

Abstract
Heart failure is a complex disease that involves genetic, environmental, and physiological factors. As a result, current medication and treatment for heart failure produces limited efficacy, and better medication is in demand. Although mammalian models exist, simple and low-cost models will be more beneficial for drug discovery and mechanistic studies of heart failure. We previously reported that aristolochic acid (AA) caused cardiac defects in zebrafish embryos that resemble heart failure. Here, we showed that cardiac troponin T and atrial natriuretic peptide were expressed at significantly higher levels in AA-treated embryos, presumably due to cardiac hypertrophy. In addition, several human heart failure drugs could moderately attenuate the AA-induced heart failure by 10%–40%, further verifying the model for drug discovery. We then developed a drug screening assay using the AA-treated zebrafish embryos and identified three compounds. Mitogen-activated protein kinase kinase inhibitor (MEK-I), an inhibitor for the MEK-1/2 known to be involved in cardiac hypertrophy and heart failure, showed nearly 60% heart failure attenuation. C25, a chalcone derivative, and A11, a phenolic compound, showed around 80% and 90% attenuation, respectively. Time course experiments revealed that, to obtain 50% efficacy, these compounds were required within different hours of AA treatment. Furthermore, quantitative polymerase chain reaction showed that C25, not MEK-I or A11, strongly suppressed inflammation. Finally, C25 and MEK-I, but not A11, could also rescue the doxorubicin-induced heart failure in zebrafish embryos. In summary, we have established two tractable heart failure models for drug discovery and three potential drugs have been identified that seem to attenuate heart failure by different mechanisms.
Introduction
Heart failure is characterized by the gradual deterioration of cardiac function, culminating in erratic heart rhythm, edema, and death. The disease is one of the leading causes of death in the United States, afflicting ∼6.6 million U.S. adults ≥18 years of age (2.8%) according to the Center for Disease Control.1 Approximately 50% of people diagnosed with heart failure will die within 5 years.1 Most heart failure is chronic and commonly results from long-term hypertension or cardiovascular diseases in the elderly.2 However, acute heart failure can occur due to sudden worsening of chronic heart failure condition, infection, or toxins, such as anthracycline3 and chemotherapeutic treatments.4 Weakening heart function often causes congestion, or fluid buildup in the lungs and other tissues, by hindering the flow of blood through the chambers of the heart. As a reflex response, several physiological systems, including the neurohormone, anti-diuresis, and rennin-angiotensin system, are triggered upon stress to the heart's normal function to compensate the insufficient cardiac output.2 These compensatory mechanisms aim at restoring the normal cardiac output by increasing (1) cardiac contractility and/or heart rate accompanied by cardiac hypertrophy, (2) blood vessel contraction, and (3) blood volume by increasing water reabsorption in kidney. These mechanisms, in turn, push the heart into a vicious cycle, which could lead to acute decompensated heart failure or sudden cardiac arrest due to cardiac overload. Recently, the inflammation system was found to be associated with and become another risk and predictive factor for heart failure progression.5 Due to the complex and still poorly understood physiological interplay preceded or triggered by heart failure, clinical treatment is still striving to find the best regime for individual patients.
To reduce the physical symptoms and protect the heart from sudden rupture, clinical interventions aim at counteracting the mentioned compensatory physiological systems while carefully balancing the cardiovascular and renal functions. As a result, patient conditions need to be evaluated and monitored frequently followed by necessary adjustments to prevent adverse effects and to achieve the best therapeutic outcome. Despite this, limitation is common in almost all the current heart failure drugs. For example, β-blocker is prescribed for heart failure patients to protect the overworking heart by counteracting the effect of neurohormone, which raises the heart rate and contractility.6 However, β-blocker decreases cardiac inotropy and is only beneficial to some forms of chronic heart failure.7–9 Inhibitors targeting the rennin-angiotensin signaling, such as angiotensin-converting enzyme inhibitor (ACE-I) and angiotensin receptor blocker (ARB), can effectively improve the heart failure symptoms but might cause hypotension and renal dysfunction due to over-diuresis.7,10 Levosimendan is a calcium sensitizer that increases cardiac contractility and quickly relieves the symptoms of acute heart failure but does not significantly reduce mortality at 180 days.11 In summary, many drugs show temporary and limited efficacy with regard to heart failure, possibly due to the specific and local effect and/or uncharacterized toxicity. Therefore, the demand of heart failure medications is still high.
Many animal models, mostly mammals, have been established to study the mechanisms of heart failure and to test the drug effect.12–14 These models, although established by diverse methods, are qualified by demonstrating the typical cardiac defects along with the specific molecular risk factors, such as elevated levels of cardiac troponin T (cTnT) and several types of natriuretic peptides.15 While these models are excellent tools for heart failure studies, a few of them are ideal for drug discovery. For example, the murine models are expensive and time and labor intensive, with some experiments taking approximately several months each.12 Recently, zebrafish embryos have shown many advantages for human disease studies and drug discovery.16 We previously reported a potential heart failure model using zebrafish embryos and aristolochic acid (AA).17 This model is low cost, easily observed, and has a very rapid experimental rate. The AA-treated zebrafish embryos developed cardiac hypertrophy, severed cardiac fibers, loss of endocardium, and gradual weakening and subsequent cessation of cardiac contractility within 2 days of treatment. Two drugs for human heart failure, metoprolol (a β-blocker)6 and captopril (an ACE-I),10 exhibited beneficial effects on the AA-induced heart failure. Furthermore, we showed that the anti-inflammation drug NS398, which selectively inhibits cyclooxygenase-2 (COX-2),18 could effectively attenuate the AA-induced heart failure. The ability of NS398 to attenuate heart failure was also reported with a mouse heart failure model.12 Thus, our AA-treated zebrafish model presents a promising avenue to drug discovery for heart failure.
Doxorubicin (DOX) is a type of anthracycline antibiotic that stops cell growth by interchelating DNA and inhibiting DNA replication. It was also found to be effective in suppressing the growth of many types of cancers and became a common agent for chemotherapy. However, DOX is found to have serious cardiotoxicity, causing acute inflammation and heart failure in humans and laboratory animals.12,19,20 With the detail characterizations of the DOX-treated animals, the DOX-induced heart failure is comparable to the chronic heart failure in humans and has been an excellent model for the studies of heart failure progression and drug testing. However, similar to other mammalian models, current DOX-induced heart failure models are costly and slow. Recently, we and other labs found that DOX also caused heart failure phenotypes in adult zebrafish and embryos.17,21
In the present study, we demonstrated the power of using the AA- and DOX-induced heart failure models using zebrafish embryos for drug discovery. We first carried out more molecular characterizations and pharmacological tests to further verify the AA model. We then developed this model into a simple drug discovery assay and attempted a small-scale survey with three small drug libraries. Very soon, three compounds, mitogen-activated protein kinase kinase inhibitor (MEK-I; 1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio] butadiene), C25 (3,5-diaryl-2-cyclohexenone), and A11 [(E)-3-methoxy-5-(2-(o-tolyloxy)vinyl)phenol], were identified with the ability to attenuate the AA-induced heart failure. They could be divided into two groups based on their toxicity and molecular characteristics. Group 1 compounds, including MEK-I and A11, cause albinism but do not suppress the inflammation significantly. Group 2 compounds, including C25 and a previously identified drug NS398, function in the opposite manner. We then used the DOX model to test these compounds. Interestingly, C25, NS398, and MEK-I, but not A11, were able to rescue both forms of heart failure, indicating that they target the common molecules involved in heart failure.
