

Abstract
Immune responses are highly energy dependent processes. Activated T cells increase glucose uptake and aerobic glycolysis to survive and function. Malnutrition and starvation limit nutrients and are associated with immune deficiency and increased susceptibility to infection. While it is clear that immunity is suppressed in times of nutrient stress, mechanisms that link systemic nutrition to T cell function are poorly understood. We show here that fasting leads to persistent defects in T cell activation and metabolism, as T cells from fasted animals had low glucose uptake and decreased ability to produce inflammatory cytokines, even when stimulated in nutrient-rich media. To explore the mechanism of this long-lasting T cell metabolic defect, we examined leptin, an adipokine reduced in fasting that regulates systemic metabolism and promotes effector T cell function. We show leptin is essential for activated T cells to upregulate glucose uptake and metabolism. This effect was cell-intrinsic and specific to activated effector T cells, as naïve T cells and Treg did not require leptin for metabolic regulation. Importantly, either leptin addition to cultured T cells from fasted animals or leptin injections to fasting animals was sufficient to rescue both T cell metabolic and functional defects. Leptin-mediated metabolic regulation was critical, as transgenic expression of the glucose transporter Glut1 rescued cytokine production of T cells from fasted mice. Together, these data demonstrate that induction of T cell metabolism upon activation is dependent on systemic nutritional status, and leptin links adipocytes to metabolically license activated T cells in states of nutritional sufficiency.
Introduction
Nutritional status is well known to regulate immune function, as obesity is associated with increased inflammation whereas malnutrition is associated with immune deficiency and increased susceptibility to infection (1-3). Although the links between nutrition and adaptive immunity remain poorly understood, systemic energy balance between the demands of the immune system and other life-critical systems such as cardiovascular, respiratory, and neurologic, must be maintained and prioritized. Immune responses can consume significant nutrients. While resting T cells utilize an oxidative metabolism primarily for ATP generation, effector T cell activation sharply increases the demand for macromolecule biosynthesis (1). To meet this need, activated effector T cells dramatically increase glucose uptake and metabolism to activate a program of aerobic glycolysis reminiscent of cancer cells (4, 5). It has recently been demonstrated that regulatory pathways controlling T cell metabolism are intimately linked to T cell function (4, 6, 7). Increased expression of the glucose transporter Glut1 is sufficient to increase T cell cytokine production and proliferation (5). Moreover, activated effector T cells rely on glucose availability, glucose uptake, and aerobic glycolysis to survive and function properly (5, 8). How T cell metabolic demands are regulated by systemic nutritional status, however, is not clear.
The adipokine, leptin, may play a key role to balance energy expenditure and nutritional status in the immune system. Leptin is secreted in proportion to adipocyte mass and is best known for its role in regulating body weight and energy expenditure via signaling in the hypothalamus, where full-length leptin receptors are highly expressed (9, 10). However, leptin is also a critical regulator of immunity and functions as a pro-inflammatory cytokine (11, 12). Leptin deficiency in both mouse and human results in immune defects characterized by decreased total T cell number, decreased CD4+ helper T cell number, and a skewing away from a Th1 and towards a Th2 phenotype, resulting in protection against certain forms of autoimmunity and increased susceptibility to intracellular infections (13-16). Both the metabolic and immune defects in leptin-deficiency are reversed following treatment with recombinant leptin protein (17-19); however, the mechanisms of leptin regulation of immunity and T cell function are uncertain (20, 21).
The leptin receptor is a member of the class I cytokine receptor family and is upregulated on T cells following activation (22, 23). Signaling via the leptin receptor results in increased phosphatidylinositol-3-kinase (PI3K)/Akt activity, Janus kinase (Jak2)/Signal Transducer and Activator of Transcription (STAT3) activation, and MAPK signaling (24-27). Leptin has also been found to activate mTORC1 in regulatory T cells (Treg) and correlate with hyporesponsiveness and decreased proliferation of Treg (28). Many of these signaling molecules, particularly PI3K/Akt and mTORC1, have been implicated in the regulation of T cell metabolism (1). Previous studies suggest that leptin exerts effects on T cell number and function both by direct signaling through leptin receptors expressed on the T cell and indirectly through influences on the T cell environment (29-33). Direct leptin signaling may enhance the production of Th1 type cells, promoting inflammation, stimulating lymphocyte proliferation, and protecting against lymphocyte apoptosis (11, 32, 34). No role for leptin in T cell metabolism, however, has been reported.
Here we show that leptin is essential to link T cell metabolism to nutritional status and balance energy expenditure and immunity. Fasting-induced hypoleptinemia led to persistent T cell metabolic and activation defects. We found leptin was required for activated effector, but not regulatory, T cells to upregulate the glucose transporter Glut1 to support glucose uptake and metabolism required for proliferation and inflammatory cytokine production. Defects in glucose metabolism and function of activated peripheral T cells from fasted mice were rescued by leptin given either in vitro to isolated T cells in culture or in vivo to fasted animals. Importantly, direct rescue of glucose uptake with a Glut1 transgene also overcame functional defects induced by fasting and hypoleptinemia. These results show that leptin provides a crucial metabolic licensing signal to allow activated T cells to increase glucose metabolism, thus linking nutritional status, cellular metabolism, and immunity.
Materials and Methods
Mice
C57BL/6J, leptin receptor mutant (db−/−), leptin receptor floxed (LepR-fl), Lck-Cre, CD4-Cre, and Rag1 knockout mice were purchased from The Jackson Laboratory (Bar Harbor, MA). Lck-Cre transgenic mice were crossed with LepR floxed mice to generate LepR-fl/fl Lck-Cre+ mutants, LepR-fl/+ Lck-Cre+ heterozygotes, and LepR+/+ Lck-Cre+ controls. CD4-Cre transgenic mice were crossed with LepR floxed mice to generate LepR-fl/fl CD4-Cre+ mutants, and LepR+/+ CD4-Cre+ controls. Glut1 transgenic mice were previously described (5). Mice were bred and housed under specific pathogen-free conditions at Duke University Medical Center. All studies were performed under protocols approved by the Institutional Animal Care and Use Committee. For fasting studies, mice were deprived of food for up to 48 hours, but had full access to water. Where indicated, recombinant leptin (R&D Systems, Minneapolis, MN) was injected at a concentration of 1 μg/g body weight, twice daily for 48 hours while fasting; phosphate buffered saline of equal volume was injected into control mice. Experiments were performed using sex-matched mice between 8 and 24 weeks of age.
Cell purification and culture
T cells were purified from spleen and mesenteric lymph nodes by negative selection (StemCell Technologies, Vancouver, BC, Canada), and cells were cultured in RPMI 1640 (Mediatech, Washington, DC) supplemented with or without 1 or 10% FBS (Gemini Bio-Products, West Sacramento, CA), L-glutamine (Invitrogen, Chicago, IL), penicillin-streptomycin (Invitrogen), HEPES buffer solution (Life Technologies, Carlsbad, CA), and 2-ME (Sigma-Aldrich, St. Louis, MO). T cell activation was induced by either culturing purified T cells on anti-CD3ε (clone 145-2C11) and anti-CD28 (clone 37.51)-coated plates (eBioscience, San Diego, CA) or co-culturing with macrophages. For macrophage T cell co-cultures, macrophages were derived from murine bone marrow and cultured in RPMI 1640 with addition of 3 ng/mL GM-CSF (eBioscience) for 7 days. Bone marrow cultures were washed and macrophages were trypsinized, replated, and treated with LPS from E. coli O127:B8 (Sigma-Aldrich) at 100 μg/mL for one hour, followed by addition of freshly purified T cells. T cell macrophage co-cultures were cultured with anti-CD3 (eBioscience) at 0.01 μg/mL for 1 day. Nonadherent cells were removed and replated for 1 hour. Cells that remained non-adherent were >90% T cells and were subject to further analysis. For Th17 and Treg subset generation, CD4+ T cells were stimulated on irradiated splenic feeder cells (3000 rad) with 2.5 μg/mL anti-CD3 Ab at a ratio of 1:5. Th17 subset was generated by culture with TGF-β (2 ng/mL; R&D Systems) and IL-6 (20 ng/mL; eBioscience); Treg subset was generated by culture with TGF-β alone (3 ng/mL). On day 3 post-stimulation, cells were split 1:2, washed, and replated with IL-2 alone for an additional 2 days. Where indicated, recombinant leptin (R&D Systems) was added into media at a concentration of 250 ng/mL (15.6 nM).
