

Abstract
Background/Purpose
Acyclic retinoid (ACR) is a promising chemopreventive agent for hepatocellular carcinoma (HCC) that selectively inhibits the growth of HCC cells (JHH7) but not normal hepatic cells (Hc). To better understand the molecular basis of the selective anti-cancer effect of ACR, we performed nuclear magnetic resonance (NMR)-based and capillary electrophoresis time-of-flight mass spectrometry (CE-TOFMS)-based metabolome analyses in JHH7 and Hc cells after treatment with ACR.
Methodology/Principal Findings
NMR-based metabolomics revealed a distinct metabolomic profile of JHH7 cells at 18 h after ACR treatment but not at 4 h after ACR treatment. CE-TOFMS analysis identified 88 principal metabolites in JHH7 and Hc cells after 24 h of treatment with ethanol (EtOH) or ACR. The abundance of 71 of these metabolites was significantly different between EtOH-treated control JHH7 and Hc cells, and 49 of these metabolites were significantly down-regulated in the ACR-treated JHH7 cells compared to the EtOH-treated JHH7 cells. Of particular interest, the increase in adenosine-5′-triphosphate (ATP), the main cellular energy source, that was observed in the EtOH-treated control JHH7 cells was almost completely suppressed in the ACR-treated JHH7 cells; treatment with ACR restored ATP to the basal levels observed in both EtOH-control and ACR-treated Hc cells (0.72-fold compared to the EtOH control-treated JHH7 cells). Moreover, real-time PCR analyses revealed that ACR significantly increased the expression of pyruvate dehydrogenase kinases 4 (PDK4), a key regulator of ATP production, in JHH7 cells but not in Hc cells (3.06-fold and 1.20-fold compared to the EtOH control, respectively).
Conclusions/Significance
The results of the present study suggest that ACR may suppress the enhanced energy metabolism of JHH7 cells but not Hc cells; this occurs at least in part via the cancer-selective enhancement of PDK4 expression. The cancer-selective metabolic pathways identified in this study will be important targets of the anti-cancer activity of ACR.
Introduction
Hepatocellular carcinoma (HCC) represents approximately 85% of all primary liver cancers and is one of the most common malignancies worldwide, especially in Eastern Asia [1]. The prognosis of HCC remains very poor; this poor prognosis is due in part to its high rate of recurrence after initial treatment, which reaches approximately 70% within 5 years [2]. Acyclic retinoid (ACR), a synthetic retinoid with a vitamin A-like structure, prevents the recurrence and development of HCC in patients after the surgical removal of primary tumors [3], [4]. ACR is currently undergoing phase II/III clinical trials (JapicCTI-121828) in Japan and is expected to become the first chemopreventive agent.
Another important characteristic of ACR is that it selectively suppresses the growth of HCC cells (JHH7 and others) but not normal hepatic cells (Hc) [5], [6]. Although the mechanism underlying this effect is not fully understood, previous basic and clinical studies by our group and others have suggested that both non-genomic and genomic signaling pathways may be responsible for the cancer-selectivity of ACR [5], [7], [8], [9], [10], [11], [12]. A typical example is the prevention by ACR of the aberrant hyper-phosphorylation and inactivation of retinoid X receptor (RXR) α that occurs during carcinogenesis in HCC [12] and the subsequent induction of apoptosis in HCC cells by the restoration of the expression of RXRα downstream genes such as p21 [11], transglutaminase 2 (TG2) [5] and more. However, to the best of our knowledge, no information is available regarding the effect of ACR on the metabolism of HCC cells.
Recently, the approach of targeting cancer metabolism to develop and improve cancer therapeutics has received a great deal of attention [13]. A distinguishing feature of cancer is that the metabolic pathways of cancer cells are adapted to support rapid and uncontrolled cell proliferation. One of the best-known alterations in cancer cell metabolism is a switch from mitochondrial oxidative phosphorylation to cytoplasmic glycolysis; this switch is known as the Warburg effect [14]. It is possible that targeting cellular metabolism may suppress cancer. In fact, several metabolism-targeting therapies have been already proven to be effective in the treatment of diverse human tumors [13], [15].
Although chronic hepatitis B virus (HBV) or hepatitis C virus (HCV) infections are believed to account for approximately 80% of HCC [16], a growing body of evidence indicates that metabolic syndrome is also a risk factor for the development of HCC [17]. Indeed, it is extremely difficult to find a single essential target for cancer therapeutics, due to the remarkable heterogeneity and adaptability of cancer cells. It is likely that further investigations into the effect of ACR on cancer cell metabolism will improve our understanding of the molecular pathways underlying the cancer-selective growth suppressive effect of ACR and benefit the development of more effective cancer drugs and therapies against HCC. To achieve this, both nuclear magnetic resonance (NMR)-based and capillary electrophoresis time-of-flight mass spectrometry (CE-TOFMS)-based metabolome analyses were performed in JHH7 and Hc cells after treatment with ACR.
Materials and Methods
Materials
ACR (NIK-333) was supplied by Kowa Co. Ltd. (Tokyo, Japan). All-trans-retinoic acid (AtRA) was purchased from Sigma-Aldrich (St Louis, MO, USA). Ethanol (EtOH) was obtained from Wako Industries (Osaka, Japan), and used as the primary solvent for all reagents. EtOH solutions were further diluted into cell culture media for treatments. The final concentration of EtOH in media used as a control was 0.05% (vol/vol).
Cell culture
The JHH7 HCC cell line was kindly supplied by Dr. Matsuura (Jikei University School of Medicine, Tokyo, Japan) [18]. The normal human hepatocyte cell line (Hc) was purchased from Cell Systems (Kirkland, WA, USA). Both cell lines were maintained in Dulbecco's Modified Eagle Medium (DMEM; Wako Industries) containing 10% fetal bovine serum (FBS, Mediatech, Herndon, VA, USA), 100 U/ml penicillin/streptomycin and 2 mmol/L L-glutamine (Mediatech, Herndon, VA, USA) and grown at 37°C in a humidified 5% CO2 incubator. For chemical treatment, the cells were cultured in serum-free media containing EtOH or ACR at the appropriate concentrations.
