

Abstract
Recurrent connections in the corticothalamic circuit underlie oscillatory behavior in this network and range from normal sleep rhythms to the abnormal spike-wave discharges seen in absence epilepsy. The propensity of thalamic neurons to fire postinhibitory rebound bursts mediated by low-threshold calcium spikes renders the circuit vulnerable to both increased excitation and increased inhibition, such as excessive excitatory cortical drive to thalamic reticular (RT) neurons or heightened inhibition of thalamocortical relay (TC) neurons by RT. In this context, a protective role may be played by group III metabotropic receptors (mGluRs), which are uniquely located in the presynaptic active zone and typically act as autoreceptors or heteroceptors to depress synaptic release. Here, we report that these receptors regulate short-term plasticity at two loci in the corticothalamic circuit in rats: glutamatergic cortical synapses onto RT neurons and GABAergic synapses onto TC neurons in somatosensory ventrobasal thalamus. The net effect of group III mGluR activation at these synapses is to suppress thalamic oscillations as assayed in vitro. These findings suggest a functional role of these receptors to modulate corticothalamic transmission and protect against prolonged activity in the network.
Introduction
The thalamus and cortex form a nested loop structure that underlies a tendency for this network to oscillate, as seen in normal sleep rhythms and the 3 Hz spike-and-wave discharges of absence epilepsy (Crunelli and Leresche, 2002; Huguenard and McCormick, 2007). Thalamocortical relay (TC) neurons in dorsal thalamus project to cortical and subcortical structures and in turn receive feedback projections from corticothalamic (CT) neurons in layer 6. Both TC and CT neurons send glutamatergic axon collaterals to the thalamic reticular nucleus (RT), a thin layer of GABAergic neurons ensheathing the dorsal thalamus and a critical regulatory center for corticothalamic activity (Steriade, 2005). During alert states, TC neurons in tonic firing mode act as a linear relay of ascending sensory information. In contrast, during sleep or seizure states, TC and RT neurons undergo out-of-phase burst firing that underlies oscillations. Despite considerable knowledge of the circuit structure and cellular physiological basis for corticothalamic oscillations, the mechanisms that prevent pathological epileptiform activity while permitting normal oscillations remain unclear.
One potential regulatory mechanism is the family of metabotropic glutamate receptors (mGluRs). In particular, group III mGluRs functioning as presynaptic autoreceptors and heteroceptors are of interest due to their involvement in short- and long-term plasticity, including in hippocampus (Scanziani et al., 1997; Pelkey et al., 2005; Kwon and Castillo, 2008) and cerebellum (Mitchell and Silver, 2000). Group III mGluRs have a characteristic localization to the presynaptic active zone, in contrast to postsynaptic group I mGluRs and extrasynaptic group II mGluRs (for review, see Niswender and Conn, 2010). Group III mGluRs are typically Gi/o coupled and inhibit presynaptic voltage-gated calcium channels, suppressing neurotransmitter release. High-affinity receptors mGluR4/8 and low-affinity mGluR7 are activated by submicromolar and near-millimolar concentrations of glutamate, respectively.
Group III mGluRs have been associated previously with epilepsy phenotypes. Disrupting mGluR7 function via uncoupling of a crucial intracellular ligand (PICK1) yields an absence seizure phenotype (Bertaso et al., 2008). Conversely, mGluR4-knock-out mice are resistant to absence-like seizures induced by GABAA receptor antagonists (Snead et al., 2000). Although such previous studies have provided intriguing evidence that dysfunction of group III mGluRs may underlie pathologies such as absence epilepsy, the precise location and function of these receptors in the corticothalamic circuit remain unclear.
Here, we examined the role of group III mGluRs in the corticothalamic circuit, focusing on glutamatergic afferent and GABAergic efferent projections of the RT nucleus and using optogenetic approaches for selective stimulation of axons. We found that group III mGluRs are located at two loci in the circuit: mGluR8 at glutamatergic corticothalamic synapses onto RT neurons and mGluR7 at GABAergic RT synapses onto TC neurons. We also tested the potential of group III mGluRs to modulate network activity and found that receptor activation causes a strong and rapid suppression of in vitro thalamic oscillations. Group III mGluRs are thus poised to gate excessive activity in the corticothalamic circuit, dampen oscillations, and modulate synaptic transmission.
Materials and Methods
Animals.
All animals were treated according to protocols approved by the Administrative Panel on Laboratory Animal Care at Stanford University.
Viral transduction.
Juvenile male Sprague Dawley rats at postnatal day 18 (P18) to P22 were anesthetized with ketamine-xylazine and placed in a stereotaxic frame. AAV5 virus bearing a pAAV-CaMKIIα-hChR2(H134R)-EYFP construct was used to drive expression in excitatory neurons in cortex and thalamus (Lee et al., 2010). Virus was loaded into a 10 μl syringe (NanoFil; WPI) connected to a 33-gauge beveled needle and mounted on a microsyringe pump (UMP-3; WPI). Virus was injected at a rate of 100–130 nl min−1 into either the right ventrobasal (VB) thalamus (lateral from midline: 2.6 mm, posterior from bregma: 2.7–2.8 mm, depth from cortical surface: 5.3 mm; 600–700 nl) or the deep layers of right barrel cortex (lateral from midline: 4.3–4.5 mm, posterior from bregma: 1.9–2.0 mm, depth from cortical surface: 2.3 mm; 800–900 nl). Optogenetic recordings were typically performed 6–12 weeks after viral injections.
Slice preparation.
Thalamic slices were prepared from male Sprague Dawley rats, either adult (P35–P174) for patch-clamp experiments or juvenile (P11–P14) for oscillation experiments (Jacobsen et al., 2001). Rats were deeply anesthetized with pentobarbital (100 mg/kg, i.p.). Brains were rapidly extracted and horizontal slices containing VB and RT nuclei were cut with a Leica VT1200 microtome in chilled (0–4°C) cutting solution containing the following (in mm): 234 sucrose, 2.5 KCl, 11 glucose, 26 NaHCO3, 1.25 NaH2PO4, 0.5 CaCl2, and 10 MgSO4. In slices for patch-clamp experiments, a cut was made between VB and RT nuclei to prevent recurrent activity. Slices were incubated at 32°C in artificial CSF (ACSF) for 45–60 min and then gradually brought to room temperature. ACSF contained the following (in mm): 126 NaCl, 2.5 KCl, 10 glucose, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, and 1 MgSO4. Both the cutting solution and ACSF were saturated with 95% O2 and 5% CO2, pH 7.4.
Oscillation recordings.
For oscillation recordings, 400 μm-thick slices from juvenile (P11–P14) rats were placed in a humidified, oxygenated interface recording chamber and perfused with oxygenated ACSF (2 ml/min) at 32–34°C. For stable perfusion, squares of lens paper were placed beneath and on top of the slice (with a small cutout in the top layer for electrode access) and fixed in place by platinum bars. l-glutamine (300 μm), the metabolic substrate of GABA, was added to the ACSF to mitigate the gradual depletion of GABA (Bryant et al., 2009). For epileptiform oscillation experiments, 50 μm picrotoxin and 1 nm apamin were added to the ACSF (Kleiman-Weiner et al., 2009). Oscillations were evoked by a square current pulse (240–260 μA, 30 μs duration) delivered once every 30 s through two parallel tungsten electrodes (50–100 kΩ, FHC) 50–100 μm apart and placed in the internal capsule, which stimulated traversing cortical and thalamic axons. Extracellular potentials were recorded with a tungsten electrode (50–100 kΩ, FHC) placed in VB. One experiment was performed per slice. To prevent a floor effect from obscuring drug-induced changes, experiments were only performed if the baseline oscillation duration was at least 2 s long. Recordings were amplified 100,000 times, digitized at 20 kHz with Digidata 1200 series, band-pass filtered 3 Hz-5 kHz, and acquired using pClamp software (Molecular Devices).
Whole-cell patch-clamp recordings.
For whole-cell patch-clamp recordings, 250 μm-thick slices from adult rats (P35–P64 for electrical stimulation experiments; P76–P174 for optogenetic experiments) were placed in a submerged recording chamber and perfused with oxygenated ACSF (2 ml/min) at 32–33°C. The chamber contained nylon netting that suspended the slice ∼1 mm from the chamber floor and enhanced slice perfusion. Slices were visualized with Dodt-contrast optics (Luigs and Newmann) on an Axioskop microscope (Zeiss). For all experiments, a single cell was recorded from each slice. Recordings were acquired using a MultiClamp 700A patch amplifier (Molecular Devices), digitized at 50 kHz with Digidata 1322A, low-pass filtered at 12 kHz, and acquired using pClamp software.
Borosilicate glass pipettes (2–4 MΩ) were pulled on a P-87 micropipette puller (Sutter Instruments). For evoked EPSC recordings, the pipette was filled with an internal solution containing the following (in mm): 120 potassium gluconate, 11 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, and 11 EGTA (ECl = −57 mV). For evoked IPSC recordings, the internal solution contained the following (in mm): 65 potassium gluconate, 66 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, and11 EGTA (ECl = −16 mV). For miniature EPSC (mEPSC) recordings, the internal solution contained the following (in mm): 130 cesium gluconate, 2 CsCl, 1 MgCl2, 10 HEPES, and 10 EGTA (ECl = −92 mV). All internal solutions were pH 7.3–7.35 and 282–286 mOsm/L.
Postsynaptic currents were evoked by electrical or optical stimulation and were recorded in TC neurons in VB thalamus or RT neurons in the portion of the RT nucleus adjacent to VB. TC neurons were distinguished from RT neurons based on location and morphological characteristics and subsequently confirmed by observing prominent sag current during hyperpolarizing steps (McCormick and Pape, 1990) and distinct firing patterns (Jahnsen and Llinás, 1984). During voltage-clamp experiments, neurons were held at −74 mV for evoked EPSC recordings or −69 mV for evoked IPSC recordings (accounting for −14 mV and −9 mV liquid junction potential, respectively). A brief, hyperpolarizing voltage pulse was applied at the beginning of every sweep to monitor series resistance (5–15 MΩ). Recordings were excluded from analysis if the series resistance changed by >20%. Paired-pulse stimulations were given with a 40 ms interval (25 Hz), which was chosen after testing a range of intervals (10–500 ms) for reliable cortico-RT facilitation (10–80 ms) and thalamo-RT depression (10–500 ms), as well as for reliable optogenetic stimulation given the limitations of ChR2 kinetics.
For all evoked EPSC recordings, 50 μm picrotoxin was added to the ACSF perfusate to block GABAA receptors, whereas for IPSC recordings, 50 μm d-AP5 and 20 μm DNQX disodium salt were added to the ACSF to block ionotropic glutamate receptors. For optical stimulation-evoked paired-pulse EPSC recordings, 473 nm light (1–3 ms) was delivered to the slice by a 300-μm-diameter fiber-optic cable (BFL37–300; Thor Laboratories) connected to a laser (30 mW 473 nm DPSS laser; OEM Laser Systems). The cable was gently placed on the slice surface at ∼30° from the horizontal plane such that light was delivered to internal capsule and RT. Light intensity was initially adjusted to find the minimal stimulation threshold for inducing EPSCs (∼50% successes) and then increased to a moderate intensity (20–200 μW/mm2 at slice plane) to excite a population of synapses (i.e., resulting in a summated EPSC with a peak amplitude 200–500% of the minimal stimulation response amplitude).
For minimal electrical stimulation recordings, brief paired-pulse current pulses (40–280 μA, 20–80 μs duration) were delivered from a stimulus isolation unit (WPI) through a pair of parallel tungsten monopolar stimulating electrodes (50–100 kΩ, FHC) 50–100 μm apart. Stimulation intensity was set slightly (10–20%) above PSC threshold. This enabled reliable single fiber stimulation, as verified by unchanged response amplitude and waveform (Dobrunz and Stevens, 1997). For EPSC recordings in RT neurons, the stimulating electrode was placed in the internal capsule. For IPSC recordings in TC neurons, the stimulating electrode was placed 100–300 μm away from the patched TC neuron. For IPSC recordings in RT neurons, the electrode was placed in RT near the RT and VB border.