Materials and Methods
Zebrafish Husbandry and In Vitro Fertilization
The zebrafish (Danio rerio) stocks used in this study are maintained following standard procedures.22 In vitro fertilization was performed to obtain synchronously fertilized zebrafish embryos as previously described.17 The embryos were kept in a 28.5±1.5°C incubator throughout the experiments. Animal experimentation has been compiled with the local and national animal use protocols.
Chemical Treatment and Scoring of Cardiac Phenotypes
The drug screening assay with AA model is illustrated in Figure 2 and Table 1. Normal zebrafish embryos of 24 hours post-fertilization (hpf) stage were arrayed into a 96-well plate with 200 μL of egg water containing 10 μM AA with or without test chemicals. For the initial survey, 10 μM of test chemical was used. The treated embryos were incubated until 55–72 hpf when nearly 100% of the embryos in the control group (treated with AA only) showed heart failure. The embryos were then dechorionated and classified carefully into four categories according to their cardiac phenotypes. The assessment of heart failure attenuation is also shown in Figure 2 and Supplementary Videos S1–S4 (Supplementary Data are available online at www.liebertpub.com/adt). Once a positive compound was identified, a series of follow-up experiments were conducted to study the dosage curve, toxicity, and functional time window by time course treatment. The chemical libraries used in this study came from (1) LOPAC (Sigma, St. Louis, MO), (2) synthetic compounds from the Chemistry Department at the University of Wisconsin–River Falls (Supplementary Fig. S1), and (3) synthetic compounds from the Chemistry Department of University of Wisconsin-La Crosse (Supplementary Fig. S2). Other compounds used in this study were furosemide, spironolactone, levosimendan, warfarin, and DOX (Sigma).
Fig. 2.
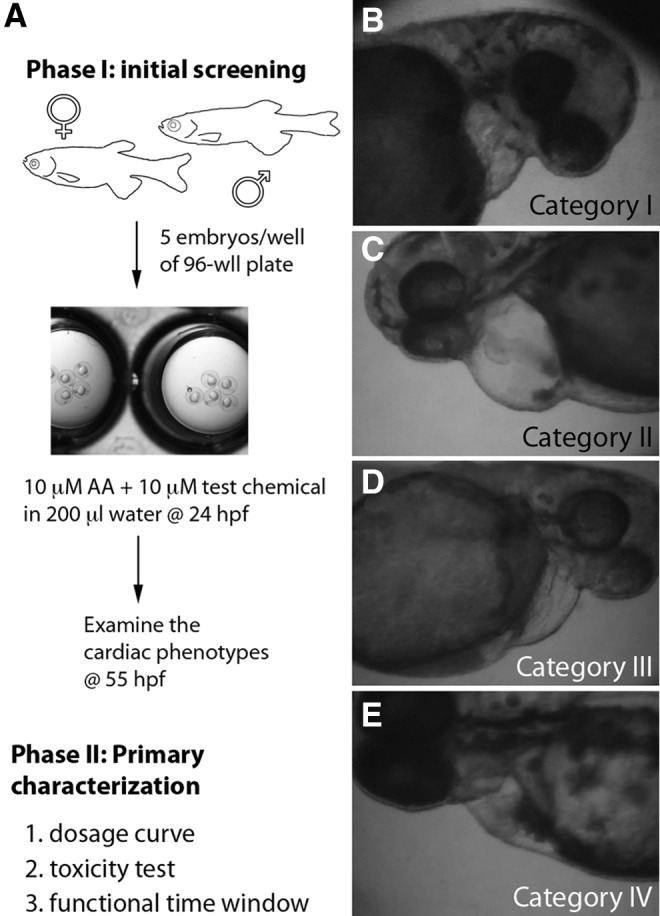
Drug discovery using the AA-induced heart failure model. (A) Protocol for drug discovery using the AA model. In phase I, zebrafish embryos were obtained by in vitro fertilization. On the next day, normal 24 hpf embryos were selected and arrayed into a 96-well plate, 5 embryos/well with AA and/or test compounds. At around 55 hpf, the cardiac function and morphology was analyzed with a dissection microscope. Positive compounds were further characterized in phase II to understand their dosage curve, toxicity, and functional time window. (B–E) Scoring for the cardiac phenotypes. A four-category scoring system was established to distinguish the severity of heart failure in the treated embryos. Category I (B): embryos show almost no contraction in the heart. Category II (C): embryos show no contraction in ventricle but weak or slow contraction in the atria. Category III (D): embryos show contraction in both atria and ventricles along with distinguishable morphological defects, including cardiac edema, enlarged hearts, blood pool, and slow circulation. Category IV (E): embryos show normal cardiac morphology and function. Video clips of each category are available in the Supplementary Data. hpf, hours post-fertilization.
Table 1.
Assay Protocol Table for Drug Testing Using Zebrafish Heart Failure Models
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 |
Fish setup |
|
Fish set up in quiet water for sexual maturation |
| 2 |
Fish breeding |
|
In vitro fertilization to generate synchronous embryos |
| 3 |
Select normal embryos |
24 hpf |
Select embryos without any morphological birth defect |
| 4 |
Array embryos |
5 embryos/well |
At least triplicate for each treatment |
| 5 |
Control |
200 μL |
10 μM AA to induce acute heart failure |
| 6 |
Library compound |
200 μL |
10 μM AA and 10 μM test compound |
| 7 |
Incubation time |
36–48 h |
28.5±1.5°C |
| 8 | Assay readout | Examination of the cardiac function with a dissection microscope |
Step Notes
1. Healthy fish were selected and set up in 1-L tanks, with three females and one male in each tank.
2. Eggs were collected on a clean 6-mm Petri dish and fertilized with sperm collected with a capillary tube.
3. Normal embryos were selected for treatment based on their morphological criteria.
4. Embryos were transferred to wells of a 96-well plate using a cut pasture pipette.
5. At least three wells of embryos were set up with 10 μM AA as the control.
6. At least three wells of embryos were treated with 10 μM AA along with 10 μM test compounds.
7. The plate was covered and incubated in a 28.5±1.5°C air incubator until analysis.
8. Embryos were transferred to a 60-mm Petri dish, dechorionated, and examined under a dissection microscope.
The readout, which consists of four categories of heart failure severity, is based on the cardiac morphology and condition.
AA, aristolochic acid; hpf, hours post fertilization.
