAAAAC |
0.4±0.2 |
0.41±0.14 |
| SIIAp2 | CAGAAAAAAAT | 0.31±0.12 | 0.29±0.19 |
SI is designed based on total energy, and SII is designed using interaction energy.
Probabilities of designed aptamers and PS within 6<dPS-lipid<16Å calculated from 10-ns simulations (top panel of Supplementary Fig. S1).
Probabilities of designed aptamers and PC within 6<dPS-lipid<16Å calculated from (lower panel of Supplementary Fig. S1). Note that the numbers inside round brackets in both PS and PC column were calculated from 20-ns simulation results (Supplementary Fig. S2). Here we picked only one sequence (the best PS binding one) as example for an extended time simulation.
dPS-lipid, PS–lipid separation distance; PC, phosphatidylcholine; PS, phosphatidylserine.
Binding affinity and specificity studies
We tested our negative control of 0% DPPS (100% DPPC) for all four SIApn and two SIIApn sequences and tested aptamer concentrations ranging from 0 to 35 μM (Fig. 2, upper right). In absence of DPPS (100% DPPC, control) the four SIApn sequences and two SIIApn sequences showed very low fluorescence levels (less than 10,000 RFU) across all the concentrations tested in the assay. These results indicate that the aptamers tested in this study have poor binding affinity for DPPC.
FIG. 2.

The upper part shows fluorescence (FL) measured in relative fluorescence units (RFU) versus aptamer concentration. Left and middle panels: selective binding of aptamer SIAp1, 3, and 4 and SIIAp1 and 2 with liposomes containing PS. Upper right panel: low non-specific binding of designed aptamer SIAp4 and SIIAp1 with liposome containing only phosphatidylcholine (PC). Note that all the data points are based on 8 independent measurements. Lower panel: FL versus ratio of 1,2-dipalmitoyl-sn-glycero-3-phospho-L-serine (sodium salt) (DPPS).
We tested all four SIApn and two SIIApn sequences to evaluate binding to 10% DPPS and aptamer sequences were evaluated over a range of concentrations (0 to 35 μM) (Fig. 2, upper left and upper middle). Our in vitro results indicate SIAp4 had the highest fluorescence level in the binding assay, followed by SIAp3 and SIAp2 respectively. SIAp 2, 3, and 4 have higher fluorescence levels than SIIApn when the concentrations of the aptamers are increased beyond 7 μM. This suggests that SIAp 2, 3, and 4 have a relatively stronger binding affinity than SIIApn, which is in agreement with the EFBA prediction. From our in vitro results it is evident that some of the aptamers also have poor binding affinity for DPPS. When evaluating affinity for PS in DPPS 10% setting, the aptamers SIAp1, SIIAp1, and SIIAp2 had low fluorescence levels (less than 20,000 RFU) even at the highest concentration of aptamer tested (35 μM) (Fig. 2).
We evaluated if the concentration of PS changes the binding affinity. We generated a series of PS/PC liposomes with increasing concentrations of DPPS and assessed the binding of aptamer SIAp4 at 20 μM (Fig. 2, lower panel). The results of this experiment clearly demonstrate that when PS concentration is at 10% or higher, fluorescence reaches a plateau and fluorescence does not increase as the concentration of DPPS is increased. It indicates a saturation of PS-SIAp4 binding.
Despite the addition of the FAM tag, the in vitro results indicate aptamer specific binding activity. This suggests that the FAM tag has a relatively insignificant effect on binding to PS. We have shown this previously with peptide sequences binding to PS. We have tested fluorescently labelled peptides binding to PS and when the same peptide sequences were instead labeled with a radioactive probe the peptides of interest retained affinity for PS (Kapty et al., 2012 and unpublished data). Therefore, this suggests the discrepancies we observed between in silico prediction and in vitro results were due to other factors.