For electrically evoked train stimulation EPSC recordings, train stimuli were composed of 10 5 Hz pulses. Stimulation intensity was set slightly (10–20%) above threshold for single fiber stimulation. Before the main experiments, we identified the effective concentration of the group III mGluR antagonist (RS)-α-methylserine-O-phosphate (MSOP) to use to test cortico-RT synaptic release. Cortico-RT synaptic responses were identified as facilitating paired-pulse responses to minimal electrical stimulation (40 ms interval). A 200 μm MSOP concentration was sufficient to reverse synaptic depression induced by 20 μm l-AP4 (n = 3), which is consistent with previous studies (Pelkey et al., 2005; Kwon and Castillo, 2008).
For mEPSC recordings, 0.5 μm TTX was added to the ACSF perfusate to block action-potential-dependent synaptic activity. RT neurons were clamped at −86 mV (accounting for −16 mV liquid junction potential) to observe mEPSCs with minimal contamination from IPSCs.
Drugs.
(S)-3,4-dicarboxyphenylglycine (DCPG), l-(+)-2-amino-4-phosphonobutyric acid (L-AP4), and MSOP were purchased from Tocris Bioscience. TTX, DNQX disodium salt, and d-AP5 were purchased from Abcam and GDP-β-S trilithium salt from Sigma-Aldrich. All stock solutions were prepared with Milli-Q water (Millipore) and stock aliquots were stored at −20°C until use.
Analysis.
Data and statistical analyses were performed using MATLAB 2011b (MathWorks) and GraphPad Prism 5 software. Oscillation spike detection and parameter analysis were performed as described previously (Sohal et al., 2003). Briefly, spikes were detected as slope deflections >3× threshold, defined as the root-mean-square of background noise of baseline sweeps. Bursts were defined as clusters of spikes (≥m spikes with <x ms interspike interval; spindle oscillations: m = 3, x = 6–10; epileptiform oscillations: m = 5, x = 10). Oscillations were defined as clusters of bursts (≥n bursts with <y s interburst interval; spindle oscillations: n = 2, y = 0.6–1; epileptiform oscillations: n = 3, y = 1). Oscillation duration was measured from stimulus onset to the final burst. Oscillation period was found by computing the autocorrelogram of the multiunit spike rate and then finding the time of the first peak of the autocorrelogram after the peak at time 0 (Sohal et al., 2003). The oscillation index (OI), a measure of synchrony (range: 0–1), was quantified by finding the mean value of the first two peaks in the autocorrelogram, subtracting the minimum value of the trough between them, and dividing by the mean value of the first two peaks (Kleiman-Weiner et al., 2009).
Evoked EPSC and IPSC traces were filtered at 1 kHz. To compute the weighted decay time constant (τwd), the decay phase of the response was first fit by a double exponential function: I = C1 e−t/τ1 + C2 e−t/τ2, where C1 and C2 are the slow and fast component coefficients and τ1 and τ2 are the slow and fast decay time constants, respectively), and then computed: τwd = (τ1 C1 + τ2 C2)/(C1 + C2). The paired-pulse ratio (PPR) for each drug epoch was calculated as the average peak amplitude of the second response divided by the average amplitude of the first response [i.e., mean(A2)/mean(A1)], rather than as the average of individual PPRs [i.e., mean(A2/A1)], which can yield spurious increases in PPR (Kim and Alger, 2001).
For mEPSC recordings, a custom program was used for automated detection of mEPSCs (Ulrich and Huguenard, 1996), which was verified by visual inspection while blinded to experimental condition. The median value of each parameter (e.g., amplitude, rise time constant) was computed for each recorded neuron. These values were then averaged across neurons. Cumulative probability distributions were created using 200 randomly selected events per neuron.
Statistical significance of experiments involving multiple drug conditions tested in two or more groups was assessed by two-way repeated-measures ANOVA (two-way rANOVA) followed by Bonferroni's post hoc test. Experiments involving multiple drug conditions tested in one group were assessed by one-way repeated-measures ANOVA (one-way rANOVA) followed by Tukey's HSD post hoc test. Experiments involving two drug conditions tested in one group or in two independent groups were assessed by the Wilcoxon signed-rank test or Wilcoxon rank-sum test, respectively. Differences in cumulative probability distributions were assessed by Kolmogorov–Smirnov test. All statistics were two-tailed with significance level α = 0.05. All values are reported as mean ± SEM.
Immunohistochemistry.
After some optogenetics experiments, slices were fixed in 4% phosphate-buffered paraformaldehyde at 4°C overnight. After cryoprotecting with 30% sucrose buffer for 30 min, slices were resectioned into 60 μm horizontal sections with a sliding freezing microtome (HM 400; Microm). Free-floating sections were incubated for 1 h in 10% normal donkey serum diluted with PBS, followed by overnight incubation at 4°C in rabbit anti-parvalbumin polyclonal antibody (1:500; Abcam) diluted with PBS containing 0.25% Triton-X. Sections were rinsed in PBS and incubated for 2 h in donkey anti-rabbit Cy3 secondary antibody (1:100; Invitrogen). Sections were rinsed in PBS, mounted with Vectashield (Vector Laboratories), and imaged with an LSM 510 confocal microscope (Zeiss).
Results
Activation of group III mGluRs suppresses thalamic network oscillations in vitro
We first investigated whether group III mGluR activation affects corticothalamic circuit function by examining the properties of evoked thalamic oscillations in vitro. Horizontal thalamic slices were obtained from P11-P14 rats because oscillations in slices from juvenile rats are typically robust and stable during long recordings (>1 h), thus serving as a reliable assay of changes in network state (Jacobsen et al., 2001). Rhythmic spiking activity was evoked by a single electrical shock to CT and TC axons in the internal capsule. Extracellular units were recorded in the somatosensory VB of the thalamus, which includes ventral posteromedial (VPM) and ventral posterolateral (VPL) nuclei (Fig. 1A).
Figure 1.
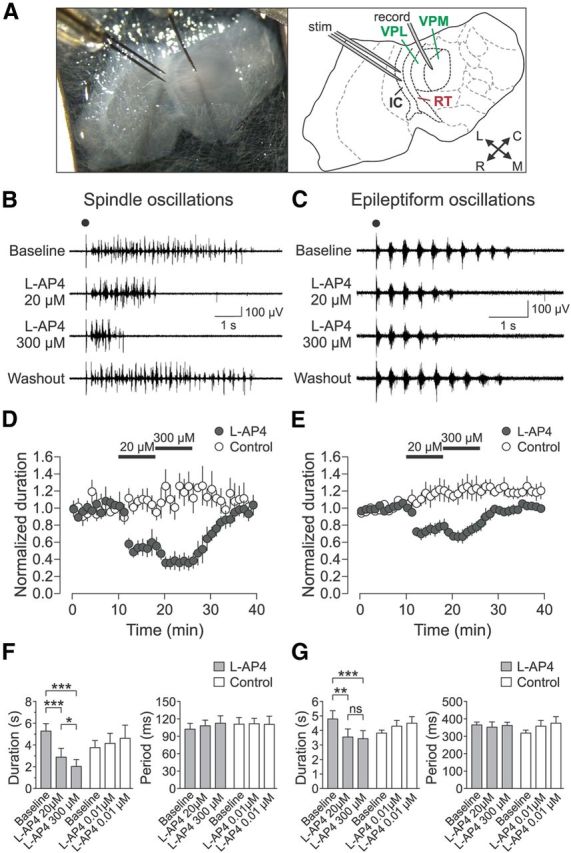
Group III mGluR activation suppresses evoked thalamic oscillations. A, Horizontal thalamic slice recording setup and schematic. IC, Internal capsule; L, lateral; M, medial; R, rostral; C, caudal. B, C, Representative oscillations, spindle (B) and epileptiform (C), during sequential activation of mGluR4/8 (by 20 μm l-AP4) and mGluR4/7/8 (by 300 μm l-AP4). Electrical stimulus is indicated by black dot. D, Spindle oscillation duration normalized to baseline for l-AP4 treated experimental (n = 5) and control (n = 4) slices. E, Epileptiform oscillation duration normalized to baseline for l-AP4-treated experimental (n = 6) and control (n = 4) slices. F, G, Summary of l-AP4 effects on oscillation duration and period. Spindle oscillations (F) showed effect of epoch on oscillation duration for the experimental group (gray bars; n = 5, F(2,8) = 75.98, p < 0.0001, one-way rANOVA), but not for the control group (white bars; n = 4, F(2,6) = 2.03, p = 0.212). Spindle oscillation duration was significantly decreased further by 300 μm l-AP4 compared with 20 μm l-AP4 (*p < 0.05, Tukey's post hoc test). Epileptiform oscillations (G) also showed an effect of epoch on oscillation duration for the experimental group (gray bars; n = 6, F(2,10) = 17.73, p = 0.0005, one-way rANOVA), but no effect for the control group (white bars; n = 4, F(2,6) = 2.69, p = 0.15). Period did not change for either spindle (experimental group: F(2,8) = 0.42, p = 0.67; control group: F(2,6) = 0.04, p = 0.97, one-way rANOVA) or epileptiform oscillations (experimental group: F(2,10) = 0.21, p = 0.81; control group: F(2,6) = 0.42, p = 0.67). Data are from last 5 min of each epoch. *p < 0.05; **p < 0.01; ***p < 0.001; NS, not significant, Tukey's post hoc test.
Two types of oscillations were examined. Spindle oscillations with ∼10 Hz multiunit burst firing with low synchrony (Fig. 1B) were evoked in drug-free ACSF. Epileptiform oscillations with well organized highly synchronous ∼3 Hz bursts (Fig. 1C) were evoked in ACSF containing picrotoxin (50 μm) and apamin (1 nm). For each slice, after a stable baseline oscillation had been established for at least 15 min, the group III mGluR-specific orthosteric agonist l-AP4 was bath-applied for two consecutive eight-minute blocks in “low-high dose” experiments (“experimental group”): first, at a low concentration (20 μm) saturating for the high-affinity receptors mGluR4 and mGluR8 and then at a high concentration (300 μm) that additionally activated the low-affinity receptor mGluR7 (Thomsen et al., 1992; Okamoto et al., 1994; Saugstad et al., 1997; Schoepp et al., 1999). Note that mGluR6 was not considered in this study because its expression is restricted to the retina (Nakajima et al., 1993). In addition, to control for subtle long-term changes in oscillation properties and procedural effects of drug application, an ultra-low, nonactivating concentration of l-AP4 (0.01 μm) was applied for two eight-minute blocks to a separate set of slices (“control group”).
Spindle oscillations (n = 9) had a duration of 4.6 ± 0.5 s and period of 105 ± 7 ms during the baseline epoch before drug application. These oscillations were highly variable in temporal structure from sweep to sweep (Fig. 1B) and exhibited a low degree of synchrony of multiunit firing within each sweep as quantified by a relatively low OI (see Materials and Methods) of 0.27 ± 0.05. For the experimental group, activation of mGluR4/8 by 20 μm l-AP4 rapidly reduced oscillation duration to 54.7 ± 15.2% of baseline, whereas additional activation of mGluR7 by 300 μm l-AP4 further reduced the duration to 38.4 ± 12.1% of baseline (Fig. 1B,D,F). This effect of drug epoch on oscillation duration was significant (n = 5; F(2,8) = 75.98, p < 0.0001, one-way rANOVA; Fig. 1F). Notably, the oscillation duration after 300 μm l-AP4 application was significantly shorter than the duration after 20 μm l-AP4 application (p < 0.05; Fig. 1F). In contrast, the oscillation duration for the control group, despite showing a slight increase over time, was not significantly different between drug epochs (p = 0.212; Fig. 1D,F). When time-matched results from the experimental and control groups were compared directly, there was a significant main effect of drug epoch on oscillation duration (F(2,14) = 6.81, p = 0.009, two-way rANOVA) and interaction of drug epoch and group (F(2,14) = 27.54, p < 0.0001), with significance reached for the pairwise comparison of baseline epoch and 300 μm l-AP4 epoch (p < 0.05, Bonferroni's post hoc test). There was no main effect of group on oscillation duration (F(1,14) = 3.15, p = 0.12). In addition, the oscillation period did not change for either the experimental group or control group (Fig. 1F). The OI also did not change for either the experimental group (baseline: 0.17 ± 0.02, 20 μm l-AP4: 0.26 ± 0.10, 300 μm l-AP4: 0.21 ± 0.05; F(2,8) = 0.90, p = 0.45, one-way rANOVA) or control group (baseline: 0.44 ± 0.06, 0.01 μm l-AP4 first block: 0.41 ± 0.07, 0.01 μm l-AP4 second block: 0.36 ± 0.06; F(2,6) = 3.45, p = 0.10). These results suggest that the strength, but not the structure, of network activity was modulated by group III mGluR activation.