Flow Cytometry and ELISA
Proliferation was determined by staining T cells with CFSE (Invitrogen) before culture and analyzed by dilution of fluorescent dye flow cytometrically. All flow cytometry was performed on a MACSQuant Analyzer (Miltenyi, Auburn, CA) and analyzed with FlowJo software (Tree Star, Ashland, OR). ELISAs were performed on supernatant from T cell cultures using rat Abs against murine IL-2 and IFN-γ (BD Pharmingen, San Jose, CA) and recombinant standards as previously described (5). Leptin ELISA was performed using Leptin Mouse Quantikine ELISA kit (R&D Systems) as per the manufacturer’s instructions.
Metabolism Assays
Assays for glucose uptake and glycolytic flux were performed as previously described (5, 6). Oxygen consumption rates (OCR) and extracellular acidification rates (ECAR) were measured with an extracellular flux analyzer (Seahorse Bioscience, North Billerica, MA).
Quantitative real-time PCR and Immunoblot Analysis
Total mRNA was isolated from mouse T cells using TRIzol (Invitrogen), and cDNA was synthesized from RNA using iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). Quantitative PCR was performed using iQ SYBR Green Supermix (Bio-Rad) and the following primers: glut1 forward, 5’-AGC CCT GCT ACA GTG TAT-3’; glut1 reverse, 5’-AGG TCT CGG GTC ACA TC-3’; lepR forward, 5’-GCT CTT CTG ATG TAT TTG GAA ATC-3’; lepR reverse, 5’-ACC TGA TAT TGA AGC GGA AAT GG-3’. All samples were normalized to beta-2-microglobulin mRNA levels. For immunoblotting, cells were lysed for one hour on ice in 1% Triton X-100 and 0.1% SDS containing protease inhibitors (BD Pharmingen). Equivalent protein concentrations were loaded on SDS-PAGE gels (BioRad) and probed with primary rabbit anti-Glut1 (Abcam, Cambridge, MA), followed by secondary anti-rabbit HRP (BD Pharmingen) and ECL (Thermo Scientific, Waltham, MA) for visualization.
Bone Marrow Transplants
Bone marrow was isolated from C57BL/6J and db−/− donor mice and injected via tail vein into Rag1 knockout recipients that had been irradiated the day prior. Transplanted mice were maintained on antibiotic (Septra) water. Following 18 weeks of reconstitution, recipient mice were sacrificed and T cells were isolated from spleen and lymph nodes, as described above.
Statistical Analysis
Data are presented as mean +/− SD and were analyzed using a two-tailed Student t test. A confidence level of p < 0.05 was considered statistically significant for all data.
Results
Fasting leads to persistent functional and metabolic T cell defects
Acute starvation and malnutrition can sharply reduce circulating leptin levels and are associated with immune defects (35-37). The mechanisms by which nutritional status is linked to energy-costly immune responses, however, are largely unknown. Effector T cells are highly dependent on glucose uptake and metabolism. To examine the role of fasting on T cell metabolism and function, wildtype C57BL/6J mice were fasted or fed ad libitum for 48 hours. As previously described (37), fasting decreased serum leptin levels (Fig. 1A) and led to lower thymocyte and peripheral total T cell numbers (Supplemental Fig. 1A,B). Remaining T cells were then isolated from control and fasted mice, cultured in complete media, and activated with antibodies to CD3 and CD28 for metabolic and functional studies.
FIGURE 1.
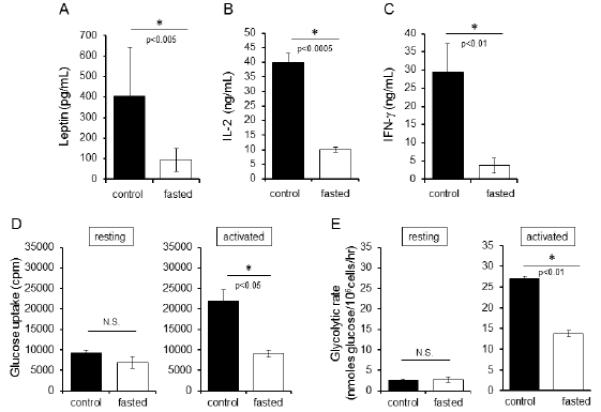
Fasting-induced hypoleptinema is associated with persistent functional and metabolic T cell defects. (A) Wildtype C57BL/6J mice were either fasted for 48 hours or allowed to feed ad libitum. All mice had full access to water. At the end of the fast, serum was collected from blood and circulating leptin levels were measured by ELISA. Data are representative of five independent experiments; three mice per group. (B and C) Peripheral T cells from control and fasted C57BL/6J mice were then cultured in complete media with full nutrients and activated with antibodies to CD3 and CD28 for 48 hours, after which IL-2 and IFN-γ were measured in culture supernatants by ELISA. Data are representative of six independent experiments, three mice per group. (D) Glucose uptake was assessed by 3[H]-2-DG uptake in both resting and activated peripheral T cells. Data are representative of two independent experiments for glycolysis, three mice per group. (E) Glycolytic flux was determined by the generation of tritiated water by enolase in both resting and activated peripheral T cells. Data are representative of two independent experiments for glycolysis, three mice per group.
Despite culture in nutrient-rich media for 48 hours, peripheral T cells isolated from fasted mice had persistent functional defects after stimulation. T cells from fasted mice had decreased ability to secrete IL-2 and IFN-γ compared to T cells isolated from fed controls stimulated under identical conditions (Fig. 1B,C). Moreover, while resting T cells from fasted or fed mice had equivalent levels of glucose uptake and glycolysis (Fig. 1D,E), activated peripheral T cells from fasted animals failed to fully increase glucose metabolism. Fasted T cells had an inability to upregulate glucose uptake (Fig. 1D) and glycolytic flux (Fig. 1E) upon activation in complete media compared to T cells isolated from fed controls. These data show that fasting leads to reduced leptin levels and a persistent T cell defect in the ability to upregulate cellular metabolism during activation, even when nutrients are abundant. Nutrient availability itself, therefore, is not the critical regulatory element in this setting to allow metabolic reprogramming for T cell activation.
Leptin is required for activated T cell proliferation and inflammatory cytokine production
Given that hypoleptinemia was associated with fasting, we sought to determine if leptin could act to directly influence T cell function and metabolism. We first examined the ability of leptin to modulate inflammatory cytokine production in peripheral T cells. Wildtype C57BL/6J CD4+ T cells were purified and activated with antibodies to CD3 and CD28 in serum-free media in the presence or absence of exogenous leptin. Full activation of T cells with anti-CD3 together with anti-CD28 co-stimulation resulted in a two-fold increase in IL-2 and IFN-γ production in the absence of leptin. While leptin had no effect on T cells stimulated only through anti-CD3, provision of exogenous leptin augmented inflammatory cytokine production of T cells fully activated with both anti-CD3 and anti-CD28 (Fig. 2A,B). These data are consistent with previous reports that leptin receptor is induced after T cell activation (22) and promotes inflammatory cytokine production. In addition, these data demonstrate that leptin requires stimulation of the TCR together with co-stimulation.