NMR-based metabolomics
For NMR analyses, cells (approximately 1×107 cells) treated with EtOH control or 10 μM ACR control for 4 h or 18 h were harvested by scraping as previously described [19]. The one-dimensional (1D) 1H spectra were measured at 500 MHz on a Varian Unity INOVA-500 spectrometer. All NMR spectra were processed using the MestReNova program (Version 5.3.0, MestRec, Santiago de Compostela, Spain). Metabolites were identified using publicly accessible databases, including BioMagRes data bank (http://www.bmrb.wisc.edu), the Metabolomics Database of Linkoping (http://www.mdl.imv.liu.se), and the Human Metabolome Data Bank (http://www.hmdb.ca). Detailed NMR methods have been described previously [19], [20].
CE-TOFMS analyses
JHH7 and Hc cells (approximately 5×106 cells) treated with EtOH control or 10 μM ACR for 24 h were washed twice with a 5% mannitol solution, and then 1,300 μL of a methanol solution containing 10 μM internal standards was added. Metabolome extraction was then performed as previously described [21]. The metabolic profiles of the cells were then measured using a CE-TOFMS-based metabolomics technique, which is a novel strategy for analyzing and differentially displaying metabolic profiles [21]. CE-TOFMS was carried out using an Agilent CE Capillary Electrophoresis System equipped with an Agilent 6210 Time-of-Flight mass spectrometer, Agilent 1100 isocratic HPLC pump, Agilent G1603A CE-MS adapter kit, and Agilent G1607A CE-ESI-MS sprayer kit (Agilent Technologies, Waldbronn, Germany).
Data analysis for CE-TOFMS and metabolite identification
The raw data obtained by CE-TOFMS were analyzed using KEIO MasterHands software exactly as previously described [22], [23]. Briefly, the injected volume for CE and the sensitivity of MS were corrected using internal standards, and then all the annotated metabolites were further corrected to the same chemicals in a standard mixture to overcome different ionization patterns. The peaks were identified based on the matched mass-to-charge ratio (m/z) values and normalized migration times of the corresponding standard compounds.
Real-time RT-PCR
For PCR analyses, RNA was isolated from each cell culture treated with EtOH, AtRA or ACR for 4 h using an RNeasy Kit (Qiagen, Valencia, CA, USA), and the amount and purity of the isolated RNA were evaluated using a NanoDrop spectrophotometer (NanoDrop products, Wilmington, DE, USA). cDNA was then synthesized using a PrimeScript RT Master Mix Kit (TaKaRa Bio, Otsu, Japan). Oligonucleotide primers were designed using OligoPerfect Designer software (Invitrogen, Carlsbad, CA, USA; http://www.tools.invitrogen.com) and synthesized by Invitrogen. The sequences of the primers and the full gene names are summarized in Table S1. PCR reactions were performed using a the Thermal Cycler Dice™ Real Time System (TP8000; Takara Bio) with SsoAdvancedTM SYBR® Green Supermix (Bio-Rad Laboratories, Hercules, CA, USA).
Western blot analysis
JHH7 and Hc cells treated with EtOH, AtRA and ACR for 24 h were lysed using RIPA buffer. After boiling at 97°C for 10 min, the protein samples were resolved by sample buffer for sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis, run on a 10% gel and transferred to a polyvinylidene difluoride membrane (Bio-Rad Laboratories). The membranes were blocked with 5% nonfat dry milk in Tris-buffered saline (TBS) and 0.1% Tween and then probed with primary antibodies against pyruvate dehydrogenase kinase 4 (PDK4; sc-14492; 1∶1,000 dilution, Santa Cruz Biotechnology, CA, USA), pyruvate dehydrogenase (lipoamide) alpha 1 (PDHA1; sc-377092; 1∶1,000 dilution, Santa Cruz Biotechnology), phospho-PDHA1 (ab92696; 1∶1,000 dilution, Abcam) or Lamin B1 (ab16048; 1∶5,000 dilution, Santa Cruz Biotechnology). The blots were then incubated with horseradish peroxidase-conjugated anti-goat, anti-mouse or anti-rabbit secondary antibodies and detected using the Amersham ECL PlusTM Western Blotting Detection System (GE Healthcare UK, Buckingham, England). Immunoreactive bands were quantified using ImageJ densitometry software (National Institutes of Health, Bethesda, MD), and normalized; the density of the corresponding band in the EtOH control was set to 1.0.
RNA interference
An siRNA targeting human PDK4 (sc-39030) and a control siRNA (sc-37007) were purchased from Santa Cruz Biotechnology. JHH7 cells were plated in either 96-well plates (1×104 cells/well) for cell proliferation analysis and RNA isolation or 60-mm dishes (3.5×105 cells/dish) for ATP assays 1 day prior to transfection. The cells were then transfected with 50 nM or 100 nM siRNAs using Lipofectamine 2000 (Life Technologies, Grand Island, NY, USA).
ATP assay
The cellular levels of ATP were measured using a firefly bioluminescence assay kit (AMERIC-ATP kit, Wako Industries) according to the manufacturer's instructions. The luciferase activity was measured using a plate reader (ARVO MX, Perkin Elmer Inc., MA, USA).
Cell viability assay
The number of viable cells was determined using the Cell Counting Kit-8 (Dojindo Molecular Technologies, Tokyo, Japan) as previously described [5].