As addressed in computational methods section, the relative entropy 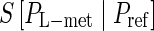 gives an approximate affinity ranking scheme of L-mer sequences. The preferred sequence is considered to interact most strongly with the target and leads to the minimum relative entropy. EFBA shows SIAp2, 3, and 4 have a lower relative entropy than SIIApn (see Fig. 1). These three are indeed the most favorable sequences.
gives an approximate affinity ranking scheme of L-mer sequences. The preferred sequence is considered to interact most strongly with the target and leads to the minimum relative entropy. EFBA shows SIAp2, 3, and 4 have a lower relative entropy than SIIApn (see Fig. 1). These three are indeed the most favorable sequences.
Based on MD simulations, we can also correlate the fluorescence level with binding free energy difference. As an example, Fig. 3 shows the separation distance dapt-lipid against time from a set of raw simulation results rather than all cases, in which five randomly generated SIAp4-PS complexes as initial structures, represented in the inset, are considered. Given this result, we evaluated the probability of a designed aptamer and PS being at a specific dapt-lipid range, which is the time duration both spent at that dapt-lipid range over the total simulation time (see Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/nat). The average probabilities of sequences with PS within 6<dapt-lipid<16 Å were calculated (see Table 1, third column) and the binding energy difference, 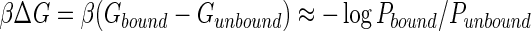 , was estimated from these probabilities. Note that Gbound is the free energy of the aptamer-PS complex in a bound state, while Gunbound corresponds to an unbound state. Figure 4 shows the fluorescence level from five PS-binding aptamers at 33 μM to be roughly linearly proportional to these average probabilities (left) and βΔG (right). This suggests that SIAp3 and 4 with the highest fluorescence levels are likely to have the highest binding energy.
, was estimated from these probabilities. Note that Gbound is the free energy of the aptamer-PS complex in a bound state, while Gunbound corresponds to an unbound state. Figure 4 shows the fluorescence level from five PS-binding aptamers at 33 μM to be roughly linearly proportional to these average probabilities (left) and βΔG (right). This suggests that SIAp3 and 4 with the highest fluorescence levels are likely to have the highest binding energy.
FIG. 3.

Ten-nanosecond (ns) molecular dynamic simulations for SIAp4–PS complex. The 10-ns molecular dynamic (MD) simulation results of SIAp4–PS complex are shown in terms of separation distance dapt-lipid versus simulation time. Five initial structures are shown in the inset.
FIG. 4.

The correlation of binding assays and simulations. Left panel: Plot of the relation between fluorescence (from the top left and top middle panels of Fig. 2 at 33 μM) and probabilities of the designed aptamers and PS within 6<dapt-lipid<16Å. Right panel: fluorescence plotted against the estimated binding free energy ΔG.
Furthermore, our binding assays (see Fig. 2) indicate that SIAp4 has a higher fluorescence level in DPPC+DPPS than in DPPC alone. Namely, SIAp4 is PS specific. However, our probability analysis based on 10-ns simulations, in which SIAp4 has a higher chance to interact with PC (18%) and not with PS within a very short distance (dapt-lipid<6 Å) seems contradictory to this result (see Supplementary Fig. S1). Note that our 10-ns simulations show SIAp4 strongly interacting with PS within 8<dapt-lipid<12 Å. We performed further 10-ns MD simulations following the first 10 ns and focused on total probabilities within 6<dapt-lipid<16 Å to investigate this issue (see Table 1). The 20-ns simulations show that the probability for SIAp4 and PC within dapt-lipid<6 Å dropped to 5% and dropped dramatically from 41 % to 30 % within 6<dapt-lipid<16 Å (see Table 1). However, the probability for SIAp4 and PS dropped slightly from 54 % to 46 % within 6<dapt-lipid<16 Å (see Table 1). Therefore, the PS specificity of SIAp4 is observed. It suggests the contradiction between 10 and 20 ns MD may simply result from insufficient simulation times.
Binding mechanisms
Figure 5 shows the binding mode of SIAp4 and PS at 20 ns from the simulation colored by cyan in Fig. 3. The surfaces of structures are colored based on partial charges (red for negative, blue for positive, and white for neutral charges). The color scale denotes the magnitude. Since there are no clear positive-negative charge domains at the interface, it suggests that the binding between SIAp4 and PS may be only slightly affected by electrostatic interactions.
