

ABSTRACT
The group of proteins known as serine protease autotransporters of Enterobacteriaceae (SPATE) is a growing family of serine proteases secreted to the external milieu by the type V secretion system. Pet toxin and some other SPATE belong to the class 1 cytotoxic SPATE, which have comparable protease strength on fodrin. Pet is internalized and is directed to its intracellular substrate by retrograde transport. However, the epithelial cell receptor for Pet has yet to be identified. We show that Pet has affinity for the epithelial cell surface until the saturation of the binding sites at 100 nM Pet. Affinity column assays and matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) analysis identified a cytokeratin (CK8) which directly binds to Pet, and both proteins colocalized on the cell surface. Interestingly, CK8 is not present in kidney cell lines, which are not susceptible to Pet. Inhibition experiments by using anti-CK8 and ck8 small interfering RNA (siRNA) blocked the cytotoxic effect induced by Pet, while exogenous CK8 expression in kidney cells made them susceptible to Pet intoxication. Recombinant CK8 showed a Pet-binding pattern similar to that seen by using fixed cells. Remarkably, Pet colocalized with CK8 and clathrin at early times (receptor-mediated endocytosis), and subsequently, Pet colocalized with CK8 and Rab5b in the early endosomes. These data support the idea that CK8 is an important receptor for Pet on epithelial cells for starting its cytotoxic effects. These data suggest that therapeutics that block Pet-CK8 interaction may improve outcome of diseases caused by Pet-secreting Enterobacteriaceae such as enteroaggregative Escherichia coli.
IMPORTANCE
Receptor-ligand binding is one mechanism by which cells sense and respond to external cues. Receptors may also be utilized by toxins to mediate their own internalization. Pet toxin is secreted by enteroaggregative Escherichia coli, an organism that causes persistent diarrhea in children, traveler’s diarrhea, and acute and persistent diarrhea in patients with HIV. Pet is a member of the family of serine protease autotransporters of Enterobacteriaceae (SPATE). SPATE in different pathogens are virulence factors, and Pet belongs to the class 1 cytotoxic SPATE, which have comparable protease strength on their biological substrate, fodrin (a cytoskeletal protein important for maintaining cell viability). To cleave fodrin, Pet enters the cells by clathrin-mediated endocytosis. This mechanism includes receptor-mediated endocytosis (a receptor-ligand complex triggers the endocytosis). We show that CK8 is an important receptor for Pet on epithelial cells and that it may be useful for identifying molecules that block the interaction of CK8 with Pet.
INTRODUCTION
In Gram-negative bacteria, the type V autotransporter secretion system is responsible for releasing a growing family of high-molecular-weight serine proteases into the external milieu (1). The type V secretion system, which includes several variants (Va, Vb, Vc, Vd, and Ve), is the most common mechanism used to release virulence factors by Gram-negative bacteria (1, 2). Proteins secreted by this system are called autotransporter proteins because they promote their own secretion through the inner and outer membranes by using two preprotein processing domains, the signal sequence and the translocation unit (2). The serine protease autotransporters from Enterobacteriaceae (SPATE) constitute a superfamily of virulence factors whose members resemble those belonging to the trypsin-like superfamily of serine proteases (2). SPATE proteins are produced by enteric pathogens, including Escherichia coli and Shigella, Salmonella, Edwardsiella, and Citrobacter species, and less frequently by commensal strains. Interestingly, SPATE have been found in all recognized E. coli pathotypes (3), as well as in extraintestinal E. coli pathogens, such as uropathogenic E. coli and septicemic E. coli, that are responsible for urinary tract infections and sepsis/meningitis, respectively (1).
The SPATE family, which includes more than 25 proteases with a range of substrates, have been phylogenetically divided into two distinct classes based on the amino acid sequence of the passenger domain: class 1 SPATE are cytotoxic, while class 2 SPATE are lectin-like immunomodulators (1–3). Interestingly, pathogens often harbor more than one SPATE protein, typically a combination of at least one SPATE from each class (1). For example, the prototype enteroaggregative E. coli (EAEC) strain 042 carries both the class 1 SPATE Pet and the class 2 SPATE Pic. It has also been found that the vast majority of EAEC genomes possess approximately 3 SPATE genes, which may include SepA, Pic, SigA, and Sat (4). Recently, it was found that the deadly German outbreak EAEC strain C227-11, which caused at least 50 deaths in Europe in 2011, carried three SPATE produced by Shigella, SigA, SepA, and Pic, a combination rarely detected in EAEC strains (5). Independent of their substrate or cleavage sites, class 1 SPATE, have a common ability to cause cytopathic effects in cultured cells and display enterotoxin activity (6–10). The Pet, Sat, and SigA class 1 SPATE show higher identity/similarity (50% to 70%) than any other members of their class, which may explain the comparable protease strength on their shared biological substrate, the actin-binding protein α-fodrin (α-spectrin) (6, 8, 11).
Pet is perhaps the most comprehensively studied class 1 SPATE (12). Initial studies identified Pet as a 108-kDa heat-labile enterotoxin (13, 14) and cytotoxin (15). Subsequently, purified Pet protein was found to induce cytopathic effects in HEp-2 and HT29 C1 cells, a phenotype dependent on the proteolytic activity mediated by the catalytic serine protease motif in the Pet passenger domain (10). This is observed as elongation, rounding, and detachment of cells from the substratum, cytoskeleton contraction, loss of actin stress fibers, and release of focal contacts, and these are the classical signals of intoxication induced by class 1 SPATE (10). Cell intoxication by Pet was associated with the ability of Pet to cleave the actin-binding protein α-fodrin, as demonstrated using in vitro and in vivo settings (11). Pet cleavage within the calmodulin-binding domain of fodrin’s 11th repetitive unit was responsible for disruption of the actin cytoskeleton; the interaction between fodrin and filamentous actin provides a degree of structural organization to the actin cytoskeleton which helps the cell withstand mechanical stress (11).
In addition to the Pet serine protease motif, Pet intoxication also requires toxin endocytosis in order to reach the intracellular target. Pet binds to the epithelial cell surface and is internalized by clathrin-coated vesicles (16). Trafficking studies have revealed that once inside the cell, Pet moves by vesicle carriers from the cell surface to endosomes, from the endosomes to the Golgi apparatus, and from the Golgi apparatus to the endoplasmic reticulum (ER). Ultimately, Pet is delivered from the ER to the cytosol, where it comes into close contact with its α-fodrin substrate (17). Importantly, Pet represents the first bacterial toxin found to target α-fodrin and the first SPATE to display enterotoxin activity (11, 14). Following the discovery of Pet, many other class 1 SPATE were found to cleave α-fodrin and to trigger similar biological effects (6, 8, 9). Despite that fact that Pet internalization requires clathrin-coated pits, suggesting a receptor-mediated endocytosis, the receptor protein has yet to be identified. In this work, we identify the Pet receptor as cytokeratin 8 (CK8) by using several approaches, including affinity experiments, inhibition assays, purified protein interactions, CK8 cell expression by ck8 knockdown, and transfection in Pet-susceptible and -unsusceptible cells.
RESULTS
Pet binds to epithelial cell plasma membrane proteins.
Our previous work suggested that Pet enters the host cell by receptor-mediated endocytosis (16). This possibility was strengthened by our observation of saturable Pet binding to the plasma membrane of cultured cells. HEp-2 cells fixed to prevent toxin endocytosis were exposed to various concentrations of Pet, which was then detected on the cell surface by enzyme-linked immunosorbent assay (ELISA). We found that 100 nM of Pet was enough to saturate the plasma membrane binding sites, and the apparent dissociation constant (Kd) of Pet binding to HEp-2 cells was determined to be 6.7 nM (see Fig. S1A in the supplemental material).
In order to identify the plasma membrane receptor(s) for Pet, an enzymatically inactive mutant, Pet-S260I, was coupled to Sepharose for affinity column chromatography. Membrane or cytoplasmic fractions from HT-29 cells were passed through the column, which captured several proteins from the membrane fraction and no proteins from the cytoplasmic fraction (see Fig. S1B in the supplemental material). Unfractionated HT-29 cell lysates produced the same pattern of eluted protein bands as the HT-29 membrane fraction (see Fig. S1B). The major protein bands were cut from the gel and identified using mass spectrometry on a matrix-assisted laser desorption ionization--time of flight mass spectrometry (MALDI-TOF/MS) system. Peptide analysis was performed with the MASCOT software from Matrix Science. We identified six bands: CK8, CK8, CK18, CK18, CK2a, and CK10 (see Fig. S1B). It is interesting that only CK8 was detected at the expected molecular mass (53 kDa), while the other protein bands appeared to be degradation products. Full-length CK8 was also the most predominant eluted protein band. Since it is well know that cytokeratins can form complexes and the affinity column assays were performed using proteins under native conditions, we decided to analyze Pet binding to these proteins using a denaturing overlay assay. Host proteins were separated by SDS-PAGE, transferred to a nitrocellulose sheet, and incubated with purified Pet-S260I. The interaction of Pet with the separated, denatured membrane proteins was subsequently detected with anti-Pet antibodies. Pet mainly interacted with four protein bands from the membrane fraction and unfractionated cell lysate, which had been identified as CK8, CK8, CK10, and CK10 (see Fig. S1C). To exclude the possible nonspecific interaction of anti-Pet antibodies with host proteins, we performed an anti-Pet Western blot assay using fractionated and unfractionated cell lysates that had not been exposed to Pet. Only the purified Pet-S260I positive control produced a signal in this Western blot, thus demonstrating that no host proteins cross-reacted with the anti-Pet antibodies (see Fig. S1D).