Quantitative Polymerase Chain Reaction
Typically, 20 embryos from each treatment were subjected to RNA extraction with Trizol following commercial instructions (Invitrogen, Carlsbad, CA). RNA concentration and quality was determined using the Genesys 10S UV-VIS spectrophotometer (Thermo Scientific, West Palm Beach, FL). Approximately 5 μg of total RNA were then used to generate cDNA library using the Superscript III kit (Invitrogen). The cDNA library was then taken to set up a quantitative polymerase chain reaction (qPCR) using the Power SYBRGreen PCR kit (Biorad, Foster City, CA) and the Mx300P QPCR thermocycler (Agilent Technologies, Santa Clara, CA). Data were analyzed with the MxPro QPCR software provided by the manufacturer to calculate the mRNA quantity of the genes of interest using the β-actin gene as a normalizer. Results of experimental groups were then compared with a control group (DMSO or AA-treated embryos) called a calibrator, whose baseline was set as 1. Primers for qPCR are as follows: β-actin L, CCT ACT AAT ACA CAG CCA TGG ATG A; β-actin R, GTC CCA TGC CAA CCA TCA C; cox-2 L, TGT TTT GAA CGA GCG GAG TT; cox-2 R, CAA AGT TGC ACA TCG ATC ACA; cTnT-L, GTC TGC ACT TCG GCG GTT ACA;23 cTnT-R, AGG TAA AAT CTA TAT TGT TCA GTG AAA TCT AAC CG;23 ANP-L, ATG GCC GGG GGA CTA ATT CT; ANP-R, AGA GTT GCA ACC GAG GGT GC.
Results
Further Validation of the AA-Induced Heart Failure Model
To validate our AA model for drug discovery, we first tested whether the current medication for human heart failure patients can improve the cardiac condition in AA-treated zebrafish embryos. Several drugs that improve heart failure symptoms in humans by targeting different mechanisms were selected, including the loop diuretic drug furosemide,24 the anti-aldosterone drug spironolactone,25 the calcium sensitizer levosimendan that increases cardiac contractility,11 and the anticoagulant warfarin which is used in patients with heart failure and atrial fibrillation.26 The zebrafish embryos exposed to AA and different concentrations of the selected human drug exhibited different degrees of cardiac improvement, ranging from 0% to nearly 40% (Fig. 1A, assessment of heart failure attenuation efficacy is described in the next section.). It is interesting to note that the human drugs at 80 μM showed no benefit to AA-treated embryos, indicating that they may pose toxicity to the developing embryos. In fact, spironolactone caused an obvious curvy body and necrosis in the tail (Fig. 1B). These results indicate that the AA-induced heart failure could be attenuated by current human heart failure medications.
Fig. 1.

Pharmacological and molecular characterizations of the AA-induced heart failure. (A) Human heart failure drugs moderately attenuate the AA-induced heart failure. Zebrafish embryos were treated with 10 μM AA with or without the selected human heart failure drugs furosemide, spironolactone, levosimendan, or warfarin at 1, 5, 20, or 80 μM. Among them, the best attenuation efficiency was seen with 20 μM of spironolactone. At 80 μM, all of them failed to attenuate the heart failure. The data presented are means±SEM of triplicate wells (n=3) and the experiments were repeated twice. (B) Spironolactone at 80 μM causes curvy body and necrotic tail (arrow) in developing embryos. (C) qPCR shows elevated expression of cTnT (∼1.8-fold, P=0.003) and ANP (∼1.4-fold, P=0.013) in AA-treated embryos than that in DMSO-treated embryos. AA, aristolochic acid; ANP, atrial natriuretic peptide; cTnT, cardiac troponin T; qPCR, quantitative polymerase chain reaction.
Next, we sought molecular evidence for the AA model. Three of the most specific diagnostic biomarkers for heart failure are atrial natriuretic peptide (ANP),27 B-type natriuretic peptide (BNP),28 and cTnT.29 Elevated protein levels of these biomarkers in patient plasma are associated with a high risk of myocardial infarction, hospitalization, and death. Due to technical difficulties, it is still impossible to obtain blood plasma from developing zebrafish embryos. In addition, the zebrafish BNP gene has not been identified by the Zebrafish Sequencing Project. Thus, we attempted to examine the transcriptional levels of ANP and cTnT genes by qPCR. Since cardiac hypertrophy is associated with heart failure, it is plausible that the expression of these two genes will increase in the AA-induced heart failure. To do that, wild-type zebrafish embryos were treated with 10 μM AA or 0.1% DMSO and harvested for RNA extraction and cDNA synthesis. The cDNA libraries of each group were then used for qPCR. The results showed that the expression of both ANP and cTnT genes was induced to a significantly high level by AA treatment, 1.4-fold for ANP (P=0.013), and 1.8-fold for cTnT (P=0.003, Fig. 1C). These results strongly argued that the AA model is molecularly and pharmacologically comparable to other forms of heart failure and should be well suited for drug discovery.
Drug Discovery Using the AA Model
We then developed the AA-treated zebrafish embryos into a simple and quick assay system for drug discovery. The protocol is illustrated in Figure 2 and Table 1, which is divided into two phases: initial testing and primary characterization. In the first phase, we obtained embryos at day 0 followed by selecting normal 24 hpf embryos for treatment with 10 μM AA alone (control) or AA with a test compound typically at 10 μM. After 30–48 h of treatment, the cardiac function of the treated embryos was classified into four categories (Fig. 2B–E and Supplementary Videos S1–S4). Category I embryos were those who lost the contractility in both atrium and ventricle. These embryos also showed signs of death, including necrosis in the tails and lack of motion on touch. Category II embryos lost their ventricular contractility, while the atrial contraction was detectable but weak. They also showed some of the distinguishable morphological phenotypes, including deformed cardiac chambers, weakening contractility, accumulation of blood cells in front of the atria, cardiac edema, and lack of circulation. Category III embryos exhibited contractility in both cardiac chambers with mild phenotypes mentioned earlier. Category IV embryos were those that showed near normal cardiac morphology and function. The percentage of Category III and IV embryos combined was calculated and referred to as the attenuation efficacy of heart failure in this project. Typically, all the AA-treated embryos showed Category I or II at 55–72 hpf, that is, 0% attenuation.
With the LOPAC library which contains diverse compounds (Sigma), we identified the MEK-I (for mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor)30 that could significantly attenuate the AA-induced heart failure. It is important to note that this result is consistent with the findings that MEK-1/2 is involved in the promotion of cardiac hypertrophy and heart failure.31,32 From two other small libraries, we identified two new compounds that can also attenuate the AA-induced heart failure: C25 and A11 (structures of the compounds can be found in Supplementary Figs. S1 and S2). After confirming their attenuation ability, these lead compounds were proceeded to phase II characterization, including dosage curve, toxicity test, and functional time window.
Characterization of MEK-I
Dosage curve
Zebrafish embryos were treated with AA alone or AA with MEK-I at 0.1–10 μM. The attenuation efficacy of MEK-I showed an interesting bell curve with the best efficacy of around 50% by 1 μM (Fig. 3A). At 10 μM, MEK-I was not able to rescue the heart effectively, suggesting the possible toxicity of MEK-I in developing heart and/or other tissues. This notion seems to match the pleiotropic role of MEK-1/2 in embryonic development.33
Fig. 3.