FIG. 5.
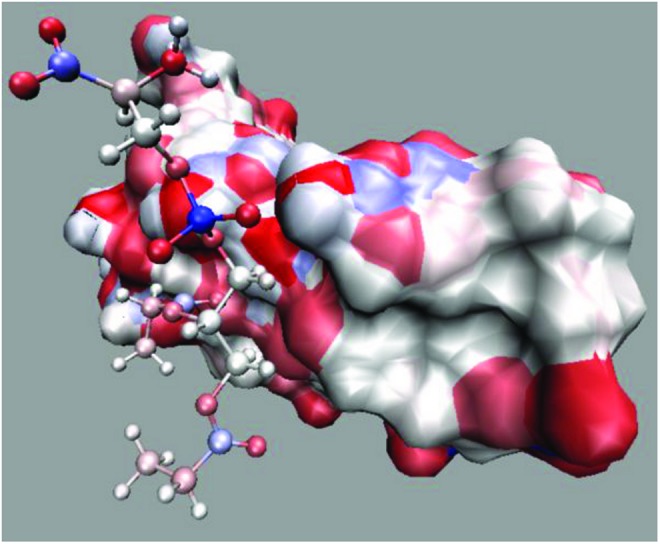
Binding mode of PS–DNA aptamer, SIAp4. The surface representations of SIAp4 at 20 ns in the simulation using initial structure marked by blue in the inset of Fig. 3 are presented. PS is plotted in the CPK (ball-and-stick) format. Red color represents negative, blue for positive, and white for neutral charge. The scale of color denotes the magnitude of charge. The plot was generated using VMD (Humphrey et al., 2006).
To quantitatively understand the effects of interactions in the aptamer-phospholipid complex, we first analyzed their ES and vdW energies and SA against dapt-lipid as shown in left panel of Fig. 6. Here we observe that SIIAp1 and 2 have relatively high positive total ES energies while all SIApn sequences are characterized by negative ES energies. Namely, there is a stronger ES repulsion between SIIAPn and PS than the one between SIApn and PS. Furthermore, the ES energies in the SIIApn case are gradually decreased when dapt-lipid is increased. This indicates that ES interactions tend to push SIIApn aptamers further away from PS. Although vdW forces attempt to pull SIIApn aptamers and PS together (negative vdW energies), their contributions are weaker than the repulsion from ES forces. Therefore, weak binding affinities are observed for SIIApn aptamers.
FIG. 6.

The left part shows total energy analyses of five aptamer–PS complexes. Mean values of total van der Waals (vdW) energy (EvdW), electrostatic (ES) energy (EES) and SA were found for five aptamer-PS complexes when the designed aptamer and PS are within a specific separation distance. The calculations are based on the 10 ns simulation results. All energy points are calculated within every consecutive 1Å interval. The right part shows the total energy analysis for the SIAp4 case. The mean values of the total vdW energy, ES energy and solvent accessible area (SA) of SIAp4-PS and SIAp4-PC complexes from 20-ns simulations are plotted.
Secondly, as shown in Fig. 6, the ES energy contribution in the SIAp4 case seems to be fluctuating around −200 kcal/mole. However, both the vdW energy and the SA area show that the SIAp4-PS complex tends to be more stable when dapt-lipid is less than about 14 Å. Consequently, the vdW and hydrophobic interactions are the primary driving forces to stabilize the SIAp4-PS complex. These charge based energetics are found to be consistent with the results of experimental observations regarding the interactions between a set of ion pores in a lipid membrane formed by chemotherapy drugs (colchicine and taxol) and lipids (PC and PS) (Ashrafuzzaman et al., 2012). It has previously been demonstrated that the distance-dependent role of ES forces (originating from the screened Coulomb charge–charge interactions) is strongly affected by the nature and distribution of localized charges on the phospholipids and the binding agents (Ashrafuzzaman and Tuszynski, 2012). In the interplay between the contributions from vdW, ES, and hydrophobic interactions, the ES therefore may not always appear to favor aptamer-phospholipid binding.
