

Abstract
Polycystic kidney disease (PKD) is a leading cause of ESRD worldwide. In PKD, excessive cell proliferation and fluid secretion, pathogenic interactions of mutated epithelial cells with an abnormal extracellular matrix and alternatively activated interstitial macrophages, and the disruption of mechanisms controlling tubular diameter contribute to cyst formation. Studies with animal models suggest that several diverse pathophysiologic mechanisms, including dysregulation of intracellular calcium levels and cAMP signaling, mediate these cystogenic mechanisms. This article reviews the evidence implicating calcium and cAMP as central players in a network of signaling pathways underlying the pathogenesis of PKD and considers the therapeutic relevance of treatment strategies targeting cAMP signaling.
Autosomal dominant polycystic kidney disease (ADPKD), the fourth leading cause of end stage kidney disease in adults, is caused by mutations in either of two genes: PKD1 (encoding polycystin-1) or PKD2 (encoding polycystin-2). Autosomal recessive polycystic kidney disease (ARPKD), an important cause of ESRD and mortality in infants and children, is caused by mutations in PKHD1 (encoding fibrocystin). In both diseases, disruption of mechanisms controlling tubular diameter, excessive cell proliferation and fluid secretion, and pathogenic interactions of mutated epithelial cells with an abnormal extracellular matrix and alternatively activated interstitial macrophages contribute to cyst formation. Numerous therapies targeting diverse, seemingly unconnected pathophysiologic mechanisms have been successful in animal models of polycystic kidney disease (PKD). This article presents a pathogenic view of PKD where calcium and cAMP play a central role in a network of signaling pathways, without denying the importance of additional pathogenic mechanisms (Figure 1), and provides an update on treatment strategies targeting cAMP signaling.
Figure 1.
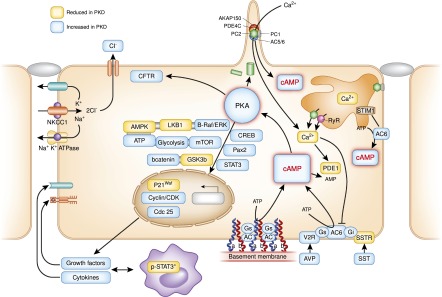
Diagram depicting putative pathways up- or downregulated in PKD. Dysregulation of [Ca2+]i and increased concentrations of cAMP play a central role. Increased accumulation of cAMP in polycystic kidneys may be explained the following hypotheses. (1) Reduced calcium activates calcium-inhibitable AC6, directly inhibits calcium/calmodulin-dependent PDE1 (by also increasing the levels of cGMP), and indirectly inhibits cGMP-inhibitable PDE3. (2) Dysfunction occurs in a ciliary protein complex (comprising A-kinase anchoring protein 150, AC5/6, polycystin-2, PDE4C, and PKA), which normally restrains cAMP signaling through inhibition of AC5/6 activity by polycystin-2–mediated calcium entry and degradation of cAMP by PDE4C transcriptionally controlled by HNF1β. (3) Depletion of the endoplasmic reticulum calcium stores trigger oligomerization and translocation of STIM1 to the plasma membrane, where it recruits and activates AC6. (4) Other contributory factors include disruption of PC1 binding to heterotrimeric G proteins, upregulation of the vasopressin V2 receptor, and increased levels of circulating vasopressin or accumulation of forskolin, lisophosphatidic acid, ATP, or other adenylyl cyclase agonists in the cyst fluid. Increased cAMP levels disrupt tubulogenesis, stimulate chloride and fluid secretion, and activate proproliferative signaling pathways, including mitogen-activated protein kinase/extracellularly-regulated kinase (in an Src- and Ras-dependent manner), mTOR, and β-catenin signaling. Activated mTOR transcriptionally stimulates aerobic glycolysis, increasing ATP synthesis and lowering AMP levels, which together with B-Raf–dependent activation of LKB1, inhibits AMPK, further enhancing mTOR activity and CFTR-driven chloride and fluid secretion. PKA signaling also activates a number of transcription factors, including STAT3. Activated STAT3 induces the transcription of cytokines, chemokines, and growth factors that, in turn, activates STAT3 on interstitial alternatively activated (M2) macrophages, which results in a feedforward loop between cyst-lining cells and M2 macrophages. Aberrant integrin–extracellular membrane interaction and cAMP signaling within focal adhesion complexes may contribute to the increased adhesion of cyst-derived cells to laminin-322 and collagen. AC-VI, adenylate cyclase 6; AMPK, AMP kinase; AVP, arginine vasopressin; B-Raf, B rapidly accelerated fibrosarcoma kinase; CDK, cyclin-dependent kinase; cGMP, cyclic guanosine monophosphate; CREB, cAMP response element binding transcription factor; ER, endoplasmic reticulum; GSK3β, glycogen synthase kinase 3β; LKB1, liver kinase B1; MAPK, mitogen-activated protein kinase; Pax2, paired box gene 2; PC1, polycystin-1; PC2, polycystin-2; SST, somatostatin; SSTR, somatostatin receptor; STIM1, stromal interaction molecule 1.
Disruption of Intracellular Calcium Homeostasis and PKD
Polycystin-1, polycystin-2, and fibrocystin, localized in the primary cilium, are required for induction of calcium transients in response to ciliary bending.1–3 The three proteins are found at low levels in tubular epithelial cells, but are abundant in urinary exosomes.4,5 Polycystin-1 resembles a receptor or adhesion protein and is also found at the plasma membrane and, possibly, in the endoplasmic reticulum.6–8 Polycystin-1 interacts with the inositol 1,4,5-trisphosphate receptor (IP3R).9,10 Polycystin-2 is a transient receptor potential (TRP) channel that is mainly located in the endoplasmic reticulum, where it functions as a calcium release channel, and, possibly, located in the plasma membrane.11,12 Polycystin-1 and fibrocystin interact with, and modulate the function of, polycystin-2.13–15 Polycystin-2 also interacts with other calcium channel proteins (IP3R, ryanodine receptor, TRPC1, TRPC4, and TRPV4).16–21 Precisely how intracellular calcium homeostasis is altered in ADPKD remains uncertain, because different experimental conditions have led to inconsistent results.22 For example, experiments in which polycystin-1 is knocked down conclude that polycystin-1 amplifies IP3-induced calcium release,23 whereas studies using heterologous overexpression of polycystin-1 reach the opposite conclusion.9,10 Nevertheless, most studies that have measured resting intracellular calcium, endoplasmic reticulum calcium stores, and store-operated calcium entry in primary cell cultures or microdissected samples from human and rodent polycystic tissues have found them to be reduced (Table 1).17,23–31
Table 1.
Intracellular calcium homeostasis in PKD microdissected tissues or cultured cells
| Author (Year) | Cells | Comparison | Resting [Ca2+]i | ER (SR) Ca2+ Store | CCE | Flow-Induced Ca2+ Entry |
|---|---|---|---|---|---|---|
| Nauli et al. (2003)1 | E15.5 kidney cells | Pkd1del34/del34 versus WT | ↔ | ↓ | ||
| Qian et al. (2003)24 | Aortic VSMC | Pkd2+/− versus WT | ↓ | ↓ | ↓ | |
| Nauli et al. (2006)2 | Cultured kidney cells | Pkd1−/− versus +/+ | ↔ | ↓ | ||
| Immortalized kidney cells | PKD1c versus WT | ↔ | ↓ | |||
| Yamaguchi et al. (2006)25 | Cultured kidney cells | ADPKD versus NHK | ↓ | |||
| ARPKD versus NHK | ↓ | |||||
| Geng et al. (2008)26 | Cultured kidney cells | Pkd1−/− versus Pkd1+/− | ↓ | |||
| Xu et al. (2007)27 | Cultured kidney cells | PKD1 versus NHK | ↔ | ↓ | ↓ | ↓ |
| Ahrabi et al. (2007)28 | Isolated collecting ducts | Pkd1+/− versus WT | ↓ | |||
| Anyatonwu et al. (2007)17 | E17.5 cardiac myocytes | Pkd2−/− versus WT | ↓ | ↓ | ||
| Morel et al. (2009)23 | Aortic VSMC | Pkd1+/− versus WT | ↓ | |||
| Banales et al. (2009)29 | Cholangiocytes | PCK versus WT | ↓ | |||
| Spirli et al. (2012)30 | Cholangiocytes | Pkd2−/− versus WT | ↓ | ↓ | ↓ | |
| Zaika et al. (2013)31 | Microdissected cysts and split-open CDs | PCK versus WT | ↓ | ↓ |
ER, endoplasmic reticulum; SR, sarcoplasmic reticulum; CCE, capacitive calcium entry; WT, wild-type; VSMC, vascular smooth muscle cell; NHK, normal human kidney; CD, collecting duct.
Increased cAMP Levels in Cystic Tissues
Tissue levels of cAMP are increased in numerous animal models of PKD not only in the kidney32–36 but also in cholangiocytes,37 vascular smooth muscle cells,38 and choroid plexus.39 Tissue levels of cAMP are determined by the activities of membrane-bound (under the positive or negative control of G protein–coupled receptors [GPCRs] and extracellular ligands) and soluble adenylyl cyclases (ACs) and cAMP phosphodiesterases (PDEs), which are also subject to complex regulatory mechanisms.
Several hypotheses may explain the increased levels of cAMP (Figure 1). (1) Reduced calcium activates calcium-inhibitable AC6, directly inhibits calcium/calmodulin-dependent PDE1 (also increasing the levels of cyclic guanosine monophosphate), and indirectly inhibits cyclic guanosine monophosphate–inhibitable PDE3.33,40 (2) Dysfunction occurs in a ciliary protein complex (comprising A-kinase anchoring protein 150, AC5/6, polycystin-2, PDE4C, and protein kinase A [PKA]), where polycystin-2–mediated calcium entry inhibits AC5/6 and activates PDE4C.41 (3) Depletion of the endoplasmic reticulum calcium stores trigger oligomerization and translocation of stromal interaction molecule 1 to the plasma membrane, where it recruits and activates AC6.30 (4) Other contributory factors include disruption of PC1 binding to heterotrimeric G proteins, upregulation of the vasopressin V2 receptor, and increased levels of circulating vasopressin or accumulation of forskolin, lisophosphatidic acid, ATP, or other adenylyl cyclase agonists in the cyst fluid.42–46 The marked amelioration of the cystic disease in collecting duct-specific Pkd1 knockout mice by a concomitant AC6 knockout provides strong support to the central role of calcium-inhibitable AC6.47
PDEs are likely important in PKD, because maximal rates of degradation by PDEs exceed by one order of magnitude those rates of synthesis by ACs and hence, control compartmentalized pools of cAMP that are likely more crucial than total intracellular cAMP. Preliminary studies, showing induction of pronephric cysts in pkd1a zebrafish morphants48 and aggravation of the cystic disease in Pkd2WS25/− by depletion of Pde3a, 49 point to the importance of these PDE isoforms in PKD. PDE3 controls a compartmentalized cAMP pool that stimulates mitogenesis in Madin-Darby canine kidney (MDCK) cells.50 A small-molecule, nonselective PDE activator lowers cAMP and inhibits the growth of MDCK cysts.51
Epithelial Tubulogenesis and Regulation of Tubular Diameter
Epithelial tubulogenesis requires canonical Wnt/β-catenin signaling at early inductive stages and noncanonical Wnt/planar cell polarity (PCP) signaling later.52 Primary cilia control the switch from canonical to noncanonical Wnt signaling. Convergent extension (process of cell intercalation by which cells elongate along an axis perpendicular to the proximal–distal axis of the tubule by actively crawling between one another to produce a narrower, longer tubule) and oriented cell division (alignment of the mitotic spindle axis and cell division with the proximal–distal axis of the tubule) establish and maintain the normal tubular diameter under the control of PCP signaling. Sustained activation of canonical Wnt signaling,53,54 downregulation of noncanonical/PCP signaling,55 and disruption of ciliogenesis or knockout of the ciliary protein inversin56 are all associated with cystic disease.