Characterization of MEK-I. (A) Dosage curve of MEK-I. The prime attenuation of heart failure is observed at 50% by 1 μM MEK-I. Higher concentration, however, reduces the attenuation efficacy. (B–G) MEK-I toxicity. Embryos treated with 10 μM MEK-I from 6 to 24 hpf (B) or from 6 to 48 hpf (C) showed albinism and body curvature with unusually large notochord vacuoles (arrows in B, C). (D, E) Later treatment of MEK-I causes weaker albinism and notochord phenotype (different degrees of kink in the tails of D, E). MEK-I, though, does not seem to affect existing melanocytes. (F, G) MEK-I-induced albinism is dosage dependent. (H) Time course experiment shows that the critical time window of MEK-I function is between 24 (∼60% attenuation) and 30 (∼30% attenuation or 50% efficacy) hpf. Paired t-test comparing 24 hpf and other time points showed P=0.072 (30 hpf), 0.017 (36 hpf), 0.008 (42 hpf), and 0.009 (48 hpf). The data presented are means±SEM of triplicate wells (n=3), and the dosage curve experiments were repeated thrice. MEK-I, mitogen-activated protein kinase kinase inhibitor.
Toxicity test
To test whether MEK-I alone causes any toxicity during embryonic development, zebrafish embryos were treated with MEK-I alone at different developmental stages starting at 6 hpf and analyzed frequently until 55 hpf. The results showed that MEK-I at 10 μM is toxic to the developing notochord and melanocytes. When added to 6 hpf embryos, MEK-I caused wavy notochord that was distinguishable starting at around 20 hpf (Fig. 3B). As development proceeded, the notochord defects worsened and by 48 hpf, the embryo's body axis was very curvy with irregular sizes of notochord vacuoles (Fig. 3C). The notochord phenotype is highly reproducible when MEK-I is added during the early stage of notochord development, but the severity gradually lessened when MEK-I was added near the end of notochord development. For example, embryos treated with MEK-I from 36 to 55 or 48 to 55 hpf developed a near normal notochord (Fig. 3D, E). The MEK-I toxicity on developing notochord is dosage dependent, as embryos treated with 2 or 5 μM MEK-I showed almost no notochord abnormality (Fig. 3F, G).
MEK-I also caused albinism in a dosage- and stage-dependent manner. Strong albinism occurred in embryos treated with 10 μM MEK-I starting at 6 hpf (Fig. 3C), suggesting a role of MEK in melanin synthesis and/or melanocyte formation. In 2 μM MEK-I, embryos were able to develop normal melanocytes (Fig. 3F). The albinism weakened when MEK-I was added at or after 24 hpf (Fig. 3D, E), indicating that MEK-I has little or no effect on existing melanocytes.
Time window of MEK-I function
To understand when MEK-I is required to attenuate the heart failure progression, we performed time course experiments. Zebrafish embryos were treated with AA alone or AA and 1 μM MEK-I, which was added at 24, 30, 36, 42, or 48 hpf and the cardiac phenotypes were scored at 55–72 hpf. The results showed that MEK-I had 60% of attenuation when started at 24 hpf but quickly dropped to 35% when added at 30 hpf and almost no attenuation occurred at and after 36 hpf (Fig. 3H). These results indicate that MEK-I is required between 24 and 30 hpf, that is, 6 h post AA treatment, in order to achieve 50% or higher of its prime attenuation efficacy. It was noted that when the later treated embryos were incubated to 72 hpf, MEK-I did not show any better attenuation (data not shown). Instead, the heart failure worsened, indicating that the lower efficacy by MEK-I in the later treated embryos is not due to insufficient incubation time.
Isolation and Characterization of C25
Dosage curve
The second library that we surveyed contains 29 chalcone-derived compounds. Two analogs, C6 and C25, showed strong attenuation efficacy (structures in Supplementary Fig. S1). The C6 is very potent, with 90% attenuation efficiency by 1 μM, nearly 50% by 0.3 μM, and 30% by 0.1 μM (Fig. 4A). However, C6 was very unstable. The attenuation could suddenly disappear after a certain period of storage, which occurred twice over a 2-year period. C25, however, is more stable than C6. Similar to C6, the attenuation of C25 is dosage dependent, with 80% by 10 μM and nearly 0% by 1 μM (Fig. 4B). We continued to characterize this group of compound using C25.
Fig. 4.

Characterization of C6 and C25. C6 and C25 are analogs (structures in Supplementary Fig. S1) with similar biological activity but different chemical stability. (A) Dosage curve of C6. The prime attenuation of heart failure is observed at 90% by 1 μM C6. (B) Dosage curve of C25. Although C25 is not as potent as C6, with the prime attenuation of heart failure at 80% by 10 μM, it is more stable than C6. (C–G) C25 toxicity. Embryos treated with 10 μM C25 show a slightly curvy tail phenotype. Similar to MEK-I, later treatment of C25 shows weaker toxicity, indicating that it targets the early developmental process. C25 does not cause albinism. (H) Time course experiment shows that the critical time window of C25 function is between 24 (80% attenuation) and 36 (40% attenuation or 50% efficacy) hpf. Paired t-test comparing 24 hpf and other time points showed P=0.12 (30 hpf), 0.074 (36 hpf), 0.016 (42 hpf), and 0.005 (48 hpf). The data presented are means±SEM of triplicate wells (n=3), and the dosage curve experiments were repeated at least thrice except for C6, which was done multiple times but not in triplicate.
Toxicity test
We performed a similar toxicity test for C25 and found that embryos treated with C25 from 6 to 55 hpf occasionally developed a hump back, otherwise normal gross morphology. The toxicity could persist when C25 was added at 42 hpf but disappeared when added at 48 hpf (Fig. 4C–G), indicating that C25 targets the development of specific tissue(s) or process(s) which takes place between 24 and 48 hpf. C25 did not show any toxicity to pigmentation, suggesting that C25 is involved in a different molecular pathway of heart failure development than MEK-I.
Time window of C25 function
Next, we tried to define the time window of C25 function during heart failure progression. The results showed that C25 could achieve 50% of its prime attenuation efficacy when added at 36 hpf, that is, ∼12 h post AA treatment (Fig. 4H). Similar to MEK-I, the embryos treated at 48 hpf did not show any better attenuation even if the incubation time was extended to 72 hpf (data not shown). These results suggest that C25 has a larger time window than MEK-I in attenuating the AA-induced heart failure.
Isolation and Characterization of A11
Dosage curve
The third library contains nearly 400 synthetic compounds and natural extracts from mushrooms. We identified a family of analogs, A6, A8, A10, and A11 (structures in Supplementary Fig. S2) that have significant yet different degrees of attenuation on AA-induced heart failure (Fig. 5A). We selected the best compound A11 for further characterization. The dosage curve of A11 showed nearly 90% attenuation at 10 μM, 50% at 5 μM, and 0% at 2 μM (Fig. 5B).