Epileptiform oscillations (n = 10) had a baseline duration (4.4 ± 0.4 s) similar to spindle oscillations, but had a baseline period (342 ± 20 ms) much slower than that of spindle oscillations and typical of oscillations involving GABAA receptor block (Huntsman et al., 1999; Kleiman-Weiner et al., 2009). These oscillations had stereotyped temporal structure across sweeps (Fig. 1C) and contained highly synchronous bursts corresponding to a high OI of 0.91 ± 0.03. For the experimental group with low-high dose l-AP4, activation of mGluR4/8 by 20 μm l-AP4 decreased oscillation duration to 74.2 ± 11.5% of baseline, whereas additional activation of mGluR7 by 300 μm l-AP4 resulted in a duration of 71.8 ± 11.4% of baseline (Fig. 1C,E,G). This effect of drug epoch on oscillation duration was significant (n = 6; F(2,10) = 17.73, p = 0.0005, one-way rANOVA; Fig. 1G). However, the effects of 20 μm l-AP4 and 300 μm l-AP4 were not significantly different (p > 0.05, Tukey's post hoc test; Fig. 1G). In addition, after the initial suppressive effect of 300 μm l-AP4, there was a slight increasing trend in oscillation duration toward the end of the wash-in epoch (Fig. 1E). This may have been due to nonspecific changes in the state of the slice or due to a gradual change in the receptor state (e.g., internalization of mGluR7) after activation of group III mGluRs by l-AP4 (Pelkey et al., 2005).
In contrast, the oscillation duration of the control group was not different between drug epochs (p = 0.15; Fig. 1E,G). When the results from the experimental and control groups were compared directly, there was a significant interaction of drug epoch and group on oscillation duration (F(2,16) = 14.84, p = 0.0002, two-way rANOVA), with significance reached for the baseline epoch versus 300 μm l-AP4 epoch (p < 0.05; Bonferroni's post hoc test). There were no main effects of drug epoch (F(2,16) = 1.22, p = 0.32) or group (F(1,16) = 1.22, p = 3.29) on oscillation duration. Oscillation period did not change for either the experimental group or control group (Fig. 1G). OI also did not change for either the experimental group (baseline: 0.91 ± 0.03, 20 μm l-AP4: 0.87 ± 0.04, 300 μm l-AP4: 0.89 ± 0.03; F(2,10) = 0.27, p = 0.77, one-way rANOVA) or control group (baseline: 0.92 ± 0.07, 0.01 μm l-AP4 first block: 0.89 ± 0.09, 0.01 μm l-AP4 second block: 0.87 ± 0.10; F(2,6) = 2.17, p = 0.20). Similar to spindle oscillations, these results suggest that the strength, but not the structure, of epileptiform network activity was modulated by group III mGluR activation. Spindle oscillation duration was more strongly suppressed than epileptiform oscillations by mGluR4/8 activation (p = 0.05, Wilcoxon rank-sum test).
In summary, the activation of group III mGluRs by l-AP4 rapidly caused a substantial, reversible suppression of evoked spindle and epileptiform thalamic oscillations in vitro, which indicates that these receptors are present in the circuit and contribute to the modulation of thalamic network activity. For both oscillation types, activation of the high-affinity receptors mGluR4/8 caused strong suppression of oscillations. Spindle oscillations were more strongly suppressed than epileptiform oscillations by mGluR4/8 activation and also were further suppressed by the additional activation of the low-affinity receptor mGluR7. These findings may reflect differences between the two oscillation types, such as the stronger output of RT cells in epileptiform oscillations (Bal and Destexhe, 2000; Blumenfeld and McCormick, 2000).
Group III mGluRs are located on cortico-RT, but not thalamo-RT, synapses
Several synapses are involved in the initiation and maintenance of oscillations in the corticothalamic circuit. These include intrathalamic synapses (e.g., between TC relay neurons and RT neurons or among RT neurons; Bal and McCormick, 1993), intracortical synapses, and synapses between cortex and thalamus. Excluding intracortical synapses not present in thalamic slices, any of these synapses could be the site of group III mGluR activation by l-AP4, which suppressed thalamic oscillations, as described previously. We began by investigating whether group III mGluRs are present at CT synapses onto RT neurons (“cortico-RT”) and/or TC synapses onto RT neurons (“thalamo-RT”). These two glutamatergic inputs drive activity in RT, which sends GABAergic projections to all dorsal thalamic nuclei and thus is a crucial modulator of the corticothalamic network state. For example, changes in RT activity can cause the circuit to switch between physiological and pathological patterns of oscillations (Bal and Destexhe, 2000; Blumenfeld and McCormick, 2000).
Because CT and TC axons have extensive spatial overlap, suprathreshold electrical stimulation of CT axons frequently also causes antidromic activation of TC collaterals. Previous studies have circumvented this difficulty by local stimulation of cortical layer 6 cells by high-K+ or AMPA-containing solutions, paired recordings of cortical and RT cells, or minimal electrical stimulation of single axons (Golshani et al., 2001; Gentet and Ulrich, 2004). Here, we selectively stimulated a pure population of thalamo-RT or cortico-RT axons by optogenetic targeting of TC neurons or layer 6 cortical neurons. After expression of ChR2 in VB thalamus (Fig. 2A) or deep layers of barrel cortex (Fig. 2B), we obtained slices and performed whole-cell patch-clamp recordings of RT neurons while optically stimulating ChR2-expressing thalamic or cortical axons. All cells were recorded in the presence of picrotoxin (50 μm) to block GABAA-receptor-mediated, disynaptic IPSCs that could arise via intraRT inhibition. In addition, a cut was made between RT and VB nuclei to prevent recurrent excitation (from the firing of TC neurons after CT axon stimulation) from contaminating the recordings of paired-pulse responses (Fig. 2A,B). We conducted a “low-high-low-dose” experiment involving three wash-in blocks: (1) 20 μm l-AP4 to activate mGluR4/8 (8 min), (2) 300 μm l-AP4 to additionally activate mGluR7 (5 min), and (3) another wash-in of 20 μm l-AP4 (8 min).
Figure 2.

Activation of group III mGluRs depresses cortico-RT synapses but not thalamo-RT synapses. A, B, Experimental schematic and examples of horizontal thalamic slices from brains with ChR2 expressed in VB thalamus (A) or barrel cortex (B). RT shows relatively dense parvalbumin expression. Dotted line denotes location of scalpel cut between RT and VB. Barrel cortex is outside of the plane of section. C, D, Representative recordings of EPSCs in patched RT neurons evoked by optical paired-pulse stimulation (25 Hz) of thalamo-RT (C) or cortico-RT (D) axons during bath application of l-AP4. Left, Time course of changes in peak amplitude of first EPSC (filled circles) and second EPSC (open circles). Right, Averaged sample traces of EPSCs from the experiment plotted on left. E, F, Summary data for effect of l-AP4 on EPSC amplitude. Left, Time course of changes in mean peak amplitude normalized to mean baseline amplitude of first EPSC. Right, Bar graphs of normalized first EPSC amplitude and PPR. Data are from the last 2 min of each drug epoch. Thalamo-RT (E) first EPSC amplitude was not affected by l-AP4 (n = 6, F(3,15) = 1.52, p = 0.25, one-way rANOVA), and PPR also was not affected (F(3,15) = 1.64, p = 0.22). Cortico-RT (F) first EPSC amplitude was decreased by l-AP4 application (n = 5, F(3,12) = 38.03, p < 0.0001, one-way rANOVA), but PPR was unaffected (F(3,12) = 2.30, p = 0.13). Baseline epoch: min 0–10. *p < 0.05; **p < 0.01, Tukey's post hoc test.
Thalamo-RT synaptic responses exhibited paired-pulse depression (baseline PPR: 0.63 ± 0.04; Fig. 2C,E), as expected from previous studies (Gentet and Ulrich, 2003). These evoked thalamo-RT EPSCs were invariably unaffected by the application of l-AP4 (baseline: 342 ± 33.4 pA, first 20 μm l-AP4: 322 ± 25.9 pA, 300 μm l-AP4: 334 ± 23.5 pA, second 20 μm l-AP4: 311 ± 19.8 pA), such that there was no significant effect of drug epoch on EPSC amplitude (p = 0.25; Fig. 2E). The PPR also did not change across drug epochs. These results indicate that thalamo-RT synapses are devoid of all group III mGluRs.
Cortico-RT synaptic responses showed paired-pulse facilitation (baseline PPR: 2.26 ± 0.17; Fig. 2D,F) similar to that reported in previous studies (Golshani et al., 2001; Granseth et al., 2002; Cruikshank et al., 2010). In marked contrast to thalamo-RT EPSCs, evoked cortico-RT EPSC amplitudes were rapidly and dramatically reduced by the application of l-AP4 (Fig. 2D,F), indicating the presence of group III mGluRs. The sequential application of low-high-low-dose l-AP4 decreased the first EPSC amplitude to 22%, 19%, and 32% of baseline, respectively (baseline: 117 ± 16.8 pA, first 20 μm l-AP4: 26.0 ± 72.2 pA, 300 μm l-AP4: 22.4 ± 4.95 pA, second 20 μm l-AP4: 37.8 ± 9.93 pA), which was a significant effect of drug epoch on EPSC amplitude (p < 0.0001; Fig. 2F). The effects of the 20 and 300 μm doses of l-AP4 were not significantly different from each other (p > 0.05 for all relevant pairwise comparisons), which may indicate a lack of mGluR7 expression at these synapses. Alternatively, mGluR7 may be present at the cortico-RT synapse, but activation of mGluR4/8 may have caused maximal EPSC suppression such that the effect of concurrent mGluR7 activation was not apparent. The PPR was not significantly different between drug epochs. There was also no significant change in input resistance from the baseline of 177 ± 14.2 MΩ (p = 0.72, one-way rANOVA).
To validate the effects of the low and high doses of l-AP4 on cortico-RT EPSCs, a separate “single-dose” experiment was conducted in which l-AP4 was applied at only one concentration (either 20 or 300 μm) for each recording (Fig. 3). As in the previous experiment, ChR2-expressing cortical axons were stimulated with paired-pulse optical stimulation. The application of l-AP4 again resulted in a rapid and substantial decrease in EPSC amplitude for both the 20 μm l-AP4 group (n = 3; Fig. 3A) and the 300 μm l-AP4 group (n = 4; Fig. 3B). The time course of recovery from suppression was similar for the two groups; for example, the first EPSC amplitude at 20 min after the end of l-AP4 application was similar between the groups (20 μm l-AP4: 46.8 ± 11.6% of baseline, 300 μm l-AP4: 51.6 ± 8.4% of baseline; p = 0.63, Wilcoxon rank-sum test). To compare the degree of synaptic depression induced by the two drug concentrations, data for the baseline and drug wash-in epochs were pooled from multiple experiments, including from the previously described low-high-low-dose experiment (Fig. 2) and an abbreviated washout experiment (data not shown). Although EPSC amplitude was significantly suppressed by both 20 and 300 μm l-AP4, there was no significant difference in the degree of suppression (Fig. 3C). There was no change in PPR after l-AP4 application (Fig. 3D).