Enhanced cAMP and PKA signaling disrupts tubulogenesis. PKA is known to enhance Wnt/β-catenin signaling through phosphorylation of glycogen synthase kinase 3β (stabilizing β-catenin)57 and phosphorylation of β-catenin (promoting its transcriptional activity) (Figure 1).58 Sustained PKA-dependent canonical Wnt signaling blocks a postepithelialization morphogenetic step (conversion of the renal vesicle to the S-shaped body) in spinal cord-induced metanephric mesenchyme, resulting in disorganized epithelial clusters and large dilations.59 Overexpression of constitutively active PKA catalytic subunits can also act as a negative regulator of PCP signaling and block convergent extension during Xenopus gastrulation.60 Thus, it is conceivable that hyperactivation of cAMP/PKA signaling may interfere with the control of tubular diameter in PKD.
Cell Proliferation
Increased cell proliferation is necessary, but not sufficient, for cystogenesis. Analysis of conditional Pkd1 models has shown that the developmental and proliferative activity of the tubular epithelial cells at the time of Pkd1 inactivation determines the rate of development and severity of the cystic disease.61 The rate of epithelial cell proliferation may also account for cyst migration in humans and animal models of PKD as the kidney develops and matures from predominately proximal to predominantly distal and collecting duct.36 Immature early tubules (S-shaped bodies) exhibit very high rates of proliferation.62 Later, when the epithelium differentiates into nephron segments recognizable by light microscopy, proliferative indices become very low in the proximal tubule but remain elevated in the distal nephron and collecting duct. In pediatric and adult kidneys, proliferative indices are very low in all tubular segments but remain higher in collecting ducts compared with those indices in proximal tubules.63
cAMP exerts opposite effects on cell proliferation in different cell types. cAMP and PKA signaling enhances several proproliferative pathways (extracellular signal-regulated kinase [ERK]) in cells derived from polycystic kidneys while inhibiting proliferation in cells derived from normal human kidney cortex.64,65 Treatment of normal human kidney or murine collecting duct cells with calcium channel blockers (gadolinium, 25 μm; nifedipine, 0.1 μm; or verapamil, 1 μm) replicates the proliferative response of the ADPKD cells to cAMP, thus linking this response to the reduction in intracellular calcium that results from disrupting the polycystin pathway.66 Conversely, treatment of ADPKD or ARPKD cyst-derived cells with calcium channel activators or calcium ionophores restores the normal antimitogenic response to cAMP. Inhibition of calcium-dependent phosphatidylinositol 3-kinase activity with downstream inhibition of Akt, allowing B rapidly accelerated fibrosarcoma (B-Raf) and ERK to be activated in a PKA-, Src-, and Ras-dependent manner, has been proposed to underlie the proliferative response to cAMP in the setting of reduced calcium (Figure 1).66 Activation of mammalian target of rapamycin (mTOR) signaling downstream from PKA through ERK-mediated phosphorylation of tuberin67,68 has been linked to transcriptional activation of aerobic glycolysis, increased levels of ATP, and together with ERK-dependent inhibition of liver kinase B1, inhibition of AMP kinase,69–71 which may further enhance mTOR signaling.72 Phosphorylation and inhibition of glycogen synthase kinase 3β57 and direct phosphorylation and stabilization of β-catenin by PKA58 enhance Wnt/β-catenin signaling. PKA-dependent upregulation of cAMP response element binding transcription factor,73 paired box gene 2 (Pax2),74 and signal transducer and activator of transcription 3 (STAT3)75–77 also contributes to the proliferative phenotype of the cystic epithelium. Pax2 haploinsufficiency inhibits renal cystogenesis in homozygous Pkd1 mutant or cpk mutant mice.78,79 The knockout of SOCS-1, a negative regulator of STAT3 expression, induces a cystic disease phenotype.80
Although the proliferative effect of cAMP on the renal cystic epithelium seems to be exclusively PKA-mediated,64 both PKA and exchange protein activated by cAMP (Epac) activations enhance cholangiocyte proliferation and hepatic cystogenesis the mitogen ERK kinase (MEK)/ERK1/2 pathway in PCK rats.29
Fluid Secretion
By the time that the renal cysts reach a diameter of approximately 2 mm, they become disconnected from the tubular segment from which they derived.81 Additional growth depends on transepithelial fluid secretion. A study in 1977 using tritiated water shows a high turnover of cyst fluid in ADPKD (>100 ml in 24 hours for a 10-ml cyst) thought to be secondary to active solute transport and therefore, susceptible to pharmacotherapy.82 Studies using polarized normal human kidney and ADPKD cells on permeable supports and microcysts in hydrated collagen confirm a model of fluid transport as previously shown in MDCK cells and other secretory epithelia.83 Intact cysts excised from ADPKD kidneys secrete fluid when treated with forskolin, confirming that intact cyst epithelia secrete fluid by mechanisms regulated by cAMP.84 The driving force is active transport of chloride from the basolateral to the apical side. The energy is generated by the sodium pump (NaKATPase) in the basolateral membrane of cyst epithelial cells (Figure 1). Chloride enters from the basolateral side through the sodium-potassium-chloride cotransporter, which uses the gradient established by the sodium pump to bring potassium and chloride into the cells.85 PKA-induced phosphorylation of the cystic fibrosis transmembrane conductance regulator (CFTR) in the apical membrane opens the channel and allows the flow of chloride ions down an electrochemical gradient into the cyst, generating increased transepithelial electron activity that, in turn, drives sodium ions through paracellular pathways. The role of CFTR in PKD is supported by whole-cell patch-clamp studies of cyst-derived epithelial cells, inhibition of fluid secretion by CFTR antisense oligonucleotides, inhibition of cyst growth in in vitro and in vivo models of cystogenesis,86–90 and the milder cystic phenotype in patients affected by both ADPKD and cystic fibrosis.91–94 However, an additional patient with coexisting ADPKD and cystic fibrosis progressed to ESRD after a lung transplant, possibly because of cyclosporin toxicity, and the disease severity of bpk CFTR double mutant mice was not less than the disease severity of bpk mice, a rapidly progressive model of PKD.95,96
Cell, Basement Membrane, and Extracellular Matrix Interactions
Evidence indicates that alterations in focal adhesion complexes, basement membranes, and extracellular matrix contribute to the pathogenesis of PKD.97 Focal adhesion complexes contain integrin αβ heterodimer receptors, which link the actin cytoskeleton to the basement membrane laminin αβγ heterotrimers and collagens and multiple structural and signaling molecules, including polycystin-1. Laminin binding integrins (α3β1, α6β1, and α6β4) are expressed in the branching ureteric bud, whereas both laminin binding and collagen binding (α1β1 and α2β1) integrins are expressed in the differentiated collecting duct.98,99 Their ligands laminin-511 (α5β1γ1 or laminin 10) and laminin-332 (α3β3γ2 or laminin-5) are also expressed in the ureteric bud and the differentiated collecting duct.98,99 Integrin β4 and -β1 are overexpressed in cyst cells and may mediate their increased adhesion to laminin-322 and collagen, respectively, which are also overexpressed.97,100,101 Periostin, an extracellular matrix protein and its receptor αv integrin,102 as well as α1 and α2 integrins are also overexpressed in cystic tissues.97,103 Laminin-322 and periostin stimulate, whereas antibodies to laminin-332 and αv integrin inhibit cyst formation in three-dimensional gel culture.101,102,104 β1 integrin knockout and laminin α5 hypomorphic mutant mice develop renal cystic disease, the latter associated with overexpression of laminin-322.105,106
The finding that cAMP signaling plays a role in the altered cell basement membrane interactions in PKD is a sound but untested hypothesis. Integrin binding to specific ligands induces clustering of integrin heterodimers and formation of a multiprotein complex, which includes G proteins.107 Although integrins are not conventional GPCRs, they interact with heterotrimeric G proteins through noncanonical pathways to activate or inhibit cAMP signaling in an extracellular matrix- and cell type–specific manner (Figure 1).108,109 Integrins may also regulate the activity of PDEs in compartmentalized cAMP pools.110 Polycystin-1 and possibly, polycystin-2 could function in the focal adhesion complexes to regulate these cAMP pools, which is described in other subcellular compartment like the cilia. In turn, PKA and Epac signaling, downstream from cAMP, modulate integrin-mediated cell adhesion and adhesion-associated events, such as actin cytoskeletal dynamics and migration, which has described in other cells.107 For example, PKA and/or Epac promote adhesion of human vascular endothelial and vascular smooth muscle cells to fibronectin in an integrin-specific manner.111,112 It is, therefore, conceivable that increased cAMP signaling contributes to the increased adhesion to laminin-322 and collagen exhibited by cyst-derived cells.
Interstitial Inflammation and Fibrosis
Almost three decades ago, the observation was made that a germ-free environment inhibits cyst development in CFWwd mice.113 This observation was confirmed in a chemical model of PKD induced by nordihydroguaiaretic acid.114 Administration of endotoxin rescued the cystic phenotype, which included marked inflammatory cell infiltrates in close opposition to the cysts. Chemokines and cytokines were found at high concentrations in cyst fluids and were found to be produced by cyst-lining epithelial cells.115 More recently, many alternatively activated macrophages aligned along cyst walls have been detected in polycystic kidneys from conditional Pkd1-knockout and Pkd2WS25/− mice.116,117 Macrophage depletion by intraperitoneal liposomal clodronate administration inhibits epithelial cell proliferation and cyst growth and improves renal function. These observations have led to the hypothesis that alternatively activated M2 macrophages contribute to cell proliferation in PKD, as has been described in cancer and during the recovery from AKI.116,117 After renal ischemia-reperfusion, macrophages infiltrate the kidney and undergo a phenotypic switch from classically activated proinflammatory M1 to alternatively activated M2 macrophages that promote tubular epithelial cell proliferation, tissue remodeling, and fibrogenesis.118–120
STAT3 activation in PKD, as previously described in cancer, plays a critical role in the development and maintenance of an inflammatory microenvironment.121–123 In cancer cells and likely, cyst-lining cells,75–77 activated STAT3 induces the transcription of cytokines, chemokines, and growth factors that, in turn, activate STAT3 on tumor-associated or M2 macrophages, which results in feed-forward loop between tumor or cyst-lining cells and tumor-associated or M2 macrophages (Figure 1). The trigger for the initial STAT3 activation in PKD is unclear. STAT3 is typically activated through phosphorylation at tyrosine 705 by intrinsic tyrosine kinase activity of activated growth factor receptors or cytokine receptor-associated Janus kinase, but GαsPCR/adenylyl cyclase/cAMP/PKA signaling can also activate STAT3.124–127 For example, G-protein α-subunit–activating mutations in sporadic benign and malignant liver tumors are characterized by an inflammatory phenotype associated with STAT3 activation.126 The vasopressin V2 receptor agonist 1-deamino-8-d-arginine vasopressin increases, whereas absence of circulating vasopressin and the V2 receptor antagonist OPC-31260 inhibits STAT3 activation and cystogenesis in rodent models of PKD (J. Talbott and T. Weimbs, personal communication and unpublished observations). Therefore, upregulation of cAMP signaling may contribute to STAT3 activation and interstitial inflammation in PKD.