Fig. 5.

Characterization of A11. (A) The A6, A8, A10, and A11 compounds are analogs (structures in Supplementary Fig. S2). They showed different degrees of heart failure attenuation in the AA model. A11 seems to be the most potent among them. (B) Dosage curve of A11. The prime attenuation of heart failure is observed at nearly 90% by 10 μM. (C–E) A11 can cause albinism throughout the pigmentation stages. Embryos treated with 10 μM A11 at the beginning (C, 24–55 hpf) or during (D, 36–55 hpf) or after (E, 48–55) pigmentation showed defects in melanocytes and different degrees of albinism. Unlike MEK-I, A11 can reduce the existing melanin and/or melanocytes. Another difference between A11 and MEK-I is that A11 does not cause any notochord defects. (F–H) A11-induced albinism is dosage dependent. (I–K) A11 analogs also show different degrees of albinism. The albinism and heart failure attenuation ability of the A11 analogs seems to show a positive correlation, as A11 with the strongest albinism and heart failure attenuation and A10 with the least in both. (L) Time course experiment shows that the critical time window of A11 function is between 24 (∼80% attenuation) and ∼40 (∼40% attenuation or 50% efficacy) hpf. Paired t-test comparing 24 hpf and other time points showed P=0.286 (30 hpf), 0.011 (36 hpf), 0.019 (42 hpf), and 0.007 (48 hpf). The data presented are means±SEM of triplicate wells (n=3), and the dosage curve experiments were repeated at least thrice.
Toxicity test
Embryos treated with 10 μM A11 at any duration showed no detectable structural or morphological abnormalities except the pigmentation (Fig. 5C–E). Treatment from 6 to 55 hpf completely block the pigmentation both in the body and in the retina, suggesting that A11 blocks the pathway(s) required for melanin and/or melanocyte formation, a characteristic similar to MEK-I. However, unlike MEK-I, embryos treated with A11 during or after pigment formation from 24, 30, 36, or 48 to 55 hpf showed spotty and fewer pigment phenotypes (Fig. 5C–E). The A11-induced albinism was dose dependent (Fig. 5F–H). Therefore, the A11 albinism seems to positively correlate with the heart failure attenuation. The same correlation was also observed among analogs, with A11 showing the strongest albinism and the best attenuation (Fig. 5A, I–K). These results suggest that, although both MEK-I and A11 showed heart failure attenuation and albinism, they are likely to have different molecular mechanisms.
Time window of A11 function
Time course experiments revealed that 10 μM A11 could attenuate the AA-induced heart failure by more than 80% when added at 24 hpf (Fig. 5L). The efficacy maintained at about 50% when A11 was added at 36 hpf. Based on the graph, the latest time for A11 to maintain 50% of its prime attenuation efficacy is estimated to be at around 40 hpf. As with MEK-I and C25, longer incubation of the 48-hpf group did not show better attenuation efficacy (data not shown), indicating that the attenuation efficacy of these drugs is highly dependent on the timing, not the duration, of treatment.
Mechanistic Studies
To address the question whether any of these potential heart failure drugs actually reduces the AA toxicity by direct binding, we performed the wash-off experiments. Zebrafish embryos were treated with 20 μM AA from 24 to 36 hpf and then washed with egg water twice followed by treatment with water or our compounds. As reported earlier, embryos treated with AA for a short time developed the same heart failure, albeit at a slower rate.17 In the absence of AA, we still observed attenuation by our compounds, with better efficacy by C25 and A11 (Fig. 6A). MEK-I only showed marginal attenuation, likely due to the early functional requirement (Fig. 3H). Likewise, the better attenuation by C25 and A11 is consistent with their late functional times (Figs. 4H and 5L).
Fig. 6.

Mechanistic studies of the potential heart failure drugs. (A) Zebrafish embryos were treated with 20 μM AA from 24 to 36 hpf, washed twice with egg water, and then replaced with H2O or the drugs. Later in the development, the H2O group showed 100% heart failure (i.e., 0% attenuation) but the lead compounds were able to attenuate the heart failure, albeit weaker, indicating that these drugs rescue the AA-induced heart failure through mechanisms other than direct binding. The data presented are means±SEM of triplicate wells (n=3), and the experiments have been repeated twice. (B) Time course experiment shows that the previously reported drug NS398 has an even wider time window than other drugs, with 50% efficacy near 48 hpf. (C) qPCR examining the COX-2 expression shows that inflammation was induced high by AA but suppressed by C25. The inflammation was not significantly suppressed by MEK-I and A11. The data presented are means±SEM of triplicate wells (n=3), and the experiments have been repeated at least twice. COX-2, cyclooxygenase-2.
Our early studies identified another compound NS398 that can also attenuate the AA-induced heart failure.17 To understand the functional time window of NS398, we performed the time course experiment for NS398. The results revealed a broader time window for NS398 than other compounds. Treatment with 10 μM NS398 at 42 hpf still showed 50% of its prime attenuation efficiency (Fig. 6B).
NS398 is a selective inhibitor for the pro-inflammation COX-2 enzyme and attenuates the AA-induced heart failure by suppressing COX-2 expression.17 To test whether any of the new drugs also suppresses inflammation, we performed qPCR to examine the expression of COX-2 in embryos on different treatments. The expression of COX-2 in the embryos treated with AA alone was five times higher than that of DMSO control (Fig. 6C). Interestingly, while the COX-2 expression in AA+C25-treated embryos was reduced to near baseline, it appeared at high levels in the AA+MEK-I and AA+A11-treated embryos. These results indicate that both NS398 and C25, but not MEK-I or A11, attenuate the AA-induced heart failure by suppressing inflammation.
Rescue of DOX-Induced Heart Failure
To test whether the compounds isolated with the AA model can also rescue other forms of heart failure, we used the DOX-induced heart failure model in zebrafish embryos.17 Titration experiments with DOX showed that zebrafish embryos required a much higher concentration and longer treatment than AA to develop heart failure. DOX caused mild cardiac defects in only 20% of the embryos at 50 μM but killed almost all the embryos at 100 μM (data not shown). At 90 μM, DOX caused heart failure in 60% of the embryos by 96 hpf, which was 24 h longer treatment than AA (Fig. 7C). DOX-induced heart failure is slightly different from the AA-induced heart failure, with more severe cardiac edema and very long-stretched heart (Fig. 7A). Interestingly, other tissues/organs seem to develop normally in the DOX-treated embryos, despite its nonspecific toxicity in cell proliferation. Next, we incubated embryos in 90 μM DOX with or without our compounds. The results were very intriguing. While C25, NS398, and MEK-I were able to attenuate the DOX-induced heart failure, A11 worsened it (Fig. 7D) by causing more severe edema in and outside the heart (Fig. 7B). These results indicate that C25, NS398, and MEK-I compounds target the molecules or pathways involved in both the AA- and DOX-induced heart failure, whereas A11 is more specific for the AA-induced heart failure. In addition, these two models revealed potential synthetic toxicity of A11 and DOX.