FIGURE 2.
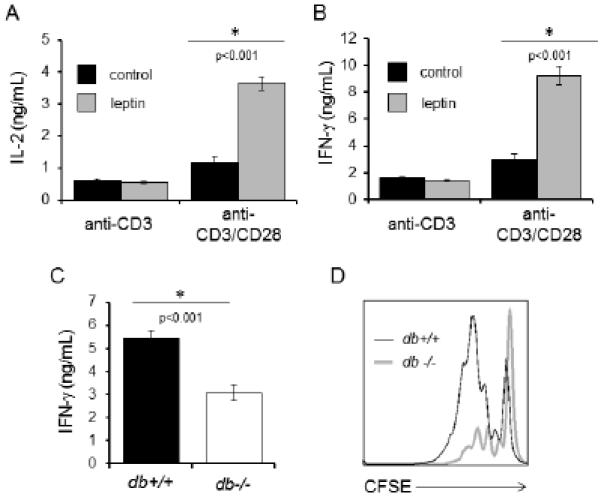
Leptin promotes activated T cell proliferation and inflammatory cytokine production. (A and B) CD4+ T cells were isolated from wildtype C57BL/6J mice and stimulated with anti-CD3 alone or in the presence of anti-CD28 in serum-free media for 24 hours. Cells were then treated with (grey bars) or without (black bars) 250 ng/mL leptin for an additional 24 hours, after which supernatants were collected and IL-2 (A) and IFN-γ (B) production in culture supernatants were measured by ELISA. Data are representative of two independent experiments. (C) CD4+ T cells were isolated from wildtype (db+/+; black bar) or leptin receptor-deficient (db−/−; white bar) mice and activated by co-culture with wildtype bone marrow-derived macrophages activated with LPS plus low-dose anti-CD3 antibody for 48 hours; production of IFN-γ in culture supernatants was measured by ELISA. Data are representative of three independent experiments. (D) CD4+ T cells from wildtype (db+/+; thin black line) or leptin receptor-deficient (db−/−; thick grey line) mice were activated with anti-CD3 and anti-CD28 antibodies for 72 hours. Proliferation was determined by staining T cells with CFSE prior to culture and analyzing dilution of the fluorescent dye flow cytometrically. Data are representative of two independent experiments.
CD4+ T cells were next isolated from wildtype (db+/+) and leptin receptor-deficient (db−/−) mice and activated in co-culture with bone marrow-derived LPS-activated macrophages to provide costimulatory signals. Leptin is available from serum in culture media in these experiments. Activation of db−/− CD4+ T cells resulted in decreased IFN-γ production as compared to db+/+ CD4+ T cells (Fig. 2C). Additionally, while activation of wildtype db+/+ CD4+ T cells resulted in robust proliferation, as indicated by dilution of carboxyfluorescein succinimidyl ester (CFSE), leptin receptor-deficient db−/− CD4+ T cells showed only modest proliferation (Fig. 2D). Thus, leptin receptor signaling is required for normal proliferation in activated peripheral T cells in vitro. Altogether, these results demonstrate that leptin promotes T cell proliferation and inflammatory cytokine production after activation.
Leptin signaling is required for normal rates of glucose uptake, glycolysis, and mitochondrial respiration in activated T cells
Activated T cells rely on increased glucose metabolism to survive, proliferate, and produce cytokines; therefore, changes in T cell metabolism can directly impact T cell function and fate (5, 6, 38, 39). Given the ability of leptin to regulate both systemic metabolism (9) and T cell function, we hypothesized that leptin may directly control peripheral T cell glucose metabolism. This role for leptin has been shown in other metabolic cells, including muscle and adipose (40, 41). To test this potential mechanism of leptin action on T cells, CD4+ T cells were isolated from wildtype (db+/+) and leptin receptor-deficient (db−/−) mice, and glucose uptake was examined in both resting T cells and T cells activated with antibodies to CD3 and CD28 in normal media with serum containing leptin. While leptin receptor deficiency (db−/−) had no impact on glucose uptake of resting T cells, T cells stimulated with both anti-CD3 and anti-CD28 were dependent on leptin receptor signaling to maximally upregulate glucose uptake following activation (Fig. 3A).
FIGURE 3.
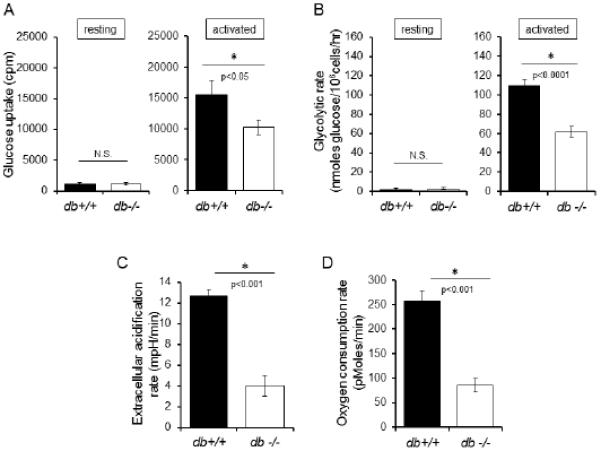
Leptin signaling is required for normal rates of glucose uptake, glycolysis, and mitochondrial respiration in activated T cells. (A and B) CD4+ T cells were isolated from db+/+ (black bar) or db−/− (white bar) mice. Glucose uptake was assessed by 3[H]-2-DG uptake (A) and glycolytic flux was determined by the generation of tritiated water by enolase (B) in both resting cells and cells activated with anti-CD3 and anti-CD28 antibodies for 48 hours, as indicated. Representative data are shown. (C and D) Extracellular acidification rates (C) and basal oxygen consumption rates (D) were measured using a Seahorse Extracellular Flux Analyzer in activated T cells isolated from wildtype (db+/+; black bar) or leptin receptor-deficient (db−/−; white bar) mice. Data are representative of two independent experiments.
Downstream of glucose uptake, glucose is metabolized to pyruvate through glycolysis, and glucose-derived pyruvate is either converted to lactate for secretion or oxidized in the mitochondria. The glycolytic rate was equivalent in resting T cells from db−/− and db+/+ mice (Fig. 3B). Consistent with reduced glucose uptake in activation, however, LepR-deficient db−/− activated T cells had decreased rates of glycolytic flux when determined by addition of 3H-glucose to measure production of 3H-H2O by enolase, the penultimate glycolytic enzyme (42) (Fig. 3B). To examine the subsequent steps in glucose metabolism through conversion of pyruvate to lactate or through entry of pyruvate into the mitochondria for oxidative metabolism, we used extracellular flux analysis to examine media acidification as an indicator of lactate production and oxygen consumption to indicate mitochondrial metabolism. In both metabolic fates for glucose-derived metabolites, activated LepR-deficient db−/− CD4+ T cells had reduced flux compared to wildtype db+/+ T cells (Fig. 3C,D). Leptin receptor, therefore, is essential to maximally upregulate glucose metabolism during T cell activation.
