

Abstract
G-protein coupled receptors (GPCRs) are typically present in a basal, inactive state, but when bound to agonist they activate downstream signaling cascades. In studying arrestin regulation of opioid receptors in dorsal root ganglia (DRG) neurons, we find that agonists of delta opioid receptors (δORs) activate cofilin through Rho-associated coiled-coiled containing protein kinase (ROCK), LIM domain kinase (LIMK) and β- arrestin 1 (β-arr1), to regulate actin polymerization. This controls receptor function, as assessed by agonist-induced inhibition of voltage-dependent Ca2+ channels in DRGs. Agonists of opioid-receptor like receptors (ORL1) similarly influence the function of this receptor through ROCK, LIMK and β-arr1. Functional evidence of this cascade was demonstrated in vivo where the behavioral effects of δOR or ORL1 agonists were enhanced in the absence of β-arr1 or prevented by inhibiting ROCK. This pathway allows δOR and ORL1 agonists to rapidly regulate receptor function.
Introduction
The neuronal cytoskeleton provides both the 3-dimensional structural stability critical for cellular function, and the infrastructure that allows the cell to respond to external stimuli. Dynamic remodeling of the cytoskeleton, particularly of the actin filaments, provides the network along which intracellular proteins may be trafficked as needed. This mechanism is important to shuttle proteins to and from the synapse, as well as allow the Golgi apparatus to sort and traffic newly synthesized proteins to the cell membrane (Lowe, 2011; Salvarezza et al., 2009). Proteins bound for the cell membrane from the Golgi may either be constitutively exported through a specific pathway with specialized secretory vesicles that are formed and move continuously along the microtubules, or secreted on demand. A dynamic actin cytoskeleton plays an important role in this process; both the actin severing protein, cofilin, and the upstream kinase, LIMK, have been shown to inhibit or facilitate the release of specific proteins from the Golgi to the cell membrane (Egea et al., 2006; Heimann et al., 1999; Salvarezza et al., 2009).
Similar to many other membrane proteins, newly synthesized G-protein coupled receptors (GPCRs) are sorted, processed, degraded or trafficked from the endoplasmic reticulum to the Golgi. Thereafter, GPCR transfer through the Golgi stack is complex and may involve several escort or chaperone proteins to release the receptor to the cell membrane. For the most part, the rate of GPCR release to the cell membrane is determined by the quality control mechanisms within the ER and once within the Golgi, GPCRs are presumed to be constitutively released to the cell membrane. By regulating the number of GPCRs released to the cell membrane, this biosynethetic pathway is able to influence receptor availability and signaling (Achour et al., 2008; Dong et al., 2007).
β-arrestin 1 or 2 recruitment to the protease-activated receptor 2 (PAR2), regulates chemotaxis by binding with and activating cofilin and associated regulatory proteins. This alters actin turnover within the leading and trailing edges and allows the cell to migrate as needed (DeFea, 2007; Xiao et al., 2010; Zoudilova et al., 2007; Zoudilova et al., 2010). In examining β-arrestin 1 (β-arr1) regulation of GPCR function in dorsal root ganglia (DRG) neurons, we have found that this arrestin isoform influences the stability of the actin cytoskeleton to rapidly control the magnitude of GPCR signaling.
Results
The primary afferent neurons of the DRGs, the first order neurons of the analgesic pathway, relay nociceptive information from the peripheral to the central nervous system. In addition to a number of Gi/o coupled GPCRs, these neurons also express the non-visual arrestins (Komori et al., 1999). Of the two isoforms, β-arrestin 2 is known to regulate the function of some Gi/o coupled GPCRs in DRG neurons (Tan et al., 2009; Walwyn et al., 2007). However, whether β-arrestin 1 regulates the function of these GPCRs has not been established.
β-arr1 modulates VDCC inhibition by some GPCRs
As agonists of Gi/o-coupled GPCRs can inhibit voltage-dependent Ca2+ channels (VDCC), the whole cell patch clamp technique was used to assess β-arr1 regulation of Gi/o-coupled GPCRs in DRG neurons. Of the 5 GPCRs examined, we found that VDCC inhibition by the δOR agonist, DPDPE (p<0.05 vs β-arr1+/+), and the ORL1 agonist, nociceptin (p<0.001 vs β-arr1+/+), was enhanced in cells lacking β-arr1. On the other hand, VDCC-inhibition by agonists of the mu opioid, GABAB and α2A adrenergic receptors was not altered (Fig 1A). Further analysis of the δOR-VDCC profile showed that, in addition to the peptidergic agonist DPDPE, the non-peptidergic and arguably more specific δOR agonist, SNC80 (1μM) also resulted in greater VDCC inhibition in medium-sized, but not small, DRG neurons lacking β-arr1 (p<0.001 vs β-arr1+/+, Fig. 1B). The concentration response curve of SNC80 in medium-sized neurons demonstrate enhanced efficacy and potency of this δOR agonist in β-arr1−/− neurons (Emax; β-arr1+/+; 32.1±3.1, β-arr1−/−; 47.7±2.5 % inhibition, p<0.001, Fig. 1C). This enhanced inhibition was receptor-specific as it was reversed by the delta antagonist naltrindole, (NTI; SNC80 and NTI applied at 1μM; β-arr1+/+; 0.1±0.6 %, β-arr1−/−; 1.1±1.8 % inhibition). It was also reversed by the ORL1 antagonist, JTC801 (JTC801 and nocicpeptin at 1μM: β-arr1+/+; 1.6±7.1 %, β-arr1−/−; 6.3±5.1 %). Medium-sized neurons from different aged mice or of different backgrounds showed the same profile of enhanced inhibition in β-arr1−/− neurons (p<0.05 vs β-arr1+/+, Fig 1D, 1E) which could not be explained by basal changes in Ca2+ channel density, the relative proportion of N, P/Q and L type Ca2+ channels present or constitutive δOR-VDCC coupling (SFig. 1).
Fig. 1. δOR and ORL1 function is enhanced in dorsal root ganglia neurons lacking β-arr1.

A. The whole cell patch clamp technique was used to examine VDCC inhibition by Go/i coupled receptors. a. DPDPE, a peptidergic δOR agonist, and nociceptin, an ORL1 agonist, demonstrated enhanced VDCC coupling in β-arr1−/− vs +/+ DRG neurons whereas VDCC inhibition by agonists of the mu receptor, DAMGO and morphine (1μM ea), the GABAB receptor, Baclofen (50μM), and the α2A receptor, Clonidine (10μM) was not different across genotypes. B. Further analysis of δOR-VDCC inhibition showed enhanced δOR inhibition of VDCCs by the non-peptidergic δOR agonist, SNC80, in medium (shown by the arrow), but not small, early post-natal medium-sized β-arr1−/− vs +/+ DRG neurons in the SvEv129SJ background, scalebar=10μm. C. The concentration response curve demonstrated enhanced efficacy and potency of SNC80 in β-arr1−/− vs +/+ neurons (p<0.001). D. Similar to neurons from mice back-crossed into the SvEv129SJ background, medium-sized DRG neurons from early post-natal β-arr1−/− in the C57Bl/6 background showed enhanced SNC80-VDCC inhibition. E. Similar to early post-natal DRG neurons, medium-sized DRG neurons from adult mice showed increased SNC80-VDCC inhibition in β-arr1−/− vs +/+ neurons. All data are shown as mean±SEM,*p<0.05 vs β-arr1+/+, **p<0.001 vs β-arr1+/+, ***p<0.001 vs β-arr1+/+, n=6–27 per data point. All exemplar currents depict the current before (1), during (2) and after agonist (3) application, and the scale bars show time, 20ms, on the x-axis, and current on the y axis; 5 or 10nA for early post-natal or adult DRG neurons respectively.