Figure 3.

Cortico-RT synaptic release is suppressed to a similar extent by separate applications of 20 or 300 μm l-AP4. A, B, Peak amplitude of paired-pulse EPSCs evoked by optical stimulation for experiments in which either 20 μm l-AP4 (A, n = 3) or 300 μm l-AP4 (B, n = 4) was applied. Amplitudes are normalized to mean baseline amplitude of first EPSC. Washout epoch: min 15–45. C, Normalized first EPSC amplitude during late baseline epoch and first drug wash-in epoch pooled from experiments in which either l-AP4 20 μm (n = 8) or l-AP4 300 μm (n = 6) were applied. EPSC amplitudes are normalized to mean baseline amplitude. There was a significant main effect of epoch (F(1,12) = 61.40, ***p < 0.001, two-way rANOVA), but no significant interaction of epoch and drug concentration (F(1,12) = 0.13, p > 0.05). D, PPR of EPSC amplitude was not affected by l-AP4. There was no main effect of epoch (F(1,12) = 1.05, p > 0.05, two-way rANOVA) or interaction of epoch and drug concentration group (F(1,12) = 0.04, p > 0.05). Bar graphs show mean amplitudes of the last 2 min of each epoch.
Finally, these optical stimulation results were reproduced in a minimal electrical stimulation experiment testing the effect of mGluR4/8 activation on thalamo-RT or cortico-RT paired-pulse responses. Depressing (PPR <1) and facilitating (PPR >1) EPSCs were categorized as putative thalamo-RT and cortico-RT responses, respectively (n = 3 for each). Thalamo-RT EPSC amplitudes were unchanged by 20 μm l-AP4 for both first EPSC (81 ± 18 to 70 ± 16 pA) and second EPSC (46 ± 9 to 35 ± 10 pA; data not shown). In contrast, cortico-RT EPSC amplitudes were strongly reduced by 20 μm l-AP4 to 18% of baseline for first EPSC (62 ± 22 to 11 ± 6 pA) and 35% for second EPSC (133 ± 27 to 47 ± 21 pA; data not shown).
Together, these results indicate that group III mGluRs are present at cortico-RT synapses but not thalamo-RT synapses. The similar degree of suppression of cortico-RT responses by 20 μm l-AP4 and 300 μm l-AP4 for both the low-high-low-dose and single-dose optogenetic experiments suggest either that mGluR7 is not expressed at these synapses or that mGluR4/8 activation exerts a maximally suppressive effect on synaptic transmission such that concurrent mGluR7 activation does not have an apparent effect.
Suppression of cortico-RT responses by mGluR4/8 activation is due to a presynaptic mechanism
The suppression of evoked cortico-RT responses by group III mGluR activation could be due to a presynaptic mechanism, such as decreased neurotransmitter release after inhibition of presynaptic voltage-gated calcium channels. Alternatively, postsynaptic mGluR activation could decrease the excitability of the postsynaptic cell, e.g., by opening potassium channels. Although group III mGluRs are typically presynaptic, postsynaptic mGluR7 has been reported in retina and locus ceruleus (Bradley et al., 1998; Koulen and Brandstatter, 2002). To distinguish between presynaptic and postsynaptic mechanisms, we recorded mEPSCs during whole-cell voltage clamp of RT neurons while applying 20 μm l-AP4 to activate mGluR4/8 (Fig. 4A–D). mEPSCs were recorded in the presence of 0.5 μm TTX to block action-potential-dependent synaptic activity and RT cells were clamped at −86 mV to observe mEPSCs without contamination from mIPSCs.
Figure 4.
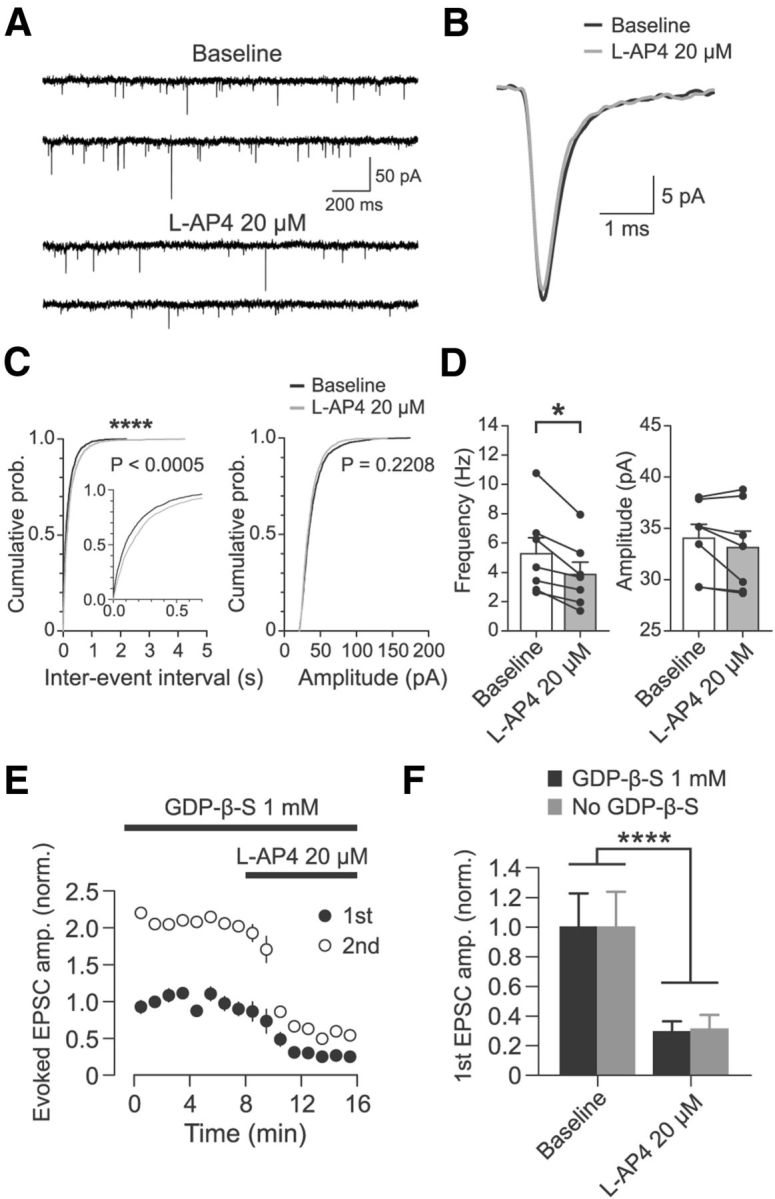
Effect of l-AP4 at cortico-RT synapses is mediated by a presynaptic mechanism. A, Representative partial recording of mEPSCs in a patched RT cell in the presence of 0.5 μm TTX before and after application of 20 μm l-AP4. B, Average mEPSC for experiment shown in A. For each epoch, 200 randomly chosen mEPSCs were peak aligned and averaged. C, Cumulative probability distributions of interevent intervals and amplitudes of mEPSCs before and after application of 20 μm l-AP4 for 200 randomly chosen mEPSCs from each experiment (n = 7, 1400 total events). Interevent intervals were significantly increased (****p < 0.0005, Kolmogorov–Smirnov test), whereas amplitudes were unchanged (p = 0.2208, Kolmogorov–Smirnov test). D, Summary data of l-AP4 resulting in decreased mEPSC frequency (n = 7, *p = 0.02, Wilcoxon signed-rank test) but no change in mEPSC amplitude (n = 7, p = 0.16, Wilcoxon signed-rank test). Individual sample points are median values of all mEPSCs from each experiment. E, Effect of 20 μm l-AP4 on optically evoked cortico-RT EPSC amplitude when postsynaptic G-protein signaling was inhibited by 1 mm GDP-β-S (n = 6). Amplitudes are normalized to mean baseline amplitude of first EPSC. F, Comparison of l-AP4 effect on cortico-RT EPSCs recorded with postsynaptic GDP-β-S (data as in E, n = 6) versus recorded without GDP-β-S (data as in Fig. 3C, n = 8). Although there was clearly a significant main effect of drug epoch (F(1,12) = 33.87, ****p < 0.0001, two-way rANOVA), there was no significant main effect of condition (F(1,12) = 0.022, p = 0.884) or a significant interaction of condition and epoch (F(1,12) = 0.07, p = 0.793). Data are from the last 2 min of baseline and min 3–5 of l-AP4 wash-in.
Activation of mGluR4/8 by 20 μm l-AP4 significantly reduced mEPSC frequency (5.3 ± 1.1 to 3.8 ± 0.8 Hz; n = 7, p = 0.02, Wilcoxon signed-rank test), but did not change mEPSC amplitude (34.0 ± 1.4 to 33.1 ± 1.6 pA; p = 0.16; Fig. 4A–D). l-AP4 also did not affect mEPSC kinetics (Fig. 4B), including: half-width (0.43 ± 0.03 to 0.45 ± 0.03 ms; n = 7, p = 0.31, Wilcoxon signed-rank test); 10–90% rise time (0.20 ± 0.01 to 0.21 ± 0.01 ms; p = 1.00); weighted decay time constant (0.81 ± 0.07 to 0.91 ± 0.08 ms; p = 0.08). The decrease in mEPSC frequency in the absence of changes in mEPSC amplitude indicates that mGluR4/8 activation affects presynaptic release mechanisms at glutamatergic synapses onto the RT cell. Although both cortico-RT and thalamo-RT synapses contributed to the mEPSCs, the majority of mEPSCs were likely from cortico-RT synapses, based on anatomical and functional studies (Ohara and Lieberman, 1985; Liu and Jones, 1999; Paz et al., 2011). Furthermore, because our previous experiments showed that thalamo-RT synapses are insensitive to l-AP4, the reduction in mEPSC frequency must have been due to presynaptic suppression of release at cortico-RT synapses.
We further confirmed the presynaptic site of action of group III mGluRs by conducting experiments in which we included a nonhydrolyzable GDP analog, GDP-β-S (1 mm), in the patch pipette to block postsynaptic G-protein signaling. If the reduction in evoked cortico-RT EPSC amplitude caused by application of the group III mGluR agonist l-AP4 (Figs. 2, 3) is at least partially dependent on activation of postsynaptic group III mGluRs, then inhibition of postsynaptic G-protein signaling with GDP-β-S should decrease the suppressive effect of l-AP4 on cortico-RT responses. As before, after expression of ChR2 in corticothalamic neurons in deep layers of barrel cortex, we performed whole-cell patch-clamp recordings of optically evoked paired-pulse cortico-RT EPSCs. Recordings were obtained using pipettes with internal solution containing GDP-β-S at a concentration (1 mm) known to block the action of postsynaptic mGluRs (Guérineau et al., 1994; Heuss et al., 1999). Before initiating recordings, the internal solution was allowed to dialyze into the patched cell for >20 min after cell break-in. We found that bath application of 20 μm l-AP4 to activate mGluR4/8 robustly suppressed evoked cortico-RT EPSC amplitudes (Fig. 4E,F). First EPSC amplitude was reduced to 24.7 ± 3.4% of baseline (168.7 ± 38.4 pA to 41.7 ± 7.3 pA; n = 6, p = 0.03; Wilcoxon signed-rank test), whereas second EPSC amplitude was reduced to 26.3 ± 3.5% of baseline (181.6 ± 44.2 pA to 53.6 ± 13.8 pA; p = 0.03). Notably, when these results were compared with the results of the experiment conducted without GDP-β-S (Fig. 3C), the magnitude of cortico-RT EPSC suppression by 20 μm l-AP4 was not significantly different (Fig. 4F). PPR was not significantly affected by l-AP4 during inhibition of postsynaptic G-protein signaling by GDP-β-S (2.4 ± 0.2 to 2.5 ± 0.3; n = 6, p = 1.00).
