

Abstract
Viridicatumtoxin (1) is a tetracycline-like fungal meroterpenoid with a unique, fused spirobicyclic ring system. Puzzlingly, no dedicated terpene cyclase is found in the gene cluster identified in Penicillium aethiopicum. The two cytochrome P450 enzymes VrtE and VrtK in the vrt gene cluster were shown to catalyze C5-hydroxylation and spirobicyclic ring formation, respectively. Feeding of acyclic previridicatumtoxin (2) to Saccharomyces cerevisiae expressing VrtK confirmed that VrtK is the sole enzyme required for cyclization of the geranyl moiety. Thus, VrtK is the first example of a P450 that can catalyze terpene cyclization, most likely via the initial oxidation of C17 to an allylic carbocation. Quantum chemical modeling revealed a possible new tertiary carbocation intermediate E that forms after the allylic carbocation formation. The intermediate E can readily undergo concerted 1,2-alkyl shift/1,3-hydride shift, either spontaneously or further aided by the active site configuration of VrtK, followed by C7 Friedel-Crafts alkylation to afford 1,. The most likely stereochemical course of the reaction was proposed based on the results of our computations.
Viridicatumtoxin (1) is a fungal meroterpenoid consists of an unusual monoterpene-derived spirobicyclic ring fused to an anhydrotetra-cycline-like, 2-carboxamide naphthacenedione scaffold.1 1 and its derivative viridicatumtoxin B exhibits anti-MRSA activity.2 Compound 1 along with a C2 acetyl analog, spirohexaline, have been demonstrated to inhibit the growth of bacteria by inhibition of the undecaprenyl diphosphate synthase,3 a potential new target for antibiotic development. The vrt gene cluster for the biosynthesis of 1 was identified in Penicilium aethiopicum using genome sequencing.4 More recently, the prenyltransferase VrtC responsible for the Friedel-Crafts alkylation at C6 of the naphthacenedione with geranyl diphosphate (GPP) to afford previridicatumtoxin (2) was characterized in detail.5 However, the mechanism for the formation of the spirobicyclic ring (Scheme 1) that is further fused to C7 of the naphthacene core, especially in the absence of a dedicated terpene cyclase encoded in the gene cluster, remains intriguing. The spirobicyclic system with a hindered C15 quaternary carbon center, is a striking structural features that sparked a recent total synthesis effort and led to structure reassignment of viridicatumtoxin B as a quinone derivative of 1.6
Scheme 1.
Spirocyclization of the geranyl moeity of 2 to afford 1.
Our initial hypothesis proposed that cyclization of the geranyl substituent of 2 could begin with the addition of a hydroxyl at the allylic C17 position, followed by protonation of the hydroxyl and formation of an allylic cation accompanied by loss of water.4 The carbocation can then initiate the cascade of cyclization reaction. We reasoned that the same enzyme that performs the C17 hydroxylation step may also orient the geranyl chain in a configuration that leads to the regiospecific C15-C20 and C7-C15 cyclization events, thereby serving the additional function as a cryptic terpene cyclase. Cyclization of terpenes catalyzed by oxygenases is exceptionally rare, one example being the oxidative cyclization of the monoterpene group of cannabigerolic-acid by a berberine bridge enzyme-like flavin-dependent monooxygenase.7
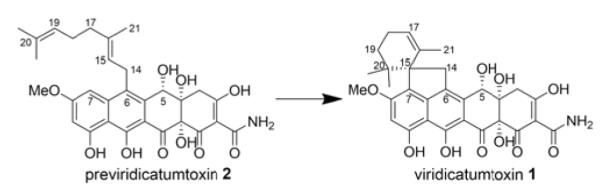
As cytochrome P450 monooxygenases are likely candidates to catalyze the proposed C17 allylic oxidation, we began the search by genetically deleting each of the two genes encoding P450 monooxygenase (vrtE and vrtK) in the vrt gene cluster in P. aethiopicum. Using a double homologous gene targeting approach described previously,4 vrtE was deleted in the P. aethiopicum ΔgsfA strain. The ΔgsfA mutant was used as a background strain due to abolishment of griseofulvin biosynthesis, which provides a cleaner chemotype for metabolite analysis. The ΔvrtE strain lost the production of 1, but accumulated a new naphthacenedione intermediate 3 (m/z 402 [M+H]+) (Figure 1 and S7). The compound was subsequently isolated and characterized by 1D and 2D-NMR. The NMR spectra of 3 comparable to that reported previously for anthrotainin9 and 5-hydroxyanthrotainin,5 except the absence of the 8-O-methyl signal. A 1,3-keto-enol tautomer can also be detected in DMSO-d6 (Table S2). Hence, the structure of 3 was assigned as the 8-O-desmethyl derivative of anthrotainin9 as shown in Figure 1D. The elucidation of the structure of 3 implies that the function of VrtE is to perform a single hydroxylation at C5 of 3. 3 is the expected product of the non-reducing PKS VrtA, which functions in collaboration with the auxiliary enzymes VrtG (dimanganese-containing Claisen Cyclase) and VrtH (C12a monooxygenase).8 The accumulation of 3 suggests downstream enzymes, including 8-O-methyltransferase (VrtD) and prenyltransferase (VrtC) require the installation of the C5-OH by VrtE for substrate recognition.
