

Abstract
Metabolic profiling of cancer cells has recently been established as a promising tool for the development of therapies and identification of cancer biomarkers. Here we characterized the metabolomic profile of human breast tumors and uncovered intrinsic metabolite signatures in these tumors using an untargeted discovery approach and validation of key metabolites. The oncometabolite 2-hydroxyglutarate (2HG) accumulated at high levels in a subset of tumors and human breast cancer cell lines. We discovered an association between increased 2HG levels and MYC pathway activation in breast cancer, and further corroborated this relationship using MYC overexpression and knockdown in human mammary epithelial and breast cancer cells. Further analyses revealed globally increased DNA methylation in 2HG-high tumors and identified a tumor subtype with high tissue 2HG and a distinct DNA methylation pattern that was associated with poor prognosis and occurred with higher frequency in African-American patients. Tumors of this subtype had a stem cell–like transcriptional signature and tended to overexpress glutaminase, suggestive of a functional relationship between glutamine and 2HG metabolism in breast cancer. Accordingly, 13C-labeled glutamine was incorporated into 2HG in cells with aberrant 2HG accumulation, whereas pharmacologic and siRNA-mediated glutaminase inhibition reduced 2HG levels. Our findings implicate 2HG as a candidate breast cancer oncometabolite associated with MYC activation and poor prognosis.
Introduction
Gene expression profiling studies of breast cancer led to the discovery of disease subtypes and expression patterns that are predictive of disease outcome (1–3). Recently, metabolomics emerged as a new discovery tool with the promise of identifying targetable metabolic dependencies of cancer cells (4, 5). Oncogenes like MYC and tumor-suppressor genes like TP53 affect cancer cell survival through regulation of cell metabolism and mitochondrial biogenesis (6–8). New essential metabolomic pathways for tumor growth have been described for breast and other cancers (4, 9–12), and “public” somatic mutations in metabolic enzymes were recently discovered. Isocitrate dehydrogenase 1 (IDH1) and IDH2 mutations are mainly restricted to gliomas and leukemias and promote tumor development by turning the 2 enzymes into catalysts for the production of oncogenic 2-hydroxyglutarate (2HG), which causes epigenetic reprogramming (13–16). These findings suggest that human tumors acquire discrete metabolic networks that define disease aggressiveness and response to therapy.
Here, using an unbiased metabolomics approach supported by validation of key metabolites, we examined metabolite signatures in breast cancer and differences between African-American (AA) and European-American (EA) patients. A highlight of our study was the finding of markedly elevated 2HG in a subgroup of breast tumors with MYC activation, a distinct DNA methylation pattern, and poor clinical outcome. We also identified breast cancer cell lines of mostly basal-like and mesenchymal origin that aberrantly accumulated 2HG, reaching levels 100-fold above those in other breast cancer cell lines and in noncancerous mammary epithelial cells. Experimentally, we linked glutamine metabolism and MYC activation to increased intracellular 2HG and provided evidence that mitochondrial enzymes are involved in the aberrant accumulation of 2HG in breast cancer.
Results
The abundance of 352 known and 184 unknown metabolites in 67 human breast tumors and 65 tumor-adjacent noncancerous tissues (Supplemental Tables 1 and 2; supplemental material available online with this article; doi: 10.1172/JCI71180DS1) was measured using an untargeted mass spectrometry–based profiling approach (discovery set). Quantitative differences for key metabolites were validated in 70 estrogen receptor–negative (ER-negative) tumors (Supplemental Table 3) and 36 adjacent noncancerous tissues (validation set) with targeted assays at 2 additional laboratories. 27 tumor samples and 19 tumor-adjacent noncancerous tissue pairs were common to the discovery and validation studies and were analyzed on all platforms. This overlap in tissues between the discovery and validation studies was chosen to evaluate possible platform-specific effects in the measurement of metabolites.
Intrinsic metabolite signatures exist in breast tumors.
Examination of the discovery set yielded 296 tissue metabolites that were detectable in more than 40% of samples. A subset of metabolites was elevated in tumors and delineated those from adjacent noncancerous tissues in both discovery and validation phases (Figure 1 and Supplemental Figure 1). Robust metabolite contrasts were seen between tumors of different grades and ER statuses (Supplemental Figures 2 and 3), while only subtle differences emerged comparing tumor metabolic profiles according to patient age, disease stage and node status, menopausal status, body mass index, or socioeconomic status (Supplemental Figure 2). Differences in tumor metabolite abundance by household income and neoadjuvant therapy were initially observed, but further analysis indicated that tumor ER status may confound this association. In an ER-stratified analysis, both household income and neoadjuvant therapy had only modest effects on the tumor metabolome. These findings are consistent with the presence of robust intrinsic metabolome signatures in primary human breast tumors that persist with disease progression and mirrors findings from the gene expression studies (17, 18). However, unlike gene expression signatures that correlate with various hormonal subtypes of breast cancer and hence distinguish them as discrete subtypes (1, 19), metabolite signatures by themselves delineated luminal A tumors from others, but did not separate HER2-positive tumors (both ER-negative and ER-positive) from HER2-negative, ER-negative tumors (Supplemental Figure 4).
Figure 1. Differences in metabolite patterns between breast tumors and adjacent noncancerous tissues and AA and EA patients.

(A) Principal component analysis for breast tumors (n = 67) and adjacent noncancerous tissues (n = 65) using the top 50 metabolites that showed the most different abundance across tissues. These metabolites were detected in >80% of all samples. (B) Z-score plots representing the deviation of the 50 metabolites in tumor (brown) and adjacent noncancerous tissue pairs (light blue; n = 65) from the average of the noncancerous tissues in linear scale. (C and D) Unsupervised hierarchical clustering of ER-negative (C, n = 34, 16 AA and 18 EA) and triple-negative (D, n = 17, 8 AA and 9 EA) breast tumors based on 296 named metabolites. Colors denote sample classes (green, AA; yellow, EA) or basal-like subtype in D (brown, n = 15). Yellow frames in C and D highlight metabolites decreased in a subset of breast tumors, representing mostly EA patients. (E) Number of differential metabolites (fold change, >2.5 or <0.4; FDR, <5%) in the comparisons in C and D.
Because our study population was ethnically diverse and race/ethnicity matched for tumor ER status and triple-negative/basal-like disease (Supplemental Table 1), we analyzed tissue metabolite differences comparing the AA patients (n = 32; average West African ancestry, 83.2%) with EA patients (n = 35; average European ancestry, 97.6%). Using unsupervised hierarchical clustering, we were able to separate both ER-negative and triple-negative tumors into distinct subclusters that were enriched for AA or EA patients (P < 0.05, AA vs. EA in ER-negative tumors, Fisher’s exact test; Figure 1, C and D). Overall, 50–80 metabolites were different between AA and EA patients in ER-negative and triple-negative tumors, whereas almost 200 metabolites distinguished triple-negative tumors from adjacent benign tissue (false discovery rate [FDR], <5%; Figure 1E and Supplemental Table 4).
2HG preferentially accumulates in ER-negative and basal-like tumors.
Several classes of metabolites were consistently elevated in ER-negative versus ER-positive tumors, including lyso(phospho)lipids, carnitines, and N-acetyl amino acids (Supplemental Figure 5 and Supplemental Table 5). ER-negative tumors also showed a robust accumulation of 2 mitochondrial metabolites, N-acetyl-aspartate (NAA) and 2HG. Both metabolites were significantly elevated in breast tumors in general (NAA, 13.4-fold vs. paired adjacent noncancerous tissue; 2HG, 4.6-fold; FDR, <1%), but most robustly in ER-negative tumors (NAA, 2-fold vs. ER-positive tumors; 2HG, 2.8-fold; FDR, <5%), and specifically in tumors of the basal-like/mesenchymal subtype (NAA, 2.4-fold vs. ER-positive; 2HG, 4.0-fold; FDR, <5%). NAA has previously been described as a brain-specific metabolite that is elevated in patients with Canavan’s disease because of deleterious aspartoacylase (ASPA) gene mutations that inhibit the hydrolysis of NAA (20). Matched microarray data from our study showed that transcript levels of ASPA were reduced in breast tumors in general (Supplemental Figure 5), but most significantly within the ER-negative tumor group (3%–4% residual expression, array-based; FDR, <1%), which suggests a possible cause for increased tumor-associated NAA.