Together, the results of the mEPSC and GDP-β-S evoked EPSC experiments suggest that mGluR4/8 have a presynaptic site of action at the cortico-RT synapse. These findings support the consensus that group III mGluRs are typically localized to presynaptic active zones, as observed in various regions such as hippocampus, cerebellum, basal ganglia, and olfactory bulb (Shigemoto et al., 1996, 1997; Wada et al., 1998; Mateos et al., 1999; Corti et al., 2002; Ferraguti et al., 2005).
mGluR8 activation suppresses cortico-RT synaptic transmission
We next investigated whether the cortico-RT synaptic depression induced by l-AP4 was primarily mediated by mGluR4, mGluR8, or both. This was done by testing the effect of a potent, selective agonist for mGluR8, (S)-3,4-DCPG (Thomas et al., 2001), on optically evoked paired-pulse cortico-RT EPSCs (Fig. 5A). Bath application of 10 μm DCPG rapidly and substantially reduced the peak amplitude of the first EPSC to 37% of baseline (266.4 ± 36.4 to 99.3 ± 18.2 pA). After the DCPG effect had reached a plateau (8 min), 20 μm l-AP4 was added to the DCPG-containing bath perfusate, which resulted in a first EPSC amplitude that was 24% of baseline (63.4 ± 8.7 pA). This effect of drug epoch on EPSC amplitude was significant (p < 0.0001; Fig. 5B,C). However, there was no difference between the effect of 10 μm DCPG alone and the combined effect of 10 μm DCPG + 20 μm l-AP4 (p > 0.05; Fig. 5C). There was no change in the PPR across epochs (Fig. 5D).
Figure 5.
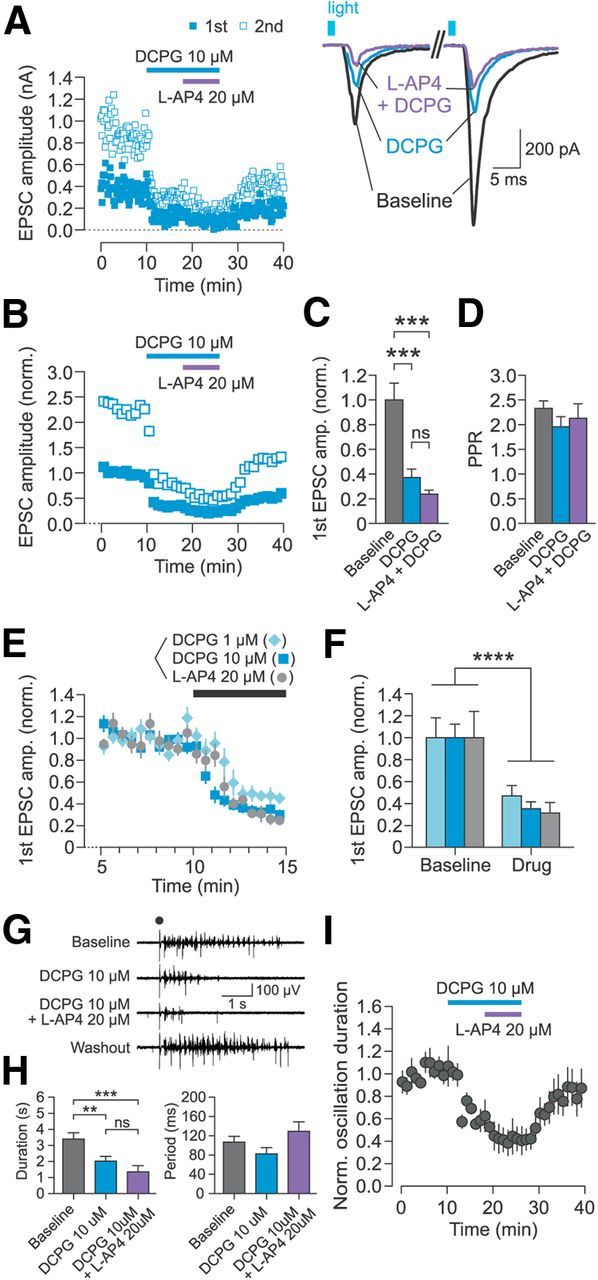
Activation of mGluR8 strongly depresses cortico-RT synaptic release and suppresses thalamic spindle oscillations. A, Representative recording of EPSCs in patched RT neurons evoked by optical paired-pulse stimulation. Left, Time course of changes in peak amplitude of first EPSC (filled squares) and second EPSC (open squares). Right, Sample traces of individual evoked EPSCs from the experiment plotted on left. B, Summary data for time course of changes in peak EPSC amplitude normalized to mean baseline amplitude of first EPSC. C, EPSC amplitude significantly differed between epochs (n = 6, F(2,10) = 46.6, p < 0.0001, one-way rANOVA), but there was no difference between the effect of 10 μm DCPG alone and 20 μm l-AP4 + 10 μm DCPG (p > 0.05, Tukey's post hoc test). D, PPR did not differ between epochs (n = 6, F(2,10) = 2.91, p = 0.10, one-way rANOVA). E, F, Comparison of EPSC amplitudes during baseline and first drug wash-in epochs for 3 experimental groups: DCPG 1 μm (n = 5), DCPG 10 μm (n = 10, including data in B), or l-AP4 20 μm (n = 8, as in Fig. 3). There was a significant main effect of epoch (F(1,20) = 83.18, p < 0.0001, two-way rANOVA), but no significant interaction of epoch and group (F(2,20) = 0.39, p > 0.05). All bar graphs represent data from the last 2 min of each epoch. G, Representative spindle oscillation recording during sequential activation of mGluR8 (by 10 μm DCPG) and mGluR4/8 (by 20 μm l-AP4 + 10 μm DCPG). Electrical stimulus is indicated by black dot. H, Drug epoch had a significant effect on oscillation duration (n = 5, F(2,8) = 29.31, p = 0.0002, one-way rANOVA), but there was no difference between the effect of 10 μm DCPG alone and 10 μm DCPG + 20 μm l-AP4 (p > 0.05, Tukey's post hoc test). There was no significant effect of drug epoch on oscillation period (n = 5, F(2,8) = 1.83, p = 0.22, one-way rANOVA). Data are from last 5 min of each epoch. I, Time course of changes in normalized spindle oscillation duration. **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant, Tukey's post hoc test.
Due to a recent report that concentrations of DCPG >1 μm may activate mGluR2 in addition to mGluR8 (but see Thomas et al., 2001; Mercier et al., 2013), we also tested the effect of 1 μm DCPG on cortico-RT release. First EPSC amplitudes of the baseline and drug wash-in epochs for 1 μm DCPG (n = 5), 10 μm DCPG (n = 10), and 20 μm l-AP4 (n = 8; same data as in Fig. 3) groups were pooled from multiple experiments and compared (Fig. 5E,F). Although there was a significant main effect of epoch on EPSC amplitude (p < 0.0001), there was no significant interaction of epoch and group (p > 0.05; Fig. 5F). Altogether, these results indicate that mGluR8 is present at the cortico-RT synapse and that selective activation of mGluR8 is sufficient to strongly suppress cortico-RT synaptic release.
To test the influence of selective mGluR8 activation on oscillatory network activity, we examined the effect of DCPG application on spindle oscillations in horizontal thalamic slices. Electrically evoked spindle oscillations had a baseline duration of 3.2 ± 0.3 s and period of 106 ± 12 ms (Fig. 5G). Bath application of 10 μm DCPG rapidly reduced oscillation duration to 59.5 ± 8.5% of baseline (Fig. 5G–I). Subsequent addition of 20 μm l-AP4 to the DCPG-containing bath perfusate reduced oscillation duration to 39.7 ± 11.6% of baseline, but there was no significant difference in duration between the two drug epochs (Fig. 5H). The strong suppression of oscillations by selective mGluR8 activation is consistent with the robust depression of cortico-RT synaptic release by mGluR8 activation.
Finally, we tested whether group III mGluRs at the cortico-RT synapse are endogenously activated by synaptically released glutamate. We used the group III mGluR-selective antagonist MSOP at 200 μm concentration, which was identified as an effective dose in previous studies (Kwon and Castillo, 2008) and which we also verified was able to reverse synaptic depression induced by 20 μm l-AP4 at cortico-RT synapses (see Materials and Methods). Given the robust presence of the high-affinity receptor mGluR8 at this synapse, we tested whether moderate frequency stimulation (5 Hz 10-pulse trains, delivered every 60 s) activated the receptors endogenously. We found no effect of 200 μm MSOP on paired-pulse ratio of evoked EPSC amplitude (baseline: 1.54 ± 0.28, MSOP: 1.64 ± 0.22; n = 5, p = 0.81, Wilcoxon signed-rank test; data not shown). There also was no effect of MSOP on the facilitation ratio, calculated as the ratio of the mean 10th EPSC amplitude to the mean first EPSC amplitude (baseline: 1.49 ± 0.32, MSOP: 1.69 ± 0.26; n = 5, p = 1.00, Wilcoxon signed-rank test; data not shown). The lack of effect of MSOP on EPSC facilitation suggests that modulation of release by group III mGluRs does not occur during short trains of cortical input, but does not rule out the possibility that stronger or more prolonged cortical activity could activate group III mGluRs.
Group III mGluRs are expressed at RT synapses onto TC neurons, but not onto other RT neurons
In addition to serving as presynaptic autoreceptors at glutamatergic synapses, group III mGluRs can function as heteroceptors at GABAergic inhibitory synapses, such as in cerebellar glomeruli and CA1 hippocampus (Mitchell and Silver, 2000; Semyanov and Kullmann, 2000). Therefore, we tested whether group III mGluRs modulate release at GABAergic reticulothalamic (RT-TC) synapses and/or reticuloreticular (RT-RT) synapses (Fig. 6). We performed whole-cell voltage clamp recordings of TC or RT neurons during minimal electrical stimulation of RT axons. Cells were patched with pipettes containing an internal solution with chloride reversal potential of −17 mV, such that IPSCs were recorded as inward currents. After cells were stably patched, d-AP5 (50 μm) and DNQX disodium salt (20 μm) were washed in to block excitatory events mediated by NMDA/kainate and AMPA receptors. Stimulation intensity was set to slightly above threshold (110–125% of threshold) such that failures were largely eliminated while evoked IPSC waveform and amplitude remained identical to the minimal stimulation response (see Materials and Methods). After a baseline epoch, l-AP4 was washed in at either 20 μm (to activate mGluR4/8) or 300 μm concentration (to activate mGluR7 as well as mGluR4/8) during RT-TC IPSC recordings. Only 300 μm l-AP4 was tested during RT-RT IPSC recordings.
Figure 6.

Expression of mGluR7 at RT synapses is target cell dependent. A, Schematic of RT-TC recording. B, C, Representative recordings of RT-TC IPSCs evoked by paired-pulse electrical stimulation. Left, Time course of IPSC amplitudes. Right, Average of six consecutive traces recorded during the last 2 min of baseline (black traces) and l-AP4 (red traces) epochs. Application of 20 μm l-AP4 did not affect peak IPSC amplitude (B), but 300 μm l-AP4 decreased IPSC amplitude (C). D, E, Summary data on the effect of 20 μm l-AP4 and 300 μm l-AP4 on evoked IPSC amplitude. Application of 20 μm l-AP4 (D) did not affect the peak amplitude of either the first or second IPSC (n = 6; first IPSC: p = 0.09, second IPSC: p = 0.69; Wilcoxon signed-rank test). In contrast, 300 μm l-AP4 (E) decreased the amplitude of both first and second IPSCs (n = 8; first IPSC: **p = 0.008, second IPSC: *p = 0.016; Wilcoxon signed-rank test). F, Schematic of RT-RT recording. G, Representative recording of RT-RT IPSCs evoked by paired-pulse electrical stimulation shows no change during bath application of 300 μm l-AP4. Left, Time course of IPSC amplitude. Right, Average of six consecutive traces recorded during the last 2 min of baseline (black traces) and l-AP4 (red traces) epochs. H, Summary data showing lack of effect of 300 μm l-AP4 on RT-RT IPSC amplitude (n = 7; first IPSC: p = 0.81, second IPSC: p = 0.16; Wilcoxon signed-rank test). All bar graphs represent data from the last 2 min of each epoch. Scale bars for IPSC traces: 20 ms, 50 pA.