β-arr1 regulation of δOR desensitization
As δORs recruit β-arr1 or 2 and show less desensitization in cells lacking β-arr1 (Qiu et al., 2007), the enhanced efficacy of δOR agonists in β-arr1−/− neurons could be a result of attenuated desensitization. This was assessed in vitro by both rapid desensitization in which an agonist is continually perfused while δOR-VDCC inhibition is monitored, and acute desensitization in which the cells are pre-incubated with SNC80, washed, and δOR-VDCC inhibition assessed. Both measures showed a genotype effect (Fig. 2A, B). Firstly, rapid desensitization occurred in both β-arr1−/− and +/+ neurons, but this was less in β-arr1−/− neurons (p<0.01 vs β-arr1+/+, Fig. 2A). Secondly, acute desensitization, involving pre-incubation with 50 or 500nM of SNC80 for 10, 30, or 60min, followed by a 1μM SNC80 test dose, showed delayed desensitization after 10min of 50nM SNC80 in βarr1−/− neurons but equivalent desensitization thereafter, and no difference in the amount or rate of desensitization induced by 500nM SNC80 (Fig. 2B). In summary, we have found that δOR desensitization was delayed in β-arr1−/− neurons. However, when compared with δOR-VDCC inhibition which reached a maximum within 20 – 40s of agonist exposure, the attenuated desensitization, seen by 120s of continual agonist perfusion, cannot explain the enhanced δOR-VDCC inhibition seen in β-arr1−/− neurons.
Fig. 2. β-arr1 regulates δOR desensitization and internalization but this does not explain the enhanced δOR-VDCC coupling in β-arr1−/− neurons.

A. Rapid desensitization: Once complete inhibition of VDCCs was obtained by SNC80, further perfusion with SNC80 desensitized this inhibition. This is shown by the decline in the peak current amplitude over 120s in the left panels. Although both β-arr1+/+ and −/− neurons desensitized, β-arr1−/− neurons showed less desensitization, **p<0.01, n=5–10. B. Acute desensitization: Whereas pre-incubation of DRGs with 50 and 500nM SNC80 for 10min desensitized δOR-VDCC inhibition at all time points in +/+ neurons, −/− neurons did not desensitize after 10min of 50nM SNC80 but desensitized thereafter and showed equivalent desensitization at all timepoints when pre-incubated in 500 nM SNC80. β-arr1+/+; a, b, c: p<0.05 vs untreated, d, e: p<0.0001 vs untreated, f: p<0.001 vs untreated, β-arr1−/−; b: p<0.001 vs untreated, c: p<0.001 vs untreated, d, e, f: p < 0.0001 vs untreated, n=6–7 per data point. Columns in shades of grey, Bi; +/+ or Bii; −/−, indicate data shown in Fig 1. C. Internalization assessed by flow cytometry in β-arr1−/− and +/+ DRG neurons using an N-terminal antibody and Allophycocynanin (APC)-conjugated secondary antibody. i The δOR-labeled non-fixed samples were initially sorted by size (FSC-H) and granularity (SSC-H) to select the medium-large DRG neurons (>25 μM in diameter Walwyn et al., 2004). APC-labeled δORs were then selected after removing non-specific background labeling and all samples of each experiment analyzed by these parameters. ii. Although basal cell surface δOR levels were unaffected by the β-arr1−/− deletion, 50 and 500, but not 1000, nM SNC80 increased cell surface δOR levels in βarr1−/− DRGs. #p<0.05 vs untreated β-arr1−/−, * p<0.05 vs β-arr1+/+, same time point, n=4–8 per data point. D. A slower rate of internalization could enhance δOR-VDCC inhibition by increasing the number of δORs on the plasma membrane. This was assessed by over-expressing Cerulean (CFP)-labeled δORs (Walwyn et al., 2009). This increased δOR-VDCC inhibition equally in both β-arr1+/+ and−/− neurons tested with either DPDPE or SNC80 (1μM ea). *p<0.05, **p<0.01 and ***p<0.001 vs β-arr1+/+, #p<0.05, ## p<0.01 vs endogenous δORs, n=6–18 per data point. All exemplar currents depict the current before, during and after agonist application (1, 2 and 3) and the scale bars show time, 20ms, and current, 5nA on the x- and y-axes respectively. Columns in dark (β-arr1+/+) or light (β-arr1−/−) shades of grey indicate data shown in Fig 1. All data are shown as mean±SEM.
β-arr1 regulation of δOR internalization
As the arrestins are known to initiate internalization, deleting β-arr1 could delay δOR internalization to increase cell surface receptor number and enhance δOR function. Internalization was examined by flow cytometry using an Allophycocyanin-labeled antibody to the N-terminus of the δOR, the specificity of which was confirmed in cells lacking δORs (SFig 2). The non-fluorescent parameters of size and granularity were then used to gate on medium-large sized neurons (Fig 2Ci, Walwyn et al., 2004). These cells showed no effect of genotype on basal cell surface receptor levels (β-arr1−/−: 89.9±4.0 of β-arr1+/+ levels). However, after 10 min of 50 and 500nM, but not 1μM, SNC80, cell surface receptor levels were increased in β-arr1−/− (p<0.05 vs untreated β-arr1−/−) but not +/+ neurons (p<0.05 vs β-arr1+/+, Fig. 2Cii). After 1h of SNC80 (1μM), virally expressed cerulean-labeled δORs expressed in β-arr1+/+ and −/− neurons, demonstrate significant internalization (SFig 3). As δORs are dynamically trafficked to and from the cell membrane, the initial increase in δORs on the cell membrane could reflect delayed internalization and/or enhanced externalization in β-arr1−/− neurons (Cahill et al., 2007; Walwyn et al., 2009).
Accumulation of δORs on the cell membrane does not account for the enhanced δOR function in β-arr1−/− neurons
Attenuated internalization or enhanced externalization would lead to an accumulation of receptors on the cell membrane, so explaining the increase in δOR-VDCC coupling in β-arr1−/− neurons. Lentiviral expression of cerulean-tagged (CFP) δORs was therefore used to over-express δORs in β-arr1−/− and +/+ neurons. This did increase δOR-VDCC inhibition, assessed by either DPDPE or SNC80, 1μM ea (p<0.05 vs endogenous levels, Fig. 2D). However, the increase occurred equally in β-arr1+/+ and −/− neurons suggesting that δOR-VDCC inhibition is sensitive to altered receptor number but that the regulatory role of β-arr1 remains even when these receptors are over-expressed (p<0.05 vs β-arr1+/+).
β-arr1 control of protein export explains the enhanced δOR-VDCC inhibition
As flow cytometric analysis showed a greater number of δORs on the cell membrane, it is possible that β-arr1 could control δOR release to the cell membrane in an agonist-dependent manner. Cells were pre-incubated with Brefeldin A, an inhibitor of protein export. This reduced δOR-VDCC inhibition in β-arr1−/− neurons to +/+ levels with no effect in +/+ neurons at this concentration (p<0.05 vs untreated β-arr1−/−, Fig. 3i). A more specific inhibitor of protein export from the Golgi, Exo1 (0.8μg/ml), reduced δOR-VDCC inhibition in β-arr1−/− neurons and, when used at a higher concentration (3.2μg/ml), reduced δOR-VDCC inhibition in both β-arr1+/+ and −/− neurons (p<0.05 vs untreated, Fig. 3ii). To probe for non-specific effects, GABAB-VDCC was examined in the presence of ExoI but no effect found (Fig. 3iii). As δOR-VDCC inhibition is closely correlated with cell surface receptor levels (Walwyn et al., 2005), these data could be interpreted as an Exo1-mediated inhibition δORs released to the cell membrane in an agonist-dependent manner.
