

Abstract
The intracellular signaling mechanisms that couple transient cerebral ischemia to cell death and neuroprotective mechanisms provide potential therapeutic targets for cardiac arrest. Protein phosphatase (PP)-1 is a major serine/threonine phosphatase that interacts with and dephosphorylates critical regulators of energy metabolism, ionic balance, and apoptosis. We report here that PP-1I, a major regulated form of PP-1, is activated in brain by approximately twofold in vivo following cardiac arrest and resuscitation in a clinically relevant pig model of transient global cerebral ischemia and reperfusion. PP-1I purified to near homogeneity from either control or ischemic pig brain consisted of the PP-1 catalytic subunit, the inhibitor-2 regulatory subunit, as well as the novel constituents 14-3-3γ, Rab GDP dissociation protein β, PFTAIRE kinase, and C-TAK1 kinase. PP-1I purified from ischemic brain contained significantly less 14-3-3γ than PP-1I purified from control brain, and purified 14-3-3γ directly inhibited the catalytic subunit of PP-1 and reconstituted PP-1I. These findings suggest that activation of brain PP-1I following global cerebral ischemia in vivo involves dissociation of 14-3-3γ, a novel inhibitory modulator of PP-1I. This identifies modulation of PP-1I by 14-3-3 in global cerebral ischemia as a potential signaling mechanism-based approach to neuroprotection.
Keywords: apoptosis, inhibitor-2, protein phosphorylation
Energy failure in global cerebral ischemia following cardiac arrest initiates massive ischemic cell injury and cell death (Siesjo 1988; Hou and MacManus 2002). Global cerebral ischemia induces irreversible cell death that results in severe brain damage and extremely poor neurological outcome following cardiac arrest that can be mitigated by prompt restoration of cerebral blood flow (Siesjo 1988; Siesjo et al. 1995; Martin et al. 1998; Lipton 1999; Liu et al. 2002; Bhardwaj et al. 2003). Ischemic cell damage continues into the post-ischemic reperfusion period following successful resuscitation as a result of reperfusion injury (Cao et al. 1988; Oliver et al. 1990; White et al. 2000). The cell signaling mechanisms that link cerebral ischemia to excitotoxicity, ionic dysregulation, cell death, and ischemic tolerance, an endogenous neuroprotective mechanism, are complex and not fully characterized. Energy depletion and membrane depolarization cause release of excitotoxic glutamate, activation of ionotropic NMDA- and α-amino-3-hydroxy-5-methylisoxazole-4-propionate-type glutamate receptors, and pathological Ca2+ influx that activates multiple cell signaling pathways involved in ischemic cell death (Siesjo 1988; Siesjo et al. 1995; Kristian and Siesjo 1998; Lipton 1999; Hou and MacManus 2002; Sugawara et al. 2004). The generation of reactive oxygen and nitrogen species upon reperfusion produces oxidative stress that is fundamental to reperfusion injury (Kontos 1985; Cao et al. 1988; Siesjo et al. 1989; Siesjo et al. 1995; Chan 2001; Oliver et al. 1990). The molecular mechanisms linking elevated Ca2+, reactive oxygen and nitrogen species and other signaling pathways to cell death and survival remain to be delineated.
Cell death in cerebral ischemia occurs by both necrotic and apoptotic mechanisms that are highly regulated by protein phosphorylation mechanisms (Wieloch et al. 1996). Protein phosphorylation is also involved in endogenous neuroprotective mechanisms such as ischemic pre-conditioning (Dirnagl et al. 2003). While considerable evidence links protein kinases to the control of ischemic cell death (Aronowski et al. 1992; Hu and Wieloch 1995; Blanck et al. 2000; Bright et al. 2004), relatively little is known about the role of protein phosphatases (PPs). PP-1, PP-2A, and PP-2B have all been implicated in the biochemical regulation of apoptosis (Klumpp and Krieglstein 2002; Garcia et al. 2003; Van Hoof and Goris 2003). The catalytic subunit of PP-1 (PP-1cat) binds and/or dephosphorylates multiple proteins involved in apoptosis, including Bad, Bcl-2, and Rb (Ayllon et al. 2001; Wang et al. 2001), and in excitotoxicity, including α-amino-3-hydroxy-5-methylisoxazole-4-propionate- and NMDA-type glutamate receptors (Greengard 2001). Based on these key connections, we hypothesized that activation of PP-1 might be a component of the cell signaling pathways that link cerebral ischemia to cell death. PP-1I, which consists of PP-1cat, inhibitor-2 (I-2), and a regulatory protein kinase, is a major form of PP-1 in brain that requires phosphorylation of T72 of the regulatory subunit I-2 for activation of catalytic activity (Yang and Fung 1985; Tung and Reed 1989; Agarwal-Mawal and Paudel 2001). Here, we show that brain PP-1I is activated following cardiac arrest in a clinically relevant pig model of cardiac arrest and resuscitation in vivo. The mechanism by which PP-1I is activated appears to involve reduced association with a novel inhibitory PP-1 regulatory protein identified as 14-3-3γ.
Experimental procedures
Materials
ATP, benzamidine, DEAE Sepharose, poly-L-lysine agarose, phenylmethylsulfonyl fluoride (PMSF), protamine, phosphorylase b, phosphorylase kinase, catalytic subunit of cAMP-dependent protein kinase (PKA), bovine serum albumin and pentobarbital were from Sigma-Aldrich (St Louis, MO, USA). Tiletamine and zolazepam were from Reading (Carros, France). Xylazine was from Bayer AG (Leverkuser-Bayerwerk, Germany). GE Healthcare (Piscataway, NJ, USA) provided [γ-32P]ATP. Anti-PP-1cat, anti-I-2, and anti-14-3-3γ were from Chemicon (Temecula, CA, USA).
Protein preparation
Full-length human dopamine- and cyclic AMP-regulated neuronal phosphoprotein Mr 32 kDa (DARPP-32), PP-1 catalytic subunit α (PP-1αcat), I-2, Bad and phosphorylase kinase γ were expressed in bacteria using the pTrcHis-Topo vector and purified by chromatography on Ni-agarose as recommended by the manufacturer (Invitrogen, Carlsbad, CA, USA). 32P-labeled phosphorylase a was prepared by phosphorylation of phosphorylase b by phosphorylase kinase as described (Cohen et al. 1988). DARPP-32 was phosphorylated by PKA as described (Hemmings et al. 1984b). 32P-labeled Bad was prepared by phosphorylation with PKA as described (Lizcano et al. 2000). PP-1I devoid of activating kinase was reconstituted by incubating purified recombinant PP-1αcat (300 μg) and I-2 (200 μg) in 50 mM imidazole-Cl pH 7.2, 0.2 mM EGTA and 0.1% (v/v) 2-mercaptothanol at 30°C for 30 min. Reconstituted PP-1I was then purified by gel filtration on Superdex 200 as described (Tung and Cohen 1984).