Network generation and pathway analyses
The Ingenuity Pathways Analysis (IPA) program (Ingenuity Systems, Mountain View, CA, USA; http://www.ingenuity.com) was used to identify networks and canonical pathways as previously described [24]. The generated biological networks were ranked by score, which is the likelihood that a set of genes is found in the networks due to random chance as measured by a Fisher's exact test. The resulting canonical pathways were ranked by P values, which were calculated using a Fisher's exact test by comparing the number of user-specified genes of interest that participate in a given function or pathway, relative to the total number of occurrences of these genes in all the functional/pathway annotations stored in the Ingenuity Pathways Knowledge Base [25].
GEO data mining
The normalized PDK4 expression from a clinical data set, which contains transcriptome profiling of 268 HCC tumor, 243 adjacent non-tumor, 40 cirrhotic and 6 healthy liver samples, was downloaded from the Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo, accession no. GSE25097).
Statistical and multivariate analyses
All the experiments in this study were performed independently two or more times to ensure the reproducibility of the results. Quantitative data were expressed as the means ± SEMs. The statistical significance of differences between values was assessed using a two-tailed Student's t-test or a Mann-Whitney U test. Values of P<0.05 were considered to indicate statistical significance. Unsupervised principal component analysis (PCA) was run using SIMCA-P+ software (Version 12.0, Umetrics, Umeå, Sweden).
Results
The effect of ACR on the metabolism of JHH7 cells detected using 1H-NMR
First, NMR-based metabolomics was performed to investigate the effect of ACR treatment on the metabolism of JHH7 cells. As shown in Figure S1, PCA analysis of the NMR spectra indicated that treatment with ACR for 4 h had a very minor effect on the metabolism of JHH7 cells, while obvious changes were observed after 18 h of ACR treatment compared to the EtOH control.
Differences between the metabolic profiles of JHH7 and Hc cells treated with EtOH and ACR detected using CE-TOFMS
To further investigate the cancer-selective effect of ACR, the metabolic profiles of JHH7 and Hc cells treated with EtOH and ACR for 24 h was measured using CE-TOFMS analysis. A total of 229 peaks (109 cationic and 120 anionic) were detected in either JHH7 or Hc cells; from these 229 peaks, 88 principal metabolites were quantified (Table S2). The metabolic pathways of all the detected metabolites are illustrated in Figure 1. These metabolites are associated with glycolysis/gluconegenesis, the pentose phosphate pathway, the tricarboxylic acid cycle, the urea cycle, pyrimidine metabolism, nicotinate and nicotinamide metabolism and amino acid metabolism. The result of the comparison of the metabolic profiles of the cells is provided in Figure 2. PCA analysis revealed a very clear distinction between the abundance of intracellular metabolites of JHH7 and Hc cells with and without ACR treatment (Figure 2A), while the first component (PC1) indicated that 67% of the total variance is due to the difference between JHH7 and Hc cells. PC2 (11.2%) indicated that the ACR-treated JHH7 cells have a metabolic profile that is similar to that of the EtOH-treated Hc cells. Furthermore, heatmap analyasis indicated that the metabolic pattern of JHH7 cells was almost completely opposite that of the Hc cells; a similar difference was observed between the ACR-treated and EtOH-treated JHH7 cells (Figure 2B). Finally, the cellular content of 71 metabolites in JHH7 and Hc cells was significantly different with P values less than 0.05 and fold changes greater than 1.2; 58 metabolites were significantly down-regulated by ACR in JHH7 cells compared to the EtOH control. Forty-nine common metabolites were shared between the two groups (Figure 2C).
Figure 1. Metabolites in the principle metabolic pathways of EtOH- or ACR-treated JHH7 and Hc cells detected by CE-TOFMS.

The relative quantities of the detected metabolites are represented as bar graphs (from left to right: EtOH-treated JHH7, ACR-treated JHH7, EtOH-treated Hc, and ACR-treated Hc). N.D., not detected.
Figure 2. A comparison of the metabolic profile of EtOH- or ACR-treated JHH7 and Hc cells determined by CE-TOFMS.

PCA scoreplot (A) and heat map (B) from metabolic data of JHH7 and Hc cells treated with EtOH and ACR (n = 3). Venn-diagrams (C) showing the number of metabolites that were significantly deregulated between the two groups.
Network generation and pathway analyses
Next, the list of the significantly different metabolites was imported into the IPA platform to investigate possible biological interactions. The biological functions of the top five IPA-generated networks and top five canonical metabolic pathways are summarized in Tables 1 and 2, respectively, and shown in Figure 3. Interestingly, IPA analysis indicated that the most highly populated biological network (“Increased Levels of Albumin, Amino Acid Metabolism, Molecular Transport”) and the top two canonical metabolic pathways (“tRNA Charging” and “Purine Nucleotides De Novo Biosynthesis II”) that were associated with the ACR-regulated metabolites by in JHH7 cells were the same as the networks that were associated with metabolic differences between JHH7 and Hc cells.
Table 1. Top five associated network functions generated by IPA.
| Top function | Score | |
| EtOH-treated JHH7 vs. Hc | Increased Levels of Albumin, Amino Acid Metabolism, Molecular Transport | 39 |
| Cellular Growth and Proliferation, Organismal Development, Cellular Compromise | 21 | |
| Cardiovascular System Development and Function, Organ Development, Carbohydrate Metabolism | 18 | |
| Cellular Growth and Proliferation, Organismal Development, Small Molecule Biochemistry | 16 | |
| Carbohydrate Metabolism, Cell Morphology, Cell-To-Cell Signaling and Interaction | 11 | |
| ACR vs. EtOH-treated JHH7 | Increased Levels of Albumin, Amino Acid Metabolism, Molecular Transport | 38 |
| Carbohydrate Metabolism, Molecular Transport, Small Molecule Biochemistry | 23 | |
| Cellular Growth and Proliferation, Organismal Development, Small Molecule Biochemistry | 16 | |
| Free Radical Scavenging, Small Molecule Biochemistry, Molecular Transport | 14 | |
| Post-Translational Modification, Cellular Assembly and Organization, Developmental Disorder | 6 |
Table 2. Top canonical pathways identified by IPA.