Fig. 3. β-arr1 control of protein export explains enhanced δOR-VDCC inhibition.

The increase in cell surface receptor number after 50 and 500nM SNC80 (Fig. 2C) could reflect β-arr1 control of δOR release to the plasma membrane. This was assessed by inhibiting protein export i. DRGs were pre-treated with Brefeldin A (Bref. A, 1μg/ml 30min pre-treatment) to inhibit protein release from the ER, and Exo1, a more specific inhibitor of protein release from the Golgi (0.8 and 3.2μg/ml, included in the intracellular recording solution). Both BrefA and Exo1 (0.8μg/ml) reduced the enhanced SNC80 inhibition of VDCCs in β-arr1−/− to +/+ levels but the higher dose of Exo1 (3.2μg/ml) reduced SNC80-inhibition in both −/− and +/+ neurons ii. Exo1 did not affect GABAB-VDCC inhibition, assessed by Baclofen (50 μM). All data are shown as mean±SEM, #p<0.05 vs untreated β-arr1+/+ or −/−. ***p<0.001 vs β-arr1+/+, n=6–12 per data point All exemplar currents depict the current before, during and after agonist application (1, 2 and 3) and the scale bars show time, 20ms, and current, 5nA on the x- and y-axes respectively. Columns in dark (β-arr1+/+) or light (β-arr1−/−) shades of grey indicate data shown in Fig 1.
δOR activation of cofilin is β-arr1 dependent
Both LIMK and cofilin are required for the export of specific proteins from the Golgi to the cell membrane (Salvarezza et al., 2009; von Blume et al., 2009). δORs could activate cofilin to regulate receptor export via this pathway, and subsequently receptor function. δOR activation of the phosphorylated, inactivated form of cofilin was assessed in mouse embryonic fibroblasts (MEFs). MEFS were chosen for this assay as they express or lack the necessary proteins of this pathway; β -arr1, ROCK, LIMK, and cofilin and have been used to study arrestin-GPCR signaling (Zoudilova et al., 2010) This pathway is also conserved in DRGs (Ahmed et al., 2011). In wildtype MEFs, we found that SNC80 (1μM) reduced the amount of phosphorylated cofilin in β-arr1 −/− (2–30 min; p<0.01 or 0.01 vs untreated β-arr1+/+) but not +/+ MEFs. Together these data show that SNC80 activates cofilin in β-arr1-dependent manner (Fig. 4A).
Fig. 4. δOR and ORL1 inhibition of VDCCs is regulated by LIMK, ROCK and actin polymerization.

δOR: A. The δOR agonist, SNC80, activates cofilin: Wild-type MEFs (wt MEFs) or β-arr1−/− MEFs were treated with SNC80 for 0–120min and lysates analyzed by Western blot. ai. Representative western blots of phosphorylated cofilin (pcof) and total cofilin (tcof) in wild type wt MEFs (upper two panels) and βarr1−/− MEFs (lower two panels) after SNC80 treatment. aii. SNC80 (1μM) decreased phospho-cofilin after 2, 5, 15 and 30min in wt MEFs (closed circles) whereas βarr1−/− MEFs (open circles) showed no change. Values on the y-axis represent phospho-cofilin normalized to basal levels at each time point shown on the x-axis. a; p<0.001, b: p<0.01, c: p<0.01 vs untreated wt MEFs. B i. The ROCK inhibitor, Y27632 (200 and 400ng/ml) reduced δOR-VDCC coupling in β-arr1−/− and +/+ neurons. Excess LIMK (2μg/ml) increased SNC80-VDCC inhibition in both β-arr1 +/+ and −/− neurons although SNC80-VDCC inhibition remained higher in β-arr1−/− neurons. LIMK phosphorylation of cofilin can be prevented by the Serine 3 peptide. This decreased SNC80-VDCC inhibition in β-arr1−/− neurons but not +/+ neurons (S3: 1.6 and 6.4ng/ml). C. i. Jasplakinolde and Thymosin B4 (200ng/ml), enhancers of actin polymerization, increased SNC80-VDCC inhibition in β-arr1+/+ to −/− levels whereas Latrunculin B (LatB: 200ng/ml), an inhibitor of actin polymerization, reversed the increased δOR-VDCC inhibition in β-arr1−/− to +/+ levels. ii. LatB did not alter GABAB-VDCC inhibition assessed by Baclofen (50μM). ORL1: D. The increase in ORL1-VDCC inhibition in β-arr1−/− DRG neurons was reversed by the ROCK inhibitor, Y27632 (400ng/ml), the LIMK inhibitory peptide, S3 (6.4ng/ml), and Latrunculin B (200ng/ml). No effect of these manipulations was seen in +/+ neurons. All data are shown as mean±SEM, *p<0.05, ***p<0.001 vs β-arr1+/+, ##, ###p<0.01, or 0.001 vs untreated, matched genotype, n=6–12 per data point. All exemplar currents depict the current before, during and after agonist application (1, 2 and 3) and the scale bars show time, 20ms, and current, 5nA on the x- and y-axes respectively. Columns in dark (β-arr1+/+) or light (β-arr1−/−) shades of grey indicate data shown in Fig 1.
ROCK, LIMK and β-arr1 regulation of δOR-VDCC coupling
As β-arr1 is known to bind with and regulate LIMK (Zoudilova et al., 2007), and LIMK has been shown to control the export of specific proteins from the Golgi (Salvarezza et al., 2009), it is possible that δOR export and therefore function may be regulated by β-arr1. We examined this possibility by inhibiting ROCK, the kinase responsible for phosphorylating and activating LIMK, in neurons containing or lacking β-arr1. The ROCK inhibitor, Y27632, decreased δOR-VDCC inhibition in both β-arr1−/− and +/+ neurons (p<0.01 vs untreated β-arr1+/+; p<0.001vs untreated β-arr1−/−, Fig. 4B) but had no effect on GABAB-VDCC coupling (Untreated: β-arr1+/+; 49.8±5.3 %, β-arr1−/−: 46.6±3.4 %, Y27632 (400ng/ml); β-arr1+/+; 49.5±2.2 %, β-arr1−/−: 51.1±2.5 %). We then used an approach designed to enhance LIMK activity by including excess LIMK (1 and 2μg/ml) within the intracellular recording solution. The 2μg/ml dose increased δOR-VDCC coupling in both β-arr1+/+ and −/− neurons (p<0.05 vs untreated β-arr1+/+ or −/−, Fig. 4B) demonstrating that LIMK may influence δOR-VDCC inhibition. As LIMK phosphorylation of cofilin can be inhibited by the Serine 3 LIMK peptide (Aizawa et al., 2001), we included this peptide in the intracellular recording solution (S3; 1.6 and 6.4μg/ml). This reduced δOR-VDCC inhibition in β-arr1−/− neurons (p<0.001 vs untreated β-arr1−/−) but not +/+ neurons (Fig. 4B). Together these data suggest that δOR-VDCC inhibition may be regulated by ROCK, LIMK and β-arr1.