Cardiac arrest model
Experiments were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals as approved by the Institutional Animal Care and Use Committee of Weill Cornell Medical College. Female Yorkshire pigs (~25 kg) were sedated with tiletamine (2.2 mg/kg i.m.), zolazepam (2.2 mg/kg i.m.), and xylazine (2.2 mg/kg i.m.), and transported to the laboratory. An intravenous catheter was placed, anesthesia was induced with pentobarbital (5 mg/kg i.v.), the trachea intubated, and mechanical ventilation initiated with room air. Anesthesia was maintained throughout the procedure by intravenous infusion of pentobarbital (8 mg/kg/h) in 0.9% NaCl at 5 mL/kg/h. Temperature was maintained at 37 ± 0.5°C by a warming blanket. Following insertion of femoral venous and arterial catheters for rapid drug administration and blood pressure monitoring, respectively, a median sternotomy was performed to expose the heart. Ventricular fibrillation was induced by rapid pacing (rate of 800, output of 16 mA) and confirmed by visual inspection, electrocardiography, and loss of pulsatile arterial pressure. Ventilation was discontinued at the onset of fibrillation. After 10 min of cardiac arrest, ventilation with 100% O2 was initiated and spontaneous cardiac function was restored by intravenous administration of lidocaine (1.5 mg/kg), epinephrine (0.5 mg), and sodium bicarbonate (10 mEq) with open chest cardiac massage at 60/min for 2 min, followed by internal defibrillation (20–50 J). After defibrillation and resumption of effective circulation, systolic blood pressure was maintained > 60 mmHg by intermittent injection of 50–100 μg epinephrine as required. Following 2 h of reperfusion and ventilation with 100% O2, animals were killed by intracardiac injection of 10 mL saturated KCl, and the brain was quickly removed (< 3 min) through a ventral approach through the palate and chilled on ice. Control animals were treated identically except that a fatal dose of KCl was injected without ventricular fibrillation and resuscitation.
Preparation of brain extracts
The rostral brain (~100 g; including cerebral cortex, hippocampus, basal ganglia, thalamus, and hypothalamus) from a control animal and an experimental animal that underwent cardiac arrest/resuscitation on the same day were treated identically for each experiment. Each brain was homogenized in five volumes of ice-cold 50 mM imidazole–Cl pH 7.3, 2 mM EDTA, 2 mM EGTA, 0.1% (v/v) 2-mercaptoethanol, 1 mM benzamidine, 0.1 mM PMSF, and 5% (v/v) glycerol using 6 × 30 s pulses at low speed in a blender. The homogenate was centrifuged at 10 000 g for 30 min at 4°C. The supernatant (soluble extract) was collected, analyzed for PP-1I activity, and used for purification of PP-1I (see below).
Assay of PP-1I
Brain PP-1I was assayed for its ability to dephosphorylate added 32P-labeled phosphorylase a following pre-incubation with ATP and Mg2+ to allow activation by endogenous protein kinase activity. PP-1I is the only known Mg2+/ATP-dependent phosphorylase a phosphatase (Cohen et al. 1988). The assay consisted of 0.01 mL of brain extract or partially purified PP-1I in Dilution Buffer (50 mM imidazole–Cl pH 7.3, 0.2 mM EGTA, 0.1% (v/v) 2-mercaptoethanol, and 1 mg/mL bovine serum albumin); 0.01 mL of Assay Buffer (50 mM imidazole–Cl pH 7.2, 0.2 mM EGTA, 0.1% (v/v) 2-mercaptoethanol, 1 mg/mL bovine serum albumin, ±0.375 mM ATP/3.75 mM MgCl2, ±300 nM phospho-T34-DARPP-32 (a specific inhibitor of PP-1; Hemmings et al. 1984a,b); and 0.01 mL of 30 μM 32P-labeled phosphorylase a in Dilution Buffer plus 75 mM caffeine. PP-1I was pre-incubated with Mg2+/ATP in Assay Buffer for 5 min at 30°C prior to initiation of the reaction with 32P-labeled phosphorylase a. Reactions proceeded for 10 min at 30°C and were terminated with 0.2 mL of 25% (v/v) trichloroacetic acid. The resulting suspension was centrifuged at 10 000 g for 5 min in a microcentrifuge, and 0.2 mL of the supernatant containing released [32P]phosphate was analyzed for Cerenkov radiation in a liquid scintillation spectrometer. One unit of PP-1I catalyzes the dephosphorylation of 1 nmol of phosphate per min at 30°C in aMg2+/ATP-dependent manner. Basal PP-1 activity was determined as above with the omission of Mg2+/ATP.
Purification of PP-1I from control and ischemic brain following cardiac arrest and resuscitation
Purification procedures were performed at 4°C. Soluble brain extract from each forebrain (100 g) was collected, diluted twofold in 25 mM imidazole–Cl pH 7.3, 0.2 mM EGTA, 0.1% (v/v) 2-mercaptoethanol, 1 mM benzamidine, 0.1 mM PMSF and 10% (v/v) glycerol (Buffer A), and loaded onto a DEAE Sepharose column (2.5 × 20 cm) equilibrated in Buffer A. The column was washed with 300 mL of Buffer A plus 50 mM NaCl and eluted with Buffer A plus 300 mM NaCl (flow rate 60 mL/h; 7 mL fractions). Active fractions of PP-1I were collected, diluted 10-fold with Buffer A, and loaded onto a poly-L-lysine column (1.5 × 10 cm) equilibrated in Buffer A. The column was washed with 150 mL of Buffer A, and eluted successively with Buffer A plus 50 mM NaCl, 100 mM NaCl, 250 mM NaCl, and 500 mM NaCl (flow rate 30 mL/h; 4.5 mL fractions). PP-1I from the 250 mM NaCl fraction was collected and PP-1I activity was determined. Total PP-1I activity of the partially purified enzyme from the poly-L-lysine agarose chromatography step was quantified as the area under the peak of activity (units). The pooled fractions were concentrated by vacuum dialysis, and separated on a Superdex 200 column (1.5 × 60 cm) equilibrated in Buffer A plus 200 mM NaCl. Active fractions from the Superdex 200 column were collected and purified on a Mono Q column (0.5 × 10 cm) using a gradient of NaCl in Buffer A as recommended by the manufacturer (GE Healthcare). Active fractions of PP-1I from the Mono Q column were collected, concentrated by vacuum dialysis, and stored at −20°C in Buffer A with 50% (v/v) glycerol.