| Top canonical pathway | P-Value | |
| EtOH-treated JHH7 vs. Hc | tRNA Charging | 6.82E-30 |
| Purine Nucleotides De Novo Biosynthesis II | 1.30E-24 | |
| Pyrimidine Ribonucleotides De Novo Biosynthesis | 9.94E-20 | |
| Superpathway of Citrulline Metabolism | 9.93E-19 | |
| Gluconeogenesis I | 8.21E-18 | |
| ACR vs. EtOH-treated JHH7 | tRNA Charging | 9.48E-30 |
| Purine Nucleotides De Novo Biosynthesis II | 2.91E-19 | |
| Arginine Biosynthesis IV | 3.10E-15 | |
| Citrulline-Nitric Oxide Cycle | 1.84E-14 | |
| NAD biosynthesis II (from tryptophan) | 5.72E-14 |
Figure 3. Network generation using Ingenuity Pathway Analysis.

The “Increased Levels of Albumin, Amino Acid Metabolism, Molecular Transport” network was associated with metabolites that were significantly different between JHH7 and Hc cells (A) and the metabolites that were differentially regulated by ACR in JHH7 cells (B). Up-regulated metabolites are indicated in red, down-regulated metabolites indicated in green, and metabolites that were not annotated in this study but are part of this network are indicated in white. Direct relationships are drawn with solid arrows, and indirect relationships are drawn with dashed arrows.
ACR inhibits the increase in adenosine-5′-triphosphate (ATP) production in JHH7 cells
A comparison of the biosynthetic metabolites (nucleotides, amino acids and lipids) in the EtOH- or ACR-treated JHH7 and Hc cells determined by CE-TOFMS is summarized in Table 3. Of particular interest, the changes in the concentrations of adenosine nucleotides are shown in Figure 4. Notably, ATP levels were 1.6-fold higher in the EtOH-treated JHH7 cells than in the EtOH-treated Hc cells; ACR suppressed this increase, nearly to the basal levels observed in Hc cells (0.72-fold and P = 0.00015 compared to the EtOH-treated JHH7 cells). In contrast, only a very minor effect of ACR was observed on the levels of adenosine diphosphate (ADP) and adenosine monophosphate (AMP) in JHH7 cells (0.84- and 0.82-fold compared to the EtOH control, respectively).
Table 3. Comparison of biosynthetic metabolites in EtOH or ACR-treated JHH7 and Hc cells determined by CE-TOFMS.
| Biosynthetic pathways | Metabolites | KEGG | Fold change | |||
| EtOH-treated JHH7 vs. Hc | ACR vs. EtOH-treated JHH7 | |||||
| Mean | SEM | Mean | SEM | |||
| Nucleotide biosynthesis | AMP | C00020 | 2.50 | 0.10 | 0.82 | 0.00 |
| ATP | C00002 | 1.64 | 0.02 | 0.72 | 0.01 | |
| CTP | C00063 | 1.84 | 0.11 | 0.74 | 0.05 | |
| dATP | C00131 | 1.26 | 0.02 | 0.47 | 0.07 | |
| dCTP | C00458 | 2.74 | 0.06 | 0.69 | 0.07 | |
| dTTP | C00459 | 1.96 | 0.04 | 0.61 | 0.06 | |
| GDP | C00035 | 2.76 | 0.15 | 0.82 | 0.02 | |
| GTP | C00044 | 1.67 | 0.11 | 0.74 | 0.04 | |
| IMP | C00130 | 2.33 | 0.11 | 0.71 | 0.08 | |
| PRPP | C00119 | 0.68 | 0.08 | 0.53 | 0.11 | |
| Ribulose 5-P | C00199 | 0.46 | 0.05 | 0.68 | 0.05 | |
| UDP | C00015 | 2.81 | 0.05 | 0.82 | 0.05 | |
| UTP | C00075 | 1.22 | 0.04 | 0.68 | 0.02 | |
| Amino acid biosynthesis | Ala | C00041 | 9.23 | 0.31 | 0.79 | 0.02 |
| Asp | C00049 | 3.23 | 0.08 | 0.77 | 0.01 | |
| Glu | C00025 | 1.38 | 0.02 | 0.76 | 0.01 | |
| Gly | C00037 | 0.64 | 0.02 | 0.82 | 0.01 | |
| Ile | C00407 | 0.80 | 0.03 | 0.79 | 0.01 | |
| Leu | C00123 | 0.73 | 0.03 | 0.78 | 0.02 | |
| Lys | C00047 | 1.94 | 0.06 | 0.77 | 0.03 | |
| Phe | C00079 | 0.69 | 0.02 | 0.78 | 0.01 | |
| Ser | C00065 | 5.31 | 0.20 | 0.81 | 0.02 | |
| Thr | C00188 | 1.94 | 0.07 | 0.77 | 0.00 | |
| Trp | C00078 | 0.06 | 0.00 | 0.78 | 0.02 | |
| Tyr | C00082 | 0.71 | 0.03 | 0.79 | 0.02 | |
| Val | C00183 | 0.83 | 0.04 | 0.78 | 0.02 | |
| Lipid biosynthesis | 3-Hydroxybutyric acid | C01089 | 1.58 | 0.03 | 0.82 | 0.03 |
| DHAP | C00111 | 0.28 | 0.03 | 0.52 | 0.03 | |
Figure 4. Levels of adenosine nucleotides in EtOH or ACR-treated JHH7 and Hc cells determined by CE-TOFMS.