Leptin regulates glucose metabolism in part by upregulation of Glut1
A family of glucose transporters mediates glucose uptake, and Glut1 is considered the primary glucose transporter expressed on the surface of activated T cells. The surface expression of Glut1 on T cells is highly regulated (43-45), and Glut1 can be a limiting component of glucose metabolism upon T cell activation (5). To test if failure to increase Glut1 expression contributed to the defective metabolic response of T cells that fail to receive leptin signals, CD4+ T cells from db−/− and db+/+ mice were activated in the presence or absence of anti-CD3 and anti-CD28 for 48 hours, and Glut1 expression was measured. Leptin is available from serum in culture media in these experiments. Leptin receptor-deficient db−/− T cells had significantly reduced induction of Glut1 RNA following activation relative to db+/+ controls (Fig. 4A). We next examined the expression of Glut1 protein in LepR-deficient (db−/−) versus wildtype (db+/+) control CD4+ T cells. Like Glut1 mRNA, total Glut1 protein induction was reduced in leptin receptor-deficient T cells following activation (Fig. 4B; note Glut1 runs as a smear due to glycosylation; quantification of Glut1 protein levels normalized to actin from duplicate experiments is shown below).
FIGURE 4.
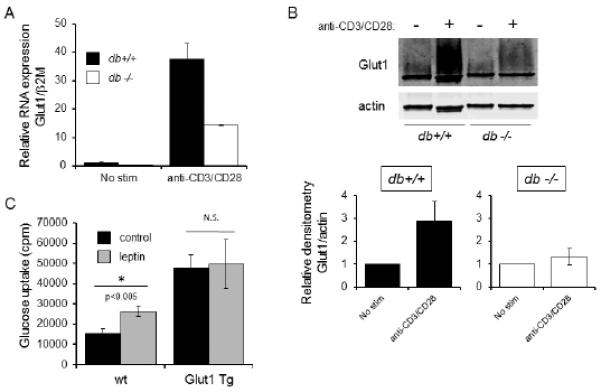
Leptin regulates glucose metabolism in part by upreglation of Glut1. (A) CD4+ T cells were isolated from wildtype (db+/+; black bar) or leptin receptor-deficient (db−/−; white bar) mice and activated with or without anti-CD3 and anti-CD28, for 48 hours. Glut1 mRNA expression was determined by qPCR. Data are expressed relative to beta-2-microglobulin RNA levels, and normalized to mRNA levels in unstimulated wt T cells. Data are representative of two independent experiments. (B) CD4+ T cells were isolated from wildtype (db+/+; black bar) or leptin receptor-deficient (db−/−; white bar) mice and activated with or without anti-CD3 and anti-CD28 antibodies, as indicated, for 48 hours. Lysates were resolved by SDS-PAGE and immunoblotted using antibodies to Glut1 and actin. Glut1 is heavily glycosylated and appears as a large smear on Western blot. Bar graphs depict densitometry of the Glut1 band, normalized to actin, in wildtype (db+/+) versus leptin receptor-deficient (db−/−) animals, as averaged from two additional independent experiments. (C) CD4+ T cells from wildtype (wt) or T cell-specific Glut1 transgenic mice (Glut1 Tg) were isolated and stimulated with anti-CD3 and anti-CD28 in serum free media for 24 hours. Cells were then treated with (grey bars) or without (black bars) 250 ng/mL leptin for an additional 24 hours, after which glucose uptake was measured. Data are representative of two independent experiments.
To determine to what extent leptin-mediated regulation of Glut1 may account for the effect of leptin on T cell metabolism, we measured the effect of leptin on T cells with transgenic expression of Glut1 (Glut1 Tg) (5). CD4+ T cells were isolated from T cell-specific Glut1 Tg animals and age-matched wildtype controls and activated with antibodies to CD3 and CD28 in serum-free media in the presence or absence of exogenous leptin. Leptin stimulation resulted in increased glucose uptake in activated wildtype T cells. As expected, glucose uptake was higher in activated Glut1 Tg T cells than in wildtype T cells. Leptin, however, did not further augment glucose uptake (Fig. 4C).
The effects of leptin on activated T cell function and metabolism are hematopoietic cell intrinsic
Leptin can act on a variety of cell types, including thymic stroma, and therefore the role of leptin in regulation of T cell number and function may be from indirect effects on T cell development and environment (30, 33). To examine whether the effects of leptin on activated peripheral T cell function and metabolism are hematopoietic cell intrinsic or extrinsic, we performed bone marrow transplants of LepR-deficient (db−/−) and wildtype (db+/+) hematopoietic cells into irradiated Rag1 knockout (LepR wildtype) recipients. Following immune reconstitution, activated CD4+ T cells from mice that received db−/− or db+/+ bone marrow were compared in vitro for production of inflammatory cytokine IFN-γ as well as glucose metabolism. T cells from recipient mice that received LepR-deficient db−/− bone marrow had defects in both IFN-γ production (Fig. 5A) and glucose uptake (Fig. 5B) following activation compared to T cells from recipient mice that received bone marrow from db+/+ controls. Similarly, recipient mice that received LepR-deficient db−/− bone marrow had defects in glycolysis following T cell activation (Fig. 5C) compared to recipient mice that received bone marrow from db+/+ controls. Importantly, resting T cells had equivalent glycolysis (Fig. 5C).
FIGURE 5.
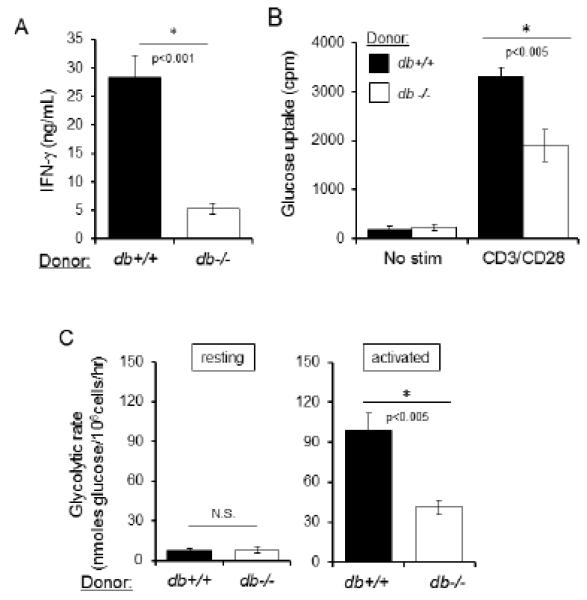
The effects of leptin on activated T cell function and metabolism are hematopoietic cell intrinsic. Irradiated Rag recipient mice were transplanted with bone marrow isolated from wildtype (db+/+; black bars) or leptin receptor-deficient (db−/−; white bars) donors. Following full immune reconstitution at 18 weeks, the following experiments were performed. (A) CD4+ T cells were isolated and activated with anti-CD3 and anti-CD28 for 48 hours, followed by measurement of IFN-γ production by ELISA. (B and C) Resting and activated CD4+ T cells were evaluated for glucose uptake; data are representative of five independent experiments (B) and glycolytic flux; data are representative of three independent experiments (C).
Leptin directly regulates T cell metabolism and function
The intrinsic role of leptin in T cell inflammatory cytokine production and metabolism was confirmed in genetic studies using mice with a conditional leptin receptor deletion. We generated conditional T cell-specific leptin receptor (LepR) knockout mice by crossing LepR floxed (LepR-fl/fl) animals with mice expressing a Cre recombinase transgene under the control of the proximal Lck promoter (Lck-Cre), and confirmed significantly lower leptin receptor expression in the conditional knockout following activation using reverse transcriptase PCR (Fig. 6A). Consistent with prior studies that showed induction of leptin receptor expression after T cell activation, leptin receptor expression was not detectable in resting T cells, (22). Activated CD4+ T cells isolated from T cell-specific LepR knockout mice secreted less IFN-γ than T cells isolated from heterozygous controls (Fig. 6B). Moreover, T cell-specific LepR deficiency reduced activated T cell glucose uptake relative to heterozygous LepR-fl/+ controls (Fig. 6C). These studies confirm a direct effect of leptin on T cell metabolism and cytokine production.