On the other hand, 2HG is a known oncometabolite in gliomas and leukemias (13–16). Our analysis showed that 2HG accumulated up to 100-fold in a subset of breast tumors compared with most adjacent noncancerous tissues, in which it was present at low levels (Figure 2A). A targeted quantitative analysis in 60 ER-negative tumors showed that this metabolite reached concentrations of 1–20 mmol/kg tissue in about one-third of these tumors (Figure 2B). These observations were further validated in 14 human breast cancer cell lines and 3 noncancerous mammary epithelial cell lines: 5 of the 14 breast cancer lines had notably high intracellular 2HG concentrations in the 2–5 mM range, about 100-fold higher than that in noncancerous lines (Figure 2B and Supplemental Table 6). Furthermore, among these 5 cell lines with high 2HG, 3 had basal-like and mesenchymal characteristics (MDA-MB-231, MDA-MB-468, and SUM159T), 1 was ER-negative with an HER2 amplification (HCC-1419), and only 1 had luminal characteristics (MDA-MB-361). Analysis of both tumors and cell lines yielded consistent results, which indicates that tumors and breast cancer cell lines with high 2HG tend to be ER-negative and frequently belong to the basal-like/mesenchymal breast cancer subtype.
Figure 2. Aberrant accumulation of 2HG in breast cancer and its relationship with genome-wide DNA methylation in breast tumors.

(A) Relative abundance of 2HG in breast tumors and adjacent noncancerous tissue for all paired samples (n = 65, 33 ER-positive and 32 ER-negative) for the discovery set. (B) 2HG quantitation for ER-negative tumor (n = 60) and adjacent noncancerous tissue (n = 29) in the validation set. P value was determined by paired t test (n = 29). Relative abundance of 2HG in 3 benign and 14 cancerous human breast cell lines is also shown. (C) Normalized genome-wide DNA methylation scores were calculated for breast tumors in the discovery set using Human Methylation 450 BeadChips data. 2HG-high tumors (median and above) had a significantly higher DNA methylation score than 2HG-low tumors (below median), as determined by Welch t test.
Highly increased 2HG has been reported from gliomas and leukemias with hotspot mutations in either the cytosolic IDH1 or the mitochondrial IDH2, which cause the enzyme to produce 2HG (13–15). Sequencing of the 67 breast tumors in the discovery set did not reveal the presence of these hotspot mutations in any of the tumors. Thus, 2HG accumulates in breast tumors in the absence of IDH mutations and reaches concentrations in the millimolar range, comparable to those found in IDH-mutant gliomas and leukemias (14).
2HG-high tumors exhibit a distinct DNA methylation pattern.
Accumulation of 2HG is known to cause the inhibition dioxygenases like the TET family of 5-methylcytosine (5mC) hydroxylases, leading to decreased levels of 5-hydroxymethylcytosine (5hmC) and a DNA hypermethylation phenotype (16, 21). We tested whether exposure to 2HG would have a similar effect in the breast cancer cell line MCF7, which has low endogenous 2HG. Addition of 2HG induced a significant shift in the 5hmC/5mC ratio in these cells (Supplemental Figure 6), consistent with decreased 5hmC levels because of TET inhibition. The finding led us to examine the relationship between genome-wide DNA methylation patterns and levels of 2HG in 62 breast tumors. We observed that 2HG-high breast tumors exhibited a genome-wide DNA hypermethylation phenotype (Figure 2C). Moreover, genome-wide DNA methylation variation classified the tumors into 3 different subgroups, of which subgroups I and III were separated by their distinct methylation patterns, whereas subgroup II showed an intermediate pattern (Figure 3A). A significant relationship emerged between these subgroups and tumor 2HG levels (Figure 3A). Subgroup III had the highest 2HG levels and predominately consisted of ER-negative tumors and tumors from AA patients, but exhibited reduced DNA methylation at the IDH2 locus and increased IDH2 expression (Supplemental Figure 7). Moreover, 2 other metabolites that have an effect on DNA methylation, S-adenosylmethionine (SAM) and S-adenosylhomocysteine (SAH), were also increased in subgroup III tumors (Figure 3B).
Figure 3. A poor outcome tumor subgroup defined by DNA methylation and metabolite profiles.

(A) Heat map representing 2,102 probes highlighting breast tumor subgroups I–III with distinct DNA methylation profiles. Colored bars below the heat map indicate sample classes: red, AA; blue, EA; brown, ER-negative. Percent AA and ER-negative cases in each subgroup is also shown. Box plots above show relative abundance of 2HG in each subgroup (box, interquartile range; line within box, median). Subgroup I is reference for fold difference. (B) Box plots representing relative abundance of SAH and SAM by subgroup. (C) Subgroup III tumors were associated with poor outcome. Kaplan-Meier curves with Cox regression analysis results are shown. HR, hazard ratio. (D) Association between subgroup III DNA methylation signature and breast cancer–specific survival in a publicly available dataset (22). The survival of patients with the subgroup III DNA methylation signature (Subgroup III signature-high) was significantly poorer than that of other patients. Kaplan-Meier curves with Cox regression analysis results are shown.
Molecular characteristics of subgroup III/2HG-high tumors are markers of poor prognosis.
To assess the clinical significance of the DNA methylation–defined subgroup III with high 2HG, we performed a survival analysis in our study population and also assessed patients in publicly available datasets using subgroup-associated DNA methylation and gene expression signatures. Patients with subgroup III tumors showed significantly decreased breast cancer–specific survival compared with patients in subgroup I (Figure 3C and Supplemental Table 7). The DNA methylation characteristics of subgroup III were also significantly associated with poor survival among 234 previously described patients from the Jules Bordet Institute in Belgium (Figure 3D, Supplemental Table 8, and ref. 22). Furthermore, gene expression characteristics of subgroup III and 2HG-high tumors were significantly associated with poor outcome in multiple datasets (Figure 4 and Supplemental Tables 9–12).
Figure 4. Gene signatures for subgroup III and 2HG-high tumors are predictors of poor survival.

(A) Association between the gene signature for subgroup III and breast cancer survival. Survival of patients with the gene expression signature of subgroup III (Subgroup III signature-high) was significantly decreased in 3 independent datasets (van de Vijver, ref. 2, n = 295; Kao, ref. 63, n = 327; Chin, Pawitan, Miller, and Desmedt, refs. 64–67, n = 710). To generate the subtype III gene expression signature, subtype III and subtype I breast tumors were compared to identify 159 and 296 genes that were consistently up- and downregulated, respectively, in subtype III tumors. (B) Association between the gene signature for 2HG-high tumors and breast cancer survival. Survival of patients with the gene expression signature of 2HG-high tumors (2HG signature-high) was significantly decreased in the 3 independent datasets shown in A. Breast tumors with high levels of 2HG (top 33%) and low levels of 2HG (lowest 33%) were compared to identify 50 and 127 genes that were consistently up- and downregulated, respectively, in 2HG-high tumors. Kaplan-Meier curves with Cox regression analysis results are shown.