An additional insight into the PS selectivity is shown in right panel of Fig. 6 where both PS and PC are subjected to similar vdW and hydrophobic interactions as those involved in SIAp4. The differences in ES interactions, which result in a more energetically stable state for SIAp4 with PS than PC, lead to the PS selectivity.
Dissociation constant of designed aptamers
To determine the dissociation constant of designed aptamers, we chose the top two candidates SIAp3 and SIAp4 for further studies. Direct comparison between the FL responses presented here with those in Fig. 2 cannot be made because the fluorescence response was recorded using a different gain (976) than that (1,626) used in Fig. 2. The use of different gains is due to the technologically achievable upper limit of response recordable in the FLUOstar OPTIMA. Figure 7 shows that the equilibrium for SIAp4 binding is achieved at∼800 μM. The second preferred aptamer SIAp3, does not reach equilibrium at a similar concentration. Note that all the data points are based on 8 independent measurements except for the case with the highest concentration due to the prohibitive cost of the fluorescent aptamer. However, based on the small standard deviation of the data point before the highest concentration (SIAp4 case), the mean value at the highest concentration will be approximated by the data point shown in the figure. Therefore, the estimation of dissociation constant in the following will be subjected to minor impacts only. Based on a single equilibrium reaction, one can derive the Hill equation[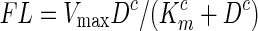 ], where the Hill coefficient (C)=1, cooperativity of the complex (Vmax) is the maximum fluorescence at equilibrium, and D is concentration (Goutelle et al., 2008). Furthermore, the parameter Km represents the aptamer concentration required to have a half of the maximum fluorescence and it is an expression of affinity (Goutelle et al., 2008). Therefore, the dissociation constant of aptamers can be estimated by Km. Fitted parameters are presented in Table 2. The models represented by red and green curves in Fig. 7 are obtained by fixing the Hill coefficient to 1, resulting in a Michaelis-Menten type equation. The best model (blue curve) has a dissociation constant around 317 μM, which is slightly less than the 373 μM estimated in the red curve. Both are far less than the value for SIAp3. Therefore, we can conclude that SIAp4 has the lowest value of the dissociation constant. It suggests that SIAp4 demonstrates weak binding.
], where the Hill coefficient (C)=1, cooperativity of the complex (Vmax) is the maximum fluorescence at equilibrium, and D is concentration (Goutelle et al., 2008). Furthermore, the parameter Km represents the aptamer concentration required to have a half of the maximum fluorescence and it is an expression of affinity (Goutelle et al., 2008). Therefore, the dissociation constant of aptamers can be estimated by Km. Fitted parameters are presented in Table 2. The models represented by red and green curves in Fig. 7 are obtained by fixing the Hill coefficient to 1, resulting in a Michaelis-Menten type equation. The best model (blue curve) has a dissociation constant around 317 μM, which is slightly less than the 373 μM estimated in the red curve. Both are far less than the value for SIAp3. Therefore, we can conclude that SIAp4 has the lowest value of the dissociation constant. It suggests that SIAp4 demonstrates weak binding.
FIG. 7.

The dissociation constant of SIAp4. FL measured in RFU versus aptamer (SIAp3 and 4) concentrations. Only PS binding is measured. The fluorescence response here was recorded using a different gain (976) than that (1626) used in the studies with all aptamer sequences presented in Fig. 2. Three models with fitted parameters as listed in Table 2 are also presented.
Table 2.
Estimated Parameters of the Hill Equation in Figure 7
| Modela | Vmax (RFU) | Km (μM) | C |
|---|---|---|---|
| green |
1.47*106 |
2.59*104 |
1 |
| red |
2.02*104 |
373.38 |
1 |
| blue | 1.87*104 | 317.71 | 1.93 |
Model green is estimated based on fluorescence study for SIAp3. Models red and blue are estimated based on SIAp4 data.
C, Hill coefficient; Km, aptamer concentration required to have one-half of the maximum fluorescence; RFU, relative fluorescence units; Vmax, maximum fluorescence at equilibrium.