Treatment Strategies Targeting cAMP
The central role of cAMP in the pathogenesis of PKD provides a strong rationale for strategies to lower its levels in cystic tissues. Clinical trials of GαsPCR antagonists (i.e., vasopressin V2 receptor antagonists) and GiPCR agonists (i.e., somatostatin analogs) have shown encouraging results.
V2 Receptor Antagonists: Rationale and Preclinical Trials
Vasopressin acting on V2 receptors is the most powerful agonist for cAMP generation in freshly isolated collecting ducts.128 Nearly exclusive localization of V2 receptor (V2R) on collecting ducts, connecting tubules, and thick ascending limbs of Henle,129,130 the main sites of cystogenesis,131 minimizes off-target size effects and improves tolerability. Vasopressin is continuously present in the circulation, likely at a higher level in PKD to compensate for a urinary concentrating defect.132
V2 receptor antagonists (mozavaptan and/or tolvaptan) attenuate the progression of PKD in cpk mice and rodent models of nephronophthisis (pcy mouse), ARPKD (PCK rat), and PKD-2 (Pkd2WS25/− mouse).33,133–135 Mozavaptan is also effective in a conditional Pkd1 knockout when treatment is started early after gene deletion.136 Cyst development is markedly inhibited in PCK rats lacking circulating vasopressin (generated by crosses of PCK and Brattleboro rats), whereas administration of the V2R agonist 1-deamino-8-d-arginine vasopressin fully rescues the cystic phenotype.137 Satavaptan (V2R antagonist) blocks tubular expression of secreted frizzled related protein 4 that is overexpressed in polycystic kidneys and promotes cystogenesis of zebrafish pronephros.138 Tolvaptan also inhibits vasopressin-induced cell proliferation, chloride secretion, and in vitro cyst growth of human ADPKD cells.139
Clinical Trials of V2R Antagonists in ADPKD
Preliminary dosing studies showed that two times per day administration of tolvaptan was necessary to maintain urine hypotonicity (a surrogate for V2R blockade) throughout a 24-hour period.140–142 Two phase 2, open-label, uncontrolled, 3-year clinical trials ascertained its long-term safety and tolerability.143 Forty-six patients in the United States were randomized to one of two daily split doses (45/15 and 60/30 mg) of tolvaptan chosen after an analysis of efficacy (Uosm persistently <300 mOsm/kg in 70% and 77% of patients, respectively) and self-reported tolerability (for rest of life in 96% and 61% of patients, respectively) during a dose escalation phase (Figure 2). Seventeen patients in Japan received a 15/15-mg split dose. Adverse events were mainly related to the aquaretic effect; 12 of 63 (19%) patients withdrew from the study, and, in six cases, the withdrawal was because of adverse events. Changes in kidney volume (determined by magnetic resonance imaging [MRI] and estimated GFR) were compared with historical controls from the Consortium of Radiologic Imaging Study of PKD and the Modification of Diet in Renal Disease studies. Kidney volume increased 5.8% versus 1.7% per year. Annualized estimated GFR declined −2.1 versus −0.71 ml/min per 1.73 m2 per year. Limitations of the study were the small number of patients and the use of noncontemporary controls with unmatched ethnicities.
Figure 2.

Tolerability and efficacy during the titration phase of TEMPO2:4. In the initial 2 months of the TEMPO2:4 study, a split-dose regimen of oral tolvaptan (8:00 AM/4:00 PM) was uptitrated (15/15, 30/15, 45/15, 60/30, and 90/30 mg/d) until tolerability was reached. Tolerability was defined as self-reported tolerance of a specific dose regimen by responding yes to the question: “Could you tolerate taking this dose of tolvaptan for the rest of your life?” Efficacy was defined by the capacity to suppress the action of vasopressin on the kidney reflected by sustained urine hypotonicity (Uosm<300 mOsm/kg). Reprinted from Higashihara et al.,143 with permission.
Because slight elevations in serum creatinine, rapidly reversible after halting tolvaptan administration, were observed in the phase 2 clinical trials, 20 ADPKD patients underwent renal clearance and MRI studies before and after the 45/15-mg split daily dose of tolvaptan for 1 week.144 Tolvaptan-induced aquaresis was accompanied by significant reduction in iothalamate clearance and increase in serum uric acid caused by decreased uric acid clearance without change in renal blood flow. Post hoc, blinded analysis of renal MRIs showed a 3.1% reduction in kidney volume and the volume of individual cysts, which was likely because of an acute effect on fluid secretion.
The results of a phase 3, global, multicenter, randomized, double-blind, placebo-controlled, parallel-arm trial of tolvaptan in ADPKD (Tolvaptan Efficacy and Safety in Management of ADPKD and its Outcomes 3:4 [TEMPO3:4]; NCT00428948) have been recently published.145,146 ADPKD subjects (n=1445) with rapid disease progression reflected by kidney volumes of at least 750 ml at a relatively young age between 18 and 50 years, but still having preserved renal function (estimated creatinine clearance>60 ml/min), were randomized two to one with tolvaptan to placebo. Daily split 45/15-mg doses were titrated at weekly intervals to 60/30 and 90/30 mg. The maximally tolerated dose was maintained for 3 years. Participants were instructed to drink enough water to prevent thirst. Serum creatinine and laboratory parameters were measured every 4 months, and renal MRIs were obtained yearly; 23% of tolvaptan-treated subjects withdrew from the trial, with 15% of subjects withdrawing because of adverse events (including aquaresis-related symptoms in 8%). Comparatively, 14% of placebo subjects withdrew from the trial, with 5% of subjects withdrawing because of adverse events (including aquaresis-related symptoms in 0.4%). Of the subjects randomized to tolvaptan completing 3 years of treatment, 24%, 21%, and 55% were tolerating 45/15, 60/30, and 90/30 mg, respectively, at the end of the study; 17% of subjects randomized to placebo were unable to tolerate the 90/30-mg dose.
Tolvaptan reduced the rate of kidney growth by 50% (from 5.5% to 2.8% per year) (Figure 3A). The treatment effect was greatest from baseline to year 1, but also, it was significant from year 1 to year 2 and from year 2 to year 3. The analysis of time to development or progression of multiple clinical events (worsening kidney function, severe kidney pain, hypertension, and albuminuria) showed fewer clinical events for tolvaptan compared with placebo, with a hazard ratio of 0.87 (95% confidence interval, 0.72 to 0.94). This result was driven by a 61% lower risk of 25% reductions in reciprocal serum creatinine and a 36% lower risk of kidney pain events (Figure 3B). Tolvaptan also reduced the rate of decline of reciprocal serum creatinine from 3.81 to 2.61 per year (Figure 3C).
Figure 3.

Effect of tolvaptan on primary (change in kidney volume) and secondary (time to multiple events of ADPKD and change in kidney function) results. (A) Slopes of total kidney volume growth (percent change from baseline). (B) Hazard ratio effects of tolvaptan on composite time to multiple events of ADPKD and its components. (C) Slopes of kidney function estimated by reciprocal serum creatinine. TOL denotes tolvaptan (blue). PBO denotes placebo (red). Reprinted from Torres et al.,145 with permission.
Frequencies of adverse events were similar in both groups; those events related to aquaresis, such as polyuria, thirst, and nocturia, were more common in the tolvaptan group, whereas those events related to ADPKD, such as kidney pain, hematuria, and urinary tract infection, were more common in the placebo group. Increases in serum sodium and uric acid were more frequently seen in tolvaptan-treated subjects. Tolvaptan-treated subjects had more frequent elevations of liver enzymes exceeding three or five times the upper limits of normal, leading to discontinuation of tolvaptan in 1.8% of subjects.
At the present time, tolvaptan is not approved for the indication of ADPKD and should not be administered outside of an approved research study. Its value in the treatment of ADPKD will depend on the balance between benefits and risks. Polyuria, thirst, and related adverse events may impact the ability of some patients to tolerate effective doses. Patients taking tolvaptan should be able to maintain adequate hydration. Levels of plasma sodium, uric acid, and liver enzymes should be monitored. Patients in TEMPO3:4 had relatively preserved renal function. Efficacy in more advanced stages of the disease has not been thoroughly ascertained.
Somatostatin Analogs: Rationale and Preclinical Trials
Somatostatin acts on five GPCRs (SSTR1 to 5). Binding to these receptors inhibits AC and mitogen-activated protein kinase, cell proliferation, and secretion of several hormones (growth hormone, insulin, glucagon, gastrin, cholecystokinin, vasoactive intestinal peptide and secretin, thyroid stimulating hormone, and adrenocorticotrophic hormone) and growth factors (IGF I and vascular endothelial growth factor).147,148 All five SSTRs are expressed in renal tubular epithelial cells and cholangiocytes. SSTR1 and 2 are expressed in the thick ascending limb of Henle, distal tubule, and collecting duct, and SSTR3, -4, and -5 are expressed in proximal tubules.149–152 Somatostatin inhibits cAMP generation in MDCK cells and rat collecting ducts and antagonizes vasopressin effects in the toad urinary bladder and dog collecting ducts.153–156 It also inhibits cAMP generation, fluid secretion, and cell proliferation in cholangiocytes and suppresses the growth of bile ducts and periportal connective tissue in rats with extrahepatic biliary obstruction.157,158
Because somatostatin has a half-life of approximately 3 minutes, more stable synthetic peptides (octreotide, lanreotide, and pasireotide) have been developed for clinical use. They differ in stability and affinity to the different SSTRs. Half-lives in the circulation are 2 hours for octreotide and lanreotide, and 12 hours for pasireotide. Octreotide and lanreotide bind with high affinity to SSTR2 and -3, bind with moderate affinity to SSTR5, and have no affinity to SSTR1 and -4. Pasireotide binds with high affinity to SSTR1, -2, -3, and -5.
In preclinical studies, octreotide and pasireotide reduce cAMP levels and proliferation of cholangiocytes in vitro, expansion of liver cysts in three-dimensional collagen culture, and development of kidney and liver cysts and fibrosis in PCK rats and Pkd2WS25/− mice.37,159 In agreement with the longer half-life and higher affinity to a broader range of SSTRs, the effects of pasireotide are consistently more potent than the effects of octreotide.
Somatostatin Analogs: Clinical Trials
Three small, randomized, placebo-controlled studies of octreotide or lanreotide (NCT00309283, NCT00426153, and NCT00565097) have been completed.160–163 Two of these studies have been extended as open-label, uncontrolled studies.164,165 The results of a fourth study comparing the effects of octreotide plus everolimus with octreotide alone (NCT01157858) have also been published. Additional clinical trials of somatostatin analogs for ADPKD and/or polycystic liver disease are currently active. The design of these clinical trials and the outcome of those trials that have been completed are summarized in Table 2. The published studies have shown similar results. Kidney growth is halted during the first year of treatment and then resumes, possibly at a lower rate than without treatment. Liver volume decreases by 4%–6% during the first year of treatment, and this reduction is sustained during the second year. Observation periods have been too short to assess an effect on renal function. The addition of everolimus to treatment with octreotide does not provide added benefit. Octreotide and lanreotide are overall well tolerated. Self-resolving abdominal cramps and loose stools are common in the first few days after the injections. Other adverse effects include injection site granuloma and pain, cholelithiasis, steatorrhea, weight loss, and rarely, hair loss. The adverse event profile of pasireotide in patients with ADPKD may include hyperglycemia, because pasireotide inhibits insulin more potently than glucagon secretion, whereas the contrary is true for octreotide and pasireotide.166 Currently, ongoing studies of longer duration with a larger number of patients are needed to better ascertain the potential long-term benefit of somatostatin analogs in ADPKD and PKD.