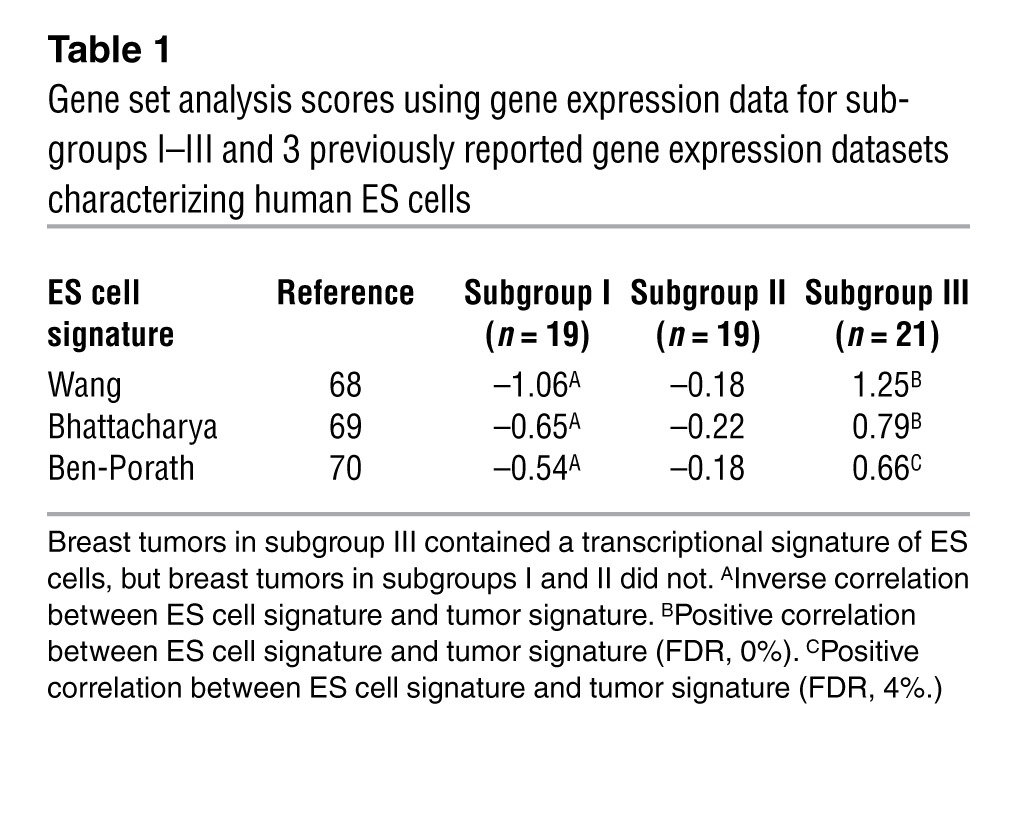
Accumulation of 2HG can induce stem cell–like features in tumors, as indicated by previous studies (16, 23). Thus, we investigated subgroup III tumors for the presence of these features and observed that most subgroup III tumors exhibited DNA hypermethylation at multiple loci encoding inhibitors of the WNT signaling pathway, with an associated decrease in expression of these genes (Figure 5). Furthermore, Kyoto Encyclopedia of Genes and Genomes–based (KEGG-based) pathway analysis ranked the WNT pathway as among those most likely affected by altered methylation in subgroup III tumors (Supplemental Table 13). In addition, breast tumors in subgroup III contained a transcriptional signature of ES cells, as revealed by a gene set analysis (Table 1).
Figure 5. WNT pathway deregulation in subgroup III tumors.

Box plots show relative gene expression levels, and bee swarm plots show DNA methylation M-values, of genes encoding various inhibitors of the WNT signaling pathway, including Wnt inhibitory factor 1 (WIF1), secreted frizzled-related protein 1 (SFRP1), SFRP2, SFRP4, Dickkopf-related protein 2 (DKK2), and DKK3, in subgroup I (n = 19) and subgroup III (n = 23). Expression of WNT pathway inhibitors was significantly reduced in subgroup III tumors (Welch test). Data were collected using GeneChip Human Gene 1.0 ST arrays and Human Methylation 450 BeadChips.
Table 1.
Gene set analysis scores using gene expression data for subgroups I–III and 3 previously reported gene expression datasets characterizing human ES cells
2HG accumulates in breast tumors with MYC pathway activation.
The absence of IDH mutations in breast tumors suggested a novel mechanism for 2HG accumulation in breast cancer. Because MYC has a dominant function in mitochondrial biogenesis and a strong influence on the cell metabolism, specifically glutamine oxidation (6, 8), we asked whether MYC activation is linked to 2HG accumulation in breast cancer. A previously described core MYC signature (24) was examined in the gene expression compendium obtained from 61 tumors in our present study in order to ascertain MYC pathway activation. Notably, 24 of 61 tumors (39%) expressed this core MYC signature (Figure 6A). Next, we assessed the association of the MYC signature with 2HG levels, DNA methylation subgroup status, and race/ethnicity. This analysis revealed a striking overlap between elevated 2HG levels in tumors and their MYC activation status (odds ratio [OR], 11.3; 95% CI, 3.4 to 37.4; i.e., an 11.3 increased odds of aberrant 2HG accumulation in a tumor with vs. without the core MYC signature; P < 10–4). Similar overlaps with MYC activation status were observed for DNA methylation subgroup III tumors (OR, 12.8; 95% CI, 3.6 to 45.5; P < 10–4) and tumors from AA patients (OR, 4.4; 95% CI, 1.3 to 16.0; P < 0.01) (Figure 6A). Importantly, the heightened prevalence of MYC activation in AA patients largely explained the observed metabolic differences between these patients and EA patients. The metabolite-based classification of ER-negative breast tumors into subclusters with predominately AA or EA patients was a classification driven by MYC pathway activation in these tumors (Supplemental Figure 8).
Figure 6. Subgroup III/2HG-high tumors are defined by a MYC activation signature.

(A) Heatmap for gene expression (374 MYC signature genes); colored bars above denote sample classes. Breast tumors were classified as having high or low MYC signature expression based on the presence or absence of a previously described core MYC gene expression signature (24). This classification revealed substantial overrepresentation of tumors from AA patients, DNA methylation subgroup III tumors, and 2HG-high tumors among the class of tumors with MYC activation. (B) Glutaminase was upregulated in 2HG-high versus 2HG-low tumors (3.4-fold; FDR, 0%) and in subgroup III versus subgroup I or II tumors (2.7-fold, subgroup III vs. subgroup I; FDR, 0%). Glutaminase protein abundance in tissue extracts was determined by mass spectrometry. Black lines in box plots denote medians. n and statistical analysis are indicated.
Having addressed the oncogenic driver for 2HG, we next sought to define the potential driver metabolite or biochemical pathway regulating 2HG levels in breast cancer. Thus, we performed a correlation analysis looking for metabolites that followed the abundance pattern of 2HG in the discovery and validation datasets. This analysis identified metabolites that followed the abundance pattern of 2HG, namely malate, glutamate, and NAA (Supplemental Figure 9). In contrast, levels of intratumor glutamine were inversely correlated with 2HG (r = –0.48; P < 0.001, Pearson correlation test). This suggested that high glutamine utilization could be linked to 2HG accumulation. In addition, MYC is known to regulate glutamine utilization and glutaminase (GLS1) protein expression through a microRNA-mediated mechanism, leading to increased glutamine consumption (25). Hence, we measured GLS1 protein abundance in tumor extracts using mass spectrometry. Tumors with high 2HG or those with subgroup III–associated DNA methylation profiles tended to overexpress GLS1 (Figure 6B), thus linking increased glutamine metabolism to 2HG accumulation in breast cancer. Accordingly, both low glutamine levels in breast tumors (lowest 25%) and their associated gene expression signature were associated with decreased patient survival (Figure 7, A and B, and Supplemental Table 14).
Figure 7. Association of tumor glutamine levels with breast cancer survival.

(A) Patients with low tumor glutamine levels (Gln-low) exhibited poor outcome in the discovery and validation sets (both with stratification at lowest 25% vs. highest 75%). (B) Survival of patients with the gene expression signature of glutamine-low tumors (Gln-low signature-high) was significantly decreased compared with patients that did not have this tumor signature in 3 publicly available datasets (van de Vijver, ref. 2; Kao, ref. 63; Chin, Pawitan, Miller, and Desmedt, refs. 64–67). Breast tumors with low and high glutamine levels were compared (lowest vs. highest quartile) to identify 8 and 25 genes that were consistently up- and downregulated, respectively, in glutamine-low tumors. This 33-gene expression signature for glutamine-low tumors was then applied. Kaplan-Meier curves with Cox regression results are shown.