During recordings of TC neurons, we observed evoked IPSCs in 52% of the neurons (38 of 73 patched TC neurons). Similarly, we observed evoked IPSCs in 50% of recorded RT neurons (20 of 40 patched RT neurons). In those TC or RT neurons for which we failed to observe evoked IPSCs, increasing the stimulation intensity either failed to elicit any response other than the stimulus artifact (though evoked EPSCs were usually observed before wash-in of d-AP5 and DNQX) or evoked direct currents that usually resulted in escape spikes. The evoked IPSCs recorded in TC and RT neurons were typical of fast GABAA-mediated responses and had weighted decay time constants of 6.3 ± 1.1 and 27.1 ± 3.7 ms, respectively. The relatively slower decay of IPSCs in RT cells compared with TC cells is consistent with previous reports, but both time constants are faster than previously reported, possibly due to differences between adult and juvenile rats (Zhang et al., 1997; Schofield and Huguenard, 2007). All IPSCs showed paired-pulse depression (recorded in TC cells: PPR = 0.65 ± 0.04, n = 14; recorded in RT cells: PPR = 0.73 ± 0.06, n = 7; Fig. 6B,C,G). Although the axonal source of the evoked IPSCs was not identified, the IPSCs recorded in TC cells and RT cells were considered to be RT-TC and RT-RT responses, respectively (see Discussion).
For RT-TC synapses, activating only the high-affinity receptors mGluR4/8 by 20 μm l-AP4 did not significantly change the evoked IPSC peak amplitude for either the first IPSC (249.4 ± 42.7 to 194.3 ± 26.5 pA; p = 0.09) or the second IPSC (155.7 ± 28.4 to 152.3 ± 31.4 pA; p = 0.69; Fig. 6B,D). In contrast, activation of all group III mGluRs by application of 300 μm l-AP4 significantly decreased the amplitude of the first IPSC to 62% of baseline (270.0 ± 72.0 to 167.5 ± 38.0 pA; p = 0.01) and the second IPSC to 66% of baseline (164.0 ± 34.8 to 107.7 ± 22.8 pA; p = 0.02; Fig. 6C,E). This dose-dependent effect of l-AP4 suggests that the modulation of RT-TC synaptic release is mainly performed by mGluR7, consistent with a previous report (Turner and Salt, 2003). We did observe that some RT-TC synapses were slightly depressed by mGluR4/8 activation, which may indicate modest, heterogeneous expression of mGluR4/8 at these synapses. Applying 300 μm l-AP4 did not change the PPR (n = 8, p = 0.20, Wilcoxon signed-rank test) or TC cell input resistance (p = 0.55).
Surprisingly, RT-RT synapses showed a different response to l-AP4 compared with RT-TC synapses. During recordings of paired-pulse RT-RT IPSCs, application of 300 μm l-AP4 failed to affect IPSC amplitude for either the first IPSC (368.3 ± 104.9 to 340.9 ± 77.3 pA; p = 0.81) or second IPSC (237.3 ± 51.2 to 207.4 ± 51.8 pA; p = 0.16; Fig. 6G,H). Because this l-AP4 dose activates mGluR7 and mGluR4/8, the insensitivity of RT-RT IPSCs to the l-AP4 application suggests that this synapse is devoid of all group III mGluRs. The results of the RT-TC and RT-RT experiments together indicate a target-cell-dependent expression of mGluR7 at RT synapses. mGluR7 located at RT terminals may function as heteroceptors such that their activation by high concentrations of glutamate (e.g., spillover glutamate from neighboring glutamatergic synapses or glial sources) results in depression of GABA release from the RT terminal, but only at those terminals synapsing onto TC neurons and not at those onto other RT neurons.
Discussion
Previous studies have associated group III mGluRs with absence seizures (Snead et al., 2000; Bertaso et al., 2008), which are aberrant oscillations in the corticothalamic circuit. Here, we investigated the distribution of group III mGluRs in the thalamic portion of the corticothalamic circuit and their contribution to network activity. We found that thalamic oscillations are strongly suppressed by activation of group III mGluRs and that these receptors modulate synaptic transmission at two loci in the circuit: glutamatergic cortico-RT and GABAergic RT-TC synapses. Our study identifies group III mGluRs as potential regulators of corticothalamic synaptic transmission that can gate excessive excitatory activity and dampen oscillations (Fig. 7).
Figure 7.

Model for activation of group III mGluRs in corticothalamic circuit. In the absence of group III mGluRs (A), cortical activity is transmitted via cortico-RT synapses to the thalamic loop, robustly initiating an oscillation. Strong inhibition at RT-TC synapses causes synchronous rebound bursting of TC neurons, perpetuating recurrent activity. When group III mGluRs are functional (B), cortical activity causes activation of mGluR4/8 at cortico-RT and cortico-TC terminals, depressing release. Glutamate spillover onto RT-TC synapses activates mGluR7, which depresses GABA release, decreases rebound bursting of TC neurons, and dampens oscillatory activity.
Presynaptic autoreceptors at cortico-RT synapses
One main finding of this study is the differential expression of group III mGluRs on glutamatergic synapses to RT. Pharmacological activation of high-affinity receptors mGluR4/8 strongly depressed cortico-RT synapses via a presynaptic mechanism. However, thalamo-RT synapses were invariably unaffected even by high agonist concentrations, indicating a lack of the low-affinity receptor mGluR7 and mGluR4/8. Overall, our findings suggest that group III mGluRs, including mGluR8, function as presynaptic autoreceptors at the cortico-RT synapse, depressing release when activated by synaptically released glutamate.
Although decreased release probability is often accompanied by an increased PPR (Zucker and Regehr, 2002), we failed to observe this in our group III mGluR activation experiments, similar to previous reports (Turner and Salt, 1999). Interestingly, corticogeniculate synapses have unusual characteristics of facilitation, in that PPR is modestly decreased when extracellular [Ca2+]o is decreased below physiological (1–1.5 mm) levels (Granseth et al., 2002). This is in contrast to other central synapses, which classically exhibit a monotonically increasing PPR as [Ca2+]o is decreased. It may be that cortico-RT synapses similarly have a weak dependence of PPR on presynaptic calcium influx, so the PPR is not noticeably affected by decreases in calcium influx via presynaptic calcium channel inhibition by group III mGluR activation. Regardless, the results of our mEPSC and GDP-β-S experiments, as well as the lack of change in input resistance in all experiments, indicate that group III mGluRs operate via a presynaptic rather than postsynaptic site of action. Given this, the question remains as to which patterns of activity release enough glutamate to activate presynaptic group III mGluRs at the cortico-RT synapse. We did not find evidence for endogenous activation by short-train stimuli, similar to a previous report that mGluR7/8 at cortico-dLGN synapses was not activated by train stimuli (Turner and Salt, 1999). This suggests that, despite its localization to active zones and high glutamate affinity, mGluR4/8 at cortico-RT synapses is not activated by glutamate released during moderate stimulation. Alternatively, the effects of group III mGluR activation may have been masked by processes such as postsynaptic receptor saturation (Sun and Beierlein, 2011).
We note that the group III mGluR agonist l-AP4 lends itself as a fast and reliable method to distinguish cortico-RT and thalamo-RT synapses. A synaptic response, initially characterized by paired-pulse facilitation or depression, could be verified at the end of the recording by briefly washing in 20 μm l-AP4 (e.g., for 5 min) and observing rapid depression (cortico-RT synapse) or no effect (thalamo-RT synapse). This is analogous to the routine use of the group II mGluR agonist DCG-IV or l-AP4 to identify responses originating from hippocampal mossy fiber synapses onto CA3 neurons, which presynaptically express group II and group III mGluRs, and distinguish them from associative-commissural synapses lacking these receptors (Salin et al., 1996; Kamiya and Ozawa, 1999; Kwon and Castillo, 2008).
Target-cell-dependent heteroceptors at RT-TC synapses
Our finding that inhibitory synapses onto TC neurons exhibit a dose-dependent sensitivity to l-AP4, indicating the expression of mGluR7 but not mGluR4/8, is consistent with the results of a previous study (Turner and Salt, 2003). Because the predominant source of inhibitory terminals in the rodent VB are RT axons (Barbaresi et al., 1986; Cox et al., 1996; for review, see Jones, 2007), the evoked IPSCs we observed during TC neuron recordings were presumably generated by RT-TC synapses. mGluR7 is likely located presynaptically at the RT-TC synapse based on its expression at other GABAergic synapses (Dalezios et al., 2002; Somogyi et al., 2003) and our observation that mGluR7 activation did not affect TC cell input resistance.
Surprisingly, we found that inhibitory synapses onto RT neurons are insensitive to high concentrations of l-AP4, indicating a complete lack of group III mGluRs. These synapses were likely axodendritic RT-RT synapses (Liu and Jones, 1999), which have previously been studied by stimulation of cortical inputs (Zhang and Jones, 2004; but see Cruikshank et al., 2010) or local excitation of RT neurons by glutamate puffing or uncaging (Shu and McCormick, 2002; Deleuze and Huguenard, 2006). However, it is possible that some synapses were from extrathalamic sources such as basal forebrain (Asanuma and Porter, 1990). Further studies selectively targeting RT synapses will elucidate this issue.
Together, our results suggest that mGluR7 exhibits target cell-dependent expression at presynaptic RT terminals, which is intriguing given a similar pattern of group III mGluR expression at glutamatergic and GABAergic synapses in hippocampus and cortex (Shigemoto et al., 1996, 1997; Corti et al., 2002; Somogyi et al., 2003). What is the endogenous agonist that activates mGluR7 at GABAergic RT-TC synapses? One possibility is glutamate from neighboring glutamatergic synapses (Mitchell and Silver, 2000; Semyanov and Kullmann, 2000). For RT-TC synapses, the two possible neuronal sources of spillover glutamate are cortical and lemniscal synapses onto TC neurons. Due to the low glutamate affinity of mGluR7, however, spillover glutamate may not reach an effective concentration to activate mGluR7 within the RT-TC synapse or may do so only during severe pathological states. Alternatively, mGluR7 may be activated by glutamate released from neighboring glia or by a nonglutamate agonist (e.g., l-serine-O-phosphate; Hampson et al., 1999) from neuronal or glial sources.
The localization of mGluR7 to the RT-TC synapse is interesting in light of previous studies associating mGluR7 with absence epilepsy. Disrupting mGluR7 function induces spontaneous absence-like seizures (Bertaso et al., 2008) and can also cause tonic-clonic seizures (Masugi et al., 1999; Sansig et al., 2001; Zhang et al., 2008). Indeed, our oscillation experiments show that activation of mGluR7 within the corticothalamic network, including at the RT-TC synapse, can repress oscillatory network activity (Fig. 1). Application of 20 μm l-AP4 reduced both spindle and epileptiform oscillation duration, which can be attributed to the activation of mGluR4/8 at cortico-RT synapses and, potentially, cortico-TC synapses (Turner and Salt, 1999). Notably, spindle oscillations were further suppressed by 300 μm l-AP4, presumably due to activation of mGluR7 at the RT-TC synapse and subsequent suppression of GABA release. Because TC neurons undergo rebound bursting after inhibition by RT (for review, see Beenhakker and Huguenard, 2009), the mGluR7-induced suppression of RT-TC GABA release likely decreased rebound bursting of TC neurons and thus dampened oscillations. We also found that epileptiform oscillations were less suppressed by mGluR8 activation compared with spindle oscillations and were not affected by additional mGluR7 activation. This may have been due to the highly amplified GABAergic output of RT cells (under conditions of increased RT cell excitability by apamin and blocked GABAAR-mediated intra-RT inhibition by picrotoxin) rendering the epileptiform oscillations less susceptible to the effects of decreased cortical glutamate release and/or decreased RT-TC GABA release.