Table 2.
Clinical trials of somatostatin analogs for PKD and/or liver disease
| Study | Drug | Entry Criteria/Sample Size | Baseline Values | Status/Outcome |
|---|---|---|---|---|
| Bergamo phase 2160 | Octreotide; R/DB/PC/CO; 6 mo | ≥18 yr; SCr 1–3 mg/dl; 14 (ADPKD) | KV; GFR; LV | KV: 71±107 ml (O) versus 162±114 ml (P), P<0.05; no change in GFR; LV: −71±57 ml (O) versus +14±85 ml (P), P<0.05 |
| LOCKCYST phases 2 and 3 (NCT00565097)162 | Lanreotidel; R/DB/PC; 6 mo | ≥18 yr; >20 liver cysts; 54 not on dialysis (32 ADPKD, 22 PLD) | LV (MRI); KV; QOL | LV: −2.9% (L) versus +1.6% (P); P=0.01; KV: −1.5% (L) versus +3.4% (P), P=0.02; improved health perception |
| LOCKCYST extension164 | Lanreotide; OL; 12 mo | Same; 41 (25 ADPKD, 16 PLD) | LV: −4%; KV: −1%; 6 mo after discontinuation of L, LV: +4%; KV: +2% | |
| Mayo PLD1 phases 2 and 3 (NCT00426153)163 | Octreotide; R/DB/PC; 24 mo | ≥18 yr; LV>4000 ml or symptoms; 42 (34 ADPKD; 8 PLD) | LV (MRI); KV; QOL | LV: −4.95% (O) versus +0.92% (P), P<0.05; KV: +0.25% (O) versus +8.61% (P), P<0.05; GFR: −5.1 (O) versus −7.1 ml/min per 1.73 m2 (P), NS; improved perception of bodily pain and physical activity scores |
| Mayo PLD1 extension165 | Octreotide; OL; ongoing | Same; 41 | On O for 2 yr; LV: −5.78%; KV: +6.49%; after change from P to O at 2 years: LV: −7.66%; KV: −0.41% | |
| ELATE phase 2 (NCT01157858)170 | Octreotide + sirolimus versus octreotide; R/OL; 12 mo | 18–70 yr; >20 liver cysts; eGFR>60 ml/min per 1.73 m2; 44 (29 PLD, 15 ADPKD) | LV (CT); QOL | LV: −3.5% (O) versus −3.8% (O + S); no difference in KV change or gastrointestinal symptoms or general heath questionnaires |
| ALADIN phase 3 (NCT00309283) | Octreotide; R/SB/PC; 36 mo | 18–75 yr; eGFR≥40 ml/min per 1.73 m2a; 78 ADPKD | KV (MRI) | Completed; results pending |
| ALADIN 2 phase 3 (NCT01377246) | Octreotide; R/DB/PC; 36 mo | 18–75 yr; eGFR=15–40 ml/min per 1.73 m2; 80 ADPKD | KV (CT); GFR | 2011–2015; recruiting |
| RESOLVE (NCT01354405) | Lanreotide; treated cohort; 6 mo | 18–70 yr; >20 liver cysts; eGFR>40 ml/min per 1.73 m2; 43 | LV (CT); KV | 2011–2012; recruiting |
| DIPAK phase 3 (NCT01616927) | Lanreotide; R/OL; 32 mo | 18–60 yr; eGFR=30–60 ml/min per 1.73 m2; 300 | eGFR; KV; LV; QOL | 2012–2017; recruiting |
| Mayo PLD2 phase 2 (NCT01670110) | Pasireotide; R/DB/PC; 36 mo | ≥18 yr; LV>4000 ml or symptoms | 48; LV (MRI); KV; QOL | 2012–2016; recruiting |
R, randomized; DB, double-blind; PC, placebo controlled; CO, crossover; SCr, serum creatinine; KV, kidney volume; LV, liver volume; O, octreotide; P, placebo; PLD, polycystic liver disease; QOL, quality of life; L, lanreotide; OL, open label; NS, not significant; eGFR, estimated GFR; CT, computed tomography; S, sirolimus.
Footnote.
High Water Intake in ADPKD
Another way to decrease the effect of arginine vasopressin (AVP) on the kidney is to suppress its secretion by increasing fluid ingestion and continuously maintaining the osmolality of the urine<250–300 mOsm/kg H2O.167 This strategy has been tested directly in the PCK rat, where high fluid intake sufficient to achieve a 3.5-fold increase in urine output by adding 5% glucose to drinking water reduced AVP excretion, renal expression of V2R, cAMP-dependent activation of the B-Raf/MEK/ERK, and attenuation of PKD.168 It must be noted that the effects of increased water intake and V2R antagonists are not equivalent, even if they both reduce cAMP in the distal nephron and collecting ducts. Increased hydration decreases, whereas the administration of a V2R antagonist increases the circulating levels of vasopressin. Compliance with increased hydration to achieve persistent urine hypotonicity is difficult to achieve, may increase the risk for hyponatremia, and may be less effective in blocking V2R-mediated signaling. However, the administration of a V2R antagonist increases the risk for dehydration, hyperuricemia, and theoretically, nonspecific effects mediated by increased endogenous vasopressin levels through V1a and V1b receptors. A small, prospective, case-controlled, 1-year clinical trial of high water intake (NCT01348035) is in progress. Lacking clinical trial results, recommendations for water intake should rest on a critical analysis of potential benefits (reduction in the rate of disease progression) and risks (hyponatremia if solute intake or GFR is too low or if drugs limiting the ability to dilute the urine are administered, as well as negative effects of polyuria or nocturia). Patients with ADPKD are capable to dilute the urine even at moderately advanced stages of the disease. Therefore, those patients with normal or moderately reduced GFR who follow a diet not severely restricted in protein (<0.6 g/kg ideal body wt per day) or sodium (<60 mEq/d), are not edematous or volume contracted, do not take medications that interfere with the reabsorption of sodium chloride in diluting segments of the nephron (e.g., loop diuretics or thiazides) or enhance the release or effect of AVP (e.g., serotonin reuptake inhibitors and tricyclic antidepressants), and have normal voiding mechanics can handle moderate increases in urine volume (4 L daily) without untoward effect. A reasonable goal is to drink fluids as evenly as possible throughout waking hours and immediately before going to bed.169 More than likely, patients will experience nocturia at least one time, and, if tolerated, water should be drunk after voiding. Monitoring plasma sodium concentration should be advised. The intake should be of nonmineralized water, with no addition of sugar or caffeine.
Other Treatment Targets in PKD
The identification of the genes mutated in ADPKD and ARPKD in 1994, 1996, and 2002 has made possible a greater understanding of the cellular pathophysiologic mechanisms and laid the foundation for potential therapies (Table 3). In addition to vasopressin V2 receptor antagonists and somatostatin analogs, mTOR (sirolimus and everolimus), Src (botusimib), and multityrosine kinase (KD019) inhibitors have been or are currently tested in clinical trials. Others have been effective in preclinical studies.
Table 3.
Therapeutic targets for PKD supported by preclinical trials
| Calcium signaling |
| Polycystin-2 channel activator |
| TRPV4 channel activator |
| Calcium-sensing receptor activator |
| cAMP signaling |
| Vasopressin V2 receptor antagonist |
| Somatostatin receptor agonist |
| PGE2 receptor (EP2 or EP4) antagonist |
| Catechol-O-methyl transferase |
| Phosphodiesterase activator |
| Cell proliferation |
| Receptor tyrosine kinase inhibitor (ErbB1, ErbB2, IGF1, VEGF, cMET) |
| Nonreceptor tyrosine kinase inhibitor (Src) |
| Serine-threonine kinase inhibitor (B-Raf, MEK1/2, p38MAPK, mTOR) or activator (AMPK) |
| Transcription factor agonists (PPARγ) or inhibitors (STAT3, STAT6) |
| Histone deacetylase (HDAC1, HDAC5, HDAC6) inhibitors |
| Cyclin-dependent kinase and Cdc25 phosphatase inhibitors |
| Apoptosis |
| Caspase inhibitors |
| Fluid secretion |
| CFTR channel blocker |
| KCa3.1 channel blocker |
| Other mechanisms |
| Proteasome inhibitors |
| Glycosyl ceramide synthase inhibitors |
| Lovastatin |
| ACE inhibitors |
| 20-HETE synthase inhibitors |
| Protein restriction |
| Soy, flax diets |
| Citrate |
References available on request. VEGF, vascular endothelial growth factor; B-Raf, B rapidly accelerated fibrosarcoma kinase; MAPK, mitogen-activated protein kinase; AMPK, AMP kinase; PPARγ, peroxisome proliferator–activated receptor γ; ACE, angiotensin-converting enzyme.
In summary, advances in molecular biology and genetics have made possible a greater understanding of cellular pathophysiologic mechanisms responsible for the development and progression of PKD and laid the foundation for the development of potential new therapies. Most therapies at the present time are aimed at delaying the growth of the cysts and associated interstitial inflammation and fibrosis by targeting tubular epithelial cell proliferation and fluid secretion by the cystic epithelium. Many therapies have proven efficacious in animal models of PKD but may be limited by toxicity. Because effective treatments for PKD are likely to be long term (possibly lifelong), low toxicity and safe profile are of the utmost importance. Therapies targeting Gs (tolvaptan) or Gi (somatostatin analogs) protein–coupled hormonal receptors with relative tissue and cell specificity have been used in clinical trials with relative safety. Repurposing of drugs with a relatively safe profile that are currently used for other indications (e.g., metformin and peroxisome proliferator–activated receptor γ agonists) deserves attention. Identification of synergisms between different classes of drugs may increase their efficiency and safety. Not all patients with PKD will require treatment. Patient selection and determination of the optimal timing for intervention deserve consideration. After the long wait, ADPKD may become a treatable disease.