We further assessed the link between glutamine and 2HG metabolism using a mass spectrometry–based flux analysis, wherein we monitored the incorporation of 13C-labeled glutamine into 2HG in MCF7 breast cancer cells (with low endogenous 2HG) and MDA-MB-231 cells (with high 2HG levels). This experiment showed significantly augmented incorporation of the C13 label from glutamine into 2HG in MDA-MB-231 cells, but not in MCF7 cells (Figure 8A). Furthermore, knockdown of GLS1 with siRNA significantly reduced intracellular 2HG in 2 breast cell lines with aberrant 2HG accumulation (MDA-MB-231 and SUM159T; Figure 8B), while inhibition of mitochondrial GLS1 using the cell-permeable GLS1 inhibitor compound 968 (26) reduced intracellular 2HG by 80%–90% (Figure 8C). Lastly, we evaluated whether MYC expression alterations influence intracellular 2HG. In a proof-of-principle experiment, we examined 2HG levels in hTERT immortalized human mammary epithelial cells with inducible upregulation of MYC (27). This experiment showed that upregulation of MYC increased intracellular 2HG (Figure 8D). In contrast, inducible knockdown of MYC using 2 different shRNAs in SUM159T cells (with aberrant 2HG accumulation) resulted in significantly reduced intracellular 2HG levels (Figure 8E). These findings are consistent with the hypothesis that aberrant 2HG accumulation in breast cancer is glutamine dependent and commonly involves MYC activation.
Figure 8. Glutamine metabolism is linked to aberrant accumulation of 2HG in breast cancer cells.

(A) Incorporation of C13-labeled glutamine into 2HG in MDA-MB-231 (2HG-high) and MCF7 (2HG-low) cells 10 seconds and 3 hours after adding C13-glutamine to the culture medium (n = 3 each). MDA-MB-231 cells, with aberrant 2HG accumulation, incorporated the C13 label into 2HG. (B) Reduced 2HG in breast cancer cells treated with GLS1 siRNA (n = 4 per group; t test). Cell pellets were harvested 48 hours after transfection. (C) Reduced 2HG and glutamate in breast cancer cells treated with the mitochondrial GLS1 inhibitor compound 968 (n = 3 per group; *P < 0.05 vs. control, t test). Cells were treated with 10 μM inhibitor for 48 hours. (D) Increased intracellular 2HG after c-Myc induction (+MYC) in human mammary epithelial cells with an inducible MYC-ER fusion transgene. Cell pellets were harvested 48 hours after induction of c-Myc (3 independent experiments; t test). (E) Knockdown of c-Myc with 2 different doxycycline-inducible shRNA expression constructs (right) caused a significant reduction of aberrantly accumulated 2HG (left) in SUM159T cells (4 independent experiments; t test). Cell pellets were harvested 3 days after shRNA induction. Values are shown normalized to internal standard. All graphs show mean ± SD. See complete unedited blots in the supplemental material.
Knockdown of mitochondrial ADHFE1 and IDH2 reduces 2HG.
We next asked whether 2HG synthesis via glutamine is regulated by mitochondrial enzymes, namely hydroxyacid-oxoacid transhydrogenase (ADHFE1) and IDH2, based on earlier reports in which these enzymes were implicated in mitochondria-associated, α-ketoglutarate–dependent (α-KG–dependent) production of D-2HG (16, 28). We targeted these enzymes with siRNA in 2HG-high cells (Supplemental Figure 10). Knockdown of these enzymes led to a marked reduction of endogenous 2HG (Figure 9, A–C). Similar results were obtained upon constitutive knockdown of ADHFE1 with shRNA in MDA-MB-231 cells (Figure 9D). ADHFE1 knockdown also resulted in moderate but significant inhibition of cell cycle kinetics and reduced migration and invasion in MDA-MB-231 and SUM159T cells (Supplemental Figure 11), suggestive of an oncogenic role for ADHFE1. Lastly, we examined the role of the mitochondrial D-2HG–dehydrogenase (D2HGDG), which can also regulate 2HG levels in mammalian cells, in aberrant 2HG accumulation. Expression of D2HGDG was significantly decreased by 4.3-fold in breast cancer cells with elevated 2HG levels (Supplemental Figure 12A), although in breast tumors, these prominent expression differences according to tumor 2HG status were not observed. We inhibited D2HGDG expression in MCF7 cells (with low 2HG levels) using siRNA, which induced a modest 1.6-fold increase of intracellular 2HG (Supplemental Figure 12B). Overall, these data are consistent with a glutamine-driven mitochondrial metabolism regulating 2HG levels in breast cancer cells and the involvement of several mitochondrial enzymes in maintaining aberrant 2HG accumulation in breast cancer cells.
Figure 9. Reduced 2HG in cells with aberrant 2HG accumulation after knockdown of ADHFE1 and IDH2 expression.

(A) Knockdown of ADHFE1 protein with siRNA. ADHFE1v, splice variant. (B) Knockdown of IDH2 protein with siRNA. (C) Reduced 2HG in breast cancer cells treated with IDH2 or ADHFE1 siRNA (n = 4 per group; *P < 0.05 vs. control, t test). 2HG measurements were performed 48 hours after siRNA transfection. (D) Reduced 2HG in MDA-MB-231 cells expressing a shRNA that targets ADHFE1 (3 independent measurements; t test). Western blot analyses were of whole cell extracts (A and B) or mitochondrial lysates (D). All graphs show mean ± SD. See complete unedited blots in the supplemental material.
Inhibition of apoptosis by 2HG.
To examine whether 2HG by itself may contribute to the pathogenesis of breast cancer, we performed additional experiments in which we exposed human mammary epithelial cells with low endogenous 2HG levels to exogenous D-2HG octylester (referred to herein as octyl-2HG). This cell-permeable ester has previously been used in studies exploring 2HG-induced phenotypes in other cancers (21, 23). Treatment of MCF7 and MCF12A cells with 1 mM octyl-2HG led to a >100-fold increased intracellular concentration of 2HG in these cells (Supplemental Figure 13), mimicking 2HG levels of human breast tumors with aberrant 2HG accumulation. Next, we tested whether this accumulation in 2HG affects cell proliferation and the apoptotic response to serum starvation/glucocorticoid withdrawal. We found that octyl-2HG–treated MCF7 cells had a modest but significant increase in cell proliferation (Supplemental Figure 14), whereas 2HG reduced the extent of apoptosis in response to serum starvation/glucocorticoid withdrawal in both MCF10A and MCF12A cells (Figure 10).
Figure 10. Inhibition of apoptosis by 2HG.

MCF10A and MCF12A mammary epithelial cells were cultured in the presence or absence of 1 mM octyl-2HG. For apoptosis induction, cells were kept in DMEM/F12 medium without horse serum and hydrocortisone (–serum). Induction of apoptosis by serum starvation/glucocorticoid withdrawal was measured after 48 and 72 hours using a caspase 3/7 activity assay (values expressed as fluorescence 499/521ex/em; n = 4). Graphs show mean ± SD.
Discussion
A highlight of our study was the finding of markedly elevated 2HG in a subgroup of breast tumors that were predominantly ER-negative and had poor clinical outcome. High 2HG was previously shown to induce DNA hypermethylation and enhance histone methylation through inhibition of 5mC hydroxylases and histone demethylases, leading to epigenetic alterations in gliomas and leukemias (16, 21, 23, 29). Consistent with these data, we observed that 2HG-high breast tumors exhibited a hypermethylation phenotype. The majority of the 2HG-high tumors were also characterized by MYC activation and a discrete DNA methylation pattern that classified them as a DNA methylation–defined molecular subgroup of breast tumors, termed subgroup III, with a stem cell–like gene expression signature. Others have described molecular subgroups in breast cancer based solely on global DNA methylation pattern (22, 30). One such report defined 3 subgroups based on their DNA methylation profiles, one of which was enriched for basal-like tumors, similar to our subgroup III (30). Here, we further described this molecular subgroup as a class of tumors that accumulated 2HG and included a disproportionately high number of AA patients. Thus, AA patients may develop a methylation-defined subgroup III tumor more frequently than EA patients, in agreement with one study that observed DNA methylation differences between these patient groups (31).