Identification of proteins by mass spectrometry
The PP-1I complex was separated on sodium dodecyl sulfate/ polyacrylamide gel electrophoresis (Laemmli 1970), and protein bands identified by Sypro Ruby staining (Molecular Probes, Portland, OR, USA) were excised and washed three times in 50 mM NH4HCO3 (pH 8.8) in 50% (v/v) acetonitrile. Gel slices were incubated at 30°C overnight with 1 μg of trypsin in 0.5 mL 50 mM NH4HCO3 and 0.05% (v/v) Zwittergent-3–16 (Sigma-Aldrich). Released tryptic peptides were dried in a rotary evaporator and reconstituted in 60% (v/v) acetonitrile and 0.1% (v/v) trifluoroacetic acid. Matrix-assisted laser desorption time of flight mass spectrometry spectra were obtained on a Voyager RP instrument (Framington, MA, USA) at The Rockefeller University Protein Core Facility (New York, NY, USA) averaged over 100–300 laser shots. The identities of peptides were determined by searching a human protein sequence database (Henzel et al. 1993; Shevchenko et al. 1996).
Miscellaneous methods
Protein concentration was determined by the method of Bradford (1976) using bovine serum albumin as standard. Immunoblotting was performed as described (Burnette 1981) except that immunoreactivity was detected with alkaline phosphatase conjugated anti-mouse or anti-rabbit secondary antibody using 5-bromo-4-chloro-3-indoyl phosphate and nitroblue tetrazolium for colorimetric detection according to the manufacturer’s instructions (Bio-Rad, Hercules, CA, USA). Gels and immunoblots were scanned on a flat bed gel scanner (PhosphorImager; Molecular Dynamics, Sunnyvale, CA, USA) and analyzed using NIH Image (http://rsb.info.nih.gov/nih-image/).
Statistical analysis
Differences between groups were determined by the Student t-test, with p < 0.05 accepted as statistically significant.
Results
PP-1I activity in pig brain
Soluble extracts of pig brain contain large amounts of spontaneously active PP activity because of PP-1 and PP-2A using phosphorylase a as a representative phosphoserine substrate (Tung and Reed 1989; Tung et al. 1997). PP-1I, also known as Mg2+/ATP-dependent PP-1, is a complex of the PP-1cat with the I-2 regulatory subunit (Hemmings et al. 1982; Tung and Reed 1989). It is activated via phosphorylation of T72 of I-2 by multiple protein kinases in vitro, although the physiologically relevant kinase(s) in vivo is unknown (Holmes et al. 1987; Wang et al. 1995; Agarwal-Mawal and Paudel 2001; Leach et al. 2003). The contribution of PP-1I to total brain phosphorylase phosphatase activity was determined as the Mg2+/ATP-dependent PP-1 activity using a maximally effective concentration of phospho-T34-DARPP-32, a specific inhibitor of PP-1 to define PP-1 activity (Hemmings et al. 1984a). PP-1I accounted for 22% of total soluble brain phosphorylase phosphatase activity (PP-1 + PP-2A) and 31% of the total PP-1 activity in control pig brain extract (Fig. 1).
Fig. 1.
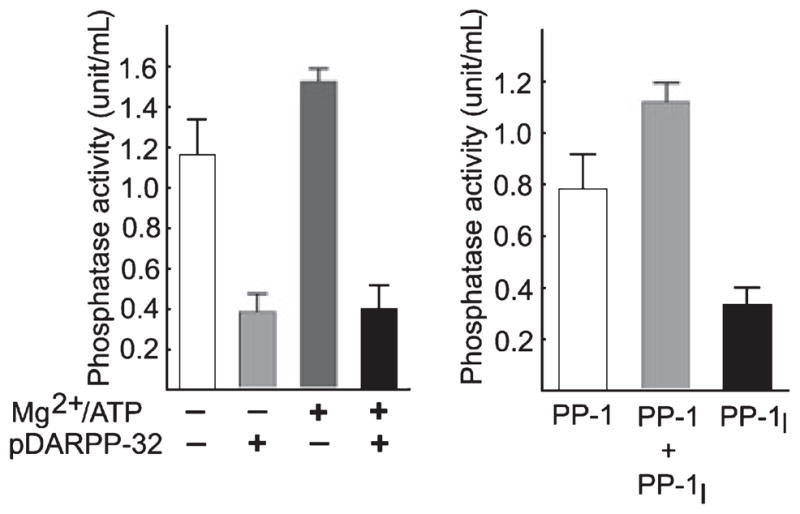
Identification of protein phosphatase (PP)-1I activity in pig brain. (a) Phosphorylase phosphatase activity was determined in soluble pig brain extracts in the absence or presence of Mg2+/ATP and phospho-T34-DARPP-32, as indicated. PP-1 activity is defined as phospho-T34-DARPP-32-sensitive phosphorylase phosphatase activity; the residual activity is primarily because of PP-2A. (b) The activity of PP-1I is defined as Mg2+/ATP-dependent PP-1 activity, or the difference between PP-1 activity measured in the absence (basal PP-1) or presence (basal PP-1 + PP-1I) of Mg2+/ATP. Mean ± SD (n = 3).
PP-1I activation by global cerebral ischemia
The activity of PP-1I was determined in control pig brain and brain from pigs subjected to 10 min of cardiac arrest followed by resuscitation and 2 h of reperfusion. Brain PP-1I was activated 1.6-fold following ischemia/reperfusion compared with control in crude extracts (Fig. 2). There was no activation of spontaneously active PP-1 (data not shown). Indirect determination of PP-1I activity in crude extracts can be inaccurate as Mg2+/ATP can inhibit free PP-1 and PP-2A (Ingebritsen and Cohen 1983). Measurement of PP-1I activity in the absence of free PP-1 and PP-2A was achieved by partial purification using DEAE Sepharose and poly-L-lysine agarose chromatography to separate interfering phosphatases and inhibitors (Yang and Fung 1985; Tung and Reed 1989; Tung et al. 1997). Total PP-1I activity, quantified as the area under the peak of Mg2+/ATP-dependent PP-1 activity eluted from poly-L-lysine agarose, was 2.2-fold greater in brain subjected to cardiac arrest and resuscitation compared with control brain (Fig. 3).