Disclosures
None.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
References
- 1.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AEH, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J: Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 33: 129–137, 2003 [DOI] [PubMed] [Google Scholar]
- 2.Nauli SM, Rossetti S, Kolb RJ, Alenghat FJ, Consugar MB, Harris PC, Ingber DE, Loghman-Adham M, Zhou J: Loss of polycystin-1 in human cyst-lining epithelia leads to ciliary dysfunction. J Am Soc Nephrol 17: 1015–1025, 2006 [DOI] [PubMed] [Google Scholar]
- 3.Wang S, Zhang J, Nauli SM, Li X, Starremans PG, Luo Y, Roberts KA, Zhou J: Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Mol Cell Biol 27: 3241–3252, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pisitkun T, Shen RF, Knepper MA: Identification and proteomic profiling of exosomes in human urine. Proc Natl Acad Sci U S A 101: 13368–13373, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hogan MC, Manganelli L, Woollard JR, Masyuk AI, Masyuk TV, Tammachote R, Huang BQ, Leontovich AA, Beito TG, Madden BJ, Charlesworth MC, Torres VE, LaRusso NF, Harris PC, Ward CJ: Characterization of PKD protein-positive exosome-like vesicles. J Am Soc Nephrol 20: 278–288, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scheffers MS, van der Bent P, Prins F, Spruit L, Breuning MH, Litvinov SV, de Heer E, Peters DJM: Polycystin-1, the product of the polycystic kidney disease 1 gene, co-localizes with desmosomes in MDCK cells. Hum Mol Genet 9: 2743–2750, 2000 [DOI] [PubMed] [Google Scholar]
- 7.Boletta A, Qian F, Onuchic LF, Bragonzi A, Cortese M, Deen PM, Courtoy PJ, Soria MR, Devuyst O, Monaco L, Germino GG: Biochemical characterization of bona fide polycystin-1 in vitro and in vivo. Am J Kidney Dis 38: 1421–1429, 2001 [DOI] [PubMed] [Google Scholar]
- 8.Newby LJ, Streets AJ, Zhao Y, Harris PC, Ward CJ, Ong ACM: Identification, characterization, and localization of a novel kidney polycystin-1-polycystin-2 complex. J Biol Chem 277: 20763–20773, 2002 [DOI] [PubMed] [Google Scholar]
- 9.Li Y, Santoso NG, Yu S, Woodward OM, Qian F, Guggino WB: Polycystin-1 interacts with inositol 1,4,5-trisphosphate receptor to modulate intracellular Ca2+ signaling with implications for polycystic kidney disease. J Biol Chem 284: 36431–36441, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santoso NG, Cebotaru L, Guggino WB: Polycystin-1, 2, and STIM1 interact with IP(3)R to modulate ER Ca release through the PI3K/Akt pathway. Cell Physiol Biochem 27: 715–726, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koulen P, Cai Y, Geng L, Maeda Y, Nishimura S, Witzgall R, Ehrlich BE, Somlo S: Polycystin-2 is an intracellular calcium release channel. Nat Cell Biol 4: 191–197, 2002 [DOI] [PubMed] [Google Scholar]
- 12.Witzgall R: Polycystin-2—an intracellular or plasma membrane channel? Naunyn Schmiedebergs Arch Pharmacol 371: 342–347, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Qian F, Germino FJ, Cai Y, Zhang X, Somlo S, Germino GG: PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat Genet 16: 179–183, 1997 [DOI] [PubMed] [Google Scholar]
- 14.Wu Y, Dai XQ, Li Q, Chen CX, Mai W, Hussain Z, Long W, Montalbetti N, Li G, Glynne R, Wang S, Cantiello HF, Wu G, Chen XZ: Kinesin-2 mediates physical and functional interactions between polycystin-2 and fibrocystin. Hum Mol Genet 15: 3280–3292, 2006 [DOI] [PubMed] [Google Scholar]
- 15.Kim I, Li C, Liang D, Chen XZ, Coffy RJ, Ma J, Zhao P, Wu G: Polycystin-2 expression is regulated by a PC2-binding domain in the intracellular portion of fibrocystin. J Biol Chem 283: 31559–31566, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Wright JM, Qian F, Germino GG, Guggino WB: Polycystin 2 interacts with type I inositol 1,4,5-trisphosphate receptor to modulate intracellular Ca2+ signaling. J Biol Chem 280: 41298–41306, 2005 [DOI] [PubMed] [Google Scholar]
- 17.Anyatonwu GI, Estrada M, Tian X, Somlo S, Ehrlich BE: Regulation of ryanodine receptor-dependent calcium signaling by polycystin-2. Proc Natl Acad Sci U S A 104: 6454–6459, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsiokas L, Arnould T, Zhu C, Kim E, Walz G, Sukhatme VP: Specific association of the gene product of PKD2 with the TRPC1 channel. Proc Natl Acad Sci U S A 96: 3934–3939, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bai CX, Giamarchi A, Rodat-Despoix L, Padilla F, Downs T, Tsiokas L, Delmas P: Formation of a new receptor-operated channel by heteromeric assembly of TRPP2 and TRPC1 subunits. EMBO Rep 9: 472–479, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Du J, Ding M, Sours-Brothers S, Graham S, Ma R: Mediation of angiotensin II-induced Ca2+ signaling by polycystin 2 in glomerular mesangial cells. Am J Physiol Renal Physiol 294: F909–F918, 2008 [DOI] [PubMed] [Google Scholar]
- 21.Köttgen M, Buchholz B, Garcia-Gonzalez MA, Kotsis F, Fu X, Doerken M, Boehlke C, Steffl D, Tauber R, Wegierski T, Nitschke R, Suzuki M, Kramer-Zucker A, Germino GG, Watnick T, Prenen J, Nilius B, Kuehn EW, Walz G: TRPP2 and TRPV4 form a polymodal sensory channel complex. J Cell Biol 182: 437–447, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mekahli D, Sammels E, Luyten T, Welkenhuyzen K, van den Heuvel LP, Levtchenko EN, Gijsbers R, Bultynck G, Parys JB, De Smedt H, Missiaen L: Polycystin-1 and polycystin-2 are both required to amplify inositol-trisphosphate-induced Ca2+ release. Cell Calcium 51: 452–458, 2012 [DOI] [PubMed] [Google Scholar]
- 23.Morel N, Vandenberg G, Ahrabi AK, Caron N, Desjardins F, Balligand JL, Horie S, Devuyst O: PKD1 haploinsufficiency is associated with altered vascular reactivity and abnormal calcium signaling in the mouse aorta. Pflugers Arch 457: 845–856, 2009 [DOI] [PubMed] [Google Scholar]
- 24.Qian Q, Hunter LW, Li M, Marin-Padilla M, Prakash YS, Somlo S, Harris PC, Torres VE, Sieck GC: Pkd2 haploinsufficiency alters intracellular calcium regulation in vascular smooth muscle cells. Hum Mol Genet 12: 1875–1880, 2003 [DOI] [PubMed] [Google Scholar]
- 25.Yamaguchi T, Hempson SJ, Reif GA, Hedge AM, Wallace DP: Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J Am Soc Nephrol 17: 178–187, 2006 [DOI] [PubMed] [Google Scholar]
- 26.Geng L, Boehmerle W, Maeda Y, Okuhara DY, Tian X, Yu Z, Choe CU, Anyatonwu GI, Ehrlich BE, Somlo S: Syntaxin 5 regulates the endoplasmic reticulum channel-release properties of polycystin-2. Proc Natl Acad Sci U S A 105: 15920–15925, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu C, Rossetti S, Jiang L, Harris PC, Brown-Glaberman U, Wandinger-Ness A, Bacallao R, Alper SL: Human ADPKD primary cyst epithelial cells with a novel, single codon deletion in the PKD1 gene exhibit defective ciliary polycystin localization and loss of flow-induced Ca2+ signaling. Am J Physiol Renal Physiol 292: F930–F945, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahrabi AK, Terryn S, Valenti G, Caron N, Serradeil-Le Gal C, Raufaste D, Nielsen S, Horie S, Verbavatz JM, Devuyst O: PKD1 haploinsufficiency causes a syndrome of inappropriate antidiuresis in mice. J Am Soc Nephrol 18: 1740–1753, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Banales JM, Masyuk TV, Gradilone SA, Masyuk AI, Medina JF, LaRusso NF: The cAMP effectors Epac and protein kinase a (PKA) are involved in the hepatic cystogenesis of an animal model of autosomal recessive polycystic kidney disease (ARPKD). Hepatology 49: 160–174, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spirli C, Locatelli L, Fiorotto R, Morell CM, Fabris L, Pozzan T, Strazzabosco M: Altered store operated calcium entry increases cyclic 3′,5′-adenosine monophosphate production and extracellular signal-regulated kinases 1 and 2 phosphorylation in polycystin-2–defective cholangiocytes. Hepatology 55: 856–868, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Zaika O, Mamenko M, Berrout J, Boukelmoune N, O’Neil RG, Pochynyuk O: TRPV4 dysfunction promotes renal cystogenesis in autosomal recessive polycystic kidney disease. J Am Soc Nephrol 24: 604–616, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamaguchi T, Nagao S, Kasahara M, Takahashi H, Grantham JJ: Renal accumulation and excretion of cyclic adenosine monophosphate in a murine model of slowly progressive polycystic kidney disease. Am J Kidney Dis 30: 703–709, 1997 [DOI] [PubMed] [Google Scholar]
- 33.Gattone VH, 2nd, Wang X, Harris PC, Torres VE: Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 9: 1323–1326, 2003 [DOI] [PubMed] [Google Scholar]
- 34.Smith LA, Bukanov NO, Husson H, Russo RJ, Barry TC, Taylor AL, Beier DR, Ibraghimov-Beskrovnaya O: Development of polycystic kidney disease in juvenile cystic kidney mice: Insights into pathogenesis, ciliary abnormalities, and common features with human disease. J Am Soc Nephrol 17: 2821–2831, 2006 [DOI] [PubMed] [Google Scholar]
- 35.Starremans PG, Li X, Finnerty PE, Guo L, Takakura A, Neilson EG, Zhou J: A mouse model for polycystic kidney disease through a somatic in-frame deletion in the 5′ end of Pkd1. Kidney Int 73: 1394–1405, 2008 [DOI] [PubMed] [Google Scholar]
- 36.Hopp K, Ward CJ, Hommerding CJ, Nasr SH, Tuan HF, Gainullin VG, Rossetti S, Torres VE, Harris PC: Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J Clin Invest 122: 4257–4273, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Masyuk TV, Masyuk AI, Torres VE, Harris PC, Larusso NF: Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3′,5′-cyclic monophosphate. Gastroenterology 132: 1104–1116, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Kip SN, Hunter LW, Ren Q, Harris PC, Somlo S, Torres VE, Sieck GC, Qian Q: [Ca2+]i reduction increases cellular proliferation and apoptosis in vascular smooth muscle cells: Relevance to the ADPKD phenotype. Circ Res 96: 873–880, 2005 [DOI] [PubMed] [Google Scholar]
- 39.Banizs B, Komlosi P, Bevensee MO, Schwiebert EM, Bell PD, Yoder BK: Altered pH(i) regulation and Na(+)/HCO3(-) transporter activity in choroid plexus of cilia-defective Tg737(orpk) mutant mouse. Am J Physiol Cell Physiol 292: C1409–C1416, 2007 [DOI] [PubMed] [Google Scholar]