FIGURE 6.
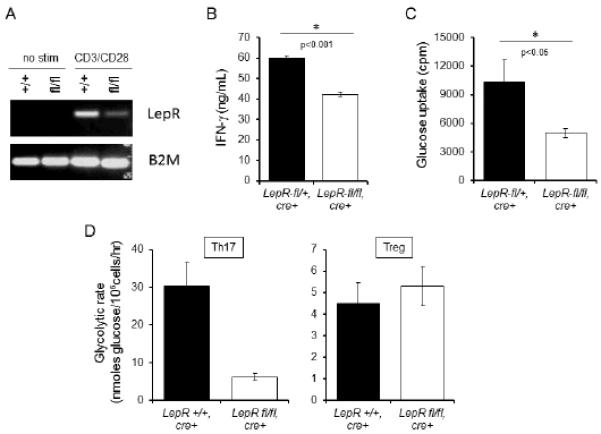
T cell-intrinsic leptin receptor signaling is required for activated T cell inflammatory cytokine production and glucose metabolism. (A) T cell-specific leptin receptor (LepR) conditional knockout was generated using cre-lox recombination. LepR expression was determined by RT-PCR using primers to LepR and beta-2-microglobulin (control). (B and C) CD4+ T cells were isolated from conditional LepR knockout (white bar) versus heterozygous control mice (black bar) and activated with anti-CD3 and anti-CD28 antibodies for 48 hours, after which glucose uptake was assessed (B) and IFN-γ concentration was measured in culture supernatants by ELISA (C). Data are representative of two independent experiments. (D) CD4+ T cells were isolated from CD4+ T cell specific conditional LepR knockout (white bar) and homozygous control mice (black bar) and induced to differentiate into Th17 and Treg populations in vitro, after which glycolytic rate was determined. Data are representative of two independent single mouse experiments.
Leptin has a known role in modulating CD4+ T cell subset differentiation. As a pro-inflammatory cytokine, leptin upregulates Th1 and Th17 cells while inhibiting Treg proliferation (46-49). To examine the direct effect of leptin on CD4+ T cell subset metabolism, we generated conditional CD4+ T cell-specific LepR knockout mice (LepR-fl/fl animals crossed with CD4-Cre). CD4+ T cells were isolated from LepR knockout and control mice and induced in vitro to differentiate into Th17 and Treg cells, as described previously (6). Following three days of in vitro cell differentiation, the cells were split and cultured for an additional two days in the presence of IL-2, after which glycolytic flux was determined. CD4+ T cell specific LepR knockout Th17 cells had significantly decreased glycolysis as compared to leptin receptor-expressing controls, whereas Treg generated from CD4+ T cell specific LepR knockout had equal glycolytic rate as leptin receptor expressing controls (Fig. 6D). This data indicates that leptin receptor signaling is critical for glucose metabolism in effector Th17 cells, but not Treg. This is consistent with previous reports that have found Th17 to be highly glycolytic and Treg to depend on an oxidative metabolism, rather than glucose uptake for energy metabolism (6, 7).
Persistent functional and metabolic defects of T cells from fasted animals can be rescued by either leptin or increased glucose uptake
In addition to reduced leptin, fasting leads to a variety of systemic hormonal and nutritional effects that could impact T cell metabolism and function (37). To examine the contribution of reduced leptin alone in fasting-induced peripheral metabolic T cell defects, T cells were isolated from control fed and fasted animals and activated in vitro in control media or with addition of exogenous leptin. Despite the presence of leptin in serum, a low concentration was required (1%) to maintain cell viability in T cells from fasted animals. Importantly, even with a low level of serum-derived leptin, provision of additional leptin was sufficient to restore glucose uptake in activated T cells from fasted mice to levels equivalent to that of control T cells isolated from animals fed ad libitum (Fig. 7A). Similarly, leptin addition restored IL-2 and IFN-γ production of T cells from fasted animals to levels at or above those of T cells isolated from fed controls (Fig. 7B,C). In these low serum conditions, additional leptin did not, however, augment IL-2 production beyond what was seen in T cells isolated from the fed controls and incubated in media with 1% serum. Exogenous leptin did indeed increase IFN-γ secretion above that seen in T cells from the fed controls. These results suggest that, while leptin is sufficient to reverse the persistent defects in T cell function and metabolism induced by fasting, the effect of leptin on IL-2 and IFN-γ production is likely concentration-dependent.
FIGURE 7.
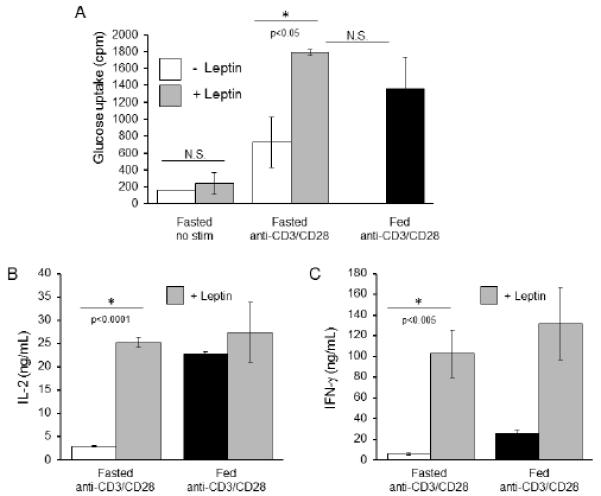
Defective glucose metabolism and inflammatory cytokine production in activated peripheral T cells from fasted mice are rescued by leptin, in vitro. T cells were isolated from control or fasted C57BL/6 mice, activated with anti-CD3 and anti-CD28 for 24 hours in low (1%) serum conditions, and then treated with or without 250 ng/mL additional leptin for an additional 24 hours, after which (A) glucose uptake was measured and (B and C) cytokine production was measured by ELISA. Data are representative of two independent experiments; three mice per experimental group.
Rescue of persistent metabolic and functional defects in T cells from fasted and hypoleptinemic animals by in vitro provision of leptin suggested that leptin provides a signal essential for T cells to upregulate glucose metabolism upon activation. The contribution of leptin to peripheral T cell function and metabolism in fasted animals was, therefore, studied in vivo. Fasted mice received either twice-daily intraperitoneal injections of phosphate buffered saline (PBS) or recombinant leptin in PBS. Fed control mice received PBS injections. Thymocytes and peripheral T cells were isolated after 48 hours of treatment. In vivo leptin administration during fasting partially rescued the cell number abnormalities acquired from fasting (37) and reduced the loss of thymocytes and peripheral T cell numbers (Supplemental Fig. 2A,B). To test if leptin altered the functional defects imparted by fasting, peripheral T cells were isolated and activated with antibodies to CD3 and CD28 in full nutrient media for an additional 48 hours in the absence of any additional leptin. Importantly, IL-2 and IFN-γ production of in vitro stimulated T cells from fasted mice was fully restored to control levels by in vivo leptin treatment (Fig. 8A,B). In addition, in vivo leptin treatment prevented the T cell metabolic defects and maintained glucose uptake of in vitro stimulated T cells from fasted mice to a level similar to that of T cells from normally fed animals (Fig. 8C).
FIGURE 8.