Conclusion
We have applied EFBA to the design of DNA aptamer templates which were expected to bind specifically to PS. Both computational and experimental studies have provided a better understanding of the properties of designed aptamers beyond those uncovered in our previous preliminary studies in Tseng et al. (Tseng et al., 2011). The results here show that SIAp4, 5′-AAAGAC-3′, binds selectively to the liposomes having a PS containing surface but show only very low and nonselective binding to liposomes containing just PC. Note that the dissociation constants for aptamers to targets of therapeutic interest are found to range from 0.0002 nM (target at Keratinocyte growth factor) to 1.1 μM (target at D-adenosine) (Keefe et al., 2010). From a therapeutic point of view, this suggests SIAp4 to have weak binding. However, a complete understanding of its interaction with lipids at the molecular level suggests a new direction for future optimization. SIAp4 can therefore be utilized as a scaffold for the development of improved constructs with stronger binding and enhanced selectivity. Practical applications of highly selective aptamer probes include their use in molecular imaging, as fluorescent or radionuclide probes, to detect apoptosis in experimental systems and in human clinical studies. These constructs also have the potential to be developed as therapeutic drugs as for example in the treatment of amyloid aggregation in TSEs.
Supplementary Material
Acknowledgments
We are grateful to Dr. J. Mane for his assistance in PS and PC modeling, Mr. D. Sharon and Ms. K. Missiaen for their help with the fluorescence determination. The simulations were conducted using our Pharmamatrix high-throughput virtual screening cluster and the facilities of the Shared Hierarchical Academic Research Computing Network (SHARCNET: www.sharcnet.ca) and Compute/Calcul Canada. MA acknowledges grant (12-MED2670-02) received from National Plan for Science, Technology, and Innovation (King Abdulaziz City for Science and Technology), Kingdom of Saudi Arabia. JAT acknowledges funding received from NSERC (Canada), Alberta Cancer Foundation, the Allard Foundation, and Alberta Advanced Education and Technology, as well as the Canadian Breast Cancer Foundation.
Author Disclosure Statement
No competing financial interests exist.
References
- ASHRAFUZZAMAN M., TSENG C.-Y., DUSZYK M., and TUSZYNSKI J. (2012). Chemotherapy drugs form ion pores in membranes due to physical interactions with lipids. Chem. Biol. Drug. Des. 80,992–1002 [DOI] [PubMed] [Google Scholar]
- ASHRAFUZZAMAN M., and TUSZYNSKI J. (2012). Regulation of channel function due to coupling with a lipid bilayer. J. Comput. Theor. Nanosci. 9,564–570 [Google Scholar]
- BLANKENBERG F.G. (2009). Imaging the molecular signatures of apoptosis and injury with radiolabeled annexin V. Proc. Am.Thorac. Soc. 6,469–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOERSMA H.H., KIETSELAER B.L., STOLK L.M., BENNAGHMOUCH A., HOFSTRA L., NARULA J., HEIDENDAL G.A., and REUTELINGSPERGER C.P. (2005). Past, present, and future of annexin A5: from protein discovery to clinical applications. J. Nucl. Med. 46,2035–2050 [PubMed] [Google Scholar]
- CAMPANA V., ZENTILIN L., MIRABILE I., KRANJC A., CASANOVA P., GIACCA M., PRUSINER S.B., LEGNAME G., and ZURZOLO C. (2009). Development of antibody fragments for immunotherapy of prion diseases. Biochem. J. 418,507–515 [DOI] [PubMed] [Google Scholar]
- CASE D.A., DARDEN T.A., CHEATHAM T.E., III, SIMMERLING C.L., WANG J., DUKE R.E., LUO R., CROWLEY M., WALKER R.C., ZHANG W., et al. (2008). AMBER 10. (University of California, San Francisco: ). [Google Scholar]