- 40.Wang X, Ward CJ, Harris PC, Torres VE: Cyclic nucleotide signaling in polycystic kidney disease. Kidney Int 77: 129–140, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi YH, Suzuki A, Hajarnis S, Ma Z, Chapin HC, Caplan MJ, Pontoglio M, Somlo S, Igarashi P: Polycystin-2 and phosphodiesterase 4C are components of a ciliary A-kinase anchoring protein complex that is disrupted in cystic kidney diseases. Proc Natl Acad Sci U S A 108: 10679–10684, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parnell SC, Magenheimer BS, Maser RL, Rankin CA, Smine A, Okamoto T, Calvet JP: The polycystic kidney disease-1 protein, polycystin-1, binds and activates heterotrimeric G-proteins in vitro. Biochem Biophys Res Commun 251: 625–631, 1998 [DOI] [PubMed] [Google Scholar]
- 43.Putnam WC, Swenson SM, Reif GA, Wallace DP, Helmkamp GM, Jr., Grantham JJ: Identification of a forskolin-like molecule in human renal cysts. J Am Soc Nephrol 18: 934–943, 2007 [DOI] [PubMed] [Google Scholar]
- 44.Hovater MB, Olteanu D, Welty EA, Schwiebert EM: Purinergic signaling in the lumen of a normal nephron and in remodeled PKD encapsulated cysts. Purinergic Signal 4: 109–124, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blazer-Yost BL, Haydon J, Eggleston-Gulyas T, Chen JH, Wang X, Gattone V, Torres VE: Pioglitazone attenuates cystic burden in the PCK rodent model of polycystic kidney disease. PPAR Res 2010: 274376, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buchholz B, Klanke B, Schley G, Bollag G, Tsai J, Kroening S, Yoshihara D, Wallace DP, Kraenzlin B, Gretz N, Hirth P, Eckardt KU, Bernhardt WM: The Raf kinase inhibitor PLX5568 slows cyst proliferation in rat polycystic kidney disease but promotes renal and hepatic fibrosis. Nephrol Dial Transplant 26: 3458–3465, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rees S, Kittikulsuth W, Roos K, Strait KA, Van Hoek A, Kohan DE: Adenylyl cyclase 6 deficiency ameliorates polycystic kidney disease [published ahead of print October 24, 2013]. J Am Soc Nephrol doi:10.1681/ASN.2013010077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sussman C, Ward C, Leightner A, Harris P, Torres V: Regulation of renal cyst formation by phosphodiesterase 1a in zebrafish [Abstract]. J Am Soc Nephrol 23: 1A, 2012 [Google Scholar]
- 49.Hong Ye, Xiaofang Wang, Caroline R. Sussman, Peter C. Harris, Vincent C. Manganielo, Christopher James Ward, Vicente E. Torres: Genetic approach to evaluate the role of the PDE3 subfamilies in polycystic kidney disease (PKD). J Am Soc Nephrol 23: 239A, 2012. 22811488 [Google Scholar]
- 50.Cheng J, Thompson MA, Walker HJ, Gray CE, Warner GM, Zhou W, Grande JP: Lixazinone stimulates mitogenesis of Madin-Darby canine kidney cells. Exp Biol Med (Maywood) 231: 288–295, 2006 [DOI] [PubMed] [Google Scholar]
- 51.Tradtrantip L, Yangthara B, Padmawar P, Morrison C, Verkman AS: Thiophenecarboxylate suppressor of cyclic nucleotides discovered in a small-molecule screen blocks toxin-induced intestinal fluid secretion. Mol Pharmacol 75: 134–142, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carroll TJ, Das A: Planar cell polarity in kidney development and disease. Organogenesis 7: 180–190, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saadi-Kheddouci S, Berrebi D, Romagnolo B, Cluzeaud F, Peuchmaur M, Kahn A, Vandewalle A, Perret C: Early development of polycystic kidney disease in transgenic mice expressing an activated mutant of the beta-catenin gene. Oncogene 20: 5972–5981, 2001 [DOI] [PubMed] [Google Scholar]
- 54.Qian CN, Knol J, Igarashi P, Lin F, Zylstra U, Teh BT, Williams BO: Cystic renal neoplasia following conditional inactivation of apc in mouse renal tubular epithelium. J Biol Chem 280: 3938–3945, 2005 [DOI] [PubMed] [Google Scholar]
- 55.Saburi S, Hester I, Fischer E, Pontoglio M, Eremina V, Gessler M, Quaggin SE, Harrison R, Mount R, McNeill H: Loss of Fat4 disrupts PCP signaling and oriented cell division and leads to cystic kidney disease. Nat Genet 40: 1010–1015, 2008 [DOI] [PubMed] [Google Scholar]
- 56.Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Krönig C, Schermer B, Benzing T, Cabello OA, Jenny A, Mlodzik M, Polok B, Driever W, Obara T, Walz G: Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet 37: 537–543, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li M, Wang X, Meintzer MK, Laessig T, Birnbaum MJ, Heidenreich KA: Cyclic AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase 3beta. Mol Cell Biol 20: 9356–9363, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Taurin S, Sandbo N, Qin Y, Browning D, Dulin NO: Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase. J Biol Chem 281: 9971–9976, 2006 [DOI] [PubMed] [Google Scholar]
- 59.Gallegos TF, Kouznetsova V, Kudlicka K, Sweeney DE, Bush KT, Willert K, Farquhar MG, Nigam SK: A protein kinase A and Wnt-dependent network regulating an intermediate stage in epithelial tubulogenesis during kidney development. Dev Biol 364: 11–21, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Song BH, Choi SC, Han JK: Local activation of protein kinase A inhibits morphogenetic movements during Xenopus gastrulation. Dev Dyn 227: 91–103, 2003 [DOI] [PubMed] [Google Scholar]
- 61.Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG: A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat Med 13: 1490–1495, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nadasdy T, Lajoie G, Laszik Z, Blick KE, Molnar-Nadasdy G, Silva FG: Cell proliferation in the developing human kidney. Pediatr Dev Pathol 1: 49–55, 1998 [DOI] [PubMed] [Google Scholar]
- 63.Nadasdy T, Laszik Z, Blick KE, Johnson LD, Silva FG: Proliferative activity of intrinsic cell populations in the normal human kidney. J Am Soc Nephrol 4: 2032–2039, 1994 [DOI] [PubMed] [Google Scholar]
- 64.Yamaguchi T, Pelling JC, Ramaswamy NT, Eppler JW, Wallace DP, Nagao S, Rome LA, Sullivan LP, Grantham JJ: cAMP stimulates the in vitro proliferation of renal cyst epithelial cells by activating the extracellular signal-regulated kinase pathway. Kidney Int 57: 1460–1471, 2000 [DOI] [PubMed] [Google Scholar]
- 65.Hanaoka K, Guggino WB: cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells. J Am Soc Nephrol 11: 1179–1187, 2000 [DOI] [PubMed] [Google Scholar]
- 66.Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP: Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 279: 40419–40430, 2004 [DOI] [PubMed] [Google Scholar]
- 67.Distefano G, Boca M, Rowe I, Wodarczyk C, Ma L, Piontek KB, Germino GG, Pandolfi PP, Boletta A: Polycystin-1 regulates extracellular signal-regulated kinase-dependent phosphorylation of tuberin to control cell size through mTOR and its downstream effectors S6K and 4EBP1. Mol Cell Biol 29: 2359–2371, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Spirli C, Okolicsanyi S, Fiorotto R, Fabris L, Cadamuro M, Lecchi S, Tian X, Somlo S, Strazzabosco M: ERK1/2-dependent vascular endothelial growth factor signaling sustains cyst growth in polycystin-2 defective mice. Gastroenterology 138: 360–371, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rowe I, Chiaravalli M, Mannella V, Ulisse V, Quilici G, Pema M, Song XW, Xu H, Mari S, Qian F, Pei Y, Musco G, Boletta A: Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med 19: 488–493, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, Vander Heiden MG, MacKeigan JP, Finan PM, Clish CB, Murphy LO, Manning BD: Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 39: 171–183, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yecies JL, Manning BD: Transcriptional control of cellular metabolism by mTOR signaling. Cancer Res 71: 2815–2820, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Takiar V, Nishio S, Seo-Mayer P, King JD, Jr., Li H, Zhang L, Karihaloo A, Hallows KR, Somlo S, Caplan MJ: Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proc Natl Acad Sci U S A 108: 2462–2467, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aguiari G, Bizzarri F, Bonon A, Mangolini A, Magri E, Pedriali M, Querzoli P, Somlo S, Harris PC, Catizone L, Del Senno L: Polycystin-1 regulates amphiregulin expression through CREB and AP1 signalling: Implications in ADPKD cell proliferation. J Mol Med (Berl) 90: 1267–1282, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Qin S, Taglienti M, Cai L, Zhou J, Kreidberg JA: c-Met and NF-κB-dependent overexpression of Wnt7a and -7b and Pax2 promotes cystogenesis in polycystic kidney disease. J Am Soc Nephrol 23: 1309–1318, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Talbot JJ, Shillingford JM, Vasanth S, Doerr N, Mukherjee S, Kinter MT, Watnick T, Weimbs T: Polycystin-1 regulates STAT activity by a dual mechanism. Proc Natl Acad Sci U S A 108: 7985–7990, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Leonhard WN, van der Wal A, Novalic Z, Kunnen SJ, Gansevoort RT, Breuning MH, de Heer E, Peters DJ: Curcumin inhibits cystogenesis by simultaneous interference of multiple signaling pathways: In vivo evidence from a Pkd1-deletion model. Am J Physiol Renal Physiol 300: F1193–F1202, 2011 [DOI] [PubMed] [Google Scholar]
- 77.Takakura A, Nelson EA, Haque N, Humphreys BD, Zandi-Nejad K, Frank DA, Zhou J: Pyrimethamine inhibits adult polycystic kidney disease by modulating STAT signaling pathways. Hum Mol Genet 20: 4143–4154, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stayner C, Iglesias DM, Goodyer PR, Ellis L, Germino G, Zhou J, Eccles MR: Pax2 gene dosage influences cystogenesis in autosomal dominant polycystic kidney disease. Hum Mol Genet 15: 3520–3528, 2006 [DOI] [PubMed] [Google Scholar]
- 79.Ostrom L, Tang MJ, Gruss P, Dressler GR: Reduced Pax2 gene dosage increases apoptosis and slows the progression of renal cystic disease. Dev Biol 219: 250–258, 2000 [DOI] [PubMed] [Google Scholar]
- 80.Metcalf D, Mifsud S, Di Rago L, Nicola NA, Hilton DJ, Alexander WS: Polycystic kidneys and chronic inflammatory lesions are the delayed consequences of loss of the suppressor of cytokine signaling-1 (SOCS-1). Proc Natl Acad Sci U S A 99: 943–948, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Grantham JJ, Geiser JL, Evan AP: Cyst formation and growth in autosomal dominant polycystic kidney disease. Kidney Int 31: 1145–1152, 1987 [DOI] [PubMed] [Google Scholar]
- 82.Jacobsson L, Lindqvist B, Michaelson G, Bjerle P: Fluid turnover in renal cysts. Acta Med Scand 202: 327–329, 1977 [DOI] [PubMed] [Google Scholar]