Fig. 2.

Activation of brain protein phosphatase (PP)-1I by global cerebral ischemia. Total activity of PP-1I was determined in soluble brain extracts from control pigs and pigs subjected to 10 min of cardiac arrest followed by resuscitation and reperfusion for 2 h. PP-1I activity was quantified as described in Fig. 1. Mean ± SD (n = 3). *p < 0.05 versus control (Student t-test).
Fig. 3.

Quantification of protein phosphatase (PP)-1I following partial purification of brain extracts from control and ischemic brain. The activity of PP-1I is defined as the activity in the presence of Mg2+/ATP minus activity in the absence of Mg2+/ATP. (a) Soluble brain extracts from a control pig and from a pig subjected to 10 min of cardiac arrest followed by resuscitation and reperfusion for 2 h were prepared as described in Experimental procedures and loaded onto a DEAE Sepharose column. PP-1I activity from control (■) and ischemic (●) brain was determined. Data are shown for a single representative experiment (n = 3). (b) Active fractions from the DEAE Sepharose column were pooled as indicated by the bar in (a) and loaded onto a poly-L-lysine agarose column. PP-1I activity from control (■) and ischemic (●) brain was determined. (c) Total PP-1I activity from the poly-L-lysine agarose chromatography step from control or ischemic brain was quantified by determining the area under the peak of activity. Mean ± SD (n = 3). *p < 0.05 versus control (Student t-test).
Purification and properties of PP-1I from control and ischemic brain
Protein phosphatase-1I was purified to near homogeneity from control and ischemic brain by successive chromatography of soluble extracts on DEAE Sepharose, poly-L-lysine agarose, Superdex 200 and Mono Q. Analysis by sodium dodecyl sulfate/polyacrylamide gel electrophoresis showed that PP-1I from control and ischemic brain contained six major proteins with apparent molecular masses 81, 53, 50, 37, 32, and 31 kDa (Fig. 4). These proteins were identified by matrix-assisted laser desorption time of flight mass spectrometry as C-TAK1 kinase (81 kDa), PFTAIRE kinase (53 kDa), Rab GDP dissociation inhibitor protein β (50 kDa), PP-1αcat (37 kDa), 14-3-3γ (32 kDa), and PP-1 I-2 (31 kDa) (Fig. 4b). All three isoforms of PP-1cat (α, β, and γ) were identified in purified PP-1I by immunoblotting with isoform-specific antibodies (data not shown). Purified PP-1I from control and ischemic brain eluted with an apparent molecular mass of 140 and 145 kDa, respectively, by gel filtration (Fig. 5). Taken together with the mass spectrometry data, the results suggest that the purified preparations of PP-1I contain several complexes. Complexes in the purified PP-1I preparation consistent with the observed molecular mass data include PP-1cat : I-2 : 14-3-3γ : PFTAIRE kinase (~140 kDa); PP-1cat : I-2 : C-TAK1 (~144 kDa); and PP-1cat : I-2 : 14-3-3γ (~95 kDa), although other combinations are possible.
Fig. 4.

Identification of protein components of purified protein phosphatase (PP)-1I by mass spectrometry. (a) Purified PP-1I from control (C) and ischemic/reperfused (IR) pig brain was subjected to sodium dodecyl sulfate/polyacrylamide gel electrophoresis. The bars on the left indicate the migration of marker proteins. The bars on the right indicate the migration of the major proteins present in purified PP-1I. (b) Protein bands were excised from the gel and digested with trypsin, and tryptic peptides were analyzed by matrix-assisted laser desorption time of flight mass spectrometry (MALDI ToF MS). The molecular masses and amino acid sequences of the tryptic peptides identified for each major protein are shown. (a and b) migrated as a doublet; analysis of both bands yielded peptides derived from C-TAK1.
Fig. 5.

Analysis of purified protein phosphatase (PP)-1I by gel filtration. Purified PP-1I from control (left) and ischemic (right) brain were analyzed on a Superdex 200 column (1.0 × 60 cm) at a flow rate of 0.75 mL/min with collection of 1.25 mL fractions. The arrows indicate the void volume (Vo) and the elution positions of the marker proteins.
Confirmation of a PP-1I : I-2 : 14-3-3γ complex by co-immunoprecipitation
The presence of PP-1αcat, I-2, and 14-3-3γ in PP-1I purified from control brain was confirmed by immunoblotting with specific antibodies (Fig. 6a). Immunoprecipitation of purified PP-1I with anti-PP-1αcat followed by immunoblotting with anti-14-3-3γ and anti-I-2, or immunoprecipitation with anti-14-3-3γ followed by immunoblotting with anti-PP-1αcat and anti-I-2 confirmed that 14-3-3γ interacts with these proteins within PP-1I (Fig. 6b and c). Various 14-3-3 isoforms can bind PP-1cat (Margolis et al. 2003; Huang et al. 2004; Pozuelo Rubio et al. 2004), and inhibit PP activity in cell extracts or purified PP-1cat in vitro (Chen and Wagner 1994; Muslin et al. 1996; Banik et al. 1997; Margolis et al. 2003). We therefore determined the effect of purified 14-3-3γ on the activities of PP-1αcat and a minimal form of PP-1I reconstituted from purified PP-1cat and I-2. 14-3-3γ inhibited both PP-1αcat and reconstituted PP-1I activity using phospho-Bad as a relevant substrate with IC50 values of ~350 nM (Fig. 7).
Fig. 6.

Identification of a protein phosphatase (PP)-1I : inhibitor-2 (I-2) : 14-3-3γ complex in control brain. (a) Purified control brain PP-1I was immunoblotted with anti-PP-1αcat (lane 1), anti-14-3-3γ (lane 2), or anti-I-2 (lane 3). (b) Purified control brain PP-1I was immunoprecipitated with anti-PP-1αcat followed by immunoblotting with anti-14-3-3γ (left) or anti-I-2 (right). (c) Purified control brain PP-1I was immunoprecipitated (IP) with anti-14-3-3γ followed by immunoblotting (IB) with anti-PP-1αcat (left) or anti-I-2 (right).