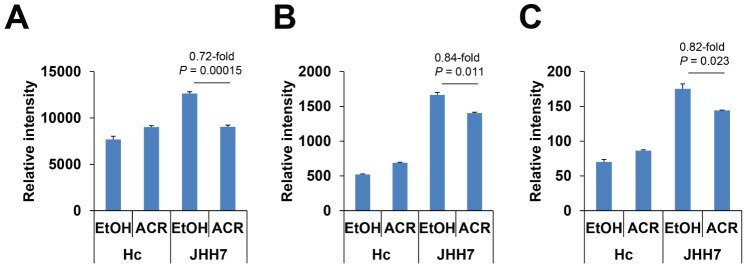
ATP (A), ADP (B) and AMP (C) levels.
ACR enhances PDK4 expression in JHH7 cells, but not in Hc cells
To further understand the cancer-selective inhibitory effect of ACR on ATP production, a set of genes that is known to be important in the regulation of energy metabolism in cancer cells was selected based on previous reports [26], [27], [28], [29], and the effect of ACR on the expression of these genes was measured using real-time PCR (Figure 5A). Of particular interest, we found that ACR significantly enhanced the expression of PDK4, an important regulator of ATP levels [30], in JHH7 cells but not in Hc cells (3.06-fold; P = 0.0033 and 1.20-fold; P = 0.062, respectively; Figure 5B). Further western blot analysis revealed a nearly 2-fold increase in PDK4 protein levels after ACR treatment, but ACR did not affect the phosphorylation of PDHA1 in JHH7 cells (Figure 5C).
Figure 5. ACR increase the expression of PDK4 in JHH7 cells but not Hc cells.

The effect of ACR on the expression of genes related to energy metabolism and ATP production in cancer cells (A). Levels of PDK4 mRNA (B) and levels of PDH4, phospho-PDHA1 and total PDHA1 protein (C) in JHH7 and Hc cells treated with EtOH, AtRA and ACR. The statistical significance of difference was evaluated using the Student's t-test.
Functional analysis of PDK4 in JHH7 cells
Furthermore, loss-of-function experiments were performed to confirm the role of PDK4 in the effect of ACR on cellular ATP levels and the proliferation of JHH7 cells. As shown in Figure 6A, treatment with an siRNA targeting PDK4 (siPDK4) caused a dose-dependent downregulation of PDK4 mRNA expression (0.57-fold and 0.41-fold compared to siControl-treated cells with 50 nM and 100 nM siPDK4, respectively). Interestingly, ACR weakly but significantly inhibited cellular ATP levels in siControl-treated JHH7 cells (0.88-fold and P = 0.042 compared with EtOH). In contrast, no significant effect was observed in siPDK4-treated JHH7 cells (1.07-fold and P = 0.42 compared with EtOH; Figure 6B). However, PDK4 knockdown did not rescue the inhibitory effect of ACR on the proliferation of JHH7 cells (Figure 6C).
Figure 6. Functional analysis of PDK4 in JHH7 cells.
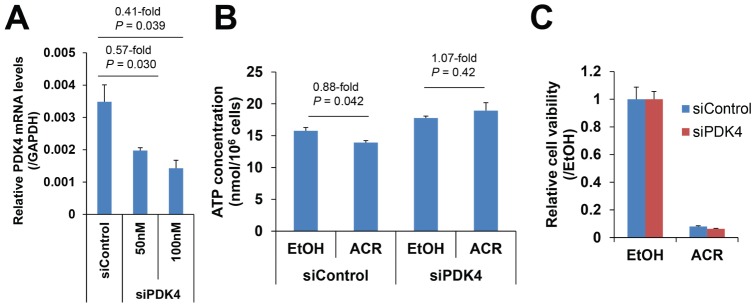
The effect of siPDK4 on PDK4 gene expression (A). The effect of 50 nM siPDK4 on the ACR-mediated cellular ATP levels (B) and the proliferation (C) of JHH7 cells.
Clinical expression levels of PDK4
The mining of microarray data from a human HCC data set revealed that PDK4 mRNA is significantly down-regulated in liver tumors compared to adjacent non-tumor liver tissues (0.66-fold, P = 3.11E-85; Figure 7A). Finally, a PDK4-dependent regulatory network that involves RXR and peroxisome proliferator-activated receptors (PPARs) and summarizes the effects of ACR on ATP production was generated using IPA (Figure 7B).
Figure 7. Clinical expression levels of PDK4.
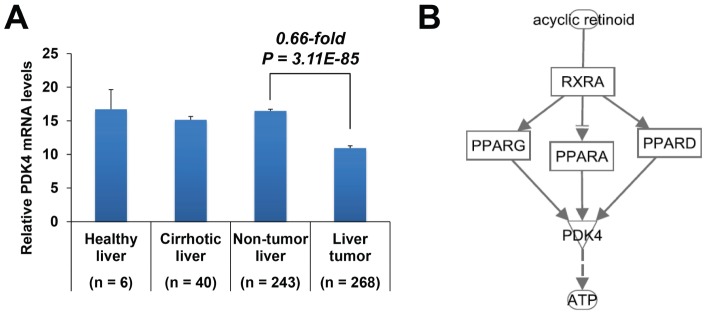
The expression of PDK4 mRNA in human liver cancer (GEO data set GSE25097) (A). Statistical significance was evaluated using the Mann-Whitney U test. A schematic model of the PDK4-denpendent regulatory network of ACR on ATP production in JHH7 cells generated using IPA (B).