Actin polymerization influences δOR-VDCC inhibition
LIMK has been shown to phosphorylate and inhibit cofilin to stabilize the actin cytoskeleton in dendritic spines (Shi et al., 2009). It is possible that LIMK, β-arr1 and cofilin could similarly regulate the stability of the actin ‘tracks’ required for the export of δORs to the cell membrane. The ability of actin polymerization to affect δOR-VDCC inhibition was therefore examined by manipulating actin polymerization minutes before assessing δOR-VDCC inhibition. Firstly we stabilized the actin cytoskeleton by including Jasplakinolide and Thymosin β4 within the intracellular recording solution (Bubb et al., 1994; Huff et al., 2001). This increased δOR-VDCC inhibition in +/+ neurons to −/− levels but had no effect in β-arr1−/− neurons (p<0.05 vs untreated β-arr1+/+, Fig. 4C). Preventing polymerization by including Latrunculin B (LatB, 200ng/ml)(Morton et al., 2000) within the intracellular recording solution reduced the enhanced δOR-VDCC inhibition in βarr1−/− neurons to +/+ levels without affecting GABAB-VDCC inhibition (p<0.05 vs untreated β-arr1−/−, Fig. 4C). Together these data demonstrate that manipulations designed to influence actin polymerization can regulate SNC80-VDCC inhibition.
ORL1-VDCC inhibition is similarly controlled by β-arr1, ROCK, LIMK and actin polymerization
The previous experiments used δORs as a model GPCR to examine each component of this regulatory pathway. As ORL1-VDCC inhibition was upregulated in β-arr1−/− neurons we next examined whether, similar to δORs, ORL1-VDCC may be controlled by this ROCK-LIMK pathway in a β-arr1 dependent manner. We assessed the effect of the following inhibitors of this pathway, Y27632 (400ng/ml), S3 (6.4μg/ml), or LatB (200nM), on ORL1-VDCC coupling in β-arr1 −/− and +/+ DRG neurons. These three compounds reversed the enhanced ORL1-VDCC inhibition in −/− neurons (p<0.001 vs untreated β-arr1−/−), but had no effect in +/+ neurons (Fig 4D). These data suggest that ORL1-VDCC coupling may be controlled by ROCK, LIMK, and actin polymerization in the absence of β-arr1−/−.
Basal F-actin incorporation appears unchanged
As deleting β-arr1 could alter basal LIMK and cofilin activity, this could affect actin turnover throughout the cell. This was assessed by the incorporation of Atto-647N Phalloidin (30nM, 10min) in living DRG neurons that were then fixed and analyzed by Stimulated Emission Depletion (STED) microscopy. No effect of genotype on Phalloidin-incorporation was observed in either the neuronal cell bodies or processes of β-arr1−/− vs +/+ neurons suggesting that basal actin polymerization was not altered in β-arr1 DRGs (sFig. 4).
Behavioral assays provide functional evidence for the role of ROCK and β-arr1 in regulating δOR and ORL1 function
δOR
Two well-known behavioral assays of δOR function, locomotion and analgesia, were used to assess the function of this pathway. In the open-field test of locomotion, SNC80 (3mg/kg s.c.) increased the locomotor activity of β-arr1+/+ mice 20min after the injection. This was reversed by both the ROCK inhibitor (Y27632; 1mg/kg i.p.) and the delta antagonist, NTI (2mg/kg s.c., p<0.001 locomotion x treatment, Fig 5Ai). In contrast, β-arr1−/− mice showed an enhanced locomotor effect of SNC80 seen both as a more rapid onset, occurring within 10min of the SNC80 injection, and lasting considerably longer than in wild type littermates (p<0.001 vs β-arr1+/+). Similar to +/+ mice, this was reversed by Y27632 and NTI (p<0.001 locomotion x treatment, Fig. 5Aii). To assess whether this inhibitory profile of Y27632 was due to non-specific suppression of locomotor behavior, we tested the effect on Y27632 on the locomotor effects of fentanyl, a specific mu opioid receptor agonist. Fentanyl (0.4mg/kg s.c.) induced an equal hyperlocomotor response in both genotypes which was not altered by Y27632 (1mg/kg i.p., sFig 5A). No gender based difference in locomotion were observed in either the SNC80 (p=0.69 locomotion x gender) or fentanyl-treated groups (p=0.54 locomotion x gender).
Fig. 5. δOR behaviors are regulated by β-arr1 and LIMK.

A. Two behavioral assays were used to assess δOR function. i. Locomotion. β-arr1+/+ mice showed a hyperlocomotor response to subcutanously administered SNC80 (3 mg/kg s.c., p<0.001, F5,28=13.125, locomotion x treatment), that was reversed by both the ROCK inhibitor (Y27632 1mg/kg i.p.) and the delta antagonist, naltrindole (2mg/kg s.c.). β-arr1−/− mice also showed a hyperlocomotor effect of SNC80 (p<0.001, F5,26=31.19 locomotion x interaction), but this was enhanced (p <0.001, F1,9=25.79 vs. β-arr1+/+), occurred more rapidly than the wildtype mice (p <0.001, F17,153=4.051 vs. β-arr1+/+) and lasted for the duration of the test, 90min. This was similarly reversed by both Y27632 and naltrindole. The data show time (min) on the x-axis and distance traveled (cm) on the y-axis. The blue arrow represents the second of the two injections. The bar graphs depict the total locomotion over time. B. Mechanical analgesia was measured by the number of responses, out of a total of 10 applications of the same von Frey filament (2g), to the plantar surface of the right hind paw. β-arr1+/+ mice showed a small effect of SNC80 (5mg/kg s.c., p<0.05, F3,39=3.388, response x treatment) at the 20min (p<0.01, F3,39=6.525), but not the 40min timepoint. β-arr1−/− mice, on the other hand, showed a greater response at both the 20 and 40min timepoints after SNC80 administration (p<0.001, F2,37=43.37, response x treatment). Y27632 (β-arr1+/+: p<0.01 vs. SNC80, β-arr1−/−: p<0.001 vs. SNC80) and naltrindole (β-arr1+/+: p<0.01 vs. SNC80, β-arr1−/−: p<0.001 vs. SNC80) reversed the antinociceptive effect of SNC80 in both β-arr1+/+ and β-arr1−/− mice. C. We have previously shown that Complete Fruend’s Adjuvant (CFA) induced chronic pain decreases the mechanical threshold in the von frey test of allodynia (p<0.001, F1,25=114.17 vs. baseline) in adult C57Bl6/J mice. This decrease is associated with a functional upregulation of δORs (Pradhan et al., 2013) as it can be reversed by SNC80 (1or 5mg/kg s.c.; p<0.001, F1,23=33.84 threshold x treatment) in wildtype mice. Administration of Y27632 (10mg/kg i.p.) prior to injecting CFA and at 12h intervals thereafter, did not alter the resultant hyperalgesia (p=0.563, F1,25=0.34 vs. vehicle), but did inhibit the effect of SNC80 (1 and 5 mg/kg; *p<0.05 vs. vehicle, ###p<0.001 vs. CFA). All data are shown as mean±SEM.
In the second behavioral test of δOR function, the analgesic effect of SNC80 was assessed by mechanical pain in which the number of paw withdrawals from 10 applications of a 2g von Frey filament was measured. We found that basal thresholds were not influenced by genotype, and that wildtype mice showed a brief analgesic effectof SNC80 at the 20 (p<0.05 response x treatment), but not 40min timepoint (5mg/kg s.c., Fig. 5Bi). In comparison, β-arr1−/− mice showed enhanced analgesia, or fewer responses 20 and 40min after SNC80 (p<0.001 response x treatment). This effect of SNC80 was reversed by Y27632 (1.5mg/kg) and NTI (4mg/kg, Fig 5Bii).