Figure 1.
Deletion of the individual P450 genes in the vrt gene cluster in P. aethiopicum. LCMS analyses of extracts from (A)ΔgsfA; (B) ΔgsfA/ΔvrtE; (C) ΔgsfA/ΔvrtK; (D) Structure of 3.
On the other hand, the vrtK deletion in the P. aethiopicum ΔgsfA strain abolished the production of 1 but accumulated a new intermediate 2 with a m/z 550 [M+H-H2O]+ and m/z 568 [M+H]+ in the culture extract (Figure 1 and S7). Comparing 2 with a standard shows that it is the uncyclized meroterpenoid 2, which is the product of the VrtC-catalyzed geranylation as shown in the previous study (Table S3).5 This confirms our hypothesis that VrtK is the P450 enzyme that initiates the cyclization of the spirobicyclic ring that eventually transforms 2 into 1. However, the genetic result alone does not exclude the possibility that other enzymes in P. aethiopicum may be involved in the cyclization step following oxidation of the terpene moiety in 2.
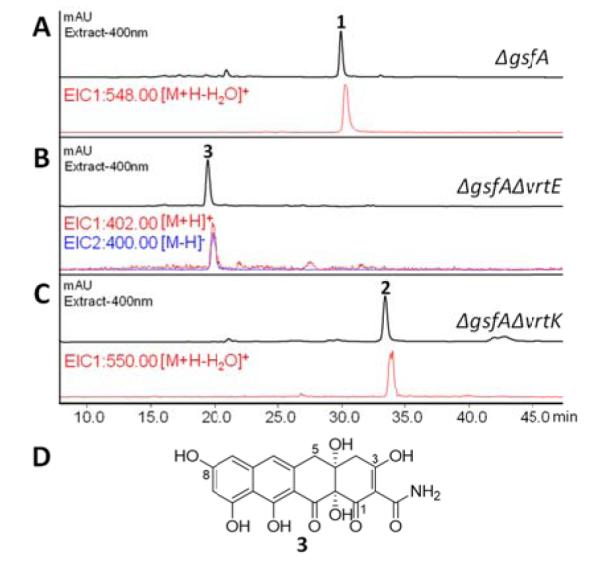
To further investigate the role of VrtK, we expressed VrtK heterologously in Saccharomyces cerevisiae BJ5464. VrtK was cloned into pESC-leu2d-AtCPR vector containing the A. terreus cytochrome P450 reductase (AtCPR) gene under the regulation of Gal10 promoter. AtCPR has been demonstrated to be able to perform the coupled reduction of fungal P450 in S. cerevisiae for biotransformations.10 The activity of VrtK was examined by feeding 1 mg of substrate 2 to 100 mL of galactose-induced yeast culture expressing AtCPR and VrtK. A slight conversion of 2 to 1 can be observed after 24 hours of incubation, while no conversion was observed in the control yeast culture expressing only AtCPR. Near complete biotransformation of 2 to 1 was obtained when 2 was fed to a culture with higher density yeast cells prepared by concentrating 500 mL of culture to 100 mL (Figure 2). No C17 hydroxylated derivative or other intermediate derived from 2 can be detected under all tested culturing conditions. The relatively slower conversion rate and the requirement of concentrated yeast cells for the biotransformation could be attributed to poor expression level of the VrtK in S. cerevisiae, the poor solubility of the substrate 2, and/or the poor entry of 2 into the yeast cells. To test if VrtK could cyclize analogs of 2 with a dimethylallyl moiety, the yeast culture expressing VrtK was fed with the dimethylallyl analog of TAN1612,5 but the substrate cannot be converted by VrtK under the same conditions (Figure S8).