Surface-localized CK8 and CK10 interact with Pet.
Since Pet interacts with CK8 and CK10 from cell lysates and membrane fractions, it was possible that these proteins serve as surface receptors for Pet. We therefore attempted to detect CK8 and CK10 on the surfaces of intact cells. HT-29, HEp-2, MDCK, and RK13 cells were fixed without permeabilization and then incubated with anti-CK8 and anti-CK10 antibodies, followed by a secondary antibody coupled to fluorescein. CK8 and CK10 were present on the surfaces of HT-29 and HEp-2 cells but were absent from the surfaces of MDCK and RK13 kidney epithelial cell (see Fig. S2 in the supplemental material). Consistent with this result, we found that Pet is unable to damage MDCK, RK13, or Vero cells but clearly causes alterations to the morphology and actin cytoskeleton of HEp-2 and HT-29 cells (see Fig. S3 in the supplemental material). This suggested that the surface expression of CK8 and/or CK10 was required for Pet intoxication.
To see if Pet bound to CK8 or CK10 on the cell surface, we performed colocalization experiments using rabbit anti-Pet antibodies and either mouse anti-CK8 or mouse anti-CK10 antibodies. These were followed by secondary antibodies against rabbit IgG labeled with rhodamine (red) and against mouse IgG labeled with fluorescein (green). To prevent Pet endocytosis, cells were slightly prefixed with 0.25% paraformaldehyde and then incubated with Pet. Confocal microscopy showed that Pet colocalized with CK8 (detected as yellow) on the surface of unpermeabilized HEp-2 and HT-29 cells. In contrast, no colocalization between Pet and CK8 was detected when MDCK or RK13 kidney cells were used; neither Pet nor CK8 was detected on the surfaces of MDCK cells, while the small number of RK13 cells with traces of CK8 and Pet did not exhibit colocalization between the two proteins (Fig. 1). Pet colocalization with CK10 could also be detected in HT-29 and Hep-2 cells, but the colocalization with CK8 was much more apparent than the colocalization with CK10 (Fig. 1). To corroborate the lack of CK8/CK10 interaction with Pet in kidney cell lines, proteins from MDCK cells and another human kidney cell line, HK-293, were passed over a Pet affinity column as described for the experiment whose results are shown in Fig. S1B in the supplemental material. The cytokeratins were again captured from HT-29 cells (used as a positive control) but not from MDCK or HK-293 cells (see Fig. S4A in the supplemental material). Moreover, Pet binding to HK-293 proteins was not seen in an overlay assay using either cell lysates or cellular fractions (see Fig. S4B). In contrast, the overlay assay was able to detect Pet binding to proteins from cell lysates and the membrane fraction of intestinal HT-29 cells (see Fig. S1C in the supplemental material). Collectively, these results indicated that the expression of CK8 and/or CK10 was required for Pet interaction with the host cell.
FIG 1 .

Pet colocalizes with CK8 and CK10 on epithelial cell membranes. HT-29, HEp-2, RK-13, and MDCK cells were slightly fixed with 0.25% paraformaldehyde (PFA) for 1 h and then were washed and treated with 37 µg/ml Pet for 30 min. After this treatment, cells were washed again and fixed properly with 4% PFA but not permeabilized. Nuclear DNA was stained with TO-PRO 3 (blue), Pet was detected using an anti-Pet polyclonal antibody followed by a secondary antibody anti-rabbit IgG coupled to rhodamine (red), and CK8 or CK10 (as appropriate) was detected using monoclonal anti-CK10 or anti-CK8 antibodies followed by a secondary anti-mouse IgG antibody coupled to fluorescein (green). The slides were observed by confocal microscopy.
Cell fractionation and coimmunoprecipitation assays were performed to further establish the interaction between Pet and CK8 on the plasma membrane. After fractionating HT-29, HEp-2, RK13, and MDCK cells into cytoplasm and membrane fractions, the quality of cell fractionation was analyzed by Western blotting to detect specific markers for each fraction. The plasma membrane Na+/K+ ATPase was found only in the membrane fraction, and the cytosolic GAPDH protein was found exclusively in the cytoplasmic fraction (Fig. 2A). These expected results demonstrated the clean separation of membrane and cytosolic fractions. After this control experiment had been run, each fraction was incubated with Pet-S260I and subsequently immunoprecipitated with either anti-CK8 or anti-Pet antibodies. Material immunoprecipitated with the anti-CK8 antibody was probed by Western blotting with the anti-Pet antibody, and material immunoprecipitated with the anti-Pet antibody was probed by Western blotting with the anti-CK8 antibody. Immunoprecipitation of the membrane fraction with the anti-CK8 antibody detected a strong interaction between CK8 and Pet in HT-29 cells, a medium interaction in HEp-2 cells, and a weak interaction in RK13 cells. No interaction between CK8 and Pet was detected from the membrane fraction of MDCK cells (Fig. 2B). A similar pattern of CK8-Pet interactions was detected with anti-Pet immunoprecipitates from membrane fractions of HT-29, HEp-2, RK13, and MDCK cells (Fig. 2B). No interactions between CK8 and Pet were detected in cytoplasm fractions immunoprecipitated with either anti-CK8 or anti-Pet antibodies (Fig. 2B).
FIG 2 .

Pet binding to CK8 on epithelial cell plasma membranes. (A) Cellular fractionation. The purity of membrane and cytoplasmic fractions from HT-29, HEp-2, RK-13, or MDCK cells was determined by Western blot analysis using specific markers for the plasma membrane (Na+/K+ ATPase β subunit) and cytoplasm (GAPDH). (B) Pet binding to cytokeratin 8 and vice versa. Membrane or cytoplasmic fractions from HT-29, HEp-2, RK-13, and MDCK cells incubated overnight with Pet-S260I were subjected to a coimmunoprecipitation assay using either anti-CK8 (left) or anti-Pet (right) antibodies. The immunocomplexes were separated by SDS-PAGE and analyzed by Western blotting using anti-Pet (left) or anti-CK8 (right) antibodies. Anti-WASP antibodies were used as a negative control for the coimmunoprecipitation step.
When similar experiments were performed for detecting the interaction between Pet and CK10, we detected weaker CK10-Pet interactions than those documented for CK8-Pet interactions. For membrane fractions immunoprecipitated with anti-Pet antibodies, CK10 binding to Pet was detected only in HT-29 cells and (weakly) HEp-2 cells, not in RK13 and MDCK cells (see Fig. S5A in the supplemental material). The interaction between Pet and CK10 could also be clearly detected when the membrane but not the cytosolic fraction of HT-29 cells was immunoprecipitated with anti-CK10 antibodies and probed with anti-Pet antibodies (see Fig. S5B in the supplemental material).
Since Pet interacts mainly with CK8 but slightly with CK10, we examined whether the three proteins form a complex. Membrane fractions incubated with Pet-S260I or with phosphate-buffered saline (PBS) as a negative control were immunoprecipitated with anti-CK10 antibodies, and the immunoprecipitated material was analyzed by Western blotting using anti-CK8 antibodies. Membrane-localized CK8 and CK10 had a very strong interaction in control HT-29 cells that were not exposed to Pet. Interestingly, the interaction between CK10 and CK8 almost disappeared in membrane fractions from HT-29 cells treated with Pet (see Fig. S5B). This suggested that Pet disrupts the CK8-CK10 complex, perhaps by competing with CK10 for binding to CK8.
CK8 is a receptor for Pet.
To further characterize Pet binding to CK8 and CK10 on the cell surface, we examined the inhibition of Pet binding to the surface of cells exposed to either anti-CK8 or anti-CK10 antibodies. HEp-2 cells were washed, fixed, blocked, and incubated with different concentrations of either anti-CK8 or anti-CK10 antibodies for 1 h. The cells were then treated with 100 nM Pet for 90 min. Toxin binding to the plasma membrane was determined by ELISA using anti-Pet antibodies. Cells incubated without anti-CK antibodies but with Pet were used as a positive control to establish the 100% signal for Pet binding. Increasing concentrations of anti-CK8 antibodies decreased the amount of Pet bound to the plasma membrane, which was statistically significant from a 1:25 dilution (P = 0.002) and reached a maximum of 80% inhibition with a 1:10 dilution of anti-CK8 antibody (P < 0.001). In contrast, 1:25 and 1:10 dilutions of anti-CK10 antibodies decreased the amount of surface-bound Pet by only ~25% (Fig. 3A). These results indicated that the presence of surface-localized CK8 was more important for Pet binding than the presence of surface-localized CK10.
FIG 3 .