- CASE D.A., DARDEN T.A., CHEATHAM T.E., III, SIMMERLING C.L., WANG J., DUKE R.E., LUO R., CROWLEY M., WALKER R.C., ZHANG W., et al. (2010). AMBER 11. (University of California, San Francisco: ) [Google Scholar]
- CATICHA A. (2004). Relative entropy and inductive inference. In: Bayesian Inference and Maximum Entropy Methods in Science and Engineering. Erickson G., Zhai Y. ed. AIP Conf. Proc. 707 (American Institute of Physics, Melville, New York: ), pp. 75–96 [Google Scholar]
- CATICHA A., and GIFFIN A. (2007). Updating probabilities. In: Bayesian Inference and Maximum Entropy Methods in Science and Engineering. Knuth K. H., Caticha A., Center J. L., Giffin A., Rodriguez C. C. ed. AIP Conf. Proc. 954 (American Institute of Physics, Melville, New York: ), pp. 74–84 [Google Scholar]
- CHEN C.-C., TSENG C.-Y., and DONG J.-J. (2007). New entropy-based method for variables selection and its application to the debris flow hazard assessment. Eng. Geol. 94,19–24 [Google Scholar]
- CHIESA R., DRISALDI B., QUAGLIO E., MIGHELI A., PICCARDO P., GHETTI B., and HARRIS D.A. (2000). Accumulation of protease-resistant prion protein (PrP) and apoptosis of cerebellar granule cells in transgenic mice expressing a PrP insertional mutation. Proc. Natl. Acad. Sci. 97,5574–5579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ELMORE S. (2007). Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35,495–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOUTELLE S., MAURIN M., ROUGIER F., BARBAUT X., BOURGUIGNON L., DUCHER M., and MAIRE P. (2008). The Hill equation: a review of its capabilities in pharmacological modeling. Fundam. Clin. Pharmacol. 22,633–648 [DOI] [PubMed] [Google Scholar]
- HAMULA C.L.A., GUTHRIE J.W., ZHANG H., LI X.F., and CHRIS LE X. (2006). Selection and analytical applications of aptamers. Trends Anal. Chem. 25,681–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEPPNER F.L., and AGUZZI A. (2004). Recent developments in prion immunotherapy. Curr. Opin. Immunol. 16,594–598 [DOI] [PubMed] [Google Scholar]
- HOPE M.J., BALLY M.B., MAYER L.D., JANOFF A.S., and CULLIS P.R. (1986). Generation of multilamellar and unilamellar phospholipid vesicles. Chem. Phys. Lipids 40,89–107 [Google Scholar]
- HUMPHREY W., DALKE A., and SCHULTEN K. (1996). VMD: Visual molecular dynamics. J. Mol. Graph 14,33–38 [DOI] [PubMed] [Google Scholar]
- JAMES W. (2000). Aptamers. In: Encyclopedia of Analytical Chemistry. Meyers R. A. ed. (John Wiley & Sons, Ltd., Chichester: ), pp. 4878–4871 [Google Scholar]
- JAMES W. (2007). Aptamers in the virologists' toolkit. J. Gen.Virol. 88,351–364 [DOI] [PubMed] [Google Scholar]
- JAYNES E.T. (1957a) Information theory and statistical mechanics. Phys. Rev. 106,620–630 [Google Scholar]
- JAYNES E.T. (1957b) Information theory and statistical mechanics II. Phys. Rev. 108,171–190 [Google Scholar]
- KAPTY J., BANMAN S., GOPING I.S., and MERCER J.R. (2012). Evaluation of Phosphatidylserine-Binding Peptides Targeting Apoptotic Cells. J. Biomol. Screen. 17,1293–1301 [DOI] [PubMed] [Google Scholar]
- KEEFE A.D., PAI S., and ELINGTON A. (2010). Aptamers as therapeutics. Nature Rev. Drug Discov. 9,537–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- KERR J.F., WYLLIE A.H., and CURRIE A.R. (1972). Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 4,239–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAO Y.H., PECK K., and CHEN L.C. (2009). Enhancement of aptamer microarray sensitivity through spacer optimization and avidity effect. Anal. Chem. 81,1747–1754 [DOI] [PubMed] [Google Scholar]