In our study, metabolome profiles robustly distinguished tumors from adjacent nontumor tissue, in agreement with a previous report (32), and yielded differences between ER-negative and -positive tumors and by disease grade. However, we could not obtain a good metabolite-based separation into the previously described subtypes with distinct gene expression profiles (1, 19). While the luminal A subtype was distinguished very well, the HER2-positive and -negative subtypes were not adequately separated. This observation was in agreement with another study that observed weak separation between HER2-negative and -positive tumors based on analysis of fatty acids (33). Perhaps a different compendium of metabolites must be profiled to achieve a metabolite-based classification that matches the gene expression–defined subtypes. Alternatively, the tumor metabolome may describe different disease traits, as previously suggested by others (34). Importantly, our finding that 2HG was high in a DNA methylation–defined subtype with poor outcome characteristics suggests that the tumor metabolome describes tumor subtypes of clinical relevance.
Because of the expected influence of body composition, lifestyle, and sex hormones on tumor metabolism, we investigated the relationships among metabolome profiles and patient age, body mass, socioeconomic status, and menopausal status. We did not find that these characteristics significantly associated with the tumor metabolome, raising the possibility that breast tumors have a robust intrinsic metabolome signature that predominates over the influence of these factors. Yet environmental exposures may affect the tumor metabolome and epigenome in more subtle ways, as was observed recently. Christensen et al. reported that global DNA methylation profiles in breast tumors are associated with alcohol and folate intake (35). Mechanistically, these nutrients are thought to influence DNA methylation through regulation of the 1-carbon metabolism and altering the availability of the universal methyl donor SAM and the methyltransferase inhibitor SAH. Still, the argument that tumor-derived mechanisms may dominate over diet-derived mechanisms in shaping the cancer metabolome is further strengthened by our observation that an intrinsic core MYC signature (24) was closely associated with 2HG accumulation in breast cancer and the global metabolite pattern in ER-negative tumors. We detected the MYC activation signature in 39% of the tumors, in agreement with previous reports of c-Myc protein overexpression in 40%–45% of human breast tumors (36). MYC is a target of the WNT signaling pathway (37, 38). MYC activation in the subgroup III/2HG-high may partly develop because of constitutive WNT signaling. This hypothesis is supported by our finding that inhibitors of WNT signaling had reduced expression in subgroup III tumors because of DNA hypermethylation at loci that encode these genes.
MYC strongly influences the metabolism of cancer cells and has been shown to regulate a transcriptional program that stimulates mitochondrial glutamine consumption (6, 8, 39). This function of c-Myc may explain why the metabolic profile of breast tumors describes tumors with a MYC activation signature as a distinct disease subtype, as shown by our data. We also observed that tumors with the core MYC signature tend to accumulate 2HG in the presence of reduced glutamine while overexpressing GLS1, a key enzyme for glutamine metabolism. Therefore, heightened glutamine consumption through MYC activation may fuel aberrant 2HG accumulation in breast tumors. Evidence exists that basal-like and mesenchymal breast cancer cells exhibit a phenotype of glutamine dependence (11). These tumors preferentially accumulate 2HG, as shown by our present data. We also observed a significant relationship between low tumor glutamine levels and poor patient survival, establishing a candidate link between increased glutaminolysis and clinical outcome in breast cancer. In this context, low tissue glutamine or increased expression of GLS1 could serve as a biomarker for breast tumors that respond favorably to GLS1 inhibition therapy, potentially providing an additional survival advantage to otherwise-refractory subtypes, such as basal-like and triple-negative disease. Previously, Seltzer et al. set precedence for inhibiting GLS1 as a therapeutic approach to slow the growth of cancer cells with mutant IDH1 and aberrant 2HG accumulation (40). Using bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES) to inhibit GLS1 activity, the authors found a preferential antitumor effect in IDH1-mutant versus wild-type glioma cells, although BPTES did not significantly alter the aberrant 2HG level in the IDH1-mutant cells under the experimental conditions. Nevertheless, the same inhibitor may preferentially slow the growth of breast cancer cells with a 2HG accumulation phenotype.
High tissue 2HG concentrations have previously been described from gliomas and leukemias with acquired IDH mutations (14–16). Our DNA sequence analysis of breast tumors did not reveal the presence of IDH mutations, consistent with other studies (41–44). In IDH-mutant gliomas, 2HG was previously shown to reach millimolar concentrations (5–35 mmol/kg tissue; ref. 14). We observed similarly high concentrations of 2HG in a subset of breast tumors and showed experimentally that aberrant 2HG synthesis in breast cancer cells was maintained by glutamine consumption. IDH2 participates in the glutamine catabolism pathway and was upregulated in subgroup III tumors. It has been shown that wild-type IDH2 produces 2HG as a byproduct in a process that can be augmented by hypoxia (45). In those studies, IDH2 modestly increased intracellular 2HG about 2- to 3-fold under normoxic conditions, with a further 3-fold augmentation under hypoxia (16, 45). Our present data showed that a subset of human breast cancer cells accumulated 2HG at concentrations 100- to 200-fold above baseline when cultured under normoxic conditions, which is rather inconsistent with a key role of IDH2 and hypoxia in this process. Nevertheless, knockdown of IDH2 markedly decreased intracellular 2HG in 2 breast cancer cell lines with aberrant 2HG accumulation. It is possible that wild-type IDH2 contributes to 2HG accumulation in breast cancer directly through its known participation in 2HG synthesis, as well as indirectly via its significant role in the TCA cycle and reductive carboxylation of glutamine-derived α-KG to maintain citrate synthesis (45, 46). Others have shown that knockdown of the enzyme D2HGDG increases intracellular 2HG in cells with wild-type and mutant IDH (47). Yet the increase of 2HG was rather modest (4- to 6-fold) in these experiments. We confirmed that D2HGDG downregulation in MCF7 breast cancer cells only modestly increased 2HG. Notably, however, D2HGDG tended to be downregulated in many of the breast cancer cells with aberrantly high endogenous 2HG, indicative of some involvement of this enzyme in maintaining 2HG levels in these cells.
Another candidate source of intracellular 2HG is the poorly studied mitochondrial enzyme ADHFE1 (28, 48). This enzyme did not seem to be elevated in breast tumors based on gene expression data. The activity of the enzyme could be altered independent of its expression, in agreement with the observation that the cancer metabolism is regulated by post-translational mechanisms, rather than by tumor-induced gene expression changes (49). We targeted this enzyme using knockdown strategies. Knockdown of ADHFE1 with siRNA reduced endogenous 2HG levels in 2 cell lines with aberrant 2HG accumulation to levels in noncancerous mammary epithelial cells. From this experiment, ADHFE1 emerged as a candidate source for 2HG in breast cancer. ADHFE1 is known both as alcohol dehydrogenase iron–containing enzyme 1 and as mitochondrial hydroxyacid-oxoacid transhydrogenase and may encode various isoforms, including 45- to 50-kDa and 27- to 32-kDa proteins (50, 51). Per assigned function, ADHFE1 metabolizes 4-hydroxybutyrate to succinic semialdehyde coupled with the reduction of α-KG to D-2HG. There is little functional description of the enzyme in cancer cells, but it is expressed in fetal tissues and has been reported to be hypermethylated and partly silenced in colorectal cancer (50–52). Thus, ADHFE1 may have an unrecognized oncogenic function in breast cancer, and future research will be needed to understand the role of ADHFE1 in 2HG accumulation in breast tumors and the regulation of this enzyme by post-translational mechanisms.