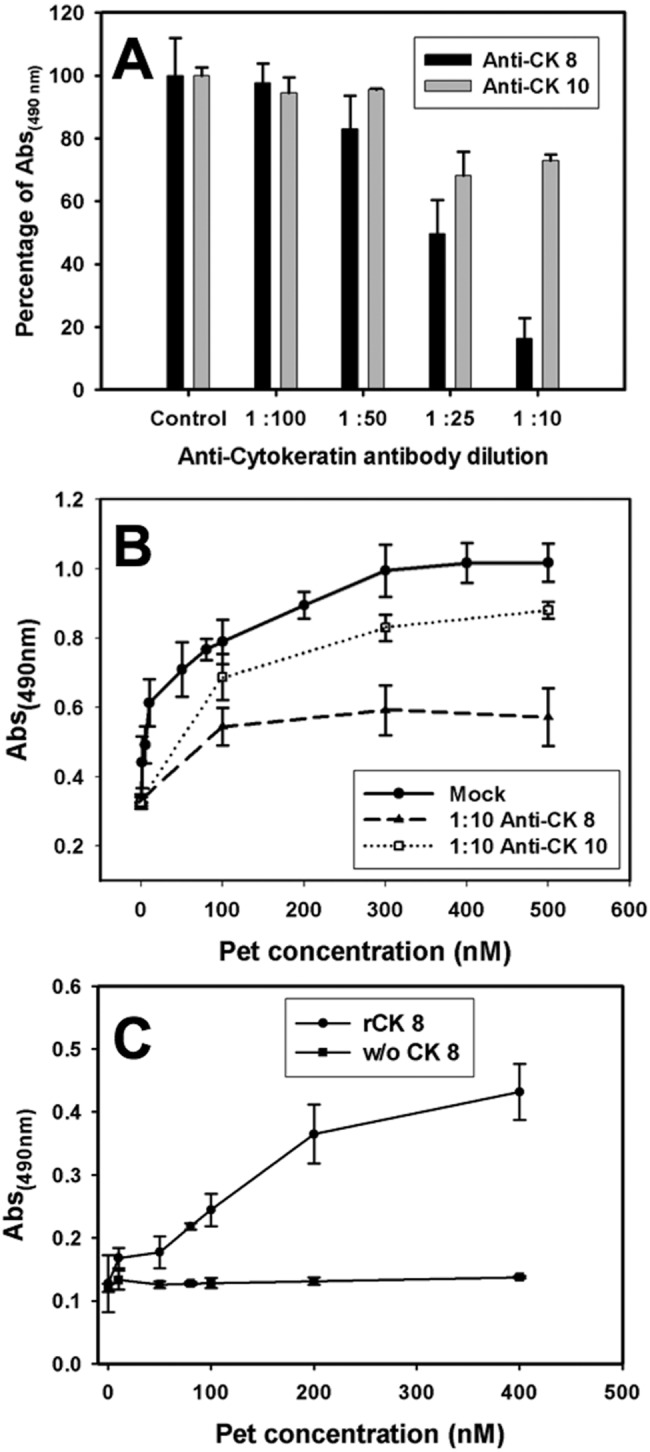
Anti-CK8 antibodies block Pet binding to plasma membrane of epithelial cells. (A) Anti-CK8 antibodies are more effective at blocking Pet binding than anti-CK10 antibodies. HEp-2 cells seeded in 96-well plates were washed, fixed (2.5% PFA), and blocked (3% BSA). Cells were incubated for 1 h with anti-CK10 or CK8 antibodies at different concentrations, and then cells were incubated with 100 nM of Pet per well at 37°C for 90 min and washed three times with PBS. The amount of membrane-bound Pet was determined by ELISA using anti-Pet antibodies followed by an anti-rabbit secondary antibody conjugated with HRP, with readings made at 490 nm. (B) Anti-CK8 antibodies inhibit Pet binding to plasma membrane. HT-29 cells were seeded, washed, and fixed as described for panel A. Cells were incubated with monoclonal anti-CK10 or anti-CK8 antibodies at a dilution of 1:10. Then, cells were incubated at 37°C for 90 min with different concentrations of Pet per well. Following 3 washes with PBS, the amount of membrane-bound Pet was determined by ELISA using rabbit anti-Pet antibodies followed by an anti-rabbit secondary antibody conjugated to HRP, with readings made at 490 nm. Values are the means of triplicate measurements from three independent experiments. (C) Pet interacts with recombinant CK8. ELISA plates coated with recombinant CK8 (20 nM) were incubated for 2 h with increasing concentrations of Pet and then washed with PBS to remove nonspecific binding. Finally, the amount of CK8-bound Pet was determined using rabbit anti-Pet antibodies followed by anti-rabbit secondary antibody conjugated to HRP, with the reading made at 490 nm. Wells without (w/o) CK8 were used as a negative control. Values are the means of triplicate measurements from three independent experiments.
To corroborate these data, we used another cell line, HT-29, which exhibited higher expression of CK8 and CK10 than HEp-2 cells (see Fig. 1). We followed the same general protocol that was used to quantify Pet binding to HEp-2 cells, but, in this case, we used a single 1:10 dilution of anti-CK8 or anti-CK10 antibody and various concentrations of Pet (100 nM to 500 nM). With this assay, we again found that the anti-CK8 antibody was more effective at inhibiting Pet binding to the cell surface (around 70%) than the anti-CK10 antibody (around 15%) (Fig. 3B). These results indicated CK8 is the predominant cell surface receptor for Pet.
To further demonstrate that CK8 directly binds to Pet, ELISA plates coated with 20 nM recombinant CK8 were incubated with different concentrations of Pet-S260I. Pet binding was then quantified using anti-Pet antibodies. As expected, Pet binding to CK8-coated wells increased with increasing Pet concentration. No Pet binding was detected at any concentration when the toxin was added to wells coated with bovine serum albumin (BSA), which demonstrated the specificity of Pet-CK8 interaction in our ELISA (Fig. 3C).
Blockage of CK8 inhibits the damage of epithelial cells induced by Pet.
In order to establish the functional role of CK8 as a receptor for Pet on the epithelial cell surface, toxicity assays were performed with cells containing inaccessible CK8. HEp-2 cells pretreated with anti-CK8 antibodies for 30 to 45 min were subsequently treated with Pet for 3 h before Pet-induced alterations to the actin cytoskeleton were examined by confocal microscopy. Cell damage was observed by detecting the F-actin cytoskeleton with rhodamine-phalloidin (red), along with TO-PRO 3 for DNA (blue) and anti-Pet antibodies followed by a secondary antibody, anti-rabbit IgG labeled with fluorescein (green). As a positive control, Pet was added to cells that had not been pretreated with anti-CK8 antibodies. Unintoxicated cells used as a negative control were seen as an intact monolayer (Fig. 4, Mock), whereas the Pet-treated positive control cells showed cell damage and were largely detached from the substratum (Fig. 4, Pet 3 h). Cells pretreated with anti-CK8 antibodies for 30 min before exposure to Pet were protected from cell damage and were, like the unintoxicated cells, observed as an intact cell monolayer (Fig. 4, anti-CK8 30 min + Pet). Interestingly, when cells were pretreated with anti-CK8 antibodies for 45 min before Pet treatment, the protection against the cell damage was less than that observed in cells pretreated with anti-CK8 antibodies for 30 min (Fig. 4, anti-CK8 45 min + Pet). It is possible that, with longer periods of pretreatment with anti-CK8 antibodies, a process of receptor recycling could generate new free CK8 receptor on the cell surface. The ability of anti-CK8 antibodies to block Pet intoxication indicates that CK8 is a functional receptor for Pet.
FIG 4 .

Anti-CK8 antibodies block Pet binding to the plasma membrane and the cell damage caused by Pet. HEp-2 cells pretreated with anti-CK8 antibody for 30 or 45 min were then washed and treated with Pet (37 µg/ml) for 3 h. Unintoxicated cells and cells intoxicated without antibody treatment were used as controls. All cells were washed, fixed, permeabilized, and stained with rhodaminated phalloidin for F actin (red), TO-PRO 3 for DNA (blue), and anti-Pet antibodies followed by a secondary anti-rabbit IgG antibody labeled with fluorescein (green). Slides were observed by confocal microscopy. When cells were incubated with the anti-CK8 antibody alone, no effects on HEp-2 cells were observed (data not shown).
Cytokeratins are part of the intermediate filament cytoskeletal system, so additional experiments using small interfering RNA (siRNA) were performed to ensure that their presence on the cell surface did not result from contamination. CK8 expression in HEp-2 cells transfected with siRNA for ck8 was analyzed by Western blot using anti-CK8 antibodies. These cells showed an ~75% decrease in CK8 expression in comparison to the untransfected control cells (Fig. 5A). The ability of Pet to damage these CK8-depleted cells was then assessed by confocal microscopy, using rhodamine-phalloidin to stain the actin cytoskeleton (red) and an anti-CK8 antibody followed by a secondary antibody coupled to Cy5 (blue). In ck8 knockdown cells, very weak staining with the anti-CK8 antibody was detected in comparison to the untransfected cells, which all expressed CK8. The lack of CK8 prevented Pet-induced damage to the actin cytoskeleton of cells treated with siRNA for ck8, whereas untransfected control cells displayed the characteristic cell damage induced by Pet: cytoskeleton disruption, cell rounding, and cell detachment (Fig. 5B). The loss of CK8 expression thus generated a toxin-resistant phenotype, thereby demonstrating a functional role for CK8 in Pet intoxication.
FIG 5 .

Knockdown of CK8 prevents cell damage induced by Pet. (A) Knockdown of CK8 by siRNA. HEp-2 cells were silenced using siRNA for ck8. Cells were lysed and analyzed by Western blotting using anti-CK8 monoclonal antibodies in comparison to normal cells. Results were analyzed by densitometry and plotted as the ratio of CK8 to actin. (B) Effect of Pet on CK8 knockdown cells. HEp-2 cells silenced using siRNA for ck8 were then treated with Pet (37 µg/ml) for 3 h, fixed, permeabilized, and stained with rhodaminated phalloidin (red). CK8 was detected using monoclonal anti-CK8 antibodies followed by a secondary anti-mouse IgG antibody labeled with Cy5 (blue). Slides were observed by confocal microscopy.