- MARTIN S.J.,; REUTELINGSPERGER C.P., MCGAHON A.J., RADER J.A., VAN SCHIE R.C., LAFACE D.M., and GREEN D.R. (1995). Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J. Exp. Med. 182,1545–1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIMJEE S.M., RUSCONI C.P., and SULLENGER B.A. (2005). Aptamers: an emerging class of therapeutics. Annu. Rev. Med. 56,555–583 [DOI] [PubMed] [Google Scholar]
- PROSKE D., GILCH S., WOPFNER F., SCHÄTZL H.M., WINNACKER E.L., and FAMULOK M. (2002). Prion-protein-specific aptamer reduces PrPSc formation. ChemBioChem 3,717–725 [DOI] [PubMed] [Google Scholar]
- QUADRIO I., PERRET-LIAUDET A., and KOVACS G.G. (2011). Molecular diagnosis of human prion disease. Expert. Opin. Med. Diag. 5,291–230 [DOI] [PubMed] [Google Scholar]
- RASTOGI R.P., RICHA , and SINHA R.P. (2009). Apoptosis: molecular mechanisms and pathogenicity EXCLI. J. 8,155–181 [Google Scholar]
- RHIE A., KIRBY L., SAYER N., WELLESLEY R., DISTERER P., SYLVESTER I., GILL A., HOPE J., JAMES W., and TAHIRI-ALAOUI A. (2003). Characterization of 2-fluoro-RNA aptamers that bind preferentially to disease-associated conformations of prion protein and inhibit conversion. J. Bio. Chem. 278,39697–39705 [DOI] [PubMed] [Google Scholar]
- ROBINSON P.J., and PINHEIRO T.J. (2010). Phospholipid composition of membranes directs prions down alternative aggregation pathways. Biophys. J. 98,1520–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHORE J.E., and JOHNSON R.W. (1980). Axiomatic derivation of the principle of maximum entropy and the principle of minimum cross entropy. IEEE Trans. Inf. Theory 26,26–37 [Google Scholar]
- SHORE J.E., and JOHNSON R.W. (1981). Properties of cross-entropy minimization. IEEE Trans. Inf. Theory 27,472–482 [Google Scholar]
- SMRZ D., LEBDUSKA P., DRÁBEROVÁ L., KORB J., and DRÁBER P. (2008). Engagement of phospholipid scramblase 1 in activated cells: implication for phosphatidylserine externalization and exocytosis. J. Biol. Chem. 283,10904–10918 [DOI] [PubMed] [Google Scholar]
- THELLUNG S., CORSARO A., VILLA V., SIMI A., VELLA S., PAGANO A., and FLORIO T. (2011). Human PrP90-231-induced cell death is associated with intracellular accumulation of insoluble and protease-resistant macroaggregates and lysosomal dysfunction. Cell Death Dis. 2, e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TSENG C.-Y. (2006). Entropic criterion for model selection. Physica A 370,530–538 [Google Scholar]
- TSENG C.-Y., and Caticha A. (2008). Using relative entropy to find optimal approximations: an application to simple fluids. Physica A 387,6759–6770 [Google Scholar]
- TSENG C.-Y., ASHRAFUZZAMAN MD, MANE J., KAPTY J., MERCER J., and TUSZYNSKI J. (2011). Entropic fragment based approach to aptamer design. Chem. Biol. Drug Design 78,1–13 [DOI] [PubMed] [Google Scholar]
- TUERK C., and GOLD L. (1990). Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249,505–510 [DOI] [PubMed] [Google Scholar]
- VAN MEER G., VOELKER D.R., and FEIGENSON G.W. (2008). Membrane lipids: where they are and how they behave. Nature Rev. Mol. Cell Biol. 9,112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG J., WOLF R.M., CALDWELL J.W., KOLLMAN P.A., and CASE D.A. (2004). Development and testing of a general AMBER force field. J. of Comp. Chem. 25,1157–1174 [DOI] [PubMed] [Google Scholar]
- WANG J., WANG W., KOLLMAN P.A., and CASE D.A. (2006). Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph Model 25,247–260 [DOI] [PubMed] [Google Scholar]
- WEINBERG R.A. (2007). The biology of cancer. (Garlan Science, New York: ) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.