Figure 2.
Biotransformation of 2 to 1 by S. cerevisiae BJ5464 co-expressing AtCPR and VrtK. The control (top) yeast harbors the pESC-leu2d-AtCPR plasmid without vrtK.
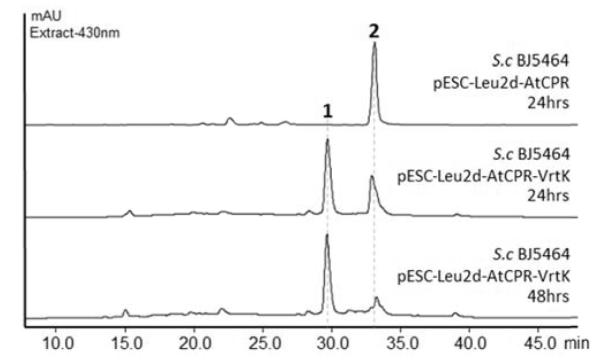
The successful biotransformation of 2 to 1 supports the proposal that VrtK is the sole enzyme required for the cyclization of the geranyl moiety of 2 to the spirobicyclic ring in 1. The cyclization could be initiated by C17-hydroxylation via the typical hydrogen abstraction/oxygen rebound mechanism followed by ionization to the allylic cation intermediate A in the presence of the Lewis acidic heme iron (Scheme 2). A similar mechanism is observed for the P450 enzyme LovA involved in lovastatin biosynthesis, whereby an allylic alcohol is first formed then dehydrates, presumably via an allylic cation intermediate, to give monacolin L by proton loss.10a However, the absence of a C-17 hydroxylated intermediate in the biotransformation assay suggests that VrtK may alternatively initiate the cyclization cascade via dehydrogenation of the allylic C17 to give a carbon radical followed by electron transfer to the iron-heme center to form the corresponding carbocation A (Scheme 2). Such a mechanism has been proposed for P450-catalyzed desaturation reactions,11 e.g. alkyl dehydrogenation/hydroxylation of capsaicinoids.12 Indeed, a homology search of VrtK indicates that the enzyme shares significant protein sequence similarity (42% identity) to the versicolorin desaturase VerB (StcL) in the aflatoxin pathway, which catalyzes desaturation of the bisfuran ring system of versicolorin B to afford versicolorin A.13
Scheme 2.
Proposed C17 carbocation formation by VrtK.
The mechanism of the geranyl cyclization was previously probed by feeding of [13C,2H] labeled mevalonates.14 The observation that two 2H atoms are present at C19 of 1 derived from feeding of (3RS)-[5-13C,4-2H2]mevalonolactone, provided evidence for a 1,3-hydride shift from C15 to C19 (corresponds to C2 and C6 of geranyl diphosphate) during the conversion of 2 to 1 (Figure 3). It was proposed that formation of the spirobicyclic ring is proceeded by the attack of C20 on C15 (Figure 3, A→B) followed by a 1,3-hydride shift to give the intermediate C. With the new biosynthetic insight, we sought to further our understanding of the mechanism of the cyclization reaction using computational quantum chemistry (mPW1PW91/6-31+G(d,p)//B3LYP/6-31+G(d,p), MPWB1K/6-31+G(d,p)//B3LYP/6-31+G(d,p) M06-2X/6-31+G(d,p)//B3LYP/6-31+G(d,p) and B3LYP/6-31+G(d,p)/6-31+G(d,p)//B3LYP/6-31+G(d,p) calculations; see Supporting Information for details and references).15 The model system shown in Figure 3 (box) was utilized to examine the inherent reactivity of putative carbocations (i.e., in the absence of the enzyme).
Figure 3.