Fig. 7.

Inhibition of protein phosphatase (PP)-1 catalytic subunit α (PP-1αcat) and reconstituted PP-1αcat : inhibitor-2 (I-2) by 14-3-3γ. Purified PP-1αcat (■) and reconstituted PP-1αcat : I-2 (●) were pre-incubated with various amounts of 14-3-3γ for 10 min and then assayed using [32P]phospho-Bad as substrate.
Mechanism of PP-1I activation in global cerebral ischemia
Having identified 14-3-3γ as a novel inhibitory modulator of native brain PP-1I, we compared the amounts of 14-3-3γ present in PP-1I purified from control versus ischemic pig brain. Densitometric scans indicated that the relative amount of 14-3-3γ was significantly reduced in PP-1I purified from ischemic versus control brain, while the relative amounts of the other identified proteins were not significantly different (Fig. 8a–c). The finding of reduced 14-3-3γ in ischemic versus control brain was confirmed by quantitative immunoblotting for 14-3-3γ (Fig. 8d–f).
Fig. 8.

Quantification of 14-3-3γ in purified protein phosphatase (PP)-1I from control and ischemic brain. (a) PP-1I was purified to near homogeneity from control (C) and ischemic/reperfused (IR) pig brain as described in Experimental procedures, analyzed by sodium dodecyl sulfate/polyacrylamide gel electrophoresis, and stained with Sypro Ruby which identified six major bands (a–f). (b) Densitometric scans of gels obtained from PP-1I purified from control (top) or ischemic (bottom) pig brain. The migration of 14-3-3γ is indicated by an asterisk. (c) Quantification of proteins present in PP-1I purified from control (C) and ischemic brain (IR) pig brain. Relative protein amounts were quantified as peak area obtained by densitometric scans of stained gels. Mean ± SD (n = 3). *p < 0.05 versus control (Student t-test). (d) Immunoblot analysis of 14-3-3γ in purified PP-1I from control (C) and ischemic (IR) brain. (e) Densitometric scans of immunoblots of purified PP-1I from control (top) or ischemic (bottom) pig brain. (f) Quantification of immunoreactive 14-3-3γ in purified PP-1I from control (C) or ischemic (IR) pig brain as peak area. Mean ± SD (n = 3). *p < 0.05 versus control (Student t-test).
Discussion
Protein phosphatase-1I is a highly regulated form of PP-1 that is involved in the regulation of multiple targets relevant to cerebral ischemia, in particular excitatory ion channels and cell death mediators (Cohen 1989; Greengard 2001; Garcia et al. 2003). We found that brain PP-1I was significantly activated by global cerebral ischemia using a clinically relevant model of ischemia/reperfusion. This is the first direct demonstration that a regulated form of PP-1 (PP-1I) is activated by transient global cerebral ischemia in vivo. As PP-1 plays such a prominent role in the dephosphorylation of several regulators of cell death (e.g. Bad, Bcl-2, and Rb) (Ayllon et al. 2001, 2002; Wang et al. 2001), our results suggest that activation of PP-1I contributes to cell death in ischemia and reperfusion. PP-1I could also dephosphorylate other phospho-proteins that contribute to neurotoxicity in ischemia and reperfusion such that PP-1I might also have a neuroprotective role. Further analysis of this signaling pathway should resolve this question. Characterization of PP-1I purified from ischemic or control brain indicates that activation of PP-1I probably involves dissociation of 14-3-3γ, a novel inhibitory modulator of PP-1 found to interact with PP-1I. Altered regulation of PP-1, a multifunctional cell signaling enzyme, represents a potentially critical event in cerebral ischemia. However, the pathophysiological significance of PP-1I activation in global cerebral ischemia remains to be tested by the application in vivo of membrane-permeable PP-1-specific inhibitors currently under development.
Previous studies have implicated PP-1 in the control of apoptosis in a number of cell types based on the protective effects of non-selective small molecule phosphatase inhibitors that do not distinguish between PP-1 and PP-2A (Morana et al. 1996; N’cho and Brahmi 1999; Chatfield and Eastman 2004). Both inhibition (Munoz et al. 2000) and activation (Yung and Tolkovsky 2003) of PP activity have been reported using in vitro models of ischemia induced by oxygen and glucose deprivation in differentiated PC12 cells and astrocytes. Initial inhibition followed by activation of PP-1 and/or PP-2A has also been reported in a rat model of transient global cerebral ischemia (Martin de la Vega et al. 2001, 2002). However, in those studies only spontaneously active forms of PP-1 and PP-2A were measured using non-selective inhibitors of PP-1 and PP-2A. Our more focused studies involving partial purification and biochemical characterization of PP-1 from brain subjected to ischemia and reperfusion in vivo provide more direct evidence that PP-1I, a major form of regulated PP-1, is activated following global cerebral ischemia with reperfusion.
Protein phosphatase-1I purified from brain subjected to global cerebral ischemia contained significantly less of the novel PP-1 inhibitor protein 14-3-3γ compared with control PP-1I. These findings suggest that dissociation of inhibitory 14-3-3γ is involved in the ischemic activation of PP-1 activity. The reduced association was specific for 14-3-3γ and was evident following 2 h of reperfusion, so it is unlikely to be an artifact of postmortem processing. Whether PP-1I activation occurs in neurons and/or glia is unknown; this issue demands higher resolution assays of PP-1I activation under development. The Ca2+-dependent protein kinases Ca2+/calmodulin-dependent protein kinase II and protein kinase Cδ have also been implicated in ischemic neuronal death (Hajimohammaddreza et al. 1995; Bright et al. 2004), and can phosphorylate 14-3-3 (Ellis et al. 2003; Hamaguchi et al. 2003). We are exploring the possibility that phosphorylation of 14-3-3γ by Ca2+/calmodulin-dependent protein kinase II and/or protein kinase Cδ induces its dissociation from PP-1I, thereby activating PP-1I and the dephosphorylation of downstream substrates involved in the regulation of cell death. Identification of ischemic activation of PP-1I and its modulation by 14-3-3γ provide the rationale for designing specific inhibitors of PP-1I activation in order to directly test the pathophysiological significance of PP-1I activation in ischemia and other neurodegenerative disorders.