A kidney cell line transfected with ck8 becomes susceptible to Pet intoxication.
To further demonstrate that the surface expression of CK8 is required for Pet uptake and cell damage, MDCK cells, which do not normally express CK8 on their surfaces (Fig. 1), were transfected with a ck8-gfp gene. Untransfected and transfected MDCK cells were assessed for CK8 expression and Pet-induced damage to the actin cytoskeleton by confocal microscopy. CK8 was detected via its green fluorescent protein (GFP) fusion partner (green), Pet was detected using anti-Pet antibodies followed by a secondary antibody labeled with Cy5 (blue), and the actin cytoskeleton was stained with rhodamine-phalloidin (red). Untransfected MDCK cells were visualized as an intact monolayer in both the absence and presence of Pet. Transfected MDCK cells, which had not been treated with Pet, were detected in green among the nontransfected cells, and their appearance was similar to that of normal cells. After a 6-h exposure to Pet, these ck8-transfected MDCK cells were damaged by the toxin and had largely detached from the substratum, leaving empty spaces among nontransfected cells (Fig. 6). The expression of CK8 thus corresponded with a gain of sensitivity to Pet intoxication.
FIG 6 .
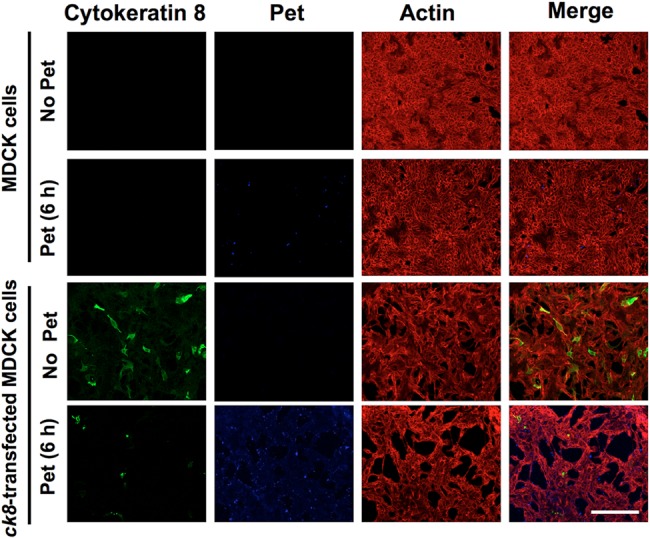
Kidney cells transfected with ck8 are susceptible to cell damage caused by Pet. At 72 h posttransfection, MDCK cells transfected with ck8-gfp were treated with Pet (37 µg/ml) for 6 h. Untransfected and transfected cells were fixed, permeabilized, and stained with rhodamine-phalloidin for F actin (red). Pet was detected using a rabbit polyclonal anti-Pet antibody followed by anti-rabbit IgG secondary antibodies labeled with Cy5 (blue). CK8 was detected using monoclonal anti-CK8 antibodies followed by a secondary anti-mouse IgG antibody labeled with fluorescein (green). Slides were observed by confocal microscopy.
Pet and CK8 undergo clathrin-mediated endocytosis.
We previously showed that intoxication of epithelial cells by Pet requires clathrin-mediated endocytosis (16). Thus, if CK8 is a Pet receptor, we predicted the Pet-CK8 complex would be endocytosed by clathrin-coated vesicles. To test this hypothesis, we performed immunofluorescence experiments using triple-staining confocal scanning microscopy to colocalize Pet, CK8, and clathrin during the endocytosis process. HEp-2 cells treated with Pet for short periods of time (1 to 8 min) were washed, fixed, and permeabilized for confocal microscopy. Antibodies against Pet, CK8, and clathrin were visualized using secondary antibodies coupled to Cy5 (blue), rhodamine (red), and fluorescein (green), respectively. At 1 min of toxin exposure, a slight colocalization between Pet and CK8 was detected (magenta). At 2 min of toxin exposure, it was possible to detect different interactions: clathrin-CK8 (aqua), Pet-CK8 (magenta), and clear triple-staining spots of Pet-CK8-clathrin (white). After 4 min, a better interaction among the three proteins was detected (white). This interaction was less apparent at 8 min; however, the interaction between Pet and CK8 (magenta) was still visible (Fig. 7). These data indicate that Pet and CK8, acting as a ligand/receptor complex, are endocytosed by clathrin-coated vesicles soon after toxin exposure. The persistent colocalization of Pet and CK8, combined with the loss of Pet-CK8-clathrin triple staining after 8 min of toxin exposure, suggests that Pet and CK8 travel as a complex to another cellular compartment such as the early endosomes.
FIG 7 .

Pet bound to CK8 undergoes clathrin-mediated endocytosis. HEp-2 cells were treated with Pet (37 µg/ml) for 1, 2, 4, or 8 min. Cells were then washed, fixed, and permeabilized. Clathrin was detected using a polyclonal anti-clathrin antibody followed by a secondary anti-goat IgG antibody coupled to fluorescein (green), CK8 was detected using a mouse monoclonal anti-CK8 antibody followed by a secondary antibody anti-mouse IgG coupled to rhodamine (red), and Pet was detected using a rabbit anti-Pet polyclonal antibody followed by a secondary anti-rabbit IgG antibody coupled to Cy5 (blue). Slides were observed by confocal microscopy.
Pet-CK8 reaches the early endosomes of epithelial cells.
We have previously documented the vesicle-mediated transport of Pet from the cell surface to the early endosomes (17). Triple-staining confocal microscopy was therefore used to determine if the Pet-CK8 complex is transported to early endosomes. To stain the early endosomes, HEp-2 cells were transduced with the CellLight early endosomes-RFP 2.0 BacMam reagent, which stains Rab5b (red), a resident protein of the early endosomes. These transduced cells were treated with Pet at different times (7, 8, 9, and 10 min) and then stained with anti-Pet and anti-CK8 antibodies followed by secondary antibodies labeled with Cy5 (blue) and fluorescein (green), respectively. At 7 min of toxin exposure, no triple colocalization of the three proteins was detected. However, Pet-CK8-Rab5b triple colocalization (white) in the Rab5b-positive early endosomes was clearly present at 8 min of toxin exposure. Interestingly, at 9 min of toxin exposure, colocalization of the three proteins started to wane. Almost all Pet-CK8-Rab5b triple colocalization was lost at 10 min of toxin exposure, and Pet-CK8 colocalization was also strongly reduced at this interval (Fig. 8). These data indicate that the Pet-CK8 complex reaches the early endosomes and then dissociates, allowing Pet to travel to the ER while CK8 possibly returns to the plasma membrane or is routed to the lysosomes for degradation.
FIG 8 .

Pet and CK8 colocalize as a ligand-receptor complex in early endosomes of epithelial cells. HEp-2 cells were treated overnight with the CellLight early endosomes-RFP BacMam 2.0 reagent to mark Rab5b, a resident protein of early endosomes (red). Cells treated with Pet for 7, 8, 9, or 10 min were then washed, fixed, and permeabilized. CK8 was detected using a mouse monoclonal anti-CK8 antibody followed by a secondary anti-mouse IgG antibody coupled to fluorescein (green), and Pet was detected using a rabbit polyclonal anti-Pet antibody followed by a secondary anti-rabbit IgG antibody coupled to Cy5 (blue). Slides were observed by confocal microscopy.
DISCUSSION
Many AB toxins move from the cell surface to the ER before accessing the host cell cytosol (18). There are a variety of retrograde trafficking pathways to the ER, and the route(s) followed by a particular toxin appears to be dictated by the association of the toxin B subunit with its specific host receptor(s). By following the intracellular trafficking and translocation of Pet, a non-AB toxin, we have shown that an AB structural organization is not required for toxin trafficking to the ER and toxin translocation to the cytosol (17). However, the epithelial surface receptor for Pet remained unknown. Here, we report that Pet does not use the classic ganglioside receptors for AB toxins but instead uses a protein, CK8, that usually forms part of the intermediate filament cytoskeleton.
We previously found that the intoxication of epithelial cells by Pet requires clathrin-mediated endocytosis (16). This suggested a receptor-mediated process, which was consistent with our current observation that Pet binding to the plasma membrane is saturable at 100 nM with a high-affinity Kd of 6.7 nM. Pet binding was also cell type specific and did not occur with kidney cell lines. These binding parameters, which suggested the presence of a specific surface receptor for Pet, are similar to those observed for other bacterial toxins: Shiga toxin (Stx) binding to its Gb3 glycolipid receptor is saturable around 100 nM, with an apparent Kd of 4.2 nM (19), and the binding of Clostridium difficile toxin A to its intestinal receptor is saturable around 90 nM, with a Kd of 60 nM (20). Stx binding is also tissue specific, with Gb3 expression playing a crucial role in determining sensitivity to Stx-induced renal disease (21).