Discussion
The war on cancer has continued for more than 40 years, but the gains have been limited. One potential reason for this limited success that typical drug development in oncology has focused on targets that are essential for the survival of all dividing cells, leading to narrow therapeutic windows [31]. Recent advances in metabolite profiling methodologies have generated an alternative window for cancer therapy in targeting cancer metabolism [13]. ACR, a very promising drug that is currently in clinical trials for HCC treatment, has been shown to markedly prevent the recurrence of HCC [3] and selectively inhibit HCC cell growth [5]. We have previously exploited the potential target molecules of ACR that are associated with the promotion of tumor cell proliferation [5] or angiogenesis [32]. In this study, we performed metabolome analyses in JHH7 and Hc cells treated with ACR using both NMR and CE-TOFMS technologies to further understand the molecular pathways that underlie the cancer-selective growth suppressive effect of ACR. We found that ACR selectively suppressed the enhanced nucleotide synthesis and energy metabolism of HCC cells, suggesting that metabolic pathways may be important targets for ACR's anti-cancer activity. The further study of these pathways will benefit the development of more effective cancer drugs and therapies against HCC.
Generally, the metabolic patterns of JHH7 and Hc cells were almost completely opposite to each other (Figure 2), which is consistent with previous reports that cancer cells exhibit considerably different metabolic requirements than most normal differentiated cells and supports the hypothesis that cancer may be a type of metabolic disease [33]. Although the primary cause of cancer is assumed to be at the level of gene expression, metabolites can be considered to be the end products of the cellular regulatory processes that underlie malignant cell growth such as genome instability and mutability [34]. Metabolomic comparison of HCC and control liver tissues have been carried out in several animal and human studies aiming to define metabolomic biomarkers for the early detection of HCC [35], [36]. In this study, the abundance of 71 metabolites was found to be significantly different between JHH7 and Hc cells; 49 of these metabolites were significantly down-regulated by ACR in JHH7 cells (Figure 2 and Table S2). It is not unexpected that an IPA analysis revealed that most of these metabolites are involved in the amino acids and nucleotide biosynthetic pathways, such as “tRNA Charging”, “Purine Nucleotides De Novo Biosynthesis II” and “Pyrimidine Ribonucleotides De Novo Biosynthesis” (Tables 2 and 3). It is well known that cancer cell metabolism must provide a large increase in lipid, protein, and nucleotide synthesis (biomass) to support their uncontrolled high rate of cell growth and proliferation [13]. Our findings indicate that ACR may exert its anti-cancer effect by blocking the biosynthetic processes of cancer cells. Interestingly, a bioinformatics-based anticancer drug screening program in Japan revealed that ACR shares similar anticancer activity pattern with the antipyrimidine drugs doxifluridine and cytarabine and an antipurine drug, 6-mercaptopurine, as assayed by growth inhibition against a panel of 39 human cancer cell lines (JFC39) [37], [38], [39].
ATP is the main energy source for the majority of cellular functions, and impaired cellular energy metabolism is the defining characteristic of nearly all cancers regardless of cellular or tissue origin [33]. Of particular interest, ACR can selectively inhibit the production of ATP in JHH7 cells but not in Hc cells (Figure 4). It has been proven that chemical depletion of ATP can inhibit the growth of HCC cells [40]. This may partially explain the cancer-selective growth suppression effect of ACR. To further understand the molecular signaling mechanisms that underlie this effect, we examined the effect of ACR on the expression of energy production-related genes and observed that the expression of PDK4 was significantly enhanced by ACR in JHH7 cells but not in Hc cells (Figure 5). PDK4 is a key regulator of tricarboxylic acid (TCA) cycle; PDK4 phosphorylates and inactivates the pyruvate dehydrogenase (PDH) complex and thereby switches the energy source for the production of ATP from glucose to fatty acids. Although the cellular pyruvate level was not detected by CE-TOFMS and no effect of ACR was found on the phosphorylation of PDHA1 by western blot analysis (Figure 5C), the knockdown of PDK4 expression using RNA interference in JHH7 cells can rescue the decreased cellular ATP levels induced by ACR (Figure 6B), suggesting that PDK4 may be an important feature of ACR's anti-cancer activity. Moreover, we performed data mining using the GEO database and found that PDK4 expression is significantly down-regulated in liver tumors compared to adjacent non-tumor liver tissues (Figure 7A). The role of PDK4 in cancer therapy is complex; the inhibition of PDK4 is sufficient to inhibit the proliferation of and induce apoptosis in lung cancer cells [41], but the overexpression of PDK4 is also able to decrease ATP levels and suppress de novo lipogenesis and proliferation in breast cancer cells [30]. The specific role of PDK4 in HCC remains to be fully determined. Our results suggest that PDK4 up-regulation has a suppressive effect on HCC. Consistent with this implication, the results of IPA analysis suggest that the cancer-selective, growth-suppressive effect of ACR in inhibiting the ATP production of HCC cells may be related to a putative PDK4-dependent molecular signaling mechanism involving RXR and PPARs, as has been reported in certain fatty acid signaling pathways [42] (Figure 7B).
Although the Warburg effect is a well-recognized hallmark of cancer metabolism, it remains controversial [43]. Warburg hypothesized that tumor cells convert most of their glucose to lactate due to mitochondrial defects. However, subsequent studies showed that most tumor mitochondria are not defective in their ability to carry out oxidative phosphorylation [43]. In fact, we propose that mitochondrial oxidative phosphorylation may be important in supporting HCC cell proliferation based on the following observations: 1) the content of lactate, the major end product of glycolysis, is lower in JHH7 cells than in Hc cells (0.40-fold, Table S2) and 2) ACR up-regulates the expression of PDK4, which attenuates the flux of glycolytic carbon into mitochondrial oxidation and can reduce the production of ATP and inhibit the growth of JHH7 cells.
In summary, our study is the first to investigate the effect of ACR on cancer cell metabolism. A comparison of the metabolic effects of ACR in JHH7 and Hc cells was performed, and a JHH7-selective inhibitory effect of ACR on the production of ATP was observed. The underlying molecular signaling mechanism may relate in part to the cancer-selective enhancement of PDK4 expression, suggesting that mitochondrial oxidative phosphorylation is important in the energy metabolism of HCC cells. However, it should be noted that although PDK4 knockdown can rescue the decreased cellular ATP levels induced by ACR, no effect was observed on the inhibitory effect of ACR on the proliferation of JHH7 cells. Further research is needed to combine the cancer-selective metabolic pathways identified in this study and other signaling pathways to increase our knowledge of ACR's selective anti-cancer activity and to develop more effective cancer drugs and therapies to help us win the war against HCC.