- 83.Mangoo-Karim R, Uchic M, Lechene C, Grantham JJ: Renal epithelial cyst formation and enlargement in vitro: Dependence on cAMP. Proc Natl Acad Sci U S A 86: 6007–6011, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ye M, Grantham JJ: The secretion of fluid by renal cysts from patients with autosomal dominant polycystic kidney disease. N Engl J Med 329: 310–313, 1993 [DOI] [PubMed] [Google Scholar]
- 85.Sullivan LP, Wallace DP, Grantham JJ: Epithelial transport in polycystic kidney disease. Physiol Rev 78: 1165–1191, 1998 [DOI] [PubMed] [Google Scholar]
- 86.Tradtrantip L, Sonawane ND, Namkung W, Verkman AS: Nanomolar potency pyrimido-pyrrolo-quinoxalinedione CFTR inhibitor reduces cyst size in a polycystic kidney disease model. J Med Chem 52: 6447–6455, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Snyder DS, Tradtrantip L, Yao C, Kurth MJ, Verkman AS: Potent, metabolically stable benzopyrimido-pyrrolo-oxazine-dione (BPO) CFTR inhibitors for polycystic kidney disease. J Med Chem 54: 5468–5477, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li H, Findlay IA, Sheppard DN: The relationship between cell proliferation, Cl- secretion, and renal cyst growth: A study using CFTR inhibitors. Kidney Int 66: 1926–1938, 2004 [DOI] [PubMed] [Google Scholar]
- 89.Magenheimer BS, St John PL, Isom KS, Abrahamson DR, De Lisle RC, Wallace DP, Maser RL, Grantham JJ, Calvet JP: Early embryonic renal tubules of wild-type and polycystic kidney disease kidneys respond to cAMP stimulation with cystic fibrosis transmembrane conductance regulator/Na(+),K(+),2Cl(-) Co-transporter-dependent cystic dilation. J Am Soc Nephrol 17: 3424–3437, 2006 [DOI] [PubMed] [Google Scholar]
- 90.Yang B, Sonawane ND, Zhao D, Somlo S, Verkman AS: Small-molecule CFTR inhibitors slow cyst growth in polycystic kidney disease. J Am Soc Nephrol 19: 1300–1310, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hanaoka K, Devuyst O, Schwiebert EM, Wilson PD, Guggino WB: A role for CFTR in human autosomal dominant polycystic kidney disease. Am J Physiol 270: C389–C399, 1996 [DOI] [PubMed] [Google Scholar]
- 92.Davidow CJ, Maser RL, Rome LA, Calvet JP, Grantham JJ: The cystic fibrosis transmembrane conductance regulator mediates transepithelial fluid secretion by human autosomal dominant polycystic kidney disease epithelium in vitro. Kidney Int 50: 208–218, 1996 [DOI] [PubMed] [Google Scholar]
- 93.O’Sullivan DA, Torres VE, Gabow PA, Thibodeau SN, King BF, Bergstralh EJ: Cystic fibrosis and the phenotypic expression of autosomal dominant polycystic kidney disease. Am J Kidney Dis 32: 976–983, 1998 [DOI] [PubMed] [Google Scholar]
- 94.Xu N, Glockner JF, Rossetti S, Babovich-Vuksanovic D, Harris PC, Torres VE: Autosomal dominant polycystic kidney disease coexisting with cystic fibrosis. J Nephrol 19: 529–534, 2006 [PubMed] [Google Scholar]
- 95.Persu A, Devuyst O, Lannoy N, Materne R, Brosnahan G, Gabow PA, Pirson Y, Verellen-Dumoulin C: CF gene and cystic fibrosis transmembrane conductance regulator expression in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 11: 2285–2296, 2000 [DOI] [PubMed] [Google Scholar]
- 96.Nakanishi K, Sweeney WE, Jr., Macrae Dell K, Cotton CU, Avner ED: Role of CFTR in autosomal recessive polycystic kidney disease. J Am Soc Nephrol 12: 719–725, 2001 [DOI] [PubMed] [Google Scholar]
- 97.Wilson PD: Polycystin: New aspects of structure, function, and regulation. J Am Soc Nephrol 12: 834–845, 2001 [DOI] [PubMed] [Google Scholar]
- 98.Chen D, Roberts R, Pohl M, Nigam S, Kreidberg J, Wang Z, Heino J, Ivaska J, Coffa S, Harris RC, Pozzi A, Zent R: Differential expression of collagen- and laminin-binding integrins mediates ureteric bud and inner medullary collecting duct cell tubulogenesis. Am J Physiol Renal Physiol 287: F602–F611, 2004 [DOI] [PubMed] [Google Scholar]
- 99.Zent R, Bush KT, Pohl ML, Quaranta V, Koshikawa N, Wang Z, Kreidberg JA, Sakurai H, Stuart RO, Nigám SK: Involvement of laminin binding integrins and laminin-5 in branching morphogenesis of the ureteric bud during kidney development. Dev Biol 238: 289–302, 2001 [DOI] [PubMed] [Google Scholar]
- 100.van Adelsberg J: Murine polycystic kidney epithelial cell lines have increased integrin-mediated adhesion to collagen. Am J Physiol 267: F1082–F1093, 1994 [DOI] [PubMed] [Google Scholar]
- 101.Joly D, Morel V, Hummel A, Ruello A, Nusbaum P, Patey N, Noël LH, Rousselle P, Knebelmann B: Beta4 integrin and laminin 5 are aberrantly expressed in polycystic kidney disease: Role in increased cell adhesion and migration. Am J Pathol 163: 1791–1800, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wallace DP, Quante MT, Reif GA, Nivens E, Ahmed F, Hempson SJ, Blanco G, Yamaguchi T: Periostin induces proliferation of human autosomal dominant polycystic kidney cells through alphaV-integrin receptor. Am J Physiol Renal Physiol 295: F1463–F1471, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Daïkha-Dahmane F, Narcy F, Dommergues M, Lacoste M, Beziau A, Gubler M-C: Distribution of alpha-integrin subunits in fetal polycystic kidney diseases. Pediatr Nephrol 11: 267–273, 1997 [DOI] [PubMed] [Google Scholar]
- 104.Joly D, Berissi S, Bertrand A, Strehl L, Patey N, Knebelmann B: Laminin 5 regulates polycystic kidney cell proliferation and cyst formation. J Biol Chem 281: 29181–29189, 2006 [DOI] [PubMed] [Google Scholar]
- 105.Wu W, Kitamura S, Truong DM, Rieg T, Vallon V, Sakurai H, Bush KT, Vera DR, Ross RS, Nigam SK: Beta1-integrin is required for kidney collecting duct morphogenesis and maintenance of renal function. Am J Physiol Renal Physiol 297: F210–F217, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shannon MB, Patton BL, Harvey SJ, Miner JH: A hypomorphic mutation in the mouse laminin alpha5 gene causes polycystic kidney disease. J Am Soc Nephrol 17: 1913–1922, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Howe AK: Regulation of actin-based cell migration by cAMP/PKA. Biochim Biophys Acta 1692: 159–174, 2004 [DOI] [PubMed] [Google Scholar]
- 108.Meyer CJ, Alenghat FJ, Rim P, Fong JH, Fabry B, Ingber DE: Mechanical control of cyclic AMP signalling and gene transcription through integrins. Nat Cell Biol 2: 666–668, 2000 [DOI] [PubMed] [Google Scholar]
- 109.Alenghat FJ, Tytell JD, Thodeti CK, Derrien A, Ingber DE: Mechanical control of cAMP signaling through integrins is mediated by the heterotrimeric Galphas protein. J Cell Biochem 106: 529–538, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.O’Connor KL, Shaw LM, Mercurio AM: Release of cAMP gating by the alpha6beta4 integrin stimulates lamellae formation and the chemotactic migration of invasive carcinoma cells. J Cell Biol 143: 1749–1760, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Netherton SJ, Sutton JA, Wilson LS, Carter RL, Maurice DH: Both protein kinase A and exchange protein activated by cAMP coordinate adhesion of human vascular endothelial cells. Circ Res 101: 768–776, 2007 [DOI] [PubMed] [Google Scholar]
- 112.Eid AH: cAMP induces adhesion of microvascular smooth muscle cells to fibronectin via an Epac-mediated but PKA-independent mechanism. Cell Physiol Biochem 30: 247–258, 2012 [DOI] [PubMed] [Google Scholar]
- 113.Werder AA, Amos MA, Nielsen AH, Wolfe GH: Comparative effects of germfree and ambient environments on the development of cystic kidney disease in CFWwd mice. J Lab Clin Med 103: 399–407, 1984 [PubMed] [Google Scholar]
- 114.Gardner KD, Jr., Reed WP, Evan AP, Zedalis J, Hylarides MD, Leon AA: Endotoxin provocation of experimental renal cystic disease. Kidney Int 32: 329–334, 1987 [DOI] [PubMed] [Google Scholar]
- 115.Gardner KD, Jr., Burnside JS, Elzinga LW, Locksley RM: Cytokines in fluids from polycystic kidneys. Kidney Int 39: 718–724, 1991 [DOI] [PubMed] [Google Scholar]
- 116.Karihaloo A, Koraishy F, Huen SC, Lee Y, Merrick D, Caplan MJ, Somlo S, Cantley LG: Macrophages promote cyst growth in polycystic kidney disease. J Am Soc Nephrol 22: 1809–1814, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Swenson-Fields KI, Vivian CJ, Salah SM, Peda JD, Davis BM, van Rooijen N, Wallace DP, Fields TA: Macrophages promote polycystic kidney disease progression. Kidney Int 83: 855–864, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lee S, Huen S, Nishio H, Nishio S, Lee HK, Choi BS, Ruhrberg C, Cantley LG: Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol 22: 317–326, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Anders HJ, Ryu M: Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int 80: 915–925, 2011 [DOI] [PubMed] [Google Scholar]
- 120.Wang Y, Harris DC: Macrophages in renal disease. J Am Soc Nephrol 22: 21–27, 2011 [DOI] [PubMed] [Google Scholar]
- 121.Mantovani A, Sica A: Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr Opin Immunol 22: 231–237, 2010 [DOI] [PubMed] [Google Scholar]
- 122.Tang X, Mo C, Wang Y, Wei D, Xiao H: Anti-tumour strategies aiming to target tumour-associated macrophages. Immunology 138: 93–104, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yu H, Pardoll D, Jove R: STATs in cancer inflammation and immunity: A leading role for STAT3. Nat Rev Cancer 9: 798–809, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liu AM, Lo RK, Wong CS, Morris C, Wise H, Wong YH: Activation of STAT3 by G alpha(s) distinctively requires protein kinase A, JNK, and phosphatidylinositol 3-kinase. J Biol Chem 281: 35812–35825, 2006 [DOI] [PubMed] [Google Scholar]
- 125.Chun KS, Lao HC, Langenbach R: The prostaglandin E2 receptor, EP2, stimulates keratinocyte proliferation in mouse skin by G protein-dependent and beta-arrestin1-dependent signaling pathways. J Biol Chem 285: 39672–39681, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nault JC, Fabre M, Couchy G, Pilati C, Jeannot E, Tran Van Nhieu J, Saint-Paul MC, De Muret A, Redon MJ, Buffet C, Salenave S, Balabaud C, Prevot S, Labrune P, Bioulac-Sage P, Scoazec JY, Chanson P, Zucman-Rossi J: GNAS-activating mutations define a rare subgroup of inflammatory liver tumors characterized by STAT3 activation. J Hepatol 56: 184–191, 2012 [DOI] [PubMed] [Google Scholar]
- 127.Pringle DR, Yin Z, Lee AA, Manchanda PK, Yu L, Parlow AF, Jarjoura D, La Perle KM, Kirschner LS: Thyroid-specific ablation of the Carney complex gene, PRKAR1A, results in hyperthyroidism and follicular thyroid cancer. Endocr Relat Cancer 19: 435–446, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yasuda G, Jeffries WB: Regulation of cAMP production in initial and terminal inner medullary collecting ducts. Kidney Int 54: 80–86, 1998 [DOI] [PubMed] [Google Scholar]