Although our results suggest that 14-3-3γ dissociation is involved in PP-1I activation in brain following ischemia/ reperfusion, the loss of 14-3-3γ by itself might not be the sole mechanism of PP-1I activation. Potential mechanisms of PP-1I activation by ischemia/reperfusion include stimulation of an activating kinase, inhibition of an inhibitory kinase, recruitment of an activator and/or loss of an inhibitor. PP-1I is activated following phosphorylation of I-2 by an unidentified endogenous protein kinase. Preliminary results indicate that this protein kinase is associated with purified PP-1I, but is not glycogen synthase kinase-3, cyclin-dependent protein kinase-5, or extracellular regulated protein kinase-1 based on the insensitivity of purified brain PP-1I activation to selective inhibitors of these kinases (data not shown). We are currently investigating the possibility that the PP-1I activating kinase is one of the kinases identified in purified PP-1I.
Acknowledgments
This work was supported by the Department of Anesthesiology of Weill Cornell Medical College, and by the Phaekhim-Sekekos Foundation Fund for Biomedical Research (New York, NY).
Abbreviations used
- I-2
inhibitor-2
- PKA
cAMP-dependent protein kinase
- DARPP
dopamine- and cyclic AMP-regulated neuronal phosphoprotein Mr 32 kDa
- PMSF
phenylmethylsulfonyl fluoride
- PP
protein phosphatase
- PP-1cat
catalytic subunit of PP-1
- PP-1αcat
PP-1 catalytic subunit α
References
- Agarwal-Mawal A, Paudel HK. Neuronal Cdc-2-like protein kinase (Cdk5/p25) is associated with protein phosphatase-1 and phosphorylates inhibitor-2. J Biol Chem. 2001;276:23712–23718. doi: 10.1074/jbc.M010002200. [DOI] [PubMed] [Google Scholar]
- Aronowski J, Grotto JC, Waxhan MN. Ischemia-induced translocation of Ca2+/calmodulin-dependent protein kinase II: potential role in neuronal damage. J Neurochem. 1992;58:1743–1753. doi: 10.1111/j.1471-4159.1992.tb10049.x. [DOI] [PubMed] [Google Scholar]
- Ayllon V, Cayla X, Garcia A, Roncal F, Fernandez R, Albar JP, Martinez A, Rebollo A. Bcl-2 targets protein phosphatase-1α to Bad. J Immunol. 2001;166:7345–7352. doi: 10.4049/jimmunol.166.12.7345. [DOI] [PubMed] [Google Scholar]
- Ayllon V, Cayla X, Garcia A, Fleisher A, Angelita R. The anti-apoptotic molecules Bcl-xL and Bcl-w target protein phosphatase 1α to Bad. Eur J Immunol. 2002;32:1847–1855. doi: 10.1002/1521-4141(200207)32:7<1847::AID-IMMU1847>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Banik U, Wang GA, Wagner PD, Kaufman S. Interaction of phosphorylated tryptophan hydroxylase with 14-3-3 proteins. J Biol Chem. 1997;272:26219–26225. doi: 10.1074/jbc.272.42.26219. [DOI] [PubMed] [Google Scholar]
- Bhardwaj A, Alkayed NJ, Kirsch JR, Hurn PD. Mechanisms of ischemic brain damage. Curr Cardiol Rep. 2003;5:160–167. doi: 10.1007/s11886-003-0085-1. [DOI] [PubMed] [Google Scholar]
- Blanck TJJ, Halle M, Xu F, Heerdt P, Beckmann J, Kang R, Adamo A, Hemmings HC., Jr Isoflurane pretreatment ameliorates postischemic neurological dysfunction and preserves hippocampal Ca2+/calmodulin-dependent protein kinase II in a canine cardiac arrest model. Anesthesiology. 2000;93:1285–1293. doi: 10.1097/00000542-200011000-00023. [DOI] [PubMed] [Google Scholar]
- Bradford AA. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;76:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Bright R, Raval AP, Dembner JM, Perez-Pinzon MA, Steinberg GK, Yenari MA, Mochly-Rosen D. Protein kinase Cδ mediates cerebral reperfusion injury in vivo. J Neurosci. 2004;24:6880–6888. doi: 10.1523/JNEUROSCI.4474-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnette WN. “Western blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified cellulose and radiographic detection with antibody and radioiodinated protein A. Anal Biochem. 1981;112:195–203. doi: 10.1016/0003-2697(81)90281-5. [DOI] [PubMed] [Google Scholar]
- Cao W, Carney JM, Duchon A, Floyd RA, Chevion M. Oxygen free radical involvement in ischemia and reperfusion injury to brain. Neurosci Lett. 1988;88:233–238. doi: 10.1016/0304-3940(88)90132-2. [DOI] [PubMed] [Google Scholar]
- Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- Chatfield K, Eastman A. Inhibitors of protein phosphatase 1 and 2A differentially prevent intrinsic and extrinsic apoptosis pathways. Biochem Biophys Res Commun. 2004;323:1313–1320. doi: 10.1016/j.bbrc.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Chen F, Wagner PD. 14-3-3 proteins bind histone and affect both histone phosphorylation and dephosphorylation. FEBS Lett. 1994;347:128–132. doi: 10.1016/0014-5793(94)00520-6. [DOI] [PubMed] [Google Scholar]
- Cohen P. The structure and regulation of protein phosphatases. Annu Rev Biochem. 1989;58:453–508. doi: 10.1146/annurev.bi.58.070189.002321. [DOI] [PubMed] [Google Scholar]
- Cohen P, Alemany S, Hemmings BA, Resink TJ, Stralfors P, Tung HYL. Protein phosphatase-1 and protein phosphatase-2A from rabbit skeletal muscle. Methods Enzymol. 1988;159:390–409. doi: 10.1016/0076-6879(88)59039-0. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Simon RP, Hallenbeck JM. Ischemic tolerance and endogenous neuroprotection. Trends Neurosci. 2003;26:248–254. doi: 10.1016/S0166-2236(03)00071-7. [DOI] [PubMed] [Google Scholar]
- Ellis JJ, Valencia TG, Zeng H, Roberts LD, Deaton RA, Grant SR. CaM kinase IIδ phosphorylation of 14-3-3b in vascular smooth muscle cells: activation of class II HDAC repression. Mol Cell Biochem. 2003;242:153–161. [PubMed] [Google Scholar]