Supporting Information
Statistical analysis of metabolites in JHH7 cells detected by 1H-NMR. PCA score plots of the NMR spectra of JHH7 cells treated with EtOH or 10 μM ACR for 4 h and 18 h (A) or only 18 h (B).
(PPTX)
The primers used in this study.
(XLSX)
Quantification of major metabolites in JHH7 and Hc cells.
(XLSX)
Funding Statement
This study was supported by the Japan Society for the Promotion of Science (JSPS) Postdoctoral Fellowship for Foreign Researchers. This study was also partly supported by the HMT Research Grant for Young Leaders in Metabolomics 2012 from Human Metabolome Technologies Inc. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Venook AP, Papandreou C, Furuse J, de Guevara LL (2010) The incidence and epidemiology of hepatocellular carcinoma: a global and regional perspective. Oncologist 15 Suppl 45–13. [DOI] [PubMed] [Google Scholar]
- 2. Llovet JM, Schwartz M, Mazzaferro V (2005) Resection and liver transplantation for hepatocellular carcinoma. Semin Liver Dis 25: 181–200. [DOI] [PubMed] [Google Scholar]
- 3. Muto Y, Moriwaki H, Ninomiya M, Adachi S, Saito A, et al. (1996) Prevention of second primary tumors by an acyclic retinoid, polyprenoic acid, in patients with hepatocellular carcinoma. Hepatoma Prevention Study Group. N Engl J Med 334: 1561–1567. [DOI] [PubMed] [Google Scholar]
- 4. Muto Y, Moriwaki H, Saito A (1999) Prevention of second primary tumors by an acyclic retinoid in patients with hepatocellular carcinoma. N Engl J Med 340: 1046–1047. [DOI] [PubMed] [Google Scholar]
- 5. Tatsukawa H, Sano T, Fukaya Y, Ishibashi N, Watanabe M, et al. (2011) Dual induction of caspase 3- and transglutaminase-dependent apoptosis by acyclic retinoid in hepatocellular carcinoma cells. Mol Cancer 10: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Obora A, Shiratori Y, Okuno M, Adachi S, Takano Y, et al. (2002) Synergistic induction of apoptosis by acyclic retinoid and interferon-beta in human hepatocellular carcinoma cells. Hepatology 36: 1115–1124. [DOI] [PubMed] [Google Scholar]
- 7. Okada H, Honda M, Campbell JS, Sakai Y, Yamashita T, et al. (2012) Acyclic retinoid targets platelet-derived growth factor signaling in the prevention of hepatic fibrosis and hepatocellular carcinoma development. Cancer Res 72: 4459–4471. [DOI] [PubMed] [Google Scholar]
- 8. Shao RX, Otsuka M, Kato N, Taniguchi H, Hoshida Y, et al. (2005) Acyclic retinoid inhibits human hepatoma cell growth by suppressing fibroblast growth factor-mediated signaling pathways. Gastroenterology 128: 86–95. [DOI] [PubMed] [Google Scholar]
- 9. Kagawa M, Sano T, Ishibashi N, Hashimoto M, Okuno M, et al. (2004) An acyclic retinoid, NIK-333, inhibits N-diethylnitrosamine-induced rat hepatocarcinogenesis through suppression of TGF-alpha expression and cell proliferation. Carcinogenesis 25: 979–985. [DOI] [PubMed] [Google Scholar]
- 10. Matsushima-Nishiwaki R, Okuno M, Takano Y, Kojima S, Friedman SL, et al. (2003) Molecular mechanism for growth suppression of human hepatocellular carcinoma cells by acyclic retinoid. Carcinogenesis 24: 1353–1359. [DOI] [PubMed] [Google Scholar]
- 11. Suzui M, Masuda M, Lim JT, Albanese C, Pestell RG, et al. (2002) Growth inhibition of human hepatoma cells by acyclic retinoid is associated with induction of p21(CIP1) and inhibition of expression of cyclin D1. Cancer Res 62: 3997–4006. [PubMed] [Google Scholar]
- 12. Matsushima-Nishiwaki R, Okuno M, Adachi S, Sano T, Akita K, et al. (2001) Phosphorylation of retinoid X receptor alpha at serine 260 impairs its metabolism and function in human hepatocellular carcinoma. Cancer Res 61: 7675–7682. [PubMed] [Google Scholar]
- 13. Vander Heiden MG (2011) Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov 10: 671–684. [DOI] [PubMed] [Google Scholar]
- 14. Koppenol WH, Bounds PL, Dang CV (2011) Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer 11: 325–337. [DOI] [PubMed] [Google Scholar]
- 15. Butler EB, Zhao Y, Munoz-Pinedo C, Lu J, Tan M (2013) Stalling the engine of resistance: targeting cancer metabolism to overcome therapeutic resistance. Cancer Res 73: 2709–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.El-Serag HB (2012) Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 142: 1264–1273 e1261. [DOI] [PMC free article] [PubMed]
- 17. Welzel TM, Graubard BI, Zeuzem S, El-Serag HB, Davila JA, et al. (2011) Metabolic syndrome increases the risk of primary liver cancer in the United States: a study in the SEER-Medicare database. Hepatology 54: 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fujise K, Nagamori S, Hasumura S, Homma S, Sujino H, et al. (1990) Integration of hepatitis B virus DNA into cells of six established human hepatocellular carcinoma cell lines. Hepatogastroenterology 37: 457–460. [PubMed] [Google Scholar]
- 19. Qin XY, Wei F, Yoshinaga J, Yonemoto J, Tanokura M, et al. (2011) siRNA-mediated knockdown of aryl hydrocarbon receptor nuclear translocator 2 affects hypoxia-inducible factor-1 regulatory signaling and metabolism in human breast cancer cells. FEBS Lett 585: 3310–3315. [DOI] [PubMed] [Google Scholar]