Proteins from the membrane fraction but not from the cytoplasm fraction were captured by the Pet affinity column and identified by MALDI-TOF as members of the cytokeratin family. Since keratin contamination is always observed in MALDI-TOF determinations, additional experiments were designed to confirm these results. Although cytokeratins are proteins of the intermediate filament cytoskeleton, they have also been located on the cell surface (22, 23). For example, surface-expressed CK8 increases the adhesion of MCF-7/MX cells (24) and is further viewed as a valuable target for cancer therapy because of its extracellular epitopes on tumor cells (25). Group B streptococci and other Gram-positive cocci bind to CK8 (26), and cytokeratins have been identified as accessory mediators of Salmonella entry into eukaryotic cells (27). Several new findings show that cytokeratins are also used as receptors for different bacterial proteins, including Tir from EPEC (28), Porphyromonas gingivalis fimbriae (29), and clumping factor B (ClfB) from Staphylococcus aureus (30). Our work has expanded this list and provides strong evidence on the role of CK8 as receptor. More experiments are needed to elucidate how CK8 and other cytokeratins are localized to the cell surface.
CK8 expression is required for Pet intoxication. We documented a direct correlation between CK8 expression and Pet activity, as kidney cells lacking CK8 were resistant to Pet. Ectopic expression of CK8 in these kidneys cells generated a toxin-sensitive phenotype. Cytokeratins are differentially expressed in tissues, with CK8 and CK18 being preferentially expressed in gastrointestinal epithelial cells (31). CK8 is the major but not sole type II keratin of the intestinal intermediate filament network (32). We found that four cytokeratins (CK2a, CK8, CK10, and CK18) were captured by our Pet affinity column, but additional overlay assays showed that only CK8 and CK10 interact directly with Pet. These results indicate that multiple cytokeratins form a complex on the cell surface (32), which is supported by our data showing the interaction of CK8 and CK10 on the plasma membrane of HT-29 cells. Pet may initially interact with this cytokeratin complex via direct binding to CK8 and may disrupt the complex through competitive binding to CK8, as suggested by the interactions between Pet, CK8, and CK10 on the surfaces of HT-29 cells. Additional work is required to understand the interactions between various surface-expressed cytokeratins.
As mentioned above, cytokeratins are detected mainly on the surfaces of cancer cells or as receptors for bacterial proteins involved in virulence, although it is thought that they are only part of the intermediate filaments (33). We accordingly hypothesize that the expression of CK8 and other cytokeratins on the cell surface is related to cell injury or stress (34). Thus, colonization of intercryptal epithelial cells (the older cell population) by bacterial aggregates could stress the cells, leading to cytokeratin expression on their surface. Indeed, in the case of the Stx receptor Gb3, the cell damage depends on the regulation of Gb3 synthesis within this tissue. Although renal glomerular endothelial cells from elderly patients are receptor negative in situ, the isolation and culture of such glomeruli results in the growth of endothelial cells which have an extremely high Gb3 content and are highly sensitive to Stx-induced cytopathology in vitro (35, 36). In the case of CK8, additional experiments are needed to support this hypothetical situation. Nevertheless, our collective evidence indicates that surface-expressed CK8 is a functional receptor for Pet.
Experiments using fixed cells showed the blockage of membrane-localized CK8 by anti-CK8 antibodies prevented Pet binding to the plasma membrane. More excitingly, the blockage of membrane-localized CK8 by anti-CK8 antibodies in living cells led to protection against the cell damage induced by Pet. This protective effect decayed over time, suggesting that a complex of CK8 and the anti-CK8 antibody could be endocytosed with free CK8 recycling to the membrane for another round of binding to Pet. Receptor recycling for toxins has been reported; for instance, a portion of Clostridium perfringens iota toxin b is recycled with its receptor back to the plasma membrane after 120 min (37). Functional experiments also showed that CK8 is an important receptor for the internalization of Pet and intoxication of epithelial cells. Knockdown experiments eliminating CK8 expression from HEp-2 cells showed that CK8 is required for epithelial cell intoxication by Pet. MDCK cells lacking endogenous expression of CK8 were resistant to Pet, but these cells shifted to a toxin-sensitive phenotype upon the ectopic expression of CK8.
Thus, we have used multiple methods to demonstrate that cells without a surface-accessible pool of CK8 are resistant to Pet, whereas the expression of CK8 in previously CK8-deficient cells results in cell damage by Pet. These data indicate that CK8 is a Pet receptor required for gastrointestinal epithelial cell damage. Furthermore, we have shown that CK8 is important for Pet uptake and trafficking to the early endosomes via the detection of a Pet-CK8 complex in this organelle. As we have previously reported, Pet delivery to the early endosomes is another requisite step for Pet retrograde transport to the ER and, ultimately, translocation to the cytosol for interaction with its intracellular target, fodrin.
MATERIALS AND METHODS
Antibodies, bacterial strains, and recombinant proteins.
Primary antibodies used in this study were rabbit anti-Pet as previously described (14); goat anti-clathrin, mouse anti-CK8 monoclonal antibodies, mouse anti-CK10 monoclonal antibodies, and rabbit anti-WASP were from Santa Cruz Biotech (Santa Cruz, CA); mouse anti-β subunit of Na+/K+ ATPase monoclonal antibodies were a gift from Ricardo Felix (Department of Cell Biology, Cinvestav, Mexico); mouse anti-GAPDH monoclonal antibodies were from Chemicon (Billerica, MA). Secondary antibodies used were donkey anti-rabbit IgG conjugated to Cy5, donkey anti-mouse IgG conjugated to Cy5, donkey anti-rabbit IgG conjugated to rhodamine, and donkey anti-mouse IgG conjugated to rhodamine were from Jackson ImmunoResearch Laboratories (West Grove, PA); donkey anti-goat IgG conjugated to fluorescein were from Rockland Antibodies and Assays (Gilbertsville, PA); goat anti-mouse IgG conjugated to fluorescein, goat anti-rabbit IgG conjugated to fluorescein, goat anti-mouse IgG conjugated to horseradish peroxidase (HRP), and goat anti-rabbit IgG conjugated to HRP were from Zymed Lab, Inc. (San Francisco, CA).
The minimal Pet clone pCEFN1 was constructed by cloning the pet gene of enteroaggregative E. coli strain 042 as previously described (13). E. coli HB101 was transformed with pCEFN1 and maintained on L agar or in L broth containing 100 µg/ml ampicillin. The pCEFN2 clone encoding the mutant Pet-S260I (serine protease mutant of Pet) was obtained by site-directed mutagenesis (10). To obtain Pet or Pet-S260I, broth cultures of HB101(pCEFN1) or HB101(pCEFN2) were incubated overnight at 37°C and then centrifuged at 7,000 × g for 15 min. The culture supernatant was filtered and concentrated 100-fold in an Ultrafree centrifugal filter device with a 100-kDa cutoff (Millipore, Bedford, MA), as previously described (10). Culture medium from non-Pet-expressing HB101(pSPORT1) was concentrated as described above and used as a negative control for immunofluorescence and toxicity assays.
Cell culture.
HEp-2 (from human larynx), HT-29 (from human colon), MDCK (from dog kidney), and RK13 (from rabbit kidney) cells were propagated in humidified 5% CO2–95% air at 37°C in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum (HyClone, Logan, UT), 1% nonessential amino acids, 5 mM l-glutamine, penicillin (100 units/ml), and streptomycin (100 µg/ml). Subcultures were serially propagated after harvesting with 10 mM EDTA and 0.25% trypsin (GIBCO BRL) in phosphate-buffered saline solution (PBS, pH 7.4). For fluorescence experiments, subconfluent cells were resuspended with EDTA-trypsin, plated in eight-well Lab-Tek slides (VWR, Bridgeport, NJ), and allowed to grow ~24 h to 70% confluence before use. For immunoprecipitation assays, cells were plated in 60-mm petri dishes and allowed to grow for ~24 h to 40 to 80% confluence before use.
For cell transfection, MDCK cells were transfected with the pCMV6-AC-GFP plasmid (1 µg per well of eight-well Lab-Tek slides) for expression of GFP-CK8 (Homo sapiens keratin 8 accession number NM_002273). Additional cells were incubated with the empty vector (OriGene) as a control. Lipofectamine 2000 (Invitrogen) was used as the transfection reagent according to the manufacturer’s protocol. Cells were examined 48 to 72 h after transfection.
Cell fractionation.
HEp-2 cells grown in 60-mm petri dishes were delicately washed three times with ice-cold PBS, pH 7.4 and scraped into a buffer consisting of Tris-HCl (2 mM) (pH 7.6), EDTA (1 mM), MgCl2 (1.5 mM), HEPES (10 mM), KCl (10 mM), and a protease inhibitor cocktail (Complete). Then, cells were lysed by three repeating sonication cycles (15 s each at intervals of 15 s). Cell lysates were ultracentrifuged at 100,000 × g for 1 h at 4°C; soluble cytoplasmic proteins were obtained from the supernatant fraction, and the membrane fraction was obtained from the pellet. Equivalent volumes were boiled for 7 min, resolved by SDS-PAGE, and electrotransferred to nitrocellulose membranes for Western blot analyses. The identity of cellular fractions was confirmed with a mouse anti-GADPH monoclonal antibody for cytosolic proteins and a mouse anti-β subunit of Na+/K+ ATPase monoclonal antibody for membrane insoluble fraction.
ELISA.