- Garcia A, Cayla X, Guergnon J, Dassauge F, Hospital V, Rebollo MP, Fleisher A, Rebollo A. Serine/threonine protein phosphatases PP1 and PP2A are key players in apoptosis. Biochimie. 2003;85:721–726. doi: 10.1016/j.biochi.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Greengard P. The neurobiology of slow synaptic transmission. Science. 2001;294:1024–1038. doi: 10.1126/science.294.5544.1024. [DOI] [PubMed] [Google Scholar]
- Hajimohammaddreza L, Probert AW, Coughenour LL, Borosky SA, Marcoux FW, Boxer PA, Wang KKW. A specific inhibitor of calcium/calmodulin-dependent protein kinase-II provides neuroprotection against NMDA- and hypoxia/hypoglycemia-induced cell death. J Neurosci. 1995;15:4093–4101. doi: 10.1523/JNEUROSCI.15-05-04093.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaguchi A, Suzuki E, Murayama K, et al. Sphingosine-dependent protein kinase-1, directed to 14-3-3, is identified as the kinase domain of protein kinase Cδ. J Biol Chem. 2003;278:41557–41565. doi: 10.1074/jbc.M305294200. [DOI] [PubMed] [Google Scholar]
- Hemmings BA, Resink TJ, Cohen P. Reconstitution of Mg-ATP dependent protein phosphatase. FEBS Lett. 1982;150:319–326. doi: 10.1016/0014-5793(82)80760-6. [DOI] [PubMed] [Google Scholar]
- Hemmings HC, Jr, Greengard P, Tung HYL, Cohen P. DARPP-32, a dopamine-regulated neuronal phosphoprotein, is a potent inhibitor of protein phosphatase-1. Nature. 1984a;310:503–505. doi: 10.1038/310503a0. [DOI] [PubMed] [Google Scholar]
- Hemmings HC, Jr, Williams KR, Konigsberg WH, Greengard P. DARPP-32, a dopamine- and adenosine 3′,5′-monophosphate-regulated neuronal phosphoprotein. I. Amino acid sequence around the phosphorylated threonine. J Biol Chem. 1984b;259:14486–14490. [PubMed] [Google Scholar]
- Henzel WJ, Billeci TM, Stults JT, Wong SC, Grimley C, Watanabe C. Identifying proteins from two-dimensional gel by molecular mass searching of peptide fragments in protein sequence databases. Proc Natl Acad Sci. 1993;90:5011–5015. doi: 10.1073/pnas.90.11.5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes CFB, Tonks NK, Major H, Cohen P. Analysis of the in vivo phosphorylation state of protein phosphatase inhibitor-2 from rabbit skeletal muscle by fast-atom bombardment mass spectrometry. Biochim Biophys Acta. 1987;929:208–219. doi: 10.1016/0167-4889(87)90178-9. [DOI] [PubMed] [Google Scholar]
- Hou SI, MacManus JP. Molecular mechanisms of cerebral ischemia-induced neuronal death. Int Rev Cytol. 2002;221:93–148. doi: 10.1016/s0074-7696(02)21011-6. [DOI] [PubMed] [Google Scholar]
- Hu BR, Wieloch T. Persistent translocation of Ca2+/calmodulin dependent protein kinase II to synaptic junctions in the vulnerable hippocampal CA1 region following transient ischemia. J Neurochem. 1995;64:277–284. doi: 10.1046/j.1471-4159.1995.64010277.x. [DOI] [PubMed] [Google Scholar]
- Huang Z, Kimberley M, Khatra B, Vijayaraghavan S. Protein 14-3-3ζ binds to protein phosphatase PP1γ2 in bovine epididymal spermatozoa. Biol Reprod. 2004;71:177–184. doi: 10.1095/biolreprod.104.027284. [DOI] [PubMed] [Google Scholar]
- Ingebritsen TS, Cohen P. The protein phosphatases involved in cellular regulation. 1 Classification and substrate specificities. Eur J Biochem. 1983;132:255–261. doi: 10.1111/j.1432-1033.1983.tb07357.x. [DOI] [PubMed] [Google Scholar]
- Klumpp S, Krieglstein J. Serine/threonine protein phosphatases in apoptosis. Curr Opin Pharmacol. 2002;2:458–462. doi: 10.1016/s1471-4892(02)00176-5. [DOI] [PubMed] [Google Scholar]
- Kontos HA. Oxygen radicals in cerebral vascular injury. Circ Res. 1985;57:508–516. doi: 10.1161/01.res.57.4.508. [DOI] [PubMed] [Google Scholar]
- Kristian T, Siesjo BK. Calcium in ischemic cell death. Stroke. 1998;29:705–718. doi: 10.1161/01.str.29.3.705. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Leach C, Shenolikar S, Brautigan DL. Phosphorylation of phosphatase inhibitor-2 at centrosomes during mitosis. J Biol Chem. 2003;278:26015–26020. doi: 10.1074/jbc.M300782200. [DOI] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Liu X, Nozari A, Basu S, Ronquist G, Rubertsson S, Wiklund L. Neurological outcome after experimental cardiopulmonary resuscitation: a result of delayed and potentially treatable neuronal injury. Acta Anaesthesiol Scand. 2002;46:537–546. doi: 10.1034/j.1399-6576.2002.460511.x. [DOI] [PubMed] [Google Scholar]
- Lizcano JM, Morrice N, Cohen P. Regulation of BAD by cAMP dependent protein kinase is mediated via phosphorylation of a novel site, ser 155. Biochem J. 2000;349:547–557. doi: 10.1042/0264-6021:3490547. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Margolis SS, Walsh S, Welser DC, Yoshida M, Shenolikar S, Kornbluth S. PP1 control of M phase entry exerted through 14-3-3-regulated Cdc25 dephosphorylation. EMBO J. 2003;22:5734–5745. doi: 10.1093/emboj/cdg545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin de la Vega C, Burda J, Salinas M. Ischemia-induced inhibition of the initiation factor 2α phosphatase activity in the rat brain. Neuroreport. 2001;12:1021–1025. doi: 10.1097/00001756-200104170-00031. [DOI] [PubMed] [Google Scholar]
- Martin de la Vega C, Burda J, Lobo MVT, Salinas M. Cerebral postischemic reperfusion-induced demethylation of the protein phosphatase-2A catalytic subunit. J Neurosci Res. 2002;69:540–549. doi: 10.1002/jnr.10306. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Al-Abdulla NA, Brambrink AM, Kirsch JR, Sieber FE, Portera-Cailliau C. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: a perspective on contributions of apoptosis and necrosis. Brain Res Bull. 1998;46:281–309. doi: 10.1016/s0361-9230(98)00024-0. [DOI] [PubMed] [Google Scholar]