- 20. Wei F, Furihata K, Hu F, Miyakawa T, Tanokura M (2010) Complex mixture analysis of organic compounds in green coffee bean extract by two-dimensional NMR spectroscopy. Magn Reson Chem 48: 857–865. [DOI] [PubMed] [Google Scholar]
- 21. Ohashi Y, Hirayama A, Ishikawa T, Nakamura S, Shimizu K, et al. (2008) Depiction of metabolome changes in histidine-starved Escherichia coli by CE-TOFMS. Mol Biosyst 4: 135–147. [DOI] [PubMed] [Google Scholar]
- 22. Sugimoto M, Hirayama A, Robert M, Abe S, Soga T, et al. (2010) Prediction of metabolite identity from accurate mass, migration time prediction and isotopic pattern information in CE-TOFMS data. Electrophoresis 31: 2311–2318. [DOI] [PubMed] [Google Scholar]
- 23. Qin XY, Akanuma H, Wei F, Nagano R, Zeng Q, et al. (2012) Effect of low-dose thalidomide on dopaminergic neuronal differentiation of human neural progenitor cells: a combined study of metabolomics and morphological analysis. Neurotoxicology 33: 1375–1380. [DOI] [PubMed] [Google Scholar]
- 24. Qin XY, Kojima Y, Mizuno K, Ueoka K, Muroya K, et al. (2012) Identification of novel low-dose bisphenol a targets in human foreskin fibroblast cells derived from hypospadias patients. PLoS One 7: e36711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bronner IF, Bochdanovits Z, Rizzu P, Kamphorst W, Ravid R, et al. (2009) Comprehensive mRNA expression profiling distinguishes tauopathies and identifies shared molecular pathways. PLoS One 4: e6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vander Heiden MG, Lunt SY, Dayton TL, Fiske BP, Israelsen WJ, et al. (2011) Metabolic pathway alterations that support cell proliferation. Cold Spring Harb Symp Quant Biol 76: 325–334. [DOI] [PubMed] [Google Scholar]
- 27. Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Levine AJ, Puzio-Kuter AM (2010) The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 330: 1340–1344. [DOI] [PubMed] [Google Scholar]
- 29. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7: 11–20. [DOI] [PubMed] [Google Scholar]
- 30. Grassian AR, Metallo CM, Coloff JL, Stephanopoulos G, Brugge JS (2011) Erk regulation of pyruvate dehydrogenase flux through PDK4 modulates cell proliferation. Genes Dev 25: 1716–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Michelakis ED, Webster L, Mackey JR (2008) Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 99: 989–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Komi Y, Sogabe Y, Ishibashi N, Sato Y, Moriwaki H, et al. (2010) Acyclic retinoid inhibits angiogenesis by suppressing the MAPK pathway. Lab Invest 90: 52–60. [DOI] [PubMed] [Google Scholar]
- 33. Seyfried TN, Shelton LM (2010) Cancer as a metabolic disease. Nutr Metab (Lond) 7: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646–674. [DOI] [PubMed] [Google Scholar]
- 35. Beyoglu D, Imbeaud S, Maurhofer O, Bioulac-Sage P, Zucman-Rossi J, et al. (2013) Tissue metabolomics of hepatocellular carcinoma: Tumor energy metabolism and the role of transcriptomic classification. Hepatology 58: 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang J, Zhang S, Li Z, Yang J, Huang C, et al. (2011) (1)H-NMR-based metabolomics of tumor tissue for the metabolic characterization of rat hepatocellular carcinoma formation and metastasis. Tumour Biol 32: 223–231. [DOI] [PubMed] [Google Scholar]
- 37. Yamori T (2004) [Chemical evaluation by cancer cell line panel and its role in molecular target-based anticancer drug screening]. Gan To Kagaku Ryoho 31: 485–490. [PubMed] [Google Scholar]
- 38. Results of molecular target antineoplastic agents screening. Gan To Kagaku Ryoho 31 Suppl 11–150. [PubMed] [Google Scholar]
- 39. Nakatsu N, Nakamura T, Yamazaki K, Sadahiro S, Makuuchi H, et al. (2007) Evaluation of action mechanisms of toxic chemicals using JFCR39, a panel of human cancer cell lines. Mol Pharmacol 72: 1171–1180. [DOI] [PubMed] [Google Scholar]
- 40.Dilip A, Cheng G, Joseph J, Kunnimalaiyaan S, Kalyanaraman B, et al.. (2013) Mitochondria-targeted antioxidant and glycolysis inhibition: synergistic therapy in hepatocellular carcinoma. Anticancer Drugs. [DOI] [PMC free article] [PubMed]
- 41. Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, et al. (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11: 37–51. [DOI] [PubMed] [Google Scholar]
- 42. Schoonjans K, Staels B, Auwerx J (1996) Role of the peroxisome proliferator-activated receptor (PPAR) in mediating the effects of fibrates and fatty acids on gene expression. J Lipid Res 37: 907–925. [PubMed] [Google Scholar]
- 43. Ward PS, Thompson CB (2012) Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 21: 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Statistical analysis of metabolites in JHH7 cells detected by 1H-NMR. PCA score plots of the NMR spectra of JHH7 cells treated with EtOH or 10 μM ACR for 4 h and 18 h (A) or only 18 h (B).
(PPTX)
The primers used in this study.
(XLSX)
Quantification of major metabolites in JHH7 and Hc cells.
(XLSX)