HEp-2 or HT-29 cells were grown at a density of 4 × 104 cell/well in 96-wells plates (Costar, Corning) and incubated to reach a confluence of 90 to 100%. Washed cells were fixed with 2.5% paraformaldehyde (PFA) for 30 min. Wells were blocked with 1% BSA-PBS, and Pet was added at 1, 5, 10, 50, 80, 100, 200, 300, 400, and 500 nM at 37°C for 90 min. Pet binding to epithelial cells was immunodetected with a standard ELISA (38) using primary antibodies against Pet and the corresponding HRP-conjugated secondary antibody (Zymed). The absorbance values for the enzymatic reactions at 490 nm were registered in an ELISA microplate reader 550 (Bio-Rad). For the inhibition experiments, before Pet incubation, cells were incubated with anti-CK8 or anti-CK10 and then incubated with Pet at different concentrations to follow the ELISA as indicated above. For Pet-rCK8 interaction, 20 nM recombinant CK8 in coating buffer (sodium bicarbonate 0.5 M and sodium carbonate 0.5 M, pH 9.5) was adsorbed to 96-well plates overnight at 4°C. Pet binding to rCK8 was detected by ELISA as described above.
Overlay assay.
Overlay assays were performed as previously reported (11). Lysed cells (30 µg), membrane fraction (30 µg), and cytoplasm fraction (30 µg) were separated by SDS-PAGE in 12% polyacrylamide gels. Proteins were transferred to nitrocellulose membranes (Bio-Rad) and then incubated for 1 h in binding buffer (20 mM Tris-HCl, 150 mM NaCl, 0.1% Tween 20, 2 mM CaCl2, and 5% bovine serum albumin) with 5 µg/ml of Pet-S260I. The membranes were washed and then incubated for 1 h in blocking buffer with rabbit polyclonal anti-Pet antibodies (1:500 dilution). Following another washing step, membranes were incubated for 1 h in blocking buffer with an HRP-conjugated goat anti-rabbit IgG antibody as indicated by the manufacturer. HRP was detected with the Western blotting reagent luminol from Santa Cruz Biotechnology.
Pet affinity column and purification.
Pet-S260I protein (5 µg) dissolved in coupling buffer (0.1 M NaHCO3 [pH 8.3], 0.5 M NaCl) was added to 100 mg of cyanogen bromide-activated Sepharose 4B (Sigma) for 2 h at ambient temperature (AT). The resin had been incubated in 8 ml of 1 mM HCl overnight at AT. The unbound sites of the Pet-bound gel were blocked by incubating the gel with 0.2 M glycine (pH 8) for 2 h at AT. The resin was washed with 0.1 M sodium acetate (pH 7), 0.5 M NaCl.
Lysates or cellular fractions (5 mg/ml) were incubated with the resin for 1 h and washed 3 times with 10 ml of Tris-buffered saline. Finally, bound proteins to the Pet affinity resin were separated using 0.1 M glycine-HCl (pH 2.8). The different elution fractions obtained by centrifugation were analyzed by SDS-PAGE.
Protein identification by MALDI-TOF.
Candidate proteins isolated by the affinity column assays were resolved by SDS-PAGE and stained with Coomassie blue. Excised gel slices were destained overnight (50% methanol, 5% acetic acid). After washing with acetonitrile, proteins in the gel slices were reduced and alkylated with 10 mM dithiothreitol (DTT) and 50 mM iodoacetamide, respectively. Gel slices were then treated with 50 mM ammonium bicarbonate and washed with acetonitrile. Protein was extracted from the gel slice using 5% formic acid and 50% acetonitrile solution. Enzymatic digestion of the extracted protein was performed with trypsin, and MALDI-TOF analysis was performed with a Voyager-DE PRO mass spectrometer, as we previously reported (39).
Fluorescence assays.
Pet or Pet-S260I was diluted directly into tissue culture medium without antibiotics or serum at a final concentration of 37 µg/ml and then added to the target cells (HEp-2, HT-29, RK-13, or MDCK cells) at a final volume of 250 µl per well in eight-well Lab-Tek slides. Following incubation for various times in a humidified atmosphere of 5% CO2–95% air at 37°C, the medium was aspirated, cells were washed twice with PBS, and 2% formalin in PBS was added for 20 min at AT. Fixed cells were either not permeabilized or permeabilized by adding 0.2% Triton X-100 in PBS for 5 min at AT.
Actin filaments were visualized in the permeabilized cells by incubation with 0.05 µg/ml tetramethyl rhodamine isothiocyanate (TRITC)-phalloidin for 30 min at AT (40). The early endosome was visualized in permeabilized cells by incubation with CellLight early endosomes conjugated to red fluorescent protein (RFP), BacMam 2.0, overnight to detect Rab5b, a resident protein of early endosomes. Proteins other than actin or Rab5b were visualized by incubation with the specified primary antibody for 1 h at AT, followed by incubation for 1 h at AT with the appropriate secondary antibody. Slides were mounted on Gelvatol, covered with a glass coverslip, and examined under a Leica TCS SP5 confocal microscope at a magnification of ×100 or ×63.
Coimmunoprecipitation assays.
Cultured HEp-2, HT-29, RK-13, or MDCK cells in 60-mm by 15-mm cell culture dishes were fractionated into cytoplasm and membrane fractions as described above. To perform the immunoprecipitation assay, the lysed cells or cellular fractions (100 µg) were incubated with Pet-S260I (10 µg) with slight agitation overnight at 4°C. Then the solution was incubated with anti-Pet, anti-CK8, or anti-CK10 antibodies (according to the specific case) for 2 h at 4°C. After that, 15 µl of protein A-agarose suspension (Roche Diagnostics, Mannheim, Germany) was added for 3 h at 4°C. The complexes were collected by centrifugation at 12,000 × g for 20 s, and the supernatant was removed. The pellet was washed five times with cold PBS containing Complete protease inhibitor. The agarose pellet was resuspended in 2× gel-loading buffer and resolved by SDS-PAGE. The resulting protein bands were transferred to nitrocellulose membranes (41), which were probed with rabbit anti-Pet antibodies at 1:500 or anti-CK8 or anti-CK10 antibodies at 1:1,000 in PBS (according to the specific case). Antigen-antibody reactions were visualized using HRP-labeled goat anti-rabbit or anti-mouse IgG antibodies and developed using the chemiluminescent reagent luminol as described above.
RNA interference.
Subconfluent cultures (~70 to 80%) of HEp-2 cells in 35-mm plates were transfected for 7 h with a mixture containing 80 pmol of synthetic double-stranded siRNA for CK8 (sc-35156; Santa Cruz Biotechnology) and 6 µl of Lipofectamine 2000 (Invitrogen) in a total volume of 1 ml of DMEM without either antibiotics or serum. After 72 h, transfected cells and untransfected controls were rinsed extensively with PBS after treatment and collected in lysis buffer using a cell scraper. Relative CK8 and actin levels were analyzed by immunoblot in 60 µg of total protein using the chemiluminescent reagent luminol, as described above. The resulting autoradiograph was scanned and quantified with the ImageJ 1.43 software (available free of charge at http://rsbweb.nih.gov/ij/), to determine the intensity. Transfected cells and untransfected controls were grown in an eight-well Lab-Tek Chamber slide, treated with Pet for 3 h, immunostained with the appropriated antibodies, and examined by confocal microscopy (Leica TCS SP2) as described above.