SNC80 produced some analgesia in naïve wildtype mice (Fig 5bi) but, as chronic inflammatory pain results in enhanced δOR analgesia (Pradhan et al., 2013), we next examined the role of ROCK in modulating δOR function in inflammatory pain. We injected saline or Y27632 (10mg/kg i.p.) prior to injecting Complete Fruend’s Adjuvant (CFA, 10μl) in the left hindpaw of wildtype mice, and at 12h intervals thereafter. After 48h we assessed the effect of SNC80 (1 and 5mg/kg s.c.) on the mechanical pain threshold in Y27632 and saline-treated groups. We found that Y27632 reduced the ability of SNC80 to relieve the mechanical hyperalgesia associated with chronic inflammatory pain (p<0.05 vs vehicle, Fig. 5C), hilighting the involvement of ROCK in the functional upregulation of δORs seen in this model.
We also assessed whether genetic background influenced the β-arr1 phenotype. Mice back-crossed to the C57Bl/6 background and lacking β-arr1 showed a similar enhanced response to SNC80 in both locomotor and acute pain studies (sFig 5B, C). Furthermore this effect of SNC80 in β-arr1−/− mice was specific to β-arr1; SNC80 induced locomotion was similar in β-arr2−/−, β-arr2+/+ or β-arr1+/+ mice (sFig 5B).
ORL1
We assessed the role of this pathway in modulating ORL1 function by measuring the locomotor effect of the ORL1 agonist, Ro 65-6570. This agonist is similar to Ro 64-6198, which has been shown to either induce hypolocomotion or have no locomotor effect in wildtype mice (Shoblock, 2007). Although wildtype mice showed no effect of Ro 65-6570 (1.5mg/kg i.p.), β-arr1−/− mice showed a hypolocomotor response (p<0.05 locomotion x treatment) which was reversed by both Y27632 (0.5mg/kg) and the ORL1 antagonist, J-113397 (p<0.05 vs Ro 65-6570, Fig 6). Together these data show enhanced ORL1 function in β-arr1−/− mice which can be reversed by inhibiting ROCK.
Fig. 6. ORL1 behavioral responses are similarly regulated by β-arr1 and LIMK.
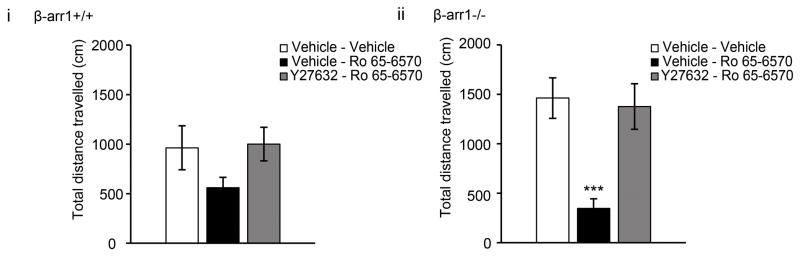
The bio-available ORL1 agonist, Ro 65-6570 (1.5mg/kg i.p.) did not alter the locomotor profile of β-arr1+/+ mice (p=0.54, F3,23=.721 locomotion x treatment) but induced a hypolocomotor response in β-arr1−/− mice. This was reversed by Y27632 (p<0.05, F3,28=3.027 locomotion x treatment). Data are shown as the total locomotor response following the injection of vehicle (vehicle), Ro 65-6570 or Y27632-Ro 65-6570 (Y27632-Ro 65) during the 45min test period. All data are shown as mean±SEM, ***p<0.001 vs. other treatment groups.
Discussion
In being able to scaffold with cofilin, the inactivating kinase, LIMK, and activating phosphatase, slingshot (SSL) or chronophin, arrestins can control the activity of cofilin and therefore actin polymerization (Condeelis, 2001; Xiao et al., 2010; Zoudilova et al., 2007; Zoudilova et al., 2010). This pathway enables a dynamic remodeling of the actin cytoskeleton in response to a chemotactic stimulus (Nishita et al., 2005) and, as we show here, modulates the function of some GPCRs; δOR and ORL1.
A model of the pathway
Based on GPCR inhibition of Ca2+ channels, a voltage-dependent and Gβγ-mediated response that modulates neurotransmitter release, we propose the following model (Fig. 7). Under normal or wildtype conditions, agonist binding to an agonist such as the δOR, activates cofilin through the RhoA-ROCK-LIMK pathway. However, as β-arr1 is associated with LIMK and one of the cofilin phosphatases such as SSL (Xiao et al., 2010; Zoudilova et al., 2010), an enhanced but local activation of cofilin occurs. We propose that this results in controlled severing of F-actin and measured actin turnover to regulate the stability of a subset of actin filaments within the Golgi. This in turn, controls the rate of export of δOR-containing cargo vesicles from the Golgi to the cell membrane to allow a limited response to a δOR agonist such as SNC80 (Fig. 7A). In the absence of β-arr1, SNC80 similarly activates LIMK through the Rho-ROCK pathway but, without the regulatory control of β-arr1, cofilin is not dephosphorylated and activated by SNC80 (Lin et al., 2003). We propose that this leaves stable actin ‘tracks’ in place resulting in enhanced export of δORs from the Golgi to the plasma membrane to enhance function (Fig 7B). This pathway can be blocked by inhibiting ROCK, the kinase responsible for phosphorylating LIMK and inactivating cofilin. In preventing agonist-induced activation of this pathway, we propose that agonist-induced turnover of a subset of actin filaments within the Golgi does not occur and additional receptors are not released to the cell membrane. Without this activation, the functional effect of the agonist is severely limited (Fig. 7C).
Fig. 7. β-arr1 regulation of GPCR function.

A. β-arr1+/+: In neurons containing β-arr1, the δOR agonist, SNC80, binds with δORs and activates ROCK. As β-arr1 is associated with LIMK and one of the phosphatases, possibly SSL, within the trans-Golgi network (TGN), this activates cofilin to enhance actin filament severing and turnover and regulate δOR release to the cell membrane. B. β-arr1−/−. Without the inhibitory control of β-arr1, LIMK phosphorylates and inactivates cofilin. This leaves stable actin ‘tracks’ in place to allow unregulated but agonist-dependent δOR release to the plasma membrane in an agonist-dependent manner. C. Y27632. Preventing ROCK phosphorylation of LIMK by applying Y27632 prevents δOR activation of the pathway and agonist-induced release of δORs to the cell membrane.
δOR trafficking and function
δOR-VDCC inhibition is a rapid, voltage-dependent cascade that can be completely reversed by a high-voltage pre-pulse (data not shown). We have previously found that δOR-VDCC inhibition is positively correlated with δOR cell surface receptor levels (Walwyn et al., 2005). Furthermore increased δOR-VDCC coupling is associated with enhanced δOR function (Pradhan et al., 2013) and, conversely, reduced δOR function is associated with reduced cell surface receptor levels (Scherrer et al., 2006). Together this suggests a close relationship between cell surface receptor levels and δOR functionality, assessed either in vitro or in vivo. The data presented here suggest that the ROCK-LIMK-β-arr1 pathway is a critical mediator of this relationship and is required to obtain an initial functional response. Thereafter, the co-operative roles of β-arr1 in both desensitizing and modulating δOR trafficking, may further regulate δOR signaling.