Proposed mechanisms for the formation of 1. Previously proposed mechanism via secondary carbocation B (in black and orange path). Newly proposed mechanism based on the results of quantum chemical calculations (black and green path). Box: model systems for computational modeling. HΔ and ● indicate the labeling of 2H and 13C on the geranyl moiety from (3RS)-[5-13C,4-2H2]-mevalonolactone.13
On the basis of the results of our calculations, we propose that the mechanism postulated by Horak et al. (Figure 3; orange path) be revised to a mechanism involving tertiary carbocation intermediate E (Figure 3, green) instead of secondary carbocation B,14 which was found not to be a minimum on the potential energy surface. Cation E is predicted to undergo ring expansion (1,2-alkyl shift) and 1,3-hydride shift through a single step reaction in which these two events are combined asynchronously.16
We examined variations on this pathway, differing in the conformations/configurations of the intermediates involved, but not in the configuration of viridicatumtoxin produced. The transition state structures involved in each of these four pathways are shown in Figure 4. We predict that all four paths are energetically accessible (see Supporting Information for all energies), but path 1 (Figure 5) does not require any conformational changes of intermediates. Thus, if the reactant is preorganized by VrtK into the conformation shown in Figure 5 for path 1, rearrangement of the active site structure to accommodate conformational changes of the substrate is not required.17 The overall barrier for path 1 is predicted to be <10 kcal/mol. For the rearrangement to follow path 2, 3 or 4, the formation of additional intermediates and/or substantial changes to substrate conformation during the cyclization/rearrangement cascade are predicted to be necessary. Thus, it is likely that the active site of VrtK provides a structural template that chaperones the reactive carbocationic intermediate A to D via path 1 in such a way that allows its inherent reactivity to be expressed while reducing the energy barrier for cyclization further.15,16
Figure 4.

Different conformations/configurations of transition state structures for reaction steps along the black/green pathway shown in Figure 3.
Figure 5.

Reaction energy diagram (M06-2X/6-31+G(d,p)//B3LYP/6-31+G(d,p)) for path 1 (Figure 4, R=OH); relative energies (kcal/mol) and selected distances (Å) are shown.
To the best our knowledge, this is the first report of a cytochrome P450 enzyme that is capable of catalyzing terpene cyclization. Interestingly, the P450 albaflavenone monooxygenase from Streptomyces coelicolor A3 was recently discovered to contain a moonlighting class I terpene synthase active site and can convert farnesyl diphosphate to farnesene isomers in addition to its original function in oxidation of epi-isozizaene to albaflavenone.18 In contrast to class I terpene cyclases, which generate an carbocation by ionization of the allylic diphosphate ester, class II terpene cyclases promote cyclization by protonation of a terminal double bond.19 VrtK is similar to class II terpene cyclases in that it does not act on a diphosphate terpene substrate; however, a carbocation is proposed to be generated by oxidation of an allylic carbon (C17), corresponding to C4 of geranyl moiety. This discovery expands our knowledge of the catalytic diversity of cytochrome P450 enzymes and provides new insights into meroterpenoid biosynthesis in fungi.
Supplementary Material
ACKNOWLEDGMENT
This work is supported by NIH 1R01GM085128 and 1DP1GM106413 to YT, NRSA GM-0846 and the UCLA Graduate Division to R.A.C, and by NSF CHE-0957416 (and supercomputing resources through a grant from the XSEDE program: CHE030089) to DJT. NMR instrumentation was supported by the NSF equipment grant CHE-1048804. Dr. Dae-Kyun Ro of University of Calgary is thanked for his generous gift of the plasmid pESC-Leu2d-AtCPR.