SUPPLEMENTAL MATERIAL
Pet binds to epithelial membrane proteins. (A) Pet binding to epithelial cells detected by ELISA. HEp-2 cells were seeded in ELISA plates of 96 wells were then washed, fixed, and blocked with BSA. Cells were incubated with Pet at different concentrations for 90 min. Pet binding was determined using anti-Pet antibody, followed by anti-rabbit IgG antibody conjugated to HRP. Symbols represent experimental data, while the line represents the fitted model for that data analyzed with Prism4 (GraphPad Software, La Jolla, CA). Values are the means for triplicate samples from three independent experiments. (B) Pet binding to epithelial cell membrane proteins detected by affinity columns. Five micrograms of Pet-S260I were coupled to a Sepharose column. HT-29 cells were fractionated to obtain membrane and cytoplasmic fractions. Either membrane or cytoplasmic fractions were passed through the column, and the proteins interacting with Pet were analyzed by SDS-PAGE. Arrows indicate bands for the following proteins: (i) CK8, (ii) CK8, (iii) CK18, (iv) CK18, (v) CK2a, and (vi) CK10. (C) Pet binding to epithelial cell membrane proteins detected by overlay. Cell lysates, membrane fractions, and cytoplasmic fractions from HT-29 cells were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and incubated with purified Pet-S260I. Pet binding to host proteins was detected using a rabbit anti-Pet antibody and a secondary anti-rabbit IgG antibody conjugated with HRP. (D) Anti-Pet antibodies do not detect any epithelial cell protein. Western blot analysis was performed to confirm the specific interaction between Pet and membrane proteins of HT-29 cells. Cell lysates, membrane fractions, and cytoplasmic fractions from HT-29 cells were separated by SDS-PAGE and transferred to a nitrocellulose membrane. Purified Pet-S260I was loaded into a fourth lane as a positive control. The membrane was exposed to a rabbit anti-Pet antibody and a secondary anti-rabbit IgG antibody conjugated with HRP. Download
CK8 and CK10 are present on epithelial cell membranes but not on kidney cells. HT-29, HEp-2, RK-13, and MDCK cells were fixed without permeabilization. Nuclei were stained with TO-PRO 3, while CK8 and CK10 were detected using mouse monoclonal anti-CK8 or anti-CK10 antibodies followed by fluorescein-labeled secondary anti-mouse IgG antibodies. The slides were observed by confocal microscopy. Download
Epithelial cells from kidney cell lines are not susceptible to Pet. MDCK, RK13, and Vero kidney cells were treated with Pet (37 µg/ml) for 6 h, and as susceptible cells, HEp-2 and HT29 epithelial cells were also treated with Pet for 4 h. Unintoxicated control cells and intoxicated cells were fixed, permeabilized, and stained with rhodamine-phalloidin (red) to detect actin cytoskeleton damage (arrows). Slides were observed by confocal microscopy. Download
Pet affinity columns do not retain cytokeratins from membrane fractions of kidney cell lines. (A) Pet affinity column. Five micrograms of Pet-S260I was coupled to a Sepharose column. MDCK (from dog kidney) and HK-293 (from human kidney) cells were fractionated to obtain cell lysates and membrane and cytoplasmic fractions. HT-29 cells were used as positive controls. Cell lysates, membrane fractions, and cytoplasmic fractions were passed through the column, and the proteins interacting with Pet were analyzed by SDS-PAGE. (B) Pet binding to kidney cell membrane proteins is not detected by overlay. Cell lysates, membrane fractions, and cytoplasmic fractions from HK-293 cells were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and incubated with Pet-S260I. Purified Pet-S260I was loaded into a fourth lane as a positive control. The membrane was exposed to a rabbit anti-Pet antibody and a secondary anti-rabbit IgG antibody conjugated with HRP. Download
Pet slightly binds to CK10 of epithelial cell plasma membrane (A). CK10 coimmunoprecipitates with Pet. Membrane or cytoplasmic fractions from HT-29, HEp-2, RK-13, or MDCK cells incubated overnight with Pet-S260I were subjected to a coimmunoprecipitation assay using anti-Pet antibody. The immunocomplexes were separated by SDS-PAGE and analyzed by Western blotting using anti-CK10 antibodies. (B) Pet or CK8 coimmunoprecipitates with CK10. Membrane or cytoplasmic fractions of HT-29 cells were incubated with Pet-S260I overnight and were then subjected to a coimmunoprecipitation assay using anti-CK10 antibodies. The immunocomplexes were transferred to a nitrocellulose membrane and analyzed by Western blotting using either anti-Pet or anti-CK8 antibodies. Download
ACKNOWLEDGMENTS
This work was supported by a grant from Consejo Nacional de Ciencia y Tecnología (CONACYT; 60714 and 128490) to FNG.
We thank Ken Teter (University of Central Florida) and Edward Dudley (Penn State University) for invaluable critical review of the manuscript and Humberto Lanz-Mendoza (Centro de Investigaciones sobre Enfermedades Infecciosas, INSP) for his help with MALDI-TOF assays. We also thank Lucia Chavez-Dueñas, Gabriela Tapia-Pastrana, and Jazmin Huerta for their technical support and Adrian Canizalez-Roman for some preliminary results.
Footnotes
Citation Nava-Acosta R, Navarro-Garcia F. 2013. Cytokeratin 8 is an epithelial cell receptor for Pet, a cytotoxic serine protease autotransporter of Enterobacteriaceae. mBio 4(6):e00838-13. doi:10.1128/mBio.00838-13.
REFERENCES
- 1. Ruiz-Perez F, Nataro JP. Bacterial serine proteases secreted by the autotransporter pathway: classification, specificity, and role in virulence. Cell. Mol. Life Sci., in press 10.1007/s00018-013-1355-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Henderson IR, Navarro-Garcia F, Desvaux M, Fernandez RC, Ala’Aldeen D. 2004. Type V protein secretion pathway: the autotransporter story. Microbiol. Mol. Biol. Rev. 68:692–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yen YT, Kostakioti M, Henderson IR, Stathopoulos C. 2008. Common themes and variations in serine protease autotransporters. Trends Microbiol. 16:370–379 [DOI] [PubMed] [Google Scholar]
- 4. Boisen N, Ruiz-Perez F, Scheutz F, Krogfelt KA, Nataro JP. 2009. Short report: high prevalence of serine protease autotransporter cytotoxins among strains of enteroaggregative Escherichia coli. Am. J. Trop. Med. Hyg. 80:294–301 [PMC free article] [PubMed] [Google Scholar]
- 5. Rasko DA, Webster DR, Sahl JW, Bashir A, Boisen N, Scheutz F, Paxinos EE, Sebra R, Chin CS, Iliopoulos D, Klammer A, Peluso P, Lee L, Kislyuk AO, Bullard J, Kasarskis A, Wang S, Eid J, Rank D, Redman JC, Steyert SR, Frimodt-Møller J, Struve C, Petersen AM, Krogfelt KA, Nataro JP, Schadt EE, Waldor MK. 2011. Origins of the E. coli strain causing an outbreak of hemolytic-uremic syndrome in Germany. N. Engl. J. Med. 365:709–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Al-Hasani K, Navarro-Garcia F, Huerta J, Sakellaris H, Adler B. 2009. The immunogenic SigA enterotoxin of Shigella flexneri 2a binds to HEp-2 cells and induces fodrin redistribution in intoxicated epithelial cells. PLoS One 4:e8223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Djafari S, Ebel F, Deibel C, Krämer S, Hudel M, Chakraborty T. 1997. Characterization of an exported protease from Shiga toxin-producing Escherichia coli. Mol. Microbiol. 25:771–784 [DOI] [PubMed] [Google Scholar]
- 8. Maroncle NM, Sivick KE, Brady R, Stokes FE, Mobley HL. 2006. Protease activity, secretion, cell entry, cytotoxicity, and cellular targets of secreted autotransporter toxin of uropathogenic Escherichia coli. Infect. Immun. 74:6124–6134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Navarro-García F, Canizalez-Roman A, Sui BQ, Nataro JP, Azamar Y. 2004. The serine protease motif of EspC from enteropathogenic Escherichia coli produces epithelial damage by a mechanism different from that of Pet toxin from enteroaggregative E. coli. Infect. Immun. 72:3609–3621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Navarro-García F, Sears C, Eslava C, Cravioto A, Nataro JP. 1999. Cytoskeletal effects induced by pet, the serine protease enterotoxin of enteroaggregative Escherichia coli. Infect. Immun. 67:2184–2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Canizalez-Roman A, Navarro-García F. 2003. Fodrin CaM-binding domain cleavage by Pet from enteroaggregative Escherichia coli leads to actin cytoskeletal disruption. Mol. Microbiol. 48:947–958 [DOI] [PubMed] [Google Scholar]
- 12. Navarro-Garcia F. 2010. Enteroaggregative Escherichia coli plasmid-encoded toxin. Future Microbiol. 5:1005–1013 [DOI] [PubMed] [Google Scholar]
- 13. Eslava C, Navarro-García F, Czeczulin JR, Henderson IR, Cravioto A, Nataro JP. 1998. Pet, an autotransporter enterotoxin from enteroaggregative Escherichia coli. Infect. Immun. 66:3155–3163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Navarro-García F, Eslava C, Villaseca JM, López-Revilla R, Czeczulin JR, Srinivas S, Nataro JP, Cravioto A. 1998. In vitro effects of a high-molecular-weight heat-labile enterotoxin from enteroaggregative Escherichia coli. Infect. Immun. 66:3149–3154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Henderson IR, Hicks S, Navarro-Garcia F, Elias WP, Philips AD, Nataro JP. 1999. Involvement of the enteroaggregative Escherichia coli plasmid-encoded toxin in causing human intestinal damage. Infect. Immun. 67:5338–5344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Navarro-García F, Canizalez-Roman A, Vidal JE, Salazar MI. 2007. Intoxication of epithelial cells by plasmid-encoded toxin requires clathrin-mediated endocytosis. Microbiology 153:2828–2838 [DOI] [PubMed] [Google Scholar]
- 17. Navarro-García F, Canizalez-Roman A, Burlingame KE, Teter K, Vidal JE. 2007. Pet, a non-AB toxin, is transported and translocated into epithelial cells by a retrograde trafficking pathway. Infect. Immun. 75:2101–2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sandvig K, van Deurs B. 2005. Delivery into cells: lessons learned from plant and bacterial toxins. Gene Ther. 12:865–872 [DOI] [PubMed] [Google Scholar]