ORL1 function
Similar to δORs, our data suggest that β-arr1 also regulates ORL1 function in a ROCK, LIMK and actin-dependent manner. However, this regulation is not evident in wildtype mice. This could be that ROCK, LIMK and β-arr1 inhibits ORL1 function in wildtype neurons and mice to non-detectable levels. This is suggested from the low levels of ORL1-VDCC coupling in wildtype neurons (Fig 1) and the lack of a locomotor response to the ORL1 agonist used (Fig 6). Interestingly ORL1 function has often been described in association with a pharmacological or behavioral stimulus (Bebawy et al., 2010; Shoblock, 2007) suggesting that this receptor, similar to δORs, may be upregulated under certain conditions.
Other receptors or channels that may be regulated by β-arr1, cofilin and LIMK
This fine-tuned control of actin polymerization to affect δOR and ORL1 function may similarly control ligand-dependent activation of other GPCRs such as dopamine receptor 1, neurotensin or PAR2 which are recruited from intracellular stores to the cell membrane ‘upon demand’ (Brismar et al., 1998; Hein et al., 1994; Perron et al., 2006). Interestingly many of these receptors share a common post-endocytic fate of being degraded once internalized suggesting that regulating the rate of receptor release may be important to allow sufficient GPCR activation. In providing a ligand-dependent spatio-temporal regulation of actin filaments, β-arr1 may also regulate the function of ligand–gated ion channels which are may be rapidly trafficked to and from the synapse upon demand.
Other regulators of the pathway
We have examined GPCR activation of this pathway but it is also possible that other receptors or molecules may be involved. For example, bradykinin or arachodonic acid has been shown to enhance δOR function (Patwardhan et al., 2005; Pettinger et al., 2013; Rowan et al., 2009). As kinins are involved in the hyperalgesic response to chronic pain (Ferreira et al., 2002), it is possible that bradykinin may activate this pathway in the CFA model of chronic pain.
Conclusion
Following agonist-receptor activation of downstream signaling cascades, GPCRs typically desensitize, becoming unable to induce the same signaling profile. In contrast, we have found that agonist binding with select GCPRs results in a functional upregulation of the receptor. This increases GPCR-Ca2+ channel inhibition and agonist-mediated behaviors demonstrating how this novel regulatory pathway allows these receptors to maintain or enhance receptor function.
Methods
Animals
All animal experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals and followed institutionally approved animal care and use protocols. Mice lacking the β-arr1 (Conner et al., 1997) or δOR (Filliol et al., 2000) and were fully back-crossed into the 129S6/SvEv or C57Bl/6 background. All animals used for the cellular experiments were bred from homozygous matings within 1 generation of heterozygous matings. For the behavioral experiments, 2–3 month equally distributed males and females were generated from heterozygous matings. Unless noted otherwise, all experiments used the 129S6/SvEv βarr1 line.
Compounds
Unless noted otherwise, all chemicals were obtained from Sigma; St Louis, MO, except for Y27632; Selleckchem Houston, TX, SNC80, Naltrindole, ICI 174–864, Nociceptin, J113397, JTC801; R&D, Minneapolis, MN, S3; Anaspec, Fremont, CA, and LIMK; Cytoskeleton, Denver, CO.
Dorsal root ganglion neuron cultures
DRGs were harvested from early postnatal (p0-1) mice or adult (6–8 wk old) mice. The DRGs were enzymatically and physically dissociated and plated in different formats and densities for different experiments. For electrophysiology experiments 5×104 dissociated cells were plated on poly-D-lysine (Sigma, MO) and laminin- (Becton Dickenson, Bedford, MA) coated coverslips (10 mm diameter) in the center of 35mm MatTek dishes (Ashland, MA). Lentiviral transduction was performed at the time of plating by adding 50ng p24/plate to each plate. For flow cytometry experiments 1×106 cells were plated in a similarly-coated 15mm MatTek dish. The cells were cultured in 2ml of Neurobasal A, B27, Glutamax (0.5mM) and antibiotic-antimycotic (12U/ml,) media (Life Technologies, Carlsbad, CA) containing NGF (10μg/ml, Roche, Indianapolis, IN) and kept at 37°C and 5% CO2. Electrophysiological assessment of endogenous δOR function was performed within 24h of plating, and those in which receptors were over-expressed, 72–96h thereafter. Flow cytometry was performed within 48h of plating.
Electrophysiology
The whole-cell patch-clamp technique was used to record voltage-dependent Ca2+ channel (VDCC) activity from DRG neurons (Axopatch 200A amplifier, Molecular Dynamics, CA) as previously described (Walwyn et al, 2007). At the time of recording the culture medium was replaced by an external solution that contained (in mM): 130 TEA-Cl, 10 CaCl2, 5 HEPES, 25 D-glucose and 2.5×10−4 tetrodotoxin at pH 7.2. Recording electrodes contained (in mM): 105 CsCl, 40 HEPES, 5 D-glucose, 2.5 MgCl2, 10 EGTA, 2 Mg2+-ATP and 0.5 Na+-GTP, pH 7.2. Ca2+ currents were activated by depolarizing neurons from −80mV to 10mV for 100ms at 20s intervals. Recordings that exhibited marked rundown were discarded. Stable recordings were fitted by a linear function to compare, by extrapolation, control current amplitude to the current amplitude recorded in the presence of the agonist. Data are expressed as mean±SEM and were compared using ANOVA with a posthoc Scheffe test.
Cofilin dephosphorylation
Wild type (wt) mouse embryonic fibroblasts (MEFs) or βarr1−/− MEFs were treated with SNC80 for 0–120 min, lysed and analyzed by SDS-PAGE followed by western blotting with antibodies to phospho-cofilin (rabbit polyclonal, Cell Signaling, 1:1000) or total cofilin (mouse monoclonal, BD Biosciences, 1:500) and analyzed as previously described (Zoudilova et al., 2007).
Statistical Analysis
The data are expressed as the mean±SEM and differences between groups determined by one-way ANOVA (Prism v5.03) with significance accepted at p<0.05.
Flow cytometry
After 48h in vitro βarr1+/+ and −/− or δOR+/+ and −/− DRG neurons were harvested and processed for δOR fluorescent labeling in non-fixed cells using a primary antibody to the N-terminus of the δOR (MBL, Woburn, MA) and an Allophycocyanin (APC)-conjugated secondary antibody (BD Biosciences, San Jose, CA) as previously described (Walwyn et al., 2009).
Statistical Analysis
The data are expressed as the mean±SEM and differences between groups determined by one-way ANOVA or paired student’s T-test with significance accepted at p<0.05 (Analyse-it Software Ltd., United Kingdom).
Behavioral tests
Two-three month old β-arr1−/− and +/+ littermates of both genders were used for the following behavioral effects.
a. Locomotion
As SNC80 is known to increase locomotor behavior, this effect was measured after 30min of habituation to the test environment and the effect of SNC80, following the injection of the δOR antagonist, naltrindole, the ROCK inhibitor Y27632 or vehicle, delivered subcutaneously (s.c.) or intraperitonealy (i.p.), measured over the following 90min. This behavior was recorded in 27×27 cm plastic boxes by live tracking using an infra-red camera and Ethovision XT (Noldus, Leesburg, VA). After 30min of habituation the mice were injected with the first of 2 injections, the second followed 15min later, and locomotion was measured for the following 90min. As ORL1 agonists can induce a hypolomotor response, this effect was tested by injecting a bio-available ORL1 agonist, Ro 65-6570 (1.5 mg/kg i.p.) after 15 min of habituation and for the following 45min. The ROCK inhibitor Y27632, the ORL1 antagonist J-113397, or vehicle, were injected 15min prior to Ro 65-6570.
b. Mechanical pain
After 2 days of habituation to the test environment, the number of responses, defined as a lifting or shaking of the paw upon stimulation of the plantar surface, out of a total of 10 repetitions, to the 2g von Frey filament was measured. Basal responses to the 2g filament were acquired prior to the injections and the effect tested 20 and 40min thereafter.