Fig. 7.

Attenuation of DOX-induced heart failure. (A) DOX-treated embryos exhibit cardiac edema and heart failure. (B) Worsening heart failure by A11 and DOX. Arrow points to the additional edema formed in the yolk. Also see (D) for quantitative result. (C) Dosage curve of DOX-induced heart failure in zebrafish embryos. The cardiac assessment was done after 96 hpf. (D) Attenuation of DOX-induced heart failure by NS398, C25, and MEK-I, but not by A11. The data presented are means±SEM of triplicate wells (n=3), and the experiments have been repeated at least thrice. DOX, doxorubicin.
Discussion
Zebrafish Models for Heart Failure Drug Discovery
Here, we report a new drug discovery assay for heart failure using two zebrafish embryo models, one with AA whose toxicological mechanism for heart failure is still not fully understood and the other with DOX whose mechanism, however, has been well studied. This approach demonstrated several significant advantages for drug discovery. First, heart failure can be induced by compounds in a very short period of time compared with the mammalian models, with less than 2 days by AA and 3–4 days by DOX. Second, while both of them will be suitable for drug screening, the AA model is quicker and cheaper because the cost and use of AA is much less than DOX. Third, the two systems allow us to cross-check and compare the drug effect. Due to the different chemical natures of AA and DOX, it is likely that these two toxins will demonstrate some unique as well as common molecular mechanisms leading to different forms of heart failure. Thus, drugs identified by one system may not work on the other. As presented in this study, after testing the new drugs with these two models, A11 appears to only rescue the AA-induced, but not the DOX-induced, heart failure. Thus, these tests may distinguish the drugs and identify those that target the common pathological mechanisms of heart failure. In the meantime, with the well-studied mechanism of DOX, comparison studies help us understand the difference between the AA and DOX toxicity.
AA is a known nephrotoxin.34,35 It was a surprise to see AA causing heart failure in zebrafish embryo. To provide a better understanding of this model, it is crucial to summarize all the characterizations reported in this as well as previous studies.17 First, the AA-treated embryos showed morphological progression of heart failure and cardiac arrest.17 Second, histological sections revealed enlarged cardiomyocytes (or cardiohypertrophy) and cardiomuscle fiber disorganization.17 Third, molecular studies showed elevated expression of cTnT, ANP (this study), and pro-inflammation genes.17 Fourth, the AA-induced heart failure is worsened by an adrenergic receptor agonist and can be attenuated by human heart failure drugs, including metoprolol (β-blocker), ACE-I, furosemide, spironolactone, levosimendan, and warfarin (this study). While these results demonstrate essential molecular and pharmacological features comparable to the human counterpart, there are limitations to the AA model. First, some cardiac physiological tests, such as cardiac output and echocardiogram that are standard diagnoses for heart failure routinely performed in humans and other mammalian model organisms, are still challenging to us due to the lack of adequate equipment, although other groups have developed techniques for these measurements using video edge detection software in zebrafish embryo.36 Second, the fast development of AA-induced cardiac failure suggests it to be a form of acute heart failure and raises the questions of the applicability of this system to the chronic heart failure, which is the most common form. Third, the toxicological mechanism of AA-induced heart failure is still not clear. To overcome these challenges, we established another model with a known heart failure inducing drug DOX. With the supporting results from DOX, we have presented evidence that strongly argue the qualification of the AA model as a suitable tool for the initial drug screening for heart failure.
The AA model demonstrated high efficiency of drug testing. In a very short period of time, we confirmed the effect of the anti-inflammation drug NS398 and identified three new compounds MEK-I, C25, and A11. The roles of NS398 and MEK-I in heart failure have been suggested by other studies.12,31,32 Thus, it is likely that C25 and A11 are truly involved in heart failure. More excitingly, C25 and A11 appear to have better attenuation efficacy than the existing human drugs, at least in our system (compare Figs. 1A, 4A, and 5A).
Pharmacological Dynamic and Efficacy of Lead Compounds
The pharmacological dynamic and efficacy of our drugs can be distinguished with the AA model due to its fast development of heart failure symptoms. First, we were able to define the functional time windows of the lead compounds: MEK-I at 24–30 hpf, C25 at 24–36 hpf, A11 at 24–40 hpf, and NS398 at 24–42 hpf (Table 2). Since cardiac hypertrophy typically occurs in the early phase of heart failure development, it makes perfect sense that the critical time of MEK-I function is early during the AA-induced heart failure (Fig. 3H). Second, these compounds apparently fall into two groups based on the findings that MEK-I and A11 cause albinism but C25 and NS398 do not and that NS398 and C25 suppress inflammation but MEK-I and A11 do not. All these suggest that the compounds are likely to regulate at least two different molecular pathways. One way to test this idea is to set up a combined treatment of two drugs at lower concentrations. If an additive or synergistic attenuation is achieved by the combination of two drugs, it suggests that they are likely in two parallel pathways. On the other hand, if a nonadditive or epistatic effect occurs, the two drugs are likely in the same pathway. Another experiment that can elucidate the mechanisms of these compounds is to examine the activity of mitogen-activated protein kinase (MAPK) signaling using the specific antibody which recognizes the activated form of MAPK. Since MEKs are upstream activators of MAPK, we anticipate less MAPK activity on MEK-I treatment. It would be very interesting to know whether NS398, C25, and A11 treatment also leads to MAPK suppression. These experiments will further help us understand the relationship between these drugs and possibly build molecular pathways of heart failure progression.
Table 2.
Comparison of Human Heart Failure Drugs and the Newly Identified Compounds on AA-Induced Heart Failure
| Optimal dose (μM) | Attenuation efficacy (%) | Functional time window (hpf) | Toxicity | |
|---|---|---|---|---|
| Human heart failure drugs | ||||
| Furosemide |
5 |
∼15 |
ND |
No |
| Levosimendan |
5 |
∼30 |
ND |
No |
| Spironolactone |
20 |
∼40 |
ND |
Body curvature, tail necrosis |
| Warfarin |
1 |
∼30 |
ND |
Necrosis |
| Newly identified compounds | ||||
| NS398 |
10 |
∼50 |
24–42 |
No |
| MEK-I |
1 |
∼60 |
24–30 |
Wavy notochord, albinisma |
| C25 |
10 |
∼80 |
24–36 |
Body curvatureb |
| A11 | 10 | ∼90 | 24–40 | Albinism |
Only in early embryos.
Only occasionally.
MEK-I, mitogen-activated protein kinase kinase inhibitor.