- Morana SJ, Wolf CM, Li J, Reynolds JE, Brown MK, Eastman A. The involvement of protein phosphatase in the activation of ICE/CED-3 protease, intracellular acidification, DNA digestion, and apoptosis. J Biol Chem. 1996;271:18263–18271. doi: 10.1074/jbc.271.30.18263. [DOI] [PubMed] [Google Scholar]
- Munoz F, Martin ME, Manso-Tomico J, Berlanga J, Salinas M, Fando JL. Ischemia-induced phosphorylation of initiation factor 2 in differentiated PC12 cells: role for initiation factor 2 phosphatase. J Neurochem. 2000;75:2335–2345. doi: 10.1046/j.1471-4159.2000.0752335.x. [DOI] [PubMed] [Google Scholar]
- Muslin A, Tanner JW, Allen PM, Shaw AS. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell. 1996;84:889–897. doi: 10.1016/s0092-8674(00)81067-3. [DOI] [PubMed] [Google Scholar]
- N’cho M, Brahmi Z. Fas-mediated apoptosis in T cells involves the dephosphorylation of the retinoblastoma protein by type 1 protein phosphatase. Hum Immunol. 1999;60:1183–1194. doi: 10.1016/s0198-8859(99)00125-1. [DOI] [PubMed] [Google Scholar]
- Oliver CN, Starke-Reed PE, Stadtman ER, Liu GJ, Carney JM, Floyd RA. Oxidative damage to brain proteins, loss of glutamine synthetase activity, and production of free radicals during ischemia/reperfusion-induced injury to gerbil brain. Proc Natl Acad Sci. 1990;87:5144–5147. doi: 10.1073/pnas.87.13.5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozuelo Rubio M, Geraghty KM, Wong BHC, Wood NT, Campbell DG, Morrice N, Mackintosh C. 14-3-3-Affinity purification of over 200 human phosphoproteins reveals new links to regulation of cellular metabolism, proliferation and trafficking. Biochem J. 2004;379:395–408. doi: 10.1042/BJ20031797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Jensen ON, Podetelejnikov AV, Saglioco F, Wilm M, Vorm O, Mortensen P, Shevchenko A, Boucherie H, Mann M. Linking genome and proteome by mass spectrometry: large-scale identification of yeast proteins from two dimensional gels. Proc Natl Acad Sci. 1996;93:14440–14445. doi: 10.1073/pnas.93.25.14440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siesjo BK. Mechanisms of ischemic brain damage. Crit Care Med. 1988;16:954–963. doi: 10.1097/00003246-198810000-00006. [DOI] [PubMed] [Google Scholar]
- Siesjo BK, Agardh CD, Bengtsson F. Free radicals and brain damage. Cerebrovasc Brain Metab Rev. 1989;1:165–211. [PubMed] [Google Scholar]
- Siesjo BK, Zhao Q, Pahlmark K, Siesjo P, Katsura K, Folbergrova J. Glutamate, calcium and free radicals as mediators of ischemic brain damage. Ann Thorac Surg. 1995;59:1316–1320. doi: 10.1016/0003-4975(95)00077-x. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Fujimura M, Noshita N, Kim GW, Saito A, Hayashi T, Narasimhan P, Maier CM, Chan PH. Neuronal death/ survival signaling pathways in cerebral ischemia. J Am Soc Exp Neurother. 2004;1:17–25. doi: 10.1602/neurorx.1.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung HYL, Cohen P. The protein phosphatases involved in cellular regulation: comparison of native and reconstituted Mg-ATP dependent protein phosphatase. Eur J Biochem. 1984;145:57–64. doi: 10.1111/j.1432-1033.1984.tb08521.x. [DOI] [PubMed] [Google Scholar]
- Tung HYL, Reed LJ. Purification and characterization of protein phosphatase-1I activating kinase from bovine brain cytosolic and particulate fractions. J Biol Chem. 1989;264:2985–2990. [PubMed] [Google Scholar]
- Tung HYL, De Rocquigny H, Zhao LJ, Cayla X, Roques BP, Ozon R. Direct activation of protein phosphatase-2A0 by HIV-1 encoded protein complex NCp7:Vpr. FEBS Lett. 1997;401:197–201. doi: 10.1016/s0014-5793(96)01470-6. [DOI] [PubMed] [Google Scholar]
- Van Hoof C, Goris J. Phosphatases in apoptosis: to be or not to be, PP2A is in the heart of the question. Biochim Biophys Acta. 2003;1640:97–104. doi: 10.1016/s0167-4889(03)00029-6. [DOI] [PubMed] [Google Scholar]
- Wang QM, Guan KL, Roach PJ, DePaoli-Roach AA. Phosphorylation and activation of the ATP-Mg-dependent protein phosphatase by the mitogen-activated protein kinase. J Biol Chem. 1995;270:18352–18358. doi: 10.1074/jbc.270.31.18352. [DOI] [PubMed] [Google Scholar]
- Wang RH, Liu CWY, Avramis VI, Berndt N. Protein phosphatase-1α-mediated stimulation of apoptosis is associated with dephosphorylation of the retinoblastoma protein. Oncogene. 2001;20:6111–6122. doi: 10.1038/sj.onc.1204829. [DOI] [PubMed] [Google Scholar]
- White BC, Sullivan JM, DeGracia DJ, O’Neil BJ, Neumar RW, Grossman LI, Rafols JA, Krause GS. Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci. 2000;179:1–33. doi: 10.1016/s0022-510x(00)00386-5. [DOI] [PubMed] [Google Scholar]
- Wieloch T, Hu BR, Boris-Moller A, Cardell M, Kamme F, Kurihara J, Sakata K. Intracellular signal transduction in the postischemic brain. Adv Neurol. 1996;71:371–387. [PubMed] [Google Scholar]
- Yang SD, Fung YL. Identification and characterization of an ATP Mg-dependent protein phosphatase from pig brain. J Biol Chem. 1985;260:13464–13470. [PubMed] [Google Scholar]
- Yung HW, Tolkovsky AM. Erasure of kinase phosphorylation in astrocytes during oxygen-glucose deprivation is controlled by ATP levels and activation of protein phosphatases. J Neurochem. 2003;86:1281–1288. doi: 10.1046/j.1471-4159.2003.01946.x. [DOI] [PubMed] [Google Scholar]