Our study describes a close relationship between the biology of aberrant MYC activation and 2HG accumulation in breast cancer. To discern the possible impact of the 2 biologies in breast cancer, we performed additional experiments to examine whether 2HG could be causally important in the pathogenesis of breast cancer. This approach led to the preliminary findings that accumulation of 2HG can increase cell proliferation and inhibit apoptosis. Increased cell proliferation after treatment with cell-permeable 2HG has also been observed in leukemogenesis (53). Future work will be needed to further define the oncogenic effects of 2HG in breast cancer and how 2HG accumulation could be targeted independent of MYC signaling. Other research may focus on 2HG as a biomarker. 2HG can be detected noninvasively by analyzing plasma samples. This has been observed in tumor-bearing mice using an animal model of azoxy-methane–induced colorectal cancer (54). Thus, blood-based 2HG is a potential biomarker for prognosis and/or diagnosis.
In summary, we have found aberrant accumulation of 2HG in a subset of human breast tumors with poor outcome and linked this observation to MYC activation and glutamine dependence. Furthermore, 2HG frequently accumulated in tumors and cell lines of the basal-like/mesenchymal subtype, reaching concentrations comparable to those in IDH-mutant gliomas and leukemias, despite the absence of IDH mutations.
Methods
Tissue collection and cell lines.
Unselected breast cancer patients at all disease stages were recruited between February 15, 1993, and August 27, 2003, into a biomarker study under a NCI contract (Resource Collection and Evaluation of Human Tissues and Cells from Donors with an Epidemiology Profile), as described previously (55). Samples of fresh-frozen, macrodissected tumor tissue and adjacent noncancerous tissue were prepared by a pathologist immediately after surgery. Clinical and pathological information was obtained from medical records and pathology reports. See Supplemental Methods for details on patient recruitment, specimen collection, and tumor classification. Human breast cancer cell lines were obtained from the American Type Culture Collection with the exception of the SUM159T cells, which were obtained from S. Lipkowitz (NCI, Bethesda, Maryland, USA).
Genetic ancestry estimation.
Genomic DNA was isolated from either noncancerous tissue or blood samples and was genotyped to determine the proportion of European, West African, and Native American genetic ancestry (see Supplemental Methods). In the discovery cohort, 105 AIMs were genotyped to determine the proportion of European, West African, and Native American genetic ancestry (56).
Metabolome analysis.
Metabolomic profiling of fresh-frozen bulk human breast tissues was performed for 2 tissue sets. Untargeted metabolic profiling of known and unknown metabolites in the discovery set was performed by Metabolon Inc. (57). Metabolic profiling in the validation set was performed at the Alkek Center for Molecular Discovery of Baylor College of Medicine and at the SAIC-NCI Laboratory of Proteomics and Analytical Technologies. For quantitation of 2HG in tissue extracts and cell lines, a serial dilution of 2HG was mixed with a fixed amount of [D27] myristic acid internal standard, and the peak area ratios of 2HG and [D27] myristic acid were plotted against the amount of 2HG injected to generate a standard curve. Tissue and cell extracts were mixed similarly with the [D27] myristic acid internal standard, analyzed by gas chromatography–coupled mass spectrometry, and estimated for normalized 2HG levels using this standard curve. See Supplemental Methods for details.
Proteome analysis.
Frozen human tissue samples were homogenized, and the extracted proteins were digested with trypsin and analyzed by mass spectrometry (see Supplemental Methods). Mass spectrometry data were searched against the UniProt Homo sapiens database (downloaded from the European Bioinformatics Institute website; http://www.ebi.ac.uk/integr8) using SEQUEST (ThermoElectron/Thermo Fisher Scientific). Normalized relative abundance of an identified protein in a sample was used for class comparison analyses.
RNA isolation and gene expression microarray analysis.
Total RNA was isolated from tissue using the TRIzol reagent. RNA integrity for each sample was confirmed with the Agilent 2100 Bioanalyzer (Agilent Technologies). 250 ng RNA was converted into cDNA, fragmented, labeled, and hybridized onto Gene Chip Human Gene 1.0 ST Arrays according to Affymetrix standard protocols. Probe cell intensity data was processed by the RMA algorithm (58). See Supplemental Methods for details. Gene expression data from this study were deposited in GEO (accession no. GSE37751).
Global DNA methylation analysis.
Global methylation profiles for breast tumors were obtained using bisulfite-converted genomic DNA that was hybridized onto Human Methylation 450 BeadChips (Illumina). See Supplemental Methods for details. DNA methylation profiles from this study were deposited in GEO (accession no. GSE37754).
Measurement of the 5hmC/5mC content of genomic DNA using liquid chromatography/mass spectrometry.
Experiments were performed as described previously (59). See Supplemental Methods for details.
Mutational analyses of IDH1 and IDH2.
Tumors were screened for IDH1 and IDH2 mutations following a previously described procedure (15). See Supplemental Methods for details.
Incorporation of 13C-labeled glutamine into 2HG.
For metabolic flux analysis, 1 × 106 MCF7 and MDA-MB-231 cells were plated overnight in 6-well plates with 2 ml DMEM supplemented with 10% FBS. Cells were then cultured for 12 hours in glutamine-depleted DMEM (devoid of glucose, glutamine, phenol red, sodium pyruvate, and sodium bicarbonate; Mediatech Inc.) supplemented with 10% FBS, 4.5 g/l glucose, and sodium pyruvate. Either 12C or universally labeled 13C glutamine (2 mM final concentration) was added to culture medium (all in triplicate), and cells were harvested immediately after addition of glutamine or after 3 hours. Cells were scraped with methanol water and frozen immediately in nitrogen. 500 μl ice-cold methanol/water (1:1 ratio) containing 20 μl spiked internal standards was added to each cell pellet. Cells were homogenized for 1 minute (2 30-second pulses), then vortex mixed in a Multi-Tube Vortexer for 10 minutes. The extract was filtered through 3-kDa molecular filters at 4°C for 90 minutes to remove proteins. The filtrate was dried at 37°C for 45 minutes in a speed vacuum prior to mass spectrometry analysis, and dried extract was derivatized using methoxamine and MSTFA plus 1% TMCS. Aliquots were analyzed using gas chromatography–coupled mass spectrometry and a single reaction monitoring strategy with electron-impact ionization. 2HG abundance was determined by mass spectrometry, and percent 13C glutamine incorporation into endogenous 2HG was estimated as follows: total 13C-2HG/(total 13C-2HG + 12C-2HG).
siRNA transfection and constitutive expression of a shRNA that targets ADHFE1.
Cells were transfected with either negative control siRNA (Silencer Select Negative Control) or ADHFE1, IDH2, GLS1, or 2DHGDG Select siRNAs (Life Technologies) following the manufacturer’s protocol for lipofectamine RNAiMAX (Life Technologies). MDA-MB-231 cells with constitutive expression of a shRNA that targets ADHFE1 were generated after infection of these cells with a lentiviral construct encoding the shRNA (CMV51eGFPsh.ADHFE1) and selection with puromycin. Control cells were infected and selected using the vector control without the shRNA.
Expression of an inducible MYC transgene in human mammary epithelial cells and knockdown of MYC with inducible shRNA expression constructs in SUM159T cells.
Human mammary epithelial cells with an inducible MYC-ER fusion transgene were cultured as described previously, and MYC was induced with tamoxifen (27). Cells were cultured in mammary epithelial growth medium (MEGM; Lonza), treated with and without 300 nM tamoxifen for 48 hours, and harvested. SUM159T cells were infected with pINDUCER lentiviral constructs containing doxycycline-inducible shRNA targeting MYC. Cells were sorted for GFP to generate fully infected populations. 1 μg/ml doxycycline was added to the culture medium for 3 days to induce knockdown of MYC. For 2HG measurements, cells were washed in PBS and pelleted, and samples were normalized to cell number.
Quantitative real-time PCR.