CFA
Wildtype mice were habituated for 3 days prior to obtaining the basal mechanical threshold by the up/down method (Chaplan et al., 1994). Saline or Y27632 (10mg/kg i.p.) was injected prior to assessing the basal response, prior to the CFA injection (10μl in the left hindpaw), and at 12h intervals thereafter. After 48h, the mechanical pain threshold was assessed and the effect of SNC80 (1 and 5mg/kg s.c.) on determined.
Statistical analysis
Data are expressed as mean±SEM and differences between groups analyzed by ANOVA with repeated measures and factorial analysis at each timepoint (Stat View v5).
Supplementary Material
Hilights.
δOR activation of the ROCK-LIMK-cofilin pathway rapidly regulates δOR function
β-arrestin1 deletion increases, while ROCK inhibition suppresses, δOR function
Activation of this pathway can alter δOR-dependent behaviors
ORL1 is similarly regulated by this ROCK-β-arrestin 1 pathway
Acknowledgments
This work was supported by the NIH grants DA05010 (CJE, WMW), DA30866 (WMW), GM066151 (KD) and DA031243(AAP) and CIHR (CMC). NM, WMW and AAP are supported in part by Hatos Scholarships and NM is supported by the Gates Millennium Scholars program. STED confocal laser scanning microscopy was performed on a Leica TCS SP5 STED confocal system at the CNSI Institute (CHE-0722519). Flow cytometry was performed in the UCLA JCCC and CAR Flow Cytometry Core Facility (NIH; CA-16042 and AI-28697. Thanks to the laboratories of Drs Lefkowitz and Seidman for the β-arrestin1 knockout mice, to Dr G. Calò for Ro65-6570, and to Dr T. Hales for critical analysis and editing of the final manuscript. There are no conflicts of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Achour L, Labbe-Jullie C, Scott MG, Marullo S. An escort for GPCRs: implications for regulation of receptor density at the cell surface. Trends Pharmacol Sci. 2008;29:528–535. doi: 10.1016/j.tips.2008.07.009. 18760490. [DOI] [PubMed] [Google Scholar]
- Ahmed Z, Douglas MR, Read ML, Berry M, Logan A. Citron kinase regulates axon growth through a pathway that converges on cofilin downstream of RhoA. Neurobiology of disease. 2011;41:421–429. doi: 10.1016/j.nbd.2010.10.012. 20971191. [DOI] [PubMed] [Google Scholar]
- Aizawa H, Wakatsuki S, Ishii A, Moriyama K, Sasaki Y, Ohashi K, Sekine-Aizawa Y, Sehara-Fujisawa A, Mizuno K, Goshima Y, Yahara I. Phosphorylation of cofilin by LIM-kinase is necessary for semaphorin 3A-induced growth cone collapse. Nature neuroscience. 2001;4:367–373. doi: 10.1038/86011. 11276226. [DOI] [PubMed] [Google Scholar]
- Bebawy D, Marquez P, Samboul S, Parikh D, Hamid A, Lutfy K. Orphanin FQ/nociceptin not only blocks but also reverses behavioral adaptive changes induced by repeated cocaine in mice. Biological psychiatry. 2010;68:223–230. doi: 10.1016/j.biopsych.2010.02.010. 20359694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brismar H, Asghar M, Carey RM, Greengard P, Aperia A. Dopamine-induced recruitment of dopamine D1 receptors to the plasma membrane. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:5573–5578. doi: 10.1073/pnas.95.10.5573. 9576924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubb MR, Senderowicz AM, Sausville EA, Duncan KL, Korn ED. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. The Journal of biological chemistry. 1994;269:14869–14871. 8195116. [PubMed] [Google Scholar]
- Cahill CM, Holdridge SV, Morinville A. Trafficking of delta-opioid receptors and other G-protein-coupled receptors: implications for pain and analgesia. Trends Pharmacol Sci. 2007;28:23–31. doi: 10.1016/j.tips.2006.11.003. 17150262. [DOI] [PubMed] [Google Scholar]
- Condeelis J. How is actin polymerization nucleated in vivo? Trends in Cell Biology. 2001;11:288–293. doi: 10.1016/s0962-8924(01)02008-6. [DOI] [PubMed] [Google Scholar]
- DeFea KA. Stop that cell! Beta-arrestin-dependent chemotaxis: a tale of localized actin assembly and receptor desensitization. Annual review of physiology. 2007;69:535–560. doi: 10.1146/annurev.physiol.69.022405.154804. 17002593. [DOI] [PubMed] [Google Scholar]
- Dong C, Filipeanu CM, Duvernay MT, Wu G. Regulation of G protein-coupled receptor export trafficking. Biochimica et biophysica acta. 2007;1768:853–870. doi: 10.1016/j.bbamem.2006.09.008. 17074298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egea G, Lazaro-Dieguez F, Vilella M. Actin dynamics at the Golgi complex in mammalian cells. Current opinion in cell biology. 2006;18:168–178. doi: 10.1016/j.ceb.2006.02.007. 16488588. [DOI] [PubMed] [Google Scholar]
- Ferreira J, Campos MM, Araujo R, Bader M, Pesquero JB, Calixto JB. The use of kinin B1 and B2 receptor knockout mice and selective antagonists to characterize the nociceptive responses caused by kinins at the spinal level. Neuropharmacology. 2002;43:1188–1197. doi: 10.1016/s0028-3908(02)00311-8. 12504926. [DOI] [PubMed] [Google Scholar]
- Heimann K, Percival JM, Weinberger R, Gunning P, Stow JL. Specific isoforms of actin-binding proteins on distinct populations of Golgi-derived vesicles. The Journal of biological chemistry. 1999;274:10743–10750. doi: 10.1074/jbc.274.16.10743. 10196146. [DOI] [PubMed] [Google Scholar]
- Hein L, Ishii K, Coughlin SR, Kobilka BK. Intracellular targeting and trafficking of thrombin receptors. A novel mechanism for resensitization of a G protein-coupled receptor. The Journal of biological chemistry. 1994;269:27719–27726. 7961693. [PubMed] [Google Scholar]
- Huff T, Muller CS, Otto AM, Netzker R, Hannappel E. beta-Thymosins, small acidic peptides with multiple functions. The international journal of biochemistry & cell biology. 2001;33:205–220. doi: 10.1016/s1357-2725(00)00087-x. 11311852. [DOI] [PubMed] [Google Scholar]
- Komori N, Matsumoto H, Cain SD, Kahn ES, Chung K. Predominant presence of beta-arrestin-1 in small sensory neurons of rat dorsal root ganglia. Neuroscience. 1999;93:1421–1426. doi: 10.1016/s0306-4522(99)00277-8. 10501467. [DOI] [PubMed] [Google Scholar]
- Lin T, Zeng L, Liu Y, DeFea K, Schwartz MA, Chien S, Shyy JY. Rho-ROCK-LIMK-cofilin pathway regulates shear stress activation of sterol regulatory element binding proteins. Circulation research. 2003;92:1296–1304. doi: 10.1161/01.RES.0000078780.65824.8B. 12775580. [DOI] [PubMed] [Google Scholar]
- Lowe M. Structural organization of the Golgi apparatus. Current opinion in cell biology. 2011;23:85–93. doi: 10.1016/j.ceb.2010.10.004. 21071196. [DOI] [PubMed] [Google Scholar]
- Morton WM, Ayscough KR, McLaughlin PJ. Latrunculin alters the actin-monomer subunit interface to prevent polymerization. Nature cell biology. 2000;2:376–378. doi: 10.1038/35014075. 10854330. [DOI] [PubMed] [Google Scholar]