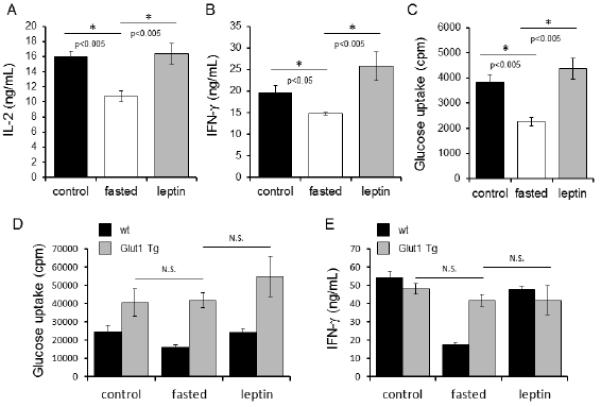
Both in vivo leptin administration and Glut1 overexpression can rescue peripheral T cell function and metabolism in fasted mice. (A-C) CD4+ T cells were isolated from control C57BL/6J mice (black bars), fasted mice (white bars), or fasted mice receiving leptin injections (grey bars), and activated with anti-CD3 and anti-CD28 for 48 hours, after which cytokine production was measured by ELISA (A and B) and glucose metabolism was analyzed by comparing relative glucose uptake (C). Data are representative of two independent experiments; three mice per experimental group. (D and E) Wildtype C57BL/6J mice (black bars) or T cell-specific Glut1 transgenic mice (Glut1 Tg; grey bars), were either fed ad libitum (control), fasted, or fasted while receiving leptin injections (leptin), as indicated, for 48 hours. Peripheral CD4+ T cells were isolated and activated with anti-CD3 and anti-CD28 for an additional 48 hours, after which glucose metabolism was analyzed by measuring glucose uptake (D) and IFN-γ production was measured in culture supernatants by ELISA (E). Data are representative of two independent experiments, three mice per group.
As leptin signal promotes Glut1 expression, and fasting is associated with both lower leptin levels and decreased glucose uptake and metabolism, we tested if independent support of glucose uptake by transgenic expression of Glut1 (Glut1 Tg) was sufficient to rescue T cells from persistent metabolic and functional defects in fasting. T cells from wildtype and Glut1 Tg fed control and fasted mice were isolated and cell counts obtained. The Glut1 transgene did not augment T cell count in either the fed control (5) or fasted condition (Supplemental Fig. 2C), demonstrating that glucose uptake alone is not sufficient to prevent T cell loss in fasting. Next, T cells from wildtype and Glut1 Tg fed control mice were compared to cells from fasted mice that received either twice-daily intraperitoneal injections of phosphate buffered saline (PBS) or recombinant leptin in PBS. T cells were isolated and stimulated in vitro with anti-CD3 and anti-CD28 for 48 hours, and glucose uptake and cytokine production were measured. Transgenic expression of Glut1 increased and maintained T cell glucose uptake even in fasting (Fig. 8D). While leptin rescued defects in T cell glucose metabolism and cytokine production in fasted wildtype mice, T cells from Glut1 Tg mice, which already resist fasting-induced defects in metabolism and function, did not further upregulate glucose uptake or IFN-γ production in response to leptin. Importantly, Glut1 Tg T cells also resisted fasting-induced dysregulation of T cell cytokine production and maintained the ability to secrete IFN-γ (Fig. 8E). Thus, persistent fasting-induced T cell defects appear due, in part, to an inability of T cells to increase glucose uptake and metabolism even when activated in the presence of abundant nutrients. We conclude that leptin acts directly on T cells to license metabolic reprogramming in activation, linking systemic nutrient status with T cell metabolism and function.
Discussion
Nutritional status is a critical regulator of overall immune function. Malnutrition reduces immunity and markedly increases the risks and mortality from severe infections (2). In contrast, obesity predisposes to systemic inflammation that promotes the development of certain forms of autoimmunity, asthma, and insulin resistance leading to type 2 diabetes (3, 50-53). There is growing evidence that adipocytes and immune cells coordinate nutritional status and immunity. One mechanism for this interaction is through secretion of adipokines, such as leptin, that can directly convey the state of nutrient availability to peripheral lymphocytes and other immune cells (10, 50). This communication of nutritional status to immune cells is critical to maintain energy balance in immunological defense, as activated effector lymphocytes have a very high metabolic demand essential for growth, proliferation, and the production of proteins required for a successful immune response (54-56). Leptin, in particular, has been shown to link adipose stores to immunity, as leptin deficiency has been associated with decreased immune reactivity in both mouse and human studies (14, 16, 19). Both neutralization of leptin in wildtype mice and genetic leptin-deficiency lead to decreased susceptibility to autoimmunity in animal models of experimental autoimmune encephalitis (EAE), glomerulonephritis, colitis, and hepatitis and increased susceptibility to intracellular infections (11). Additionally, calorie-restriction has been shown to attenuate autoimmune diseases such as EAE in mice (57), and fasting-induced hypoleptinemia was seen to expand regulatory T cells in a lupus-prone mouse model (58). Our study now shows leptin acts directly on activated effector T cells to allow glucose uptake and metabolic reprogramming that links systemic nutrient availability to metabolic licensing of T cells essential for activation.
Given the dual roles of leptin in regulating both overall body metabolism and T cell function, we hypothesized that leptin signaling may control T cell activation in part through direct regulation of lymphocyte metabolism. This role for leptin has been established in metabolic cells such as muscle and adipose (40, 41). In support of this role for leptin in T cells, recent publications have highlighted the ability of leptin to activate mTOR, which is a signaling molecule well known to affect T cell metabolism and function (59). Indeed, we found leptin receptor signal was required for increased Glut1 expression in peripheral T cells and upregulation of glucose uptake and glucose metabolism following T cell activation. This leptin-mediated increase in glucose metabolism was observed in effector Th17 cells but not Treg, which is consistent with findings that leptin promotes Th17 proliferation while inhibiting proliferation of Treg (47, 60). Therefore, a key role for leptin in T cell activation may be to allow effector T cells to increase Glut1 expression to fuel the demands of T cell growth, cytokine production, and proliferation. To test if directly augmenting T cell glucose metabolism bypassed the leptin requirement, we used T cell-specific Glut1 overexpressing mice. These studies showed that Glut1 overexpression was sufficient to maintain normal glucose uptake and rescue inflammatory cytokine production in T cells from fasted animals. The ability of Glut1 and glucose uptake to overcome persistent fasting-induced T cell dysfunction demonstrates that the ability of leptin to upregulate glucose metabolism is a key mechanism by which leptin directly enhances or supports T cell activation.
This ability of leptin to upregulate T cell Glut1 expression and glucose metabolism was observed only in lymphocytes activated through TCR together with co-stimulation. Resting T cells or T cells activated with anti-CD3 alone did not increase glucose metabolism following leptin therapy in vitro or in vivo. This finding correlates with the upregulation of leptin receptor expression on T cells following full activation (22). This is also consistent with previous reports that CD28 co-stimulation is required for increased glycolysis in activation (61). Further, these data suggest that leptin, which is secreted in proportion to fat mass and is therefore a surrogate for nutritional sufficiency, is not required for resting T cell metabolism but is selective to license T cells to increase metabolism to support activation. Therefore, in the absence of adequate nutritional stores, T cells are unable to upregulate glucose metabolism and cannot become fully activated.
Mice fasted for 48 hours were expected to have a rapid and significant drop in both thymocyte and T cell numbers (29, 37). Fasting has a number of important systemic effects, including increasing circulating levels of glucocorticoids (62, 63), which have potent effects on lymphocyte survival and function. Rising glucocorticoids in fasting, therefore, may contribute significantly to decreased overall thymocyte and T cell numbers (64). Injection of leptin alone during fasting only partially rescued thymocyte and T cell numbers. Moreover, Glut1 overexpression did not rescue T cell numbers in fasting, suggesting that the ability of leptin to partially rescue T cells numbers in fasting is via a mechanism independent of increased glucose uptake. Leptin may rescue cell number by opposing the apoptotic actions of glucocorticoids on both thymocytes and peripheral lymphocytes, as there is evidence in the literature for such as role (34). Whereas, in vivo injection of leptin alone during fasting fully rescued the persistent T cell defects in cytokine production and glucose metabolism following activation. Moreover, addition of leptin to cultured peripheral T cells from fasted animals in vitro rescued cytokine production and glucose metabolism to levels at or above the fed control. These data suggest a critical role of leptin in directly regulating activated T cell function and metabolism during states of malnutrition. However, the effects of leptin on activated peripheral T cell metabolism and function likely differ from other indirect mechanisms by which leptin affects resting T cell number and development in the thymus.