- 19. Gallegos KM, Conrady DG, Karve SS, Gunasekera TS, Herr AB, Weiss AA. 2012. Shiga toxin binding to glycolipids and glycans. PLoS One 7:e30368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pothoulakis C, Gilbert RJ, Cladaras C, Castagliuolo I, Semenza G, Hitti Y, Montcrief JS, Linevsky J, Kelly CP, Nikulasson S, Desai HP, Wilkins TD, LaMont JT. 1996. Rabbit sucrase-isomaltase contains a functional intestinal receptor for Clostridium difficile toxin A. J. Clin. Invest. 98:641–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu XH, Lingwood CA, Ray PE. 1999. Recruitment of renal tubular epithelial cells expressing verotoxin-1 (Stx1) receptors in HIV-1 transgenic mice with renal disease. Kidney Int. 55:554–561 [DOI] [PubMed] [Google Scholar]
- 22. Hembrough TA, Vasudevan J, Allietta MM, Glass WF, Gonias SL. 1995. A cytokeratin 8-like protein with plasminogen-binding activity is present on the external surfaces of hepatocytes, HepG2 cells and breast carcinoma cell lines. J. Cell Sci. 108:1071–1082 [DOI] [PubMed] [Google Scholar]
- 23. Wells MJ, Hatton MW, Hewlett B, Podor TJ, Sheffield WP, Blajchman MA. 1997. Cytokeratin 18 is expressed on the hepatocyte plasma membrane surface and interacts with thrombin-antithrombin complexes. J. Biol. Chem. 272:28574–28581 [DOI] [PubMed] [Google Scholar]
- 24. Liu F, Chen Z, Wang J, Shao X, Cui Z, Yang C, Zhu Z, Xiong D. 2008. Overexpression of cell surface cytokeratin 8 in multidrug-resistant MCF-7/MX cells enhances cell adhesion to the extracellular matrix. Neoplasia 10:1275–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gires O, Andratschke M, Schmitt B, Mack B, Schaffrik M. 2005. Cytokeratin 8 associates with the external leaflet of plasma membranes in tumour cells. Biochem. Biophys. Res. Commun. 328:1154–1162 [DOI] [PubMed] [Google Scholar]
- 26. Tamura GS, Nittayajarn A. 2000. Group B streptococci and other gram-positive cocci bind to cytokeratin 8. Infect. Immun. 68:2129–2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Carlson SA, Omary MB, Jones BD. 2002. Identification of cytokeratins as accessory mediators of Salmonella entry into eukaryotic cells. Life Sci. 70:1415–1426 [DOI] [PubMed] [Google Scholar]
- 28. Batchelor M, Guignot J, Patel A, Cummings N, Cleary J, Knutton S, Holden DW, Connerton I, Frankel G. 2004. Involvement of the intermediate filament protein cytokeratin-18 in actin pedestal formation during EPEC infection. EMBO Rep. 5:104–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sojar HT, Sharma A, Genco RJ. 2002. Porphyromonas gingivalis fimbriae bind to cytokeratin of epithelial cells. Infect. Immun. 70:96–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Haim M, Trost A, Maier CJ, Achatz G, Feichtner S, Hintner H, Bauer JW, Onder K. 2010. Cytokeratin 8 interacts with clumping factor B: a new possible virulence factor target. Microbiology 156:3710–3721 [DOI] [PubMed] [Google Scholar]
- 31. Ku NO, Zhou X, Toivola DM, Omary MB. 1999. The cytoskeleton of digestive epithelia in health and disease. Am. J. Physiol. 277:G1108–G1137 [DOI] [PubMed] [Google Scholar]
- 32. Moll R, Franke WW, Schiller DL, Geiger B, Krepler R. 1982. The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cell 31:11–24 [DOI] [PubMed] [Google Scholar]
- 33. Riopel CL, Butt I, Omary MB. 1993. Method of cell handling affects leakiness of cell surface labeling and detection of intracellular keratins. Cell Motil. Cytoskeleton 26:77–87 [DOI] [PubMed] [Google Scholar]
- 34. Gonias SL, Braud LL, Geary WA, VandenBerg SR. 1989. Plasminogen binding to rat hepatocytes in primary culture and to thin slices of rat liver. Blood 74:729–736 [PubMed] [Google Scholar]
- 35. Lingwood CA. 1999. Verotoxin/globotriaosyl ceramide recognition: angiopathy, angiogenesis and antineoplasia. Biosci. Rep. 19:345–354 [DOI] [PubMed] [Google Scholar]
- 36. Obrig TG, Louise CB, Lingwood CA, Boyd B, Barley-Maloney L, Daniel TO. 1993. Endothelial heterogeneity in Shiga toxin receptors and responses. J. Biol. Chem. 268:15484–15488 [PubMed] [Google Scholar]
- 37. Nagahama M, Umezaki M, Tashiro R, Oda M, Kobayashi K, Shibutani M, Takagishi T, Ishidoh K, Fukuda M, Sakurai J. 2012. Intracellular trafficking of Clostridium perfringens iota-toxin b. Infect. Immun. 80:3410–3416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Salazar-Gonzalez H, Navarro-Garcia F. 2011. Intimate adherence by enteropathogenic Escherichia coli modulates TLR5 localization and proinflammatory host response in intestinal epithelial cells. Scand. J. Immunol. 73:268–283 [DOI] [PubMed] [Google Scholar]
- 39. Tapia-Pastrana G, Chavez-Dueñas L, Lanz-Mendoza H, Teter K, Navarro-García F. 2012. VirK is a periplasmic protein required for efficient secretion of plasmid-encoded toxin from enteroaggregative Escherichia coli. Infect. Immun. 80:2276–2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Knutton S, Phillips AD, Smith HR, Gross RJ, Shaw R, Watson P, Price E. 1991. Screening for enteropathogenic Escherichia coli in infants with diarrhea by the fluorescent-actin staining test. Infect. Immun. 59:365–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Towbin H, Staehelin T, Gordon J. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. U. S. A. 76:4350–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Pet binds to epithelial membrane proteins. (A) Pet binding to epithelial cells detected by ELISA. HEp-2 cells were seeded in ELISA plates of 96 wells were then washed, fixed, and blocked with BSA. Cells were incubated with Pet at different concentrations for 90 min. Pet binding was determined using anti-Pet antibody, followed by anti-rabbit IgG antibody conjugated to HRP. Symbols represent experimental data, while the line represents the fitted model for that data analyzed with Prism4 (GraphPad Software, La Jolla, CA). Values are the means for triplicate samples from three independent experiments. (B) Pet binding to epithelial cell membrane proteins detected by affinity columns. Five micrograms of Pet-S260I were coupled to a Sepharose column. HT-29 cells were fractionated to obtain membrane and cytoplasmic fractions. Either membrane or cytoplasmic fractions were passed through the column, and the proteins interacting with Pet were analyzed by SDS-PAGE. Arrows indicate bands for the following proteins: (i) CK8, (ii) CK8, (iii) CK18, (iv) CK18, (v) CK2a, and (vi) CK10. (C) Pet binding to epithelial cell membrane proteins detected by overlay. Cell lysates, membrane fractions, and cytoplasmic fractions from HT-29 cells were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and incubated with purified Pet-S260I. Pet binding to host proteins was detected using a rabbit anti-Pet antibody and a secondary anti-rabbit IgG antibody conjugated with HRP. (D) Anti-Pet antibodies do not detect any epithelial cell protein. Western blot analysis was performed to confirm the specific interaction between Pet and membrane proteins of HT-29 cells. Cell lysates, membrane fractions, and cytoplasmic fractions from HT-29 cells were separated by SDS-PAGE and transferred to a nitrocellulose membrane. Purified Pet-S260I was loaded into a fourth lane as a positive control. The membrane was exposed to a rabbit anti-Pet antibody and a secondary anti-rabbit IgG antibody conjugated with HRP. Download
CK8 and CK10 are present on epithelial cell membranes but not on kidney cells. HT-29, HEp-2, RK-13, and MDCK cells were fixed without permeabilization. Nuclei were stained with TO-PRO 3, while CK8 and CK10 were detected using mouse monoclonal anti-CK8 or anti-CK10 antibodies followed by fluorescein-labeled secondary anti-mouse IgG antibodies. The slides were observed by confocal microscopy. Download
Epithelial cells from kidney cell lines are not susceptible to Pet. MDCK, RK13, and Vero kidney cells were treated with Pet (37 µg/ml) for 6 h, and as susceptible cells, HEp-2 and HT29 epithelial cells were also treated with Pet for 4 h. Unintoxicated control cells and intoxicated cells were fixed, permeabilized, and stained with rhodamine-phalloidin (red) to detect actin cytoskeleton damage (arrows). Slides were observed by confocal microscopy. Download
Pet affinity columns do not retain cytokeratins from membrane fractions of kidney cell lines. (A) Pet affinity column. Five micrograms of Pet-S260I was coupled to a Sepharose column. MDCK (from dog kidney) and HK-293 (from human kidney) cells were fractionated to obtain cell lysates and membrane and cytoplasmic fractions. HT-29 cells were used as positive controls. Cell lysates, membrane fractions, and cytoplasmic fractions were passed through the column, and the proteins interacting with Pet were analyzed by SDS-PAGE. (B) Pet binding to kidney cell membrane proteins is not detected by overlay. Cell lysates, membrane fractions, and cytoplasmic fractions from HK-293 cells were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and incubated with Pet-S260I. Purified Pet-S260I was loaded into a fourth lane as a positive control. The membrane was exposed to a rabbit anti-Pet antibody and a secondary anti-rabbit IgG antibody conjugated with HRP. Download
Pet slightly binds to CK10 of epithelial cell plasma membrane (A). CK10 coimmunoprecipitates with Pet. Membrane or cytoplasmic fractions from HT-29, HEp-2, RK-13, or MDCK cells incubated overnight with Pet-S260I were subjected to a coimmunoprecipitation assay using anti-Pet antibody. The immunocomplexes were separated by SDS-PAGE and analyzed by Western blotting using anti-CK10 antibodies. (B) Pet or CK8 coimmunoprecipitates with CK10. Membrane or cytoplasmic fractions of HT-29 cells were incubated with Pet-S260I overnight and were then subjected to a coimmunoprecipitation assay using anti-CK10 antibodies. The immunocomplexes were transferred to a nitrocellulose membrane and analyzed by Western blotting using either anti-Pet or anti-CK8 antibodies. Download