Footnotes
Supporting Information Experimental methods, computational details and NMR spectroscopic data. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1) (a).Hutchison RD, Steyn PS, Van Rensburg SJ. Toxicol. Appl. Pharmacol. 1973;24:507. doi: 10.1016/0041-008x(73)90057-4. [DOI] [PubMed] [Google Scholar]; (b) Kabuto C, Silverton J, Akiyama T, Sankawa U, Hutchison RD, Steyn PS, Vleggaar R. J. Chem. Soc, Chem. Comm. 1976;728 [Google Scholar]
- (2).Zheng CJ, Yu HE, Kim EH, Kim WG. J. Antiboot. 2008;61:633. doi: 10.1038/ja.2008.84. [DOI] [PubMed] [Google Scholar]
- (3).Inokoshi J, Nakamura Y, Zhang H, Uchida R, Nonaka K, Masuma R, Tomoda H. J. Antiboot. 2013;66:37. doi: 10.1038/ja.2012.83. [DOI] [PubMed] [Google Scholar]
- (4).Chooi YH, Cacho R, Tang Y. Chem. Biol. 2010;17:483. doi: 10.1016/j.chembiol.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Chooi YH, Wang P, Fang J, Li Y, Wu K, Tang Y. J. Am. Chem. Soc. 2012;134:9428. doi: 10.1021/ja3028636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Nicolaou KC, Nilewski C, Hale CRH, Ioannidou HA, ElMarrouni A, Koch LG. Angew. Chem. Int. Ed. 2013;52:8736. doi: 10.1002/anie.201304691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7) (a).Shoyama Y, Tamada T, Kurihara K, Takeuchi A, Taura F, Arai S, Blaber M, Shoyama Y, Morimoto S, Kuroki R. J Mol Biol. 2012;423:96. doi: 10.1016/j.jmb.2012.06.030. [DOI] [PubMed] [Google Scholar]; (b) Taura F, Sirikantaramas S, Shoyama Y, Yoshikai K, Shoyama Y, Morimoto S. FEBS Lett. 2007;581:2929. doi: 10.1016/j.febslet.2007.05.043. [DOI] [PubMed] [Google Scholar]
- (8).Li Y, Chooi Y-H, Sheng Y, Valentine JS, Tang Y. J. Am. Chem. Soc. 2011;133:15773. doi: 10.1021/ja206906d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wong S-M, Kullnig R, Dedinas J, Appell KC, Kydd GC, Gillum AM, Cooper R, Moore R. J. Antibiot. 1993;46:214. doi: 10.7164/antibiotics.46.214. [DOI] [PubMed] [Google Scholar]
- (10) (a).Barriuso J, Nguyen DT, Li JWH, Roberts JN, MacNevin G, Chaytor JL, Marcus SL, Vederas JC, Ro DK. J. Am. Chem. Soc. 2011;133:8078. doi: 10.1021/ja201138v. [DOI] [PubMed] [Google Scholar]; (b) Cacho T, Chooi YH, Zhou H, Tang Y. ACS Chem. Bool. 2013;8:2322. doi: 10.1021/cb400541z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11) (a).de Montellano PRO, Nelson SD. Arch. Biochem. Biophys. 2011;507:95. doi: 10.1016/j.abb.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Meunier B, deVisser SP, Shaik S. Chem. Rev. 2004;104:3947. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- (12).Reilly CA, Yost GS. Drug Metab. Dispos. 2005;33:530. doi: 10.1124/dmd.104.001214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kelkar HS, Skloss TW, Haw JF, Keller NP, Adams TH. J. Bool. Chem. 1997;272:1589. doi: 10.1074/jbc.272.3.1589. [DOI] [PubMed] [Google Scholar]
- (14).Horak RM, Maharaj VJ, Marais SF, van Heerden FR, Vleggaar R. J. Chem. Soc., Chem. Comm. 1988;23:1562. [Google Scholar]
- (15) (a).Tantillo DJ. Nat. Prod. Rep. 2013;30:1079. doi: 10.1039/c3np70028c. [DOI] [PubMed] [Google Scholar]; (b) Tantillo DJ. Nat. Prod. Rep. 2011;28:1035. doi: 10.1039/c1np00006c. [DOI] [PubMed] [Google Scholar]
- (16) (a).Tantillo DJ. Chem. Soc. Rev. 2010;39:2847–2854. doi: 10.1039/b917107j. [DOI] [PubMed] [Google Scholar]; (b) Tantillo DJ. J. Phys. Org. Chem. 2008;21:561. [Google Scholar]
- (17) (a).Carpenter BK. Annu. Rev. Phys. Chem. 2005;56:57. doi: 10.1146/annurev.physchem.56.092503.141240. [DOI] [PubMed] [Google Scholar]; (b) Hong YJ, Tantillo DJ. J. Am. Chem. Soc. 2009;131:7999. doi: 10.1021/ja9005332. [DOI] [PubMed] [Google Scholar]
- (18).Zhao B, Lei L, Vassylyev DG, Lin X, Cane DE, Kelly SL, Yuan H, Lamb DC, Waterman MR. J. Biol. Chem. 2009;284:36711. doi: 10.1074/jbc.M109.064683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19) (a).Christianson DW. Chem. Rev. 2006;106:3412. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]; (b) Gao Y, Honzatko RB, Peters RJ. Nat Prod Rep. 2012;29:1153. doi: 10.1039/c2np20059g. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.