The relationship between leptin-deficiency and immunodeficiency is complex. While multiple studies have shown direct effects of leptin on T cell function (29, 31, 32), others have demonstrated that leptin may influence immune cell number and function indirectly through regulation of stromal cells or other hormones (30, 65, 66). More recent reports suggest leptin may also mediate its effects on immune cell populations via central signaling; in one study, intracerebroventricular injections of leptin were sufficient to rescue B cell numbers in fasted mice (67). However, the results from our studies, using both bone marrow transplant of leptin receptor-deficient db−/− donors into Rag1 knockout recipients and the T cell-specific conditional leptin receptor knockout mouse, support a direct and cell-intrinsic role for leptin in modulating peripheral T cell metabolism during activation. Leptin, therefore, links nutritional status to multiple levels of the immune system.
Collectively, our studies indicate that leptin serves as a nutritional rheostat that can communicate energy stores to peripheral lymphocytes undergoing activation. As lymphocyte activation is energetically costly, it is important to limit immunity in times of nutritional deficiency. Decreased leptin that occurs in malnutrition thus limits the capacity of T cells to consume the limiting availability of nutrients. Upregulation of leptin receptor following peripheral T cell activation allows leptin signal to metabolically license T cells for activation when nutritional stores are at sufficient levels, thus providing a key link between systemic and cellular metabolism in the immune system. Evolutionarily, this response may be critical to balance the energy costs of immunity with more immediately critical systems, such as neurologic, cardiovascular, respiratory, and musculoskeletal.
Supplementary Material
Acknowledgements
We thank M. Freemark for critical reading of the manuscript and members of the Rathmell Laboratory for helpful discussion and review of data. This work was supported in part by NIH K08-DK087944 (N.J.M.), R01HL108006 (J.C.R.), The Leukemia and Lymphoma Society (J.C.R.), The Hartwell Foundation (N.J.M.), The Pediatric Endocrine Society (N.J.M.), and the Children’s Miracle Network (N.J.M.).
References
- 1.MacIver NJ, Michalek RD, Rathmell JC. Metabolic Regulation of T Lymphocytes. Annual review of immunology. 2013 doi: 10.1146/annurev-immunol-032712-095956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schlaudecker EP, Steinhoff MC, Moore SR. Interactions of diarrhea, pneumonia, and malnutrition in childhood: recent evidence from developing countries. Curr Opin Infect Dis. 2011;24:496–502. doi: 10.1097/QCO.0b013e328349287d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annual review of immunology. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 4.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, Green DR. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacobs SR, Herman CE, MacIver NJ, Wofford JA, Wieman HL, Hammen JJ, Rathmell JC. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. Journal of immunology. 2008;180:4476–4486. doi: 10.4049/jimmunol.180.7.4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. Journal of immunology. 2011;186:3299–3303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi. H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. The Journal of experimental medicine. 2011;208:1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cham CM, Driessens G, O'Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. European journal of immunology. 2008;38:2438–2450. doi: 10.1002/eji.200838289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, Sewter CP, Digby JE, Mohammed SN, Hurst JA, Cheetham CH, Earley AR, Barnett AH, Prins JB, O'Rahilly S. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–908. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- 10.Mantzoros CS, Magkos F, Brinkoetter M, Sienkiewicz E, Dardeno TA, Kim SY, Hamnvik OP, Koniaris A. Leptin in human physiology and pathophysiology. Am J Physiol Endocrinol Metab. 2011;301:E567–584. doi: 10.1152/ajpendo.00315.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Procaccini C, Jirillo E, Matarese G. Leptin as an immunomodulator. Mol Aspects Med. 2012;33:35–45. doi: 10.1016/j.mam.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 12.Fernandez-Riejos P, Najib S, Santos-Alvarez J, Martin-Romero C, Perez-Perez A, Gonzalez-Yanes C, Sanchez-Margalet V. Role of leptin in the activation of immune cells. Mediators Inflamm. 20102010:568343. doi: 10.1155/2010/568343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ozata M, Ozdemir IC, Licinio J. Human leptin deficiency caused by a missense mutation: multiple endocrine defects, decreased sympathetic tone, and immune system dysfunction indicate new targets for leptin action, greater central than peripheral resistance to the effects of leptin, and spontaneous correction of leptin-mediated defects. The Journal of clinical endocrinology and metabolism. 1999;84:3686–3695. doi: 10.1210/jcem.84.10.5999. [DOI] [PubMed] [Google Scholar]
- 14.Mandel MA, Mahmoud AA. Impairment of cell-mediated immunity in mutation diabetic mice (db/db) Journal of immunology. 1978;120:1375–1377. [PubMed] [Google Scholar]
- 15.Farooqi IS, Wangensteen T, Collins S, Kimber W, Matarese G, Keogh JM, Lank E, Bottomley B, Lopez-Fernandez J, Ferraz-Amaro I, Dattani MT, Ercan O, Myhre AG, Retterstol L, Stanhope R, Edge JA, McKenzie S, Lessan N, Ghodsi M, De Rosa V, Perna F, Fontana S, Barroso I, Undlien DE, O'Rahilly S. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. The New England journal of medicine. 2007;356:237–247. doi: 10.1056/NEJMoa063988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chandra RK. Cell-mediated immunity in genetically obese C57BL/6J ob/ob) mice. Am J Clin Nutr. 1980;33:13–16. doi: 10.1093/ajcn/33.1.13. [DOI] [PubMed] [Google Scholar]
- 17.Paz-Filho GJ, Delibasi T, Erol HK, Wong ML, Licinio J. Cellular immunity before and after leptin replacement therapy. J Pediatr Endocrinol Metab. 2009;22:1069–1074. doi: 10.1515/jpem.2009.22.11.1069. [DOI] [PubMed] [Google Scholar]
- 18.Paz-Filho G, Mastronardi C, Delibasi T, Wong ML, Licinio J. Congenital leptin deficiency: diagnosis and effects of leptin replacement therapy. Arq Bras Endocrinol Metabol. 2010;54:690–697. doi: 10.1590/s0004-27302010000800005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, Sanna V, Jebb SA, Perna F, Fontana S, Lechler RI, DePaoli AM, O'Rahilly S. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002;110:1093–1103. doi: 10.1172/JCI15693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Procaccini C, Lourenco EV, Matarese G, La Cava A. Leptin signaling: A key pathway in immune responses. Curr Signal Transduct Ther. 2009;4:22–30. doi: 10.2174/157436209787048711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fantuzzi G. Three questions about leptin and immunity. Brain Behav Immun. 2009;23:405–410. doi: 10.1016/j.bbi.2008.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papathanassoglou E, El-Haschimi K, Li XC, Matarese G, Strom T, Mantzoros C. Leptin receptor expression and signaling in lymphocytes: kinetics during lymphocyte activation, role in lymphocyte survival, and response to high fat diet in mice. Journal of immunology. 2006;176:7745–7752. doi: 10.4049/jimmunol.176.12.7745. [DOI] [PubMed] [Google Scholar]