Inflammation and Heart Failure
Inflammation is known to adversely associate with many cardiovascular disease conditions.5 It is interesting to find that both NS398 and C25 attenuate the AA-induced heart failure by suppressing inflammation. However, C25 is required at a much earlier stage to rescue the heart failure, suggesting that C25 may function early in a pathway which is linked to inflammation. If this is true, the combined treatment of C25 and NS398 will likely produce the same efficacy as C25 lone treatment. By contrast, MEK-I and A11 treatments showed minimum suppression of COX-2 expression (Fig. 6C), suggesting that MEK-I and A11 attenuate the AA-induced heart failure primarily through mechanisms other than inflammation, and the slight suppression of COX-2 expression is likely to be secondary. Furthermore, both NS398 and C25 attenuate the DOX-induced heart failure, indicating inflammation is common in both forms of heart failure.
Due to the close relationship between the kidney and heart in regulating hemodynamic, many cardiac conditions can result in kidney malfunction and vice versa.37 In zebrafish embryo, the cardiac contraction can be detected as early as 24 hpf, whereas the glomerular filtration starts at 48 hpf or even as early as 40 hpf, as suggested by the elaborated foot processes and the unfolding of the glomerular basement membrane.38 The AA-induced heart failure phenotypes typically happen between 48 and 72 hpf when both the heart and kidney are functioning. This might explain why diuretic drugs, such as furosemide, show some degree of attenuation. Recently, AA is also found to cause kidney abnormalities in developing zebrafish embryo.39 However, the timing and relation of AA toxicity in the kidney and heart in zebrafish embryos is still not clear. Nevertheless, the AA model might identify drugs that attenuate heart failure by targeting the cardiac or kidney or both. With the many available tissue-specific transgenic zebrafish lines, such as the Tg(myl7:EGFP) that labels myocardium,40 TG(fli1:EGFP) that labels endocardium,41 Tg(wt1b:GFP) that labels kidney,42 and Tg(mpx:GFP) that labels neutrophils,43 it becomes much easier to study the drug mechanisms using the zebrafish models.
Toxicology and Teratology of Heart Failure Drugs
Since current heart failure drugs usually target the cardiovascular, neurohormonal, and renal systems, it is plausible that overdose and long-term usage can cause toxicity in adults and/or developing embryos. Our results provide supporting evidence to this hypothesis. The four human heart failure drugs tested in this study showed reduced cardiac protection at high concentrations. Spironolactone, an aldosterone receptor antagonist, apparently causes additional toxicity to zebrafish embryos. Warfarin also caused necrosis in developing embryos (not shown). Two other drugs furosemide and levosimendan did not cause obvious morphological defects but failed to rescue the heart failure at high concentrations, suggesting adverse effects in the heart and/or other tissues. Similar concerns have been raised for these drugs by other studies.2 Furthermore, it is important to note that although A11 rescues the AA-induced heart failure, it causes synthetic toxicity with DOX. Thus, our zebrafish models provide a great tool for pharmaco-toxicity/teratology tests.
Pigmentation and Heart Failure
Melanin and neurohormones, such as epinephrine, are biochemically related because they are derivatives of tyrosine through branched biochemical pathways.44 Even though the enzymatic synthesis of both have been well studied, the relationship between pigmentation and cardiac function is still not clear. Our study identified MEK-I and A11 that attenuate heart failure and interfere with pigmentation simultaneously but in slightly different manners. While MEK-I only causes albinism when applied before melanin synthesis, A11 can cause albinism before and after melanin synthesis. Thus, it is likely that they function in different pathways of melanin synthesis. It is noted that the albinism by both compounds persists for approximately 5 days post-fertilization (unpublished results), indicating that the albinism is not due to developmental delay. Furthermore, the albinism and heart failure attenuation by A11 are positively correlated, suggesting its role at the merging point of the two pathways. It will be very interesting to identify the molecular target of A11.
Supplementary Material
Abbreviations Used
- AA
aristolochic acid
- ACE-I
angiotensin converting enzyme inhibitor
- ANP
atrial natriuretic peptide
- BNP
brain-derived natriuretic peptide/B-type natriuretic peptide
- COX-2
cyclooxygenase-2
- cTnT
cardiac troponin T
- DOX
doxorubicin
- hpf
hours post-fertilization
- MAPK
mitogen-activated protein kinase
- MEK-I
mitogen-activated protein kinase kinase inhibitor
- qPCR
quantitative polymerase chain reaction
Acknowledgments
The authors would like to thank John Yu at the Institute of Cellular and Organismic Biology, Academia Sinica, Taipei, Taiwan, for providing the LOPAC library. Thanks are due to Timothy Lyden for comments and suggestions and also to the undergraduate students at UW-RF who helped with this project over the years. This project is supported by a grant from the University of Wisconsin System (106-SYS-06-8000-4).
Disclosure Statement
The authors declare no conflicts of interest.
References
- 1.The American Heart Association Statistics Committee and Stroke Statistics Subcommittee: Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation 2012;125:e2–e220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McMurray J, Pfeffer M: Heart failure. Lancet 2005;365:1877–1889 [DOI] [PubMed] [Google Scholar]
- 3.Feenstra J, Grobbee D, Remme W, Stricker B: Drug-induced heart failure. J Am Coll Cardiol 1999;33:1152–1162 [DOI] [PubMed] [Google Scholar]
- 4.Gharib M, Burnett K: Chemotherapy-induced cardiotoxicity: current practice and prospects of prophylaxis. Eur J Heart Fail 2002;4:235–242 [DOI] [PubMed] [Google Scholar]
- 5.Anker S, von Haehling S: Inflammatory mediators in chronic heart failure: an overview. Heart 2004;90:464–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MERIT-HF Investigators: Effect of metoprolol CR/XL in chronic heart failure: metoprolol CR/XL randomized intervention trial in congestive heart failure (MERIT-HF). Lancet 1999;353:9–13 [PubMed] [Google Scholar]
- 7.SOLVD Investigators: Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congested heart failure. N Engl J Med 1991;325:293–302 [DOI] [PubMed] [Google Scholar]
- 8.Ko D, Hebert P, Coffey C, et al. : Adverse effects of β-blocker therapy for patients with heart failure: a quantitative overview of randomized trials. Arch Intern Med 2004;164:1389–1394 [DOI] [PubMed] [Google Scholar]