Total RNA was isolated from cell lines using the QIAGEN RNeasy method (QIAGEN). qRT-PCR was performed in triplicate using the TaqMan ASPA (aspartoacylase; EC 3.5.1.15), IDH2 (isocitrate dehydrogenase 2, mitochondrial; EC 1.1.1.42), ADHFE1 (alcohol dehydrogenase iron containing 1 or hydroxyacid-oxoacid transhydrogenase, mitochondrial; EC 1.1.99.24), GLS1 (glutaminase; EC 3.5.1.2), and D2HGDG (2-D-hydroxyglutarate dehydrogenase; EC 1.1.99.2) expression assays (Applied Biosystems), which included preoptimized probes and primer sets for these genes.
Western blot analysis.
Western blot analysis was performed according to standard procedures. The following antibodies were used at manufacturer-recommended concentrations to detect the membrane-bound proteins: rabbit polyclonal anti-ADHFE1 (H-240, catalog no. sc-292533; Santa Cruz Biotechnology), rabbit polyclonal anti-ADHFE1 (catalog no. ab102600; Abcam), mouse monoclonal anti-vinculin (catalog no. V9131; Sigma-Aldrich), mouse monoclonal anti–c-Myc (catalog no. M4439; Sigma-Aldrich), mouse monoclonal anti-IDH2 (catalog no. ab55271; Abcam).
Proliferation assay and cell cycle analysis.
Cell proliferation was assessed using the CellTiter-Blue Cell Viability Assay kit (G8081; Promega). For cell cycle analysis, cells were transfected with siRNA targeting ADHEF1, harvested by trypsinization, washed 3 times with PBS, fixed with 70% cold ethanol, and then stained with 50 μg/ml 7-amino-actinocycin D (Invitrogen) for 30 min. At least 10,000 events were analyzed per sample using a FACSCalibur cytometer (BD Biosciences), and cell phase percentages were determined using FlowJo software (Ashland).
Apoptosis assay.
Immortalized nonmalignant MCF10A and MCF12A cells were seeded in black-coated standard 96-well culture plates (5,000 cells/well) in DMEM/F12 (1:1 ratio) medium (complete medium containing 5% heat-inactivated horse serum, 10 μg/ml insulin, 20 ng/ml EGF, 500 ng/ml hydrocortisone, and 100 ng/ml cholera toxin). For induction of apoptosis in response to serum starvation/glucocorticoid withdrawal, cells were kept in complete DMEM/F12 medium, but without horse serum and hydrocortisone, and cultured in the presence or absence of 1 mM octyl-2HG ([2R]-2-hydroxyglutaric acid octyl ester; SLR Biosciences). Apoptosis was measured using the ApoOne Homogenous Caspase 3/7 Assay according to the manufacturer’s protocol (Promega). Fluorescence for each treatments was measured at 499/521ex/em with a FLUOstar Omega reader (BMG Labtech).
Migration and invasion assay.
Migration and invasion were examined using xCelligence System Technology (Roche Diagnostics) for real-time monitoring of cellular processes with electronic cell sensor array technology, according to the manufacturer’s instructions. See Supplemental Methods for details.
Statistics.
All statistical tests were 2-sided, and an association was considered statistically significant with P < 0.05. Statistical analyses were performed using R software, developed by R Development Core Team at R Foundation for Statistical Computing and packages in Bioconductor (60). Unconditional logistic regression was used to calculate ORs and 95% CIs. The abundance of metabolites and transcripts was analyzed globally using significance analysis of microarrays (61). Heatmaps were drawn using dChip software (62). Survival analysis was performed using the survival package of R. 6 publicly available datasets of gene expression microarray were included in the survival analysis. The van de Vijver dataset (2) was obtained from Netherlands Cancer Institute. The Kao, Pawitan, Miller, and Desmedt datasets (63–66) were obtained from GEO (accession nos. GSE20685, GSE1456, GSE3494, and GSE7390, respectively). The Chin dataset (67) was obtained from EMBL-EBI by European Bioinformatics Institute (accession no. E-TABM-158). The Pawitan, Miller, Desmedt, and Chin datasets (64–67) were combined into a single matrix. See Supplemental Methods for details.
Study approval.
The collection of biospecimens and the clinical and pathological information was approved by the University of Maryland Institutional Review Board for the participating institutions (UMD protocol no. 0298229). IRB approval of this protocol was then obtained at all institutions (Veterans Affairs Medical Center, Union Memorial Hospital, Mercy Medical Center, and Sinai Hospital, Baltimore, Maryland, USA). The research was also reviewed and approved by the NIH Office of Human Subjects Research Protections (OHSRP no. 2248). All patients signed a consent form and completed an interviewer-administered questionnaire.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, NCI, CCR and by a NCI Director’s Innovation Award to S. Ambs. A. Sreekumar acknowledges the following grant support: 1RO1CA133458-01, NIH SPORE CA58183, Susan Komen Foundation grant KG110818, U01 CA167234, DMS-1161759, RP120092, and funds from the Alkek Center for Molecular Discovery (ASK). The authors thank Raymond Jones, Audrey Salabes, Leoni Leondaridis, Glennwood Trivers, Elise Bowman, and personnel at the University of Maryland and the Baltimore Veterans Administration and the Surgery and Pathology Departments at the University of Maryland Medical Center, Baltimore Veterans Affairs Medical Center, Union Memorial Hospital, Mercy Medical Center, and Sinai Hospital for their contributions in patient recruitment.
Footnotes
Conflict of interest: Edward D. Karoly and Jacob Wulff are employees of Metabolon, Inc., a fee-for-service metabolomics provider.
Citation for this article: J Clin Invest. 2014;124(1):398–412. doi:10.1172/JCI71180.