Conclusions for corticothalamic processing and epilepsy
We can anticipate the net functional role of group III mGluRs for the two major modes of operation of the corticothalamic circuit. In awake states, the thalamus is a high-fidelity relay of ascending sensory input to cortex and other subcortical structures, whereas corticothalamic inputs provide highly processed feedback which influences the tuning of TC neurons (for review, see Briggs and Usrey, 2008). Group III mGluRs located at cortico-RT synapses may act as a low-pass filter of this corticothalamic feedback.
During sleep states or seizures, an oscillation initiated by focal cortical activity (Meeren et al., 2002; Polack et al., 2007) is amplified by recruitment of RT and subsequent postinhibitory rebound bursting of TC neurons. The circuit is thus susceptible to both excessive excitation (i.e., intense cortico-RT activity) and inhibition (i.e., strong RT-TC drive). In this context, group III mGluRs may act to suppress thalamocortical seizures (Fig. 7). Activation of presynaptic mGluRs at cortico-RT synapses, and potentially cortico-TC synapses (Turner and Salt, 1999), would attenuate glutamate release and RT recruitment. In addition, glutamate released from initial intense activity of cortico-TC synapses may spill over onto neighboring GABAergic RT-TC terminals and activate presynaptic mGluR7, depressing GABA release and decreasing synchronous rebound bursting of TC neurons. Meanwhile, RT-RT synapses lacking group III mGluRs would not undergo depression, thus retaining an important desynchronizing mechanism for oscillatory network activity (Huntsman et al., 1999; Sohal and Huguenard, 2003).
Given that various genetic and pathophysiological mechanisms are associated with absence epilepsy, our data suggest that dampening oscillations by augmenting thalamic group III mGluR activity could potentially be a general therapeutic approach for absence seizures. Clearly, the modulatory role of group III mGluRs on synapses in the corticothalamic circuit must be examined in parallel with other receptors. An instructive example is the hippocampal mossy fiber synapse to the CA3 pyramidal neuron, in which glutamate release suppression by presynaptic mGluRs dominates over augmentation by kainate receptors (Kwon and Castillo, 2008). In the thalamus, presynaptic kainate receptors and presynaptic group II mGluRs are present at cortico-RT synapses (Alexander and Godwin, 2006; Miyata and Imoto, 2009) and postsynaptic group I and group II mGluRs are expressed in RT neurons (Cox and Sherman, 1999). Further studies are needed to compare the functional contributions of these glutamate receptors at cortico-RT and RT-TC synapses and to determine the potential for selective activation of group III mGluRs to dampen or prevent seizures while preserving normal sensory processing in the corticothalamic circuit.
Footnotes
This work was supported by the National Institutes of Health (Grant NS034774), the Stanford Medical Scientist Training Program, and a National Science Foundation Integrative Graduate Education and Research Traineeship/Stanford Center for Mind, Brain and Computation. We thank Isabel Parada for assistance with histological preparations; Julia Brill for insight from preliminary work and assistance with virus injections; Anne Herbert, Sha Jin, and Jessica McKercher for animal husbandry; Karl Deisseroth for supplying virus; and Kwabena Boahen, Michael Gutnick, Jeanne Paz, Jongmin Kim, Mark Sellmyer, Hanmi Lee, Sung-Yon Kim, and members of the Huguenard and Prince laboratories for helpful discussions and comments on the manuscript.
The authors declare no competing financial interests.
References
- Alexander GM, Godwin DW. Unique presynaptic and postsynaptic roles of Group II metabotropic glutamate receptors in the modulation of thalamic network activity. Neuroscience. 2006;141:501–513. doi: 10.1016/j.neuroscience.2006.03.060. [DOI] [PubMed] [Google Scholar]
- Asanuma C, Porter LL. Light and electron microscopic evidence for a GABAergic projection from the caudal basal forebrain to the thalamic reticular nucleus in rats. J Comp Neurol. 1990;302:159–172. doi: 10.1002/cne.903020112. [DOI] [PubMed] [Google Scholar]
- Bal T, Debay D, Destexhe A. Cortical feedback controls the frequency and synchrony of oscillations in the visual thalamus. J Neurosci. 2000;20:7478–7488. doi: 10.1523/JNEUROSCI.20-19-07478.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal T, McCormick DA. Mechanisms of oscillatory activity in guinea-pig nucleus reticularis thalami in vitro: a mammalian pacemaker. J Physiol. 1993;468:669–691. doi: 10.1113/jphysiol.1993.sp019794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbaresi P, Spreafico R, Frassoni C, Rustioni A. GABAergic neurons are present in the dorsal column nuclei but not in the ventroposterior complex of rats. Brain Res. 1986;382:305–326. doi: 10.1016/0006-8993(86)91340-5. [DOI] [PubMed] [Google Scholar]
- Beenhakker MP, Huguenard JR. Neurons that fire together also conspire together: is normal sleep circuitry hijacked to generate epilepsy? Neuron. 2009;62:612–632. doi: 10.1016/j.neuron.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertaso F, Zhang C, Scheschonka A, de Bock F, Fontanaud P, Marin P, Huganir RL, Betz H, Bockaert J, Fagni L, Lerner-Natoli M. PICK1 uncoupling from mGluR7a causes absence-like seizures. Nat Neurosci. 2008;11:940–948. doi: 10.1038/nn.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenfeld H, McCormick DA. Corticothalamic inputs control the pattern of activity generated in thalamocortical networks. J Neurosci. 2000;20:5153–5162. doi: 10.1523/JNEUROSCI.20-13-05153.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley SR, Rees HD, Yi H, Levey AI, Conn PJ. Distribution and developmental regulation of metabotropic glutamate receptor 7a in rat brain. J Neurochem. 1998;71:636–645. doi: 10.1046/j.1471-4159.1998.71020636.x. [DOI] [PubMed] [Google Scholar]
- Briggs F, Usrey WM. Emerging views of corticothalamic function. Curr Opin Neurobiol. 2008;18:403–407. doi: 10.1016/j.conb.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant AS, Li B, Beenhakker MP, Huguenard JR. Maintenance of thalamic epileptiform activity depends on the astrocytic glutamate-glutamine cycle. J Neurophysiol. 2009;102:2880–2888. doi: 10.1152/jn.00476.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti C, Aldegheri L, Somogyi P, Ferraguti F. Distribution and synaptic localisation of the metabotropic glutamate receptor 4 (mGluR4) in the rodent CNS. Neuroscience. 2002;110:403–420. doi: 10.1016/S0306-4522(01)00591-7. [DOI] [PubMed] [Google Scholar]
- Cox CL, Sherman SM. Glutamate inhibits thalamic reticular neurons. J Neurosci. 1999;19:6694–6699. doi: 10.1523/JNEUROSCI.19-15-06694.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CL, Huguenard JR, Prince DA. Heterogeneous axonal arborizations of rat thalamic reticular neurons in the ventrobasal nucleus. J Comp Neurol. 1996;366:416–430. doi: 10.1002/(SICI)1096-9861(19960311)366:3<416::AID-CNE4>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Urabe H, Nurmikko AV, Connors BW. Pathway-specific feedforward circuits between thalamus and neocortex revealed by selective optical stimulation of axons. Neuron. 2010;65:230–245. doi: 10.1016/j.neuron.2009.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V, Leresche N. Childhood absence epilepsy: genes, channels, neurons and networks. Nat Rev Neurosci. 2002;3:371–382. doi: 10.1038/nrn811. [DOI] [PubMed] [Google Scholar]
- Dalezios Y, Luján R, Shigemoto R, Roberts JD, Somogyi P. Enrichment of mGluR7a in the presynaptic active zones of GABAergic and non-GABAergic terminals on interneurons in the rat somatosensory cortex. Cereb Cortex. 2002;12:961–974. doi: 10.1093/cercor/12.9.961. [DOI] [PubMed] [Google Scholar]
- Deleuze C, Huguenard JR. Distinct electrical and chemical connectivity maps in the thalamic reticular nucleus: potential roles in synchronization and sensation. J Neurosci. 2006;26:8633–8645. doi: 10.1523/JNEUROSCI.2333-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/S0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Ferraguti F, Klausberger T, Cobden P, Baude A, Roberts JD, Szucs P, Kinoshita A, Shigemoto R, Somogyi P, Dalezios Y. Metabotropic glutamate receptor 8-expressing nerve terminals target subsets of GABAergic neurons in the hippocampus. J Neurosci. 2005;25:10520–10536. doi: 10.1523/JNEUROSCI.2547-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentet LJ, Ulrich D. Strong, reliable and precise synaptic connections between thalamic relay cells and neurones of the nucleus reticularis in juvenile rats. J Physiol. 2003;546:801–811. doi: 10.1113/jphysiol.2002.032730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentet LJ, Ulrich D. Electrophysiological characterization of synaptic connections between layer VI cortical cells and neurons of the nucleus reticularis thalami in juvenile rats. Eur J Neurosci. 2004;19:625–633. doi: 10.1111/j.1460-9568.2004.03168.x. [DOI] [PubMed] [Google Scholar]
- Golshani P, Liu XB, Jones EG. Differences in quantal amplitude reflect GluR4- subunit number at corticothalamic synapses on two populations of thalamic neurons. Proc Natl Acad Sci U S A. 2001;98:4172–4177. doi: 10.1073/pnas.061013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granseth B, Ahlstrand E, Lindström S. Paired pulse facilitation of corticogeniculate EPSCs in the dorsal lateral geniculate nucleus of the rat investigated in vitro. J Physiol. 2002;544:477–486. doi: 10.1113/jphysiol.2002.024703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guérineau NC, Gähwiler BH, Gerber U. Reduction of resting K+ current by metabotropic glutamate and muscarinic receptors in rat CA3 cells: mediation by G-proteins. J Physiol. 1994;474:27–33. doi: 10.1113/jphysiol.1994.sp019999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampson DR, Huang XP, Pekhletski R, Peltekova V, Hornby G, Thomsen C, Thøgersen H. Probing the ligand-binding domain of the mGluR4 subtype of metabotropic glutamate receptor. J Biol Chem. 1999;274:33488–33495. doi: 10.1074/jbc.274.47.33488. [DOI] [PubMed] [Google Scholar]
- Heuss C, Scanziani M, Gähwiler BH, Gerber U. G-protein-independent signaling mediated by metabotropic glutamate receptors. Nat Neurosci. 1999;2:1070–1077. doi: 10.1038/15996. [DOI] [PubMed] [Google Scholar]
- Huguenard JR, McCormick DA. Thalamic synchrony and dynamic regulation of global forebrain oscillations. Trends Neurosci. 2007;30:350–356. doi: 10.1016/j.tins.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Huntsman MM, Porcello DM, Homanics GE, DeLorey TM, Huguenard JR. Reciprocal inhibitory connections and network synchrony in the mammalian thalamus. Science. 1999;283:541–543. doi: 10.1126/science.283.5401.541. [DOI] [PubMed] [Google Scholar]
- Jacobsen RB, Ulrich D, Huguenard JR. GABA(B) and NMDA receptors contribute to spindle-like oscillations in rat thalamus in vitro. J Neurophysiol. 2001;86:1365–1375. doi: 10.1152/jn.2001.86.3.1365. [DOI] [PubMed] [Google Scholar]
- Jahnsen H, Llinás R. Electrophysiological properties of guinea-pig thalamic neurones: an in vitro study. J Physiol. 1984;349:205–226. doi: 10.1113/jphysiol.1984.sp015153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EG. The thalamus. Ed 2. Cambridge: Cambridge UP; 2007. [Google Scholar]