- 9.Zafrir B, Amir O: Beta blocker therapy, decompensated heart failure, and inotropic interactios: current perspectives. Isr Med Assoc J 2012;14:184–189 [PubMed] [Google Scholar]
- 10.Cohn J, Tognoni G: Valsartan heart failure trial investigators. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N Engl J Med 2001;345:1667–1675 [DOI] [PubMed] [Google Scholar]
- 11.Follath F, Cleland J, Just H, et al. : Efficacy and safety of intravenous levosimendan compared with dobutamine in severe low-output heart failure (the LIDO study): a randomized double-blind trial. Lancet 2002;360:196–202 [DOI] [PubMed] [Google Scholar]
- 12.Delgado R, Nawar M, Zewail A, et al. : Cyclooxygenase-2 inhibitor treatment improves left ventricular function and mortality in a murine model of doxorubicin-induced heart failure. Circulation 2004;109:1428–1433 [DOI] [PubMed] [Google Scholar]
- 13.Simunek T, Klimtova I, Kaplanova J, et al. : Rabbit model for in vivo study of anthracycline-induced heart failure and for the evaluation of protective agents. Eur J Heart Fail 2004;6:377–387 [DOI] [PubMed] [Google Scholar]
- 14.Dixon J, Spinale F: Large animal models of heart failure: a critical link in the translation of basic science to clinical practice. Circ Heart Fail 2009;1:262–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Braunwald E: Biomarkers in heart failure. N Engl J Med 2008;358:2148–2159 [DOI] [PubMed] [Google Scholar]
- 16.Zon L, Peterson R: In vivo drug discovery in the zebrafish. Nat Rev Drug Dis 2005;4:35–44 [DOI] [PubMed] [Google Scholar]
- 17.Huang C, Chen P, Huang C, Yu J: Aristolochic acid induces heart failure in zebrafish embryos that is mediated by inflammation. Toxicol Sci 2007;100:486–494 [DOI] [PubMed] [Google Scholar]
- 18.Futaki N, Takahashi S, Yokoyama M, Arai I, Higuchi S, Otomo S: NS-398, a new anti-inflammatory agent, selectively inhibits prostaglandin G/H synthase/cyclooxygenase (COX-2) activity in vitro. Prostaglandins 1994;47:55–59 [DOI] [PubMed] [Google Scholar]
- 19.Ferreira A, Matsubara L, Matsubara B: Anthracycline-induced cardiotoxicity. Cardiovasc Hematol Agents Med Chem 2008;6:278–281 [DOI] [PubMed] [Google Scholar]
- 20.van Acker S, Kramer K, Voest E, et al. : Doxorubicin-induced cardiotoxicity monitored by ECG in freely moving mice. A new model to test potential protectors. Cancer Chemother Pharmacol 1996;38:95–101 [DOI] [PubMed] [Google Scholar]
- 21.Ding Y, Sun X, Huang W, et al. : Haploinsufficiency of target of rapamycin attenuates cardiomyopathies in adult zebrafish. Circ Res 2011;109:658–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Westerfield M: The Zebrafish Book A Guide for the Laboratory Use of Zebrafish (Danio rerio). University of Oregon Press, Eugene, OR, 2000 [Google Scholar]
- 23.Hsiao C, Tsai W, Horng L, Tsai H: Molecular structure and developmental expression of three muscle-type troponin T genes in zebrafish. Dev Dyn 2003;227:266–279 [DOI] [PubMed] [Google Scholar]
- 24.Gerlag P, van Meijel J: High-dose furosemide in the treatment of refractory congestive heart failure. Arch Intern Med 1988;148:286–291 [PubMed] [Google Scholar]
- 25.Pitt B, Zannad F, Remme W, et al. : The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med 1999;341:709–717 [DOI] [PubMed] [Google Scholar]
- 26.Cokkinos D, Haralabopoulos G, Kostis J, Toutouzas P, HELAS Investigators: Efficacy of antithrombotic therapy in chronic heart failure: the HELAS study. Eur J Heart Fail 2006;8:428–432 [DOI] [PubMed] [Google Scholar]
- 27.von Haehling S, Jankowska E, Morgenthaler N, et al. : Comparison of midregional proatrial natriuretic peptide with N-terminal pro-B-type natriuretic peptide in predicting survival in patients with chronic heart failure. J Am Coll Cardiol 2007;50:1973–1980 [DOI] [PubMed] [Google Scholar]
- 28.Mueller C, Scholer A, Laule-Kilian K, et al. : Use of B-type natriuretic peptide in the evaluation and management of acute dyspnea. N Engl J Med 2004;350:647–654 [DOI] [PubMed] [Google Scholar]
- 29.Hudson M, O'Connor C, Gattis W, et al. : Implications of elevated cardiac troponin T in ambulatory patients with heart failure: a prospective analysis. Am Heart J 2004;147:546–552 [DOI] [PubMed] [Google Scholar]
- 30.Favata M, Horiuchi K, Manos E, et al.: Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem 1998;273:18623–18632 [DOI] [PubMed] [Google Scholar]
- 31.Bueno O, Molkentin J: Involvement of extracellular signal-regulated kinases 1/2 in cardiac hypertrophy and cell death. Circ Res 2002;91:776–781 [DOI] [PubMed] [Google Scholar]
- 32.Lorenz K, Schmitt J, Schmitteckert E, Lohse M: A new type of ERK1/2 autophosphorylation causes cardiac hypertrophy. Nat Med 2009;15:75–83 [DOI] [PubMed] [Google Scholar]
- 33.Craig E, Stevens M, Vaillancourt R, Camenisch T: MAP3Ks as central regulators of cell fate during development. Dev Dyn 2008;237:3102–3114 [DOI] [PubMed] [Google Scholar]
- 34.Vanhaelen M, Vanhaelen-Fastre R, But P, Vanherweghem J: Identification of aristolochic acid in Chinese herbs. Lancet 1994;343:174. [DOI] [PubMed] [Google Scholar]
- 35.Vanherweghem J, Depierreux M, Tielemans C, et al. : Rapidly progressive interstitial renal fibrosis in young women: association with slimming regimen including Chinese herbs. Lancet 1993;341:387–391 [DOI] [PubMed] [Google Scholar]
- 36.Denvir M, Tucker C, Mullins J: Systolic and diastolic ventricular function in zebrafish embryos: influence of norepinephrine, MS-222 and temperature. BMC Biotechnol 2008;8:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iwanaga Y, Miyazaki S: Heart failure, chronic kidney disease, and biomarker. Circ J 2010;74:1274–1282 [DOI] [PubMed] [Google Scholar]
- 38.Drummond I, Majumdar A, Hentschel H, et al. : Early development of the zebrafish pronephros and analysis of mutations affecting pronephric function. Development 1998;125:4655–4667 [DOI] [PubMed] [Google Scholar]
- 39.Ding Y, Chen Y: Developmental netphtrotoxicity of aristolochic acid in a zebrafish model. Toxicol Appl Pharmacol 2012;261:50–65 [DOI] [PubMed] [Google Scholar]
- 40.Huang C, Tu C, Hsiao D, Hsieh F, Tsai H: Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Dev Dyn 2003;228:30–40 [DOI] [PubMed] [Google Scholar]
- 41.Lawson N, Weinstein B: In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev Biol 2002;248:307–318 [DOI] [PubMed] [Google Scholar]
- 42.Perner B, Englert C, Bollig F: The Wilms tumor genes wt1a and wt1b control different steps during formation of the zebrafish pronephros. Dev Biol 2006:309;87–96 [DOI] [PubMed] [Google Scholar]
- 43.Renshaw S, Loynes C, Trushell D, Elworthy S, Ingham P, Whyte M: A transgenic zebrafish model of neutrophilic inflammation. Blood 2006;108:3976–3978 [DOI] [PubMed] [Google Scholar]
- 44.Krzysciak W: Activity of selected aromatic amino acids in biological systems. Acta Biochim Pol 2011;58:461–466 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.