References
- 1.Sorlie T, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van de Vijver MJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347(25):1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 3.Paik S, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351(27):2817–2826. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 4.Sreekumar A, et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature. 2009;457(7231):910–914. doi: 10.1038/nature07762. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 5.Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. 2011;10(9):671–684. doi: 10.1038/nrd3504. [DOI] [PubMed] [Google Scholar]
- 6.Li F, et al. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol. 2005;25(14):6225–6234. doi: 10.1128/MCB.25.14.6225-6234.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bensaad K, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126(1):107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 8.Wise DR, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. 2008;105(48):18782–18787. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christofk HR, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452(7184):230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 10.Possemato R, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476(7360):346–350. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kung HN, Marks JR, Chi JT. Glutamine synthetase is a genetic determinant of cell type-specific glutamine independence in breast epithelia. PLoS Genet. 2011;7(8):e1002229. doi: 10.1371/journal.pgen.1002229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang WC, et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell. 2012;148(1–2):259–272. doi: 10.1016/j.cell.2011.11.050. [DOI] [PubMed] [Google Scholar]
- 13.Parsons DW, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dang L, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ward PS, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17(3):225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Figueroa ME, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma XJ, et al. Gene expression profiles of human breast cancer progression. Proc Natl Acad Sci U S A. 2003;100(10):5974–5979. doi: 10.1073/pnas.0931261100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sotiriou C, et al. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc Natl Acad Sci U S A. 2003;100(18):10393–10398. doi: 10.1073/pnas.1732912100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perou CM, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 20.Madhavarao CN, et al. Defective N-acetylaspartate catabolism reduces brain acetate levels and myelin lipid synthesis in Canavan’s disease. Proc Natl Acad Sci U S A. 2005;102(14):5221–5226. doi: 10.1073/pnas.0409184102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu W, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dedeurwaerder S, et al. DNA methylation profiling reveals a predominant immune component in breast cancers. EMBO Mol Med. 2011;3(12):726–741. doi: 10.1002/emmm.201100801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu C, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chandriani S, et al. A core MYC gene expression signature is prominent in basal-like breast cancer but only partially overlaps the core serum response. PLoS One. 2009;4(8):e6693. doi: 10.1371/journal.pone.0006693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gao P, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458(7239):762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang JB, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010;18(3):207–219. doi: 10.1016/j.ccr.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kessler JD, et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science. 2012;335(6066):348–353. doi: 10.1126/science.1212728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Struys EA, Verhoeven NM, Ten Brink HJ, Wickenhagen WV, Gibson KM, Jakobs C. Kinetic characterization of human hydroxyacid-oxoacid transhydrogenase: relevance to D-2-hydroxyglutaric and gamma-hydroxybutyric acidurias. J Inherit Metab Dis. 2005;28(6):921–930. doi: 10.1007/s10545-005-0114-x. [DOI] [PubMed] [Google Scholar]
- 29.Turcan S, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483(7390):479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holm K, et al. Molecular subtypes of breast cancer are associated with characteristic DNA methylation patterns. Breast Cancer Res. 2010;12(3):R36. doi: 10.1186/bcr2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mehrotra J, et al. Estrogen receptor/progesterone receptor-negative breast cancers of young African-American women have a higher frequency of methylation of multiple genes than those of Caucasian women. Clin Cancer Res. 2004;10(6):2052–2057. doi: 10.1158/1078-0432.CCR-03-0514. [DOI] [PubMed] [Google Scholar]
- 32.Budczies J, et al. Remodeling of central metabolism in invasive breast cancer compared to normal breast tissue - a GC-TOFMS based metabolomics study. BMC Genomics. 2012;13:334. doi: 10.1186/1471-2164-13-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hilvo M, et al. Novel theranostic opportunities offered by characterization of altered membrane lipid metabolism in breast cancer progression. Cancer Res. 2011;71(9):3236–3245. doi: 10.1158/0008-5472.CAN-10-3894. [DOI] [PubMed] [Google Scholar]
- 34.Borgan E, et al. Merging transcriptomics and metabolomics - advances in breast cancer profiling. BMC Genomics. 2010;10:628. doi: 10.1186/1471-2164-10-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Christensen BC, et al. Breast cancer DNA methylation profiles are associated with tumor size and alcohol and folate intake. PLoS Genet. 2010;6(7):e1001043. doi: 10.1371/journal.pgen.1001043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Y, Olopade OI. MYC in breast tumor progression. Expert Rev Anticancer Ther. 2008;8(10):1689–1698. doi: 10.1586/14737140.8.10.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He TC, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281(5382):1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 38.Yoon JC, Ng A, Kim BH, Bianco A, Xavier RJ, Elledge SJ. Wnt signaling regulates mitochondrial physiology and insulin sensitivity. Genes Dev. 2010;24(14):1507–1518. doi: 10.1101/gad.1924910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res. 2009;15(21):6479–6483. doi: 10.1158/1078-0432.CCR-09-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seltzer MJ, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010;70(22):8981–8987. doi: 10.1158/0008-5472.CAN-10-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kang MR, et al. Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int J Cancer. 2009;125(2):353–355. doi: 10.1002/ijc.24379. [DOI] [PubMed] [Google Scholar]
- 42.Bleeker FE, et al. IDH1 mutations at residue p.R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat. 2009;30(1):7–11. doi: 10.1002/humu.20937. [DOI] [PubMed] [Google Scholar]
- 43.Banerji S, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486(7403):405–409. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wise DR, et al. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad U S A. 2011;108(49):19611–19616. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Le A, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15(1):110–121. doi: 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matsunaga H, Futakuchi-Tsuchida A, Takahashi M, Ishikawa T, Tsuji M, Ando O. IDH1 and IDH2 have critical roles in 2-hydroxyglutarate production in D-2-hydroxyglutarate dehydrogenase depleted cells. Biochem Biophys Res Commun. 2012;423(3):553–556. doi: 10.1016/j.bbrc.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 48.Kaufman EE, Nelson T, Fales HM, Levin DM. Isolation and characterization of a hydroxyacid-oxoacid transhydrogenase from rat kidney mitochondria. J Biol Chem. 1988;263(32):16872–16879. [PubMed] [Google Scholar]
- 49.Hu J, et al. Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat Biotechnol. 2013;31(6):522–529. doi: 10.1038/nbt.2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deng Y, et al. Cloning and characterization of a novel human alcohol dehydrogenase gene (ADHFe1). DNA Seq. 2002;13(5):301–306. doi: 10.1080/1042517021000011636. [DOI] [PubMed] [Google Scholar]
- 51.Lyon RC, Johnston SM, Panopoulos A, Alzeer S, McGarvie G, Ellis EM. Enzymes involved in the metabolism of γ-hydroxybutyrate in SH-SY5Y cells: identification of an iron-dependent alcohol dehydrogenase ADHFe1. Chem Biol Interact. 2009;178(1–3):283–287. doi: 10.1016/j.cbi.2008.10.025. [DOI] [PubMed] [Google Scholar]
- 52.Oster B, et al. Identification and validation of highly frequent CpG island hypermethylation in colorectal adenomas and carcinomas. Int J Cancer. 2011;129(12):2855–2866. doi: 10.1002/ijc.25951. [DOI] [PubMed] [Google Scholar]
- 53.Losman JA, et al. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339(6127):1621–1625. doi: 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Montrose DC, et al. Metabolic profiling, a noninvasive approach for the detection of experimental colorectal neoplasia. Cancer Prev Res (Phila). 2012;5(12):1358–1367. doi: 10.1158/1940-6207.CAPR-12-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boersma BJ, et al. Association of breast cancer outcome with status of p53 and MDM2 SNP309. J Natl Cancer Inst. 2006;98(13):911–919. doi: 10.1093/jnci/djj245. [DOI] [PubMed] [Google Scholar]
- 56.Worsham MJ, Divine G, Kittles RA. Race as a social construct in head and neck cancer outcomes. Otolaryngol Head Neck Surg. 2011;144(3):381–389. doi: 10.1177/0194599810393884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Evans AM, DeHaven CD, Barrett T, Mitchell M, Milgram E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal Chem. 2009;81(16):6656–6667. doi: 10.1021/ac901536h. [DOI] [PubMed] [Google Scholar]
- 58.Irizarry RA, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 59.Friso S, Choi SW, Dolnikowski GG, Selhub J. A method to assess genomic DNA methylation using high-performance liquid chromatography/electrospray ionization mass spectrometry. Anal Chem. 2002;74(17):4526–4531. doi: 10.1021/ac020050h. [DOI] [PubMed] [Google Scholar]
- 60.Gentleman RC, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad U S A. 2001;98(9):5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad U S A. 2001;98(1):31–36. doi: 10.1073/pnas.98.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kao KJ, Chang KM, Hsu HC, Huang AT. Correlation of microarray-based breast cancer molecular subtypes and clinical outcomes: implications for treatment optimization. BMC Cancer. 2011;11:143. doi: 10.1186/1471-2407-11-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pawitan Y, et al. Gene expression profiling spares early breast cancer patients from adjuvant therapy: derived and validated in two population-based cohorts. Breast Cancer Res. 2005;7(6):R953–R964. doi: 10.1186/bcr1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miller WR, et al. Changes in breast cancer transcriptional profiles after treatment with the aromatase inhibitor, letrozole. Pharmacogenet Genomics. 2007;17(10):813–826. doi: 10.1097/FPC.0b013e32820b853a. [DOI] [PubMed] [Google Scholar]
- 66.Desmedt C, et al. Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series. Clin Cancer Res. 2007;13(11):3207–3214. doi: 10.1158/1078-0432.CCR-06-2765. [DOI] [PubMed] [Google Scholar]
- 67.Chin K, et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell. 2006;10(6):529–541. doi: 10.1016/j.ccr.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 68.Wang X. Computational analysis of expression of human embryonic stem cell-associated signatures in tumors. BMC Res Notes. 2011;4:471. doi: 10.1186/1756-0500-4-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bhattacharya B, et al. Gene expression in human embryonic stem cell lines: unique molecular signature. Blood. 2004;103(8):2956–2964. doi: 10.1182/blood-2003-09-3314. [DOI] [PubMed] [Google Scholar]
- 70.Ben-Porath I, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40(5):499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.