- 23.Sanchez-Margalet V, Martin-Romero C, Gonzalez-Yanes C, Goberna R, Rodriguez-Bano J, Muniain MA. Leptin receptor (Ob-R) expression is induced in peripheral blood mononuclear cells by in vitro activation and in vivo in HIV-infected patients. Clinical and experimental immunology. 2002;129:119–124. doi: 10.1046/j.1365-2249.2002.01900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hegyi K, Fulop K, acs K. Ko, Toth S, Falus A. Leptin-induced signal transduction pathways. Cell biology international. 2004;28:159–169. doi: 10.1016/j.cellbi.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 25.Martin-Romero C, Sanchez-Margalet V. Human leptin activates PI3K and MAPK pathways in human peripheral blood mononuclear cells: possible role of Sam68. Cell Immunol. 2001;212:83–91. doi: 10.1006/cimm.2001.1851. [DOI] [PubMed] [Google Scholar]
- 26.Sanchez-Margalet V, Martin-Romero C. Human leptin signaling in human peripheral blood mononuclear cells: activation of the JAK-STAT pathway. Cell Immunol. 2001;211:30–36. doi: 10.1006/cimm.2001.1815. [DOI] [PubMed] [Google Scholar]
- 27.Sanchez-Margalet V, Martin-Romero C, Santos-Alvarez J, Goberna R, Najib S, Gonzalez-Yanes C. Role of leptin as an immunomodulator of blood mononuclear cells: mechanisms of action. Clinical and experimental immunology. 2003;133:11–19. doi: 10.1046/j.1365-2249.2003.02190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Procaccini C, De Rosa V, Galgani M, Abanni L, Cali G, Porcellini A, Carbone F, Fontana S, Horvath TL, La Cava A, Matarese G. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity. 2010;33:929–941. doi: 10.1016/j.immuni.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394:897–901. doi: 10.1038/29795. [DOI] [PubMed] [Google Scholar]
- 30.Palmer G, Aurrand-Lions M, Contassot E, Talabot-Ayer D, Ducrest-Gay D, Vesin C, Chobaz-Peclat V, Busso N, Gabay C. Indirect effects of leptin receptor deficiency on lymphocyte populations and immune response in db/db mice. Journal of immunology. 2006;177:2899–2907. doi: 10.4049/jimmunol.177.5.2899. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez L, Graniel J, Ortiz R. Effect of leptin on activation and cytokine synthesis in peripheral blood lymphocytes of malnourished infected children. Clinical and experimental immunology. 2007;148:478–485. doi: 10.1111/j.1365-2249.2007.03361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin-Romero C, Santos-Alvarez J, Goberna R, Sanchez-Margalet V. Human leptin enhances activation and proliferation of human circulating T lymphocytes. Cell Immunol. 2000;199:15–24. doi: 10.1006/cimm.1999.1594. [DOI] [PubMed] [Google Scholar]
- 33.Gove ME, Sherry CL, Pini M, Fantuzzi G. Generation of leptin receptor bone marrow chimeras: recovery from irradiation, immune cellularity, cytokine expression, and metabolic parameters. Obesity (Silver Spring) 2010;18:2274–2281. doi: 10.1038/oby.2010.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujita Y, Murakami M, Ogawa Y, Masuzaki H, Tanaka M, Ozaki S, Nakao K, Mimori T. Leptin inhibits stress-induced apoptosis of T lymphocytes. Clinical and experimental immunology. 2002;128:21–26. doi: 10.1046/j.1365-2249.2002.01797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boden G, Chen X, Mozzoli M, Ryan I. Effect of fasting on serum leptin in normal human subjects. The Journal of clinical endocrinology and metabolism. 1996;81:3419–3423. doi: 10.1210/jcem.81.9.8784108. [DOI] [PubMed] [Google Scholar]
- 36.Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, Flier JS. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–252. doi: 10.1038/382250a0. [DOI] [PubMed] [Google Scholar]
- 37.Howard JK, Lord GM, Matarese G, Vendetti S, Ghatei MA, Ritter MA, Lechler RI, Bloom SR. Leptin protects mice from starvation-induced lymphoid atrophy and increases thymic cellularity in ob/ob mice. J Clin Invest. 1999;104:1051–1059. doi: 10.1172/JCI6762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacIver NJ, Jacobs SR, Wieman HL, Wofford JA, Coloff JL, Rathmell JC. Glucose metabolism in lymphocytes is a regulated process with significant effects on immune cell function and survival. Journal of leukocyte biology. 2008;84:949–957. doi: 10.1189/jlb.0108024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cham CM, Gajewski TF. Glucose availability regulates IFN-gamma production and p70S6 kinase activation in CD8+ effector T cells. Journal of immunology. 2005;174:4670–4677. doi: 10.4049/jimmunol.174.8.4670. [DOI] [PubMed] [Google Scholar]
- 40.Ceddia RB, William WN, Jr., Curi. R. Comparing effects of leptin and insulin on glucose metabolism in skeletal muscle: evidence for an effect of leptin on glucose uptake and decarboxylation. International journal of obesity and related metabolic disorders : journal of the International Association for the Study of Obesity. 1999;23:75–82. doi: 10.1038/sj.ijo.0800762. [DOI] [PubMed] [Google Scholar]
- 41.Ceddia RB. Direct metabolic regulation in skeletal muscle and fat tissue by leptin: implications for glucose and fatty acids homeostasis. International journal of obesity. 2005;29:1175–1183. doi: 10.1038/sj.ijo.0803025. [DOI] [PubMed] [Google Scholar]
- 42.Vander Heiden MG, Plas DR, Rathmell JC, Fox CJ, Harris MH, Thompson CB. Growth factors can influence cell growth and survival through effects on glucose metabolism. Molecular and cellular biology. 2001;21:5899–5912. doi: 10.1128/MCB.21.17.5899-5912.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell. 2007;18:1437–1446. doi: 10.1091/mbc.E06-07-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wofford JA, Wieman HL, Jacobs SR, Zhao Y, Rathmell JC. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood. 2008;111:2101–2111. doi: 10.1182/blood-2007-06-096297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Swainson L, Kinet S, Manel N, Battini JL, Sitbon M, Taylor N. Glucose transporter 1 expression identifies a population of cycling CD4+ CD8+ human thymocytes with high CXCR4-induced chemotaxis. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:12867–12872. doi: 10.1073/pnas.0503603102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Rosa V, Procaccini C, La Cava A, Chieffi P, Nicoletti GF, Fontana S, Zappacosta S, Matarese G. Leptin neutralization interferes with pathogenic T cell autoreactivity in autoimmune encephalomyelitis. J Clin Invest. 2006;116:447–455. doi: 10.1172/JCI26523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu Y, Liu Y, Shi FD, Zou H, Matarese G, La Cava A. Cutting edge: Leptin-induced RORgammat expression in CD4+ T cells promotes Th17 responses in systemic lupus erythematosus. Journal of immunology. 2013;190:3054–3058. doi: 10.4049/jimmunol.1203275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deng J, Liu Y, Yang M, Wang S, Zhang M, Wang X, Ko KH, Hua Z, Sun L, Cao X, Lu L. Leptin exacerbates collagen-induced arthritis via enhancement of Th17 cell response. Arthritis and rheumatism. 2012;64:3564–3573. doi: 10.1002/art.34637. [DOI] [PubMed] [Google Scholar]
- 49.Wang S, Baidoo SE, Liu Y, Zhu C, Tian J, Ma J, Tong J, Chen J, Tang X, Xu H, Lu L. T cell-derived leptin contributes to increased frequency of T helper type 17 cells in female patients with Hashimoto's thyroiditis. Clinical and experimental immunology. 2013;171:63–68. doi: 10.1111/j.1365-2249.2012.04670.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11:85–97. doi: 10.1038/nri2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ong KK, Kuh D, Pierce