- 129.Mutig K, Paliege A, Kahl T, Jöns T, Müller-Esterl WP, Bachmann S: Vasopressin V2 receptor expression along rat, mouse, and human renal epithelia with focus on TAL. Am J Physiol Renal Physiol 293: F1166–F1177, 2007 [DOI] [PubMed] [Google Scholar]
- 130.Carmosino M, Brooks HL, Cai Q, Davis LS, Opalenik S, Hao C, Breyer MD: Axial heterogeneity of vasopressin-receptor subtypes along the human and mouse collecting duct. Am J Physiol Renal Physiol 292: F351–F360, 2007 [DOI] [PubMed] [Google Scholar]
- 131.Verani RR, Silva FG: Histogenesis of the renal cysts in adult (autosomal dominant) polycystic kidney disease: A histochemical study. Mod Pathol 1: 457–463, 1988 [PubMed] [Google Scholar]
- 132.Bankir L, Bichet DG: Polycystic kidney disease: An early urea-selective urine-concentrating defect in ADPKD. Nat Rev Nephrol 8: 437–439, 2012 [DOI] [PubMed] [Google Scholar]
- 133.Gattone VH, 2nd, Maser RL, Tian C, Rosenberg JM, Branden MG: Developmental expression of urine concentration-associated genes and their altered expression in murine infantile-type polycystic kidney disease. Dev Genet 24: 309–318, 1999 [DOI] [PubMed] [Google Scholar]
- 134.Torres VE, Wang X, Qian Q, Somlo S, Harris PC, Gattone VH, 2nd: Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat Med 10: 363–364, 2004 [DOI] [PubMed] [Google Scholar]
- 135.Wang X, Gattone V, 2nd, Harris PC, Torres VE: Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol 16: 846–851, 2005 [DOI] [PubMed] [Google Scholar]
- 136.Meijer E, Gansevoort RT, de Jong PE, van der Wal AM, Leonhard WN, de Krey SR, van den Born J, Mulder GM, van Goor H, Struck J, de Heer E, Peters DJ: Therapeutic potential of vasopressin V2 receptor antagonist in a mouse model for autosomal dominant polycystic kidney disease: Optimal timing and dosing of the drug. Nephrol Dial Transplant 26: 2445–2453, 2011 [DOI] [PubMed] [Google Scholar]
- 137.Wang X, Wu Y, Ward CJ, Harris PC, Torres VE: Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol 19: 102–108, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Romaker D, Puetz M, Teschner S, Donauer J, Geyer M, Gerke P, Rumberger B, Dworniczak B, Pennekamp P, Buchholz B, Neumann HP, Kumar R, Gloy J, Eckardt KU, Walz G: Increased expression of secreted frizzled-related protein 4 in polycystic kidneys. J Am Soc Nephrol 20: 48–56, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Reif GA, Yamaguchi T, Nivens E, Fujiki H, Pinto CS, Wallace DP: Tolvaptan inhibits ERK-dependent cell proliferation, Cl⁻ secretion, and in vitro cyst growth of human ADPKD cells stimulated by vasopressin. Am J Physiol Renal Physiol 301: F1005–F1013, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Grantham JJ, Chapman AB, Torres VE, Ouyang J, Shoaf SE, Czerwiec FS: Acute and chronic osmostasis after vassopressin V2 receptor inhibiton with Tolvaptan in ADPKD [Abstract]. J Am Soc Nephrol 16: 361A, 2005 [Google Scholar]
- 141.Chapman AB, Torres VE, Grantham JJ, Shoaf SS, Ouyang JJ, Czerwiec FS: A phase IIB pilot study of the safety and efficacy of tolvaptan, a vasopressin V2 receptor antagonist (V2RA) in paitents with ADPKD. J Am Soc Nephrol 16: 68A, 2005. 15537870 [Google Scholar]
- 142.Torres VE, Wang X, Ward CJ, Grantham JJ, Chapman AB, Ouyang J, Shoaf SE, Czerwiec FS: Urine aquaporin 2 and cyclic AMP responses to tolvaptan administration in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 16: 361A, 2005 [Google Scholar]
- 143.Higashihara E, Torres VE, Chapman AB, Grantham JJ, Bae K, Watnick TJ, Horie S, Nutahara K, Ouyang J, Krasa HB, Czerwiec FS, TEMPOFormula and 156-05-002 Study Investigators : Tolvaptan in autosomal dominant polycystic kidney disease: Three years’ experience. Clin J Am Soc Nephrol 6: 2499–2507, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Irazabal MV, Torres VE, Hogan MC, Glockner J, King BF, Ofstie TG, Krasa HB, Ouyang J, Czerwiec FS: Short-term effects of tolvaptan on renal function and volume in patients with autosomal dominant polycystic kidney disease. Kidney Int 80: 295–301, 2011 [DOI] [PubMed] [Google Scholar]
- 145.Torres VE, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, Perrone RD, Krasa HB, Ouyang J, Czerwiec FS, TEMPO 3:4 Trial Investigators : Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med 367: 2407–2418, 2012. 23121377 [Google Scholar]
- 146.Torres VE, Meijer E, Bae KT, Chapman AB, Devuyst O, Gansevoort RT, Grantham JJ, Higashihara E, Perrone RD, Krasa HB, Ouyang JJ, Czerwiec FS: Rationale and design of the TEMPO (Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and its Outcomes) 3-4 Study. Am J Kidney Dis 57: 692–699, 2011 [DOI] [PubMed] [Google Scholar]
- 147.Grozinsky-Glasberg S, Shimon I, Korbonits M, Grossman AB: Somatostatin analogues in the control of neuroendocrine tumours: Efficacy and mechanisms. Endocr Relat Cancer 15: 701–720, 2008 [DOI] [PubMed] [Google Scholar]
- 148.Pyronnet S, Bousquet C, Najib S, Azar R, Laklai H, Susini C: Antitumor effects of somatostatin. Mol Cell Endocrinol 286: 230–237, 2008 [DOI] [PubMed] [Google Scholar]
- 149.Balster DA, O’Dorisio MS, Summers MA, Turman MA: Segmental expression of somatostatin receptor subtypes sst(1) and sst(2) in tubules and glomeruli of human kidney. Am J Physiol Renal Physiol 280: F457–F465, 2001 [DOI] [PubMed] [Google Scholar]
- 150.Bates CM, Kegg H, Grady S: Expression of somatostatin in the adult and developing mouse kidney. Kidney Int 66: 1785–1793, 2004 [DOI] [PubMed] [Google Scholar]
- 151.Bates CM, Kegg H, Grady S: Expression of somatostatin receptors 1 and 2 in the adult mouse kidney. Regul Pept 119: 11–20, 2004 [DOI] [PubMed] [Google Scholar]
- 152.Bhandari S, Watson N, Long E, Sharpe S, Zhong W, Xu SZ, Atkin SL: Expression of somatostatin and somatostatin receptor subtypes 1-5 in human normal and diseased kidney. J Histochem Cytochem 56: 733–743, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Winkler SN, Torikai S, Levine BS, Kurokawa K: Effect of somatostatin on vasopressin-induced antidiuresis and renal cyclic AMP of rats. Miner Electrolyte Metab 7: 8–14, 1982 [PubMed] [Google Scholar]
- 154.Friedlander G, Amiel C: Somatostatin and alpha 2-adrenergic agonists selectively inhibit vasopressin-induced cyclic AMP accumulation in MDCK cells. FEBS Lett 198: 38–42, 1986 [DOI] [PubMed] [Google Scholar]
- 155.Forrest JN, Jr., Reichlin S, Goodman DB: Somatostatin: An endogenous peptide in the toad urinary bladder inhibits vasopressin-stimulated water flow. Proc Natl Acad Sci U S A 77: 4984–4987, 1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mountokalakis T, Levy M: Effect of somatostatin on renal water handling in the dog. Can J Physiol Pharmacol 60: 655–664, 1982 [DOI] [PubMed] [Google Scholar]
- 157.Tan CK, Podila PV, Taylor JE, Nagorney DM, Wiseman GA, Gores GJ, LaRusso NF: Human cholangiocarcinomas express somatostatin receptors and respond to somatostatin with growth inhibition. Gastroenterology 108: 1908–1916, 1995 [DOI] [PubMed] [Google Scholar]
- 158.Tracy TF, Jr., Tector AJ, Goerke ME, Kitchen S, Lagunoff D: Somatostatin analogue (octreotide) inhibits bile duct epithelial cell proliferation and fibrosis after extrahepatic biliary obstruction. Am J Pathol 143: 1574–1578, 1993 [PMC free article] [PubMed] [Google Scholar]
- 159.Masyuk TV, Radtke BN, Stroope AJ, Banales JM, Gradilone SA, Huang B, Masyuk AI, Hogan MC, Torres VE, Larusso NF: Pasireotide is more effective than octreotide in reducing hepatorenal cystogenesis in rodents with polycystic kidney and liver diseases. Hepatology 58: 409–421, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ruggenenti P, Remuzzi A, Ondei P, Fasolini G, Antiga L, Ene-Iordache B, Remuzzi G, Epstein FH: Safety and efficacy of long-acting somatostatin treatment in autosomal-dominant polycystic kidney disease. Kidney Int 68: 206–216, 2005 [DOI] [PubMed] [Google Scholar]
- 161.Caroli A, Antiga L, Cafaro M, Fasolini G, Remuzzi A, Remuzzi G, Ruggenenti P: Reducing polycystic liver volume in ADPKD: Effects of somatostatin analogue octreotide. Clin J Am Soc Nephrol 5: 783–789, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.van Keimpema L, Nevens F, Vanslembrouck R, van Oijen MG, Hoffmann AL, Dekker HM, de Man RA, Drenth JP: Lanreotide reduces the volume of polycystic liver: A randomized, double-blind, placebo-controlled trial. Gastroenterology 137: 1661–1668, 2009 [DOI] [PubMed] [Google Scholar]
- 163.Hogan MC, Masyuk TV, Page LJ, Kubly VJ, Bergstralh EJ, Li X, Kim B, King BF, Glockner J, Holmes DR, 3rd, Rossetti S, Harris PC, LaRusso NF, Torres VE: Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J Am Soc Nephrol 21: 1052–1061, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chrispijn M, Nevens F, Gevers TJ, Vanslembrouck R, van Oijen MG, Coudyzer W, Hoffmann AL, Dekker HM, de Man RA, van Keimpema L, Drenth JP: The long-term outcome of patients with polycystic liver disease treated with lanreotide. Aliment Pharmacol Ther 35: 266–274, 2012 [DOI] [PubMed] [Google Scholar]
- 165.Hogan MC, Masyuk TV, Page L, Holmes DR, 3rd, Li X, Bergstralh EJ, Irazabal MV, Kim B, King BF, Glockner JF, Larusso NF, Torres VE: Somatostatin analog therapy for severe polycystic liver disease: Results after 2 years. Nephrol Dial Transplant 27: 3532–3539, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Schmid HA, Brueggen J: Effects of somatostatin analogs on glucose homeostasis in rats. J Endocrinol 212: 49–60, 2012 [DOI] [PubMed] [Google Scholar]
- 167.Torres VE, Bankir L, Grantham JJ: A case for water in the treatment of polycystic kidney disease. Clin J Am Soc Nephrol 4: 1140–1150, 2009 [DOI] [PubMed] [Google Scholar]
- 168.Nagao S, Nishii K, Katsuyama M, Kurahashi H, Marunouchi T, Takahashi H, Wallace DP: Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J Am Soc Nephrol 17: 2220–2227, 2006 [DOI] [PubMed] [Google Scholar]
- 169.Wang CJ, Creed C, Winklhofer FT, Grantham JJ: Water prescription in autosomal dominant polycystic kidney disease: A pilot study. Clin J Am Soc Nephrol 6: 192–197, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Chrispijn M, Gevers TJ, Hol JC, Monshouwer R, Dekker HM, Drenth JP: Everolimus does not further reduce polycystic liver volume when added to long acting octreotide: Results from a randomized controlled trial. J Hepatol 59: 153–159, 2013 [DOI] [PubMed] [Google Scholar]