- Nishita M, Tomizawa C, Yamamoto M, Horita Y, Ohashi K, Mizuno K. Spatial and temporal regulation of cofilin activity by LIM kinase and Slingshot is critical for directional cell migration. The Journal of cell biology. 2005;171:349–359. doi: 10.1083/jcb.200504029. 16230460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan AM, Berg KA, Akopain AN, Jeske NA, Gamper N, Clarke WP, Hargreaves KM. Bradykinin-induced functional competence and trafficking of the delta-opioid receptor in trigeminal nociceptors. J Neurosci. 2005;25:8825–8832. doi: 10.1523/JNEUROSCI.0160-05.2005. 16192372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perron A, Sharif N, Gendron L, Lavallee M, Stroh T, Mazella J, Beaudet A. Sustained neurotensin exposure promotes cell surface recruitment of NTS2 receptors. Biochemical and biophysical research communications. 2006;343:799–808. doi: 10.1016/j.bbrc.2006.03.047. 16564027. [DOI] [PubMed] [Google Scholar]
- Pettinger L, Gigout S, Linley JE, Gamper N. Bradykinin controls pool size of sensory neurons expressing functional delta-opioid receptors. J Neurosci. 2013;33:10762–10771. doi: 10.1523/JNEUROSCI.0123-13.2013. 23804098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan A, Smith M, McGuire B, Evans C, Walwyn W. Chronic inflammatory injury results in increased coupling of delta opioid receptors to voltage-gated Ca2+ channels. Molecular pain. 2013;9:8. doi: 10.1186/1744-8069-9-8. 23497324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y, Loh HH, Law PY. Phosphorylation of the delta-opioid receptor regulates its beta-arrestins selectivity and subsequent receptor internalization and adenylyl cyclase desensitization. The Journal of biological chemistry. 2007;282:22315–22323. doi: 10.1074/jbc.M611258200. 17565992. [DOI] [PubMed] [Google Scholar]
- Rowan MP, Ruparel NB, Patwardhan AM, Berg KA, Clarke WP, Hargreaves KM. Peripheral delta opioid receptors require priming for functional competence in vivo. European journal of pharmacology. 2009;602:283–287. doi: 10.1016/j.ejphar.2008.11.028. 19063879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvarezza SB, Deborde S, Schreiner R, Campagne F, Kessels MM, Qualmann B, Caceres A, Kreitzer G, Rodriguez-Boulan E. LIM kinase 1 and cofilin regulate actin filament population required for dynamin-dependent apical carrier fission from the trans-Golgi network. Molecular biology of the cell. 2009;20:438–451. doi: 10.1091/mbc.E08-08-0891. 18987335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherrer G, Tryoen-Toth P, Filliol D, Matifas A, Laustriat D, Cao YQ, Basbaum AI, Dierich A, Vonesh JL, Gaveriaux-Ruff C, Kieffer BL. Knockin mice expressing fluorescent delta-opioid receptors uncover G protein-coupled receptor dynamics in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:9691–9696. doi: 10.1073/pnas.0603359103. 16766653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Pontrello CG, DeFea KA, Reichardt LF, Ethell IM. Focal adhesion kinase acts downstream of EphB receptors to maintain mature dendritic spines by regulating cofilin activity. J Neurosci. 2009;29:8129–8142. doi: 10.1523/JNEUROSCI.4681-08.2009. 19553453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoblock JR. The pharmacology of Ro 64-6198, a systemically active, nonpeptide NOP receptor (opiate receptor-like 1, ORL-1) agonist with diverse preclinical therapeutic activity. CNS drug reviews. 2007;13:107–136. doi: 10.1111/j.1527-3458.2007.00007.x. 17461893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Walwyn WM, Evans CJ, Xie CW. p38 MAPK and beta-arrestin 2 mediate functional interactions between endogenous micro-opioid and alpha2A-adrenergic receptors in neurons. The Journal of biological chemistry. 2009;284:6270–6281. doi: 10.1074/jbc.M806742200. 19126537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Blume J, Duran JM, Forlanelli E, Alleaume AM, Egorov M, Polishchuk R, Molina H, Malhotra V. Actin remodeling by ADF/cofilin is required for cargo sorting at the trans-Golgi network. The Journal of cell biology. 2009;187:1055–1069. doi: 10.1083/jcb.200908040. 20026655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walwyn W, Evans CJ, Hales TG. Beta-arrestin2 and c-Src regulate the constitutive activity and recycling of mu opioid receptors in dorsal root ganglion neurons. J Neurosci. 2007;27:5092–5104. doi: 10.1523/JNEUROSCI.1157-07.2007. 17494695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walwyn W, John S, Maga M, Evans CJ, Hales TG. Delta receptors are required for full inhibitory coupling of mu-receptors to voltage-dependent Ca(2+) channels in dorsal root ganglion neurons. Molecular pharmacology. 2009;76:134–143. doi: 10.1124/mol.109.055913. 19357247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walwyn W, Maidment NT, Sanders M, Evans CJ, Kieffer BL, Hales TG. Induction of delta opioid receptor function by up-regulation of membrane receptors in mouse primary afferent neurons. Molecular pharmacology. 2005;68:1688–1698. doi: 10.1124/mol.105.014829. 16135785. [DOI] [PubMed] [Google Scholar]
- Walwyn WM, Keith DE, Jr, Wei W, Tan AM, Xie CW, Evans CJ, Kieffer BL, Maidment NT. Functional coupling, desensitization and internalization of virally expressed mu opioid receptors in cultured dorsal root ganglion neurons from mu opioid receptor knockout mice. Neuroscience. 2004;123:111–121. doi: 10.1016/j.neuroscience.2003.08.060. 14667446. [DOI] [PubMed] [Google Scholar]
- Xiao K, Sun J, Kim J, Rajagopal S, Zhai B, Villen J, Haas W, Kovacs JJ, Shukla AK, Hara MR, et al. Global phosphorylation analysis of beta-arrestin-mediated signaling downstream of a seven transmembrane receptor (7TMR) Proceedings of the National Academy of Sciences of the United States of America. 2010;107:15299–15304. doi: 10.1073/pnas.1008461107. 20686112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoudilova M, Kumar P, Ge L, Wang P, Bokoch GM, DeFea KA. Beta-arrestin-dependent regulation of the cofilin pathway downstream of protease-activated receptor-2. The Journal of biological chemistry. 2007;282:20634–20646. doi: 10.1074/jbc.M701391200. 17500066. [DOI] [PubMed] [Google Scholar]
- Zoudilova M, Min J, Richards HL, Carter D, Huang T, DeFea KA. beta-Arrestins scaffold cofilin with chronophin to direct localized actin filament severing and membrane protrusions downstream of protease-activated receptor-2. The Journal of biological chemistry. 2010;285:14318–14329. doi: 10.1074/jbc.M109.055806. 20207744. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.