- Kamiya H, Ozawa S. Dual mechanism for presynaptic modulation by axonal metabotropic glutamate receptor at the mouse mossy fibre-CA3 synapse. J Physiol. 1999;518:497–506. doi: 10.1111/j.1469-7793.1999.0497p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Alger BE. Random response fluctuations lead to spurious paired-pulse facilitation. J Neurosci. 2001;21:9608–9618. doi: 10.1523/JNEUROSCI.21-24-09608.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiman-Weiner M, Beenhakker MP, Segal WA, Huguenard JR. Synergistic roles of GABAA receptors and SK channels in regulating thalamocortical oscillations. J Neurophysiol. 2009;102:203–213. doi: 10.1152/jn.91158.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koulen P, Brandstätter JH. Pre- and postsynaptic sites of action of mGluR8a in the mammalian retina. Invest Ophthalmol Vis Sci. 2002;43:1933–1940. [PubMed] [Google Scholar]
- Kwon HB, Castillo PE. Role of glutamate autoreceptors at hippocampal mossy fiber synapses. Neuron. 2008;60:1082–1094. doi: 10.1016/j.neuron.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Durand R, Gradinaru V, Zhang F, Goshen I, Kim DS, Fenno LE, Ramakrishnan C, Deisseroth K. Global and local fMRI signals driven by neurons defined optogenetically by type and wiring. Nature. 2010;465:788–792. doi: 10.1038/nature09108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XB, Jones EG. Predominance of corticothalamic synaptic inputs to thalamic reticular nucleus neurons in the rat. J Comp Neurol. 1999;414:67–79. doi: 10.1002/(SICI)1096-9861(19991108)414:1<67::AID-CNE6>3.0.CO%3B2-Z. [DOI] [PubMed] [Google Scholar]
- Masugi M, Yokoi M, Shigemoto R, Muguruma K, Watanabe Y, Sansig G, van der Putten H, Nakanishi S. Metabotropic glutamate receptor subtype 7 ablation causes deficit in fear response and conditioned taste aversion. J Neurosci. 1999;19:955–963. doi: 10.1523/JNEUROSCI.19-03-00955.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateos JM, Elezgarai I, Benítez R, Osorio A, Bilbao A, Azkue JJ, Kuhn R, Knöpfel T, Grandes P. Clustering of the group III metabotropic glutamate receptor 4a at parallel fiber synaptic terminals in the rat cerebellar molecular layer. Neurosci Res. 1999;35:71–74. doi: 10.1016/S0168-0102(99)00066-8. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Pape HC. Properties of a hyperpolarization-activated cation current and its role in rhythmic oscillation in thalamic relay neurones. J Physiol. 1990;431:291–318. doi: 10.1113/jphysiol.1990.sp018331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeren HK, Pijn JP, Van Luijtelaar EL, Coenen AM, Lopes da Silva FH. Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats. J Neurosci. 2002;22:1480–1495. doi: 10.1523/JNEUROSCI.22-04-01480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier MS, Lodge D, Fang G, Nicolas CS, Collett VJ, Jane DE, Collingridge GL, Bortolotto ZA. Characterisation of an mGlu8 receptor-selective agonist and antagonist in the lateral and medial perforant path inputs to the dentate gyrus. Neuropharmacology. 2013;67:294–303. doi: 10.1016/j.neuropharm.2012.11.020. [DOI] [PubMed] [Google Scholar]
- Mitchell SJ, Silver RA. Glutamate spillover suppresses inhibition by activating presynaptic mGluRs. Nature. 2000;404:498–502. doi: 10.1038/35006649. [DOI] [PubMed] [Google Scholar]
- Miyata M, Imoto K. Contrary roles of kainate receptors in transmitter release at corticothalamic synapses onto thalamic relay and reticular neurons. J Physiol. 2009;587:999–1012. doi: 10.1113/jphysiol.2008.164996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima Y, Iwakabe H, Akazawa C, Nawa H, Shigemoto R, Mizuno N, Nakanishi S. Molecular characterization of a novel retinal metabotropic glutamate receptor mGluR6 with a high agonist selectivity for l-2-amino-4-phosphonobutyrate. J Biol Chem. 1993;268:11868–11873. [PubMed] [Google Scholar]
- Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohara PT, Lieberman AR. The thalamic reticular nucleus of the adult rat: experimental anatomical studies. J Neurocytol. 1985;14:365–411. doi: 10.1007/BF01217752. [DOI] [PubMed] [Google Scholar]
- Okamoto N, Hori S, Akazawa C, Hayashi Y, Shigemoto R, Mizuno N, Nakanishi S. Molecular characterization of a new metabotropic glutamate receptor mGluR7 coupled to inhibitory cyclic AMP signal transduction. J Biol Chem. 1994;269:1231–1236. [PubMed] [Google Scholar]
- Paz JT, Bryant AS, Peng K, Fenno L, Yizhar O, Frankel WN, Deisseroth K, Huguenard JR. A new mode of corticothalamic transmission revealed in the Gria4(-/-) model of absence epilepsy. Nat Neurosci. 2011;14:1167–1173. doi: 10.1038/nn.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkey KA, Lavezzari G, Racca C, Roche KW, McBain CJ. mGluR7 is a metaplastic switch controlling bidirectional plasticity of feedforward inhibition. Neuron. 2005;46:89–102. doi: 10.1016/j.neuron.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Polack PO, Guillemain I, Hu E, Deransart C, Depaulis A, Charpier S. Deep layer somatosensory cortical neurons initiate spike-and-wave discharges in a genetic model of absence seizures. J Neurosci. 2007;27:6590–6599. doi: 10.1523/JNEUROSCI.0753-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salin PA, Scanziani M, Malenka RC, Nicoll RA. Distinct short-term plasticity at two excitatory synapses in the hippocampus. Proc Natl Acad Sci U S A. 1996;93:13304–13309. doi: 10.1073/pnas.93.23.13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansig G, Bushell TJ, Clarke VR, Rozov A, Burnashev N, Portet C, Gasparini F, Schmutz M, Klebs K, Shigemoto R, Flor PJ, Kuhn R, Knoepfel T, Schroeder M, Hampson DR, Collett VJ, Zhang C, Duvoisin RM, Collingridge GL, van Der Putten H. Increased seizure susceptibility in mice lacking metabotropic glutamate receptor 7. J Neurosci. 2001;21:8734–8745. doi: 10.1523/JNEUROSCI.21-22-08734.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saugstad JA, Kinzie JM, Shinohara MM, Segerson TP, Westbrook GL. Cloning and expression of rat metabotropic glutamate receptor 8 reveals a distinct pharmacological profile. Mol Pharmacol. 1997;51:119–125. doi: 10.1124/mol.51.1.119. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Salin PA, Vogt KE, Malenka RC, Nicoll RA. Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature. 1997;385:630–634. doi: 10.1038/385630a0. [DOI] [PubMed] [Google Scholar]
- Schoepp DD, Jane DE, Monn JA. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/S0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- Schofield CM, Huguenard JR. GABA affinity shapes IPSCs in thalamic nuclei. J Neurosci. 2007;27:7954–7962. doi: 10.1523/JNEUROSCI.0377-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semyanov A, Kullmann DM. Modulation of GABAergic signaling among interneurons by metabotropic glutamate receptors. Neuron. 2000;25:663–672. doi: 10.1016/S0896-6273(00)81068-5. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Kulik A, Roberts JD, Ohishi H, Nusser Z, Kaneko T, Somogyi P. Target-cell-specific concentration of a metabotropic glutamate receptor in the presynaptic active zone. Nature. 1996;381:523–525. doi: 10.1038/381523a0. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, Flor PJ, Neki A, Abe T, Nakanishi S, Mizuno N. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci. 1997;17:7503–7522. doi: 10.1523/JNEUROSCI.17-19-07503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y, McCormick DA. Inhibitory interactions between ferret thalamic reticular neurons. J Neurophysiol. 2002;87:2571–2576. doi: 10.1152/jn.00850.2001. [DOI] [PubMed] [Google Scholar]
- Snead OC, 3rd, Banerjee PK, Burnham M, Hampson D. Modulation of absence seizures by the GABA(A) receptor: a critical role for metabotropic glutamate receptor 4 (mGluR4) J Neurosci. 2000;20:6218–6224. doi: 10.1523/JNEUROSCI.20-16-06218.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal VS, Huguenard JR. Inhibitory interconnections control burst pattern and emergent network synchrony in reticular thalamus. J Neurosci. 2003;23:8978–8988. doi: 10.1523/JNEUROSCI.23-26-08978.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal VS, Keist R, Rudolph U, Huguenard JR. Dynamic GABA(A) receptor subtype-specific modulation of the synchrony and duration of thalamic oscillations. J Neurosci. 2003;23:3649–3657. doi: 10.1523/JNEUROSCI.23-09-03649.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi P, Dalezios Y, Luján R, Roberts JD, Watanabe M, Shigemoto R. High level of mGluR7 in the presynaptic active zones of select populations of GABAergic terminals innervating interneurons in the rat hippocampus. Eur J Neurosci. 2003;17:2503–2520. doi: 10.1046/j.1460-9568.2003.02697.x. [DOI] [PubMed] [Google Scholar]
- Steriade M. Sleep, epilepsy and thalamic reticular inhibitory neurons. Trends Neurosci. 2005;28:317–324. doi: 10.1016/j.tins.2005.03.007. [DOI] [PubMed] [Google Scholar]
- Sun YG, Beierlein M. Receptor saturation controls short-term synaptic plasticity at corticothalamic synapses. J Neurophysiol. 2011;105:2319–2329. doi: 10.1152/jn.00942.2010. [DOI] [PubMed] [Google Scholar]
- Thomas NK, Wright RA, Howson PA, Kingston AE, Schoepp DD, Jane DE. (S)-3,4-DCPG, a potent and selective mGlu8a receptor agonist, activates metabotropic glutamate receptors on primary afferent terminals in the neonatal rat spinal cord. Neuropharmacology. 2001;40:311–318. doi: 10.1016/S0028-3908(00)00169-6. [DOI] [PubMed] [Google Scholar]
- Thomsen C, Kristensen P, Mulvihill E, Haldeman B, Suzdak PD. L-2-Aminno-4-phosphonobutyrate (L-AP4) is an agonist at the type IV metabotropic glutamate receptor which is negatively coupled to adenylate cyclase. Eur J Pharmacol. 1992;227:361–362. doi: 10.1016/0922-4106(92)90018-Q. [DOI] [PubMed] [Google Scholar]
- Turner JP, Salt TE. Group III metabotropic glutamate receptors control corticothalamic synaptic transmission in the rat thalamus in vitro. J Physiol. 1999;519:481–491. doi: 10.1111/j.1469-7793.1999.0481m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JP, Salt TE. Group II and III metabotropic glutamate receptors and the control of the nucleus reticularis thalami input to rat thalamocortical neurones in vitro. Neuroscience. 2003;122:459–469. doi: 10.1016/j.neuroscience.2003.08.014. [DOI] [PubMed] [Google Scholar]
- Ulrich D, Huguenard JR. GABAB receptor-mediated responses in GABAergic projection neurones of rat nucleus reticularis thalami in vitro. J Physiol. 1996;493:845–854. doi: 10.1113/jphysiol.1996.sp021427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada E, Shigemoto R, Kinoshita A, Ohishi H, Mizuno N. Metabotropic glutamate receptor subtypes in axon terminals of projection fibers from the main and accessory olfactory bulbs: a light and electron microscopic immunohistochemical study in the rat. J Comp Neurol. 1998;393:493–504. doi: 10.1002/(SICI)1096-9861(19980420)393:43.0.CO%3B2-W. [DOI] [PubMed] [Google Scholar]
- Zhang CS, Bertaso F, Eulenburg V, Lerner-Natoli M, Herin GA, Bauer L, Bockaert J, Fagni L, Betz H, Scheschonka A. Knock-in mice lacking the PDZ-ligand motif of mGluR7a show impaired PKC-dependent autoinhibition of glutamate release, spatial working memory deficits, and increased susceptibility to pentylenetetrazol. J Neurosci. 2008;28:8604–8614. doi: 10.1523/JNEUROSCI.0628-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Jones EG. Corticothalamic inhibition in the thalamic reticular nucleus. J Neurophysiol. 2004;91:759–766. doi: 10.1152/jn.00624.2003. [DOI] [PubMed] [Google Scholar]
- Zhang SJ, Huguenard JR, Prince DA. GABAA receptor-mediated Cl- currents in rat thalamic reticular and relay neurons. J Neurophysiol. 1997;78:2280–2286. doi: 10.1152/jn.1997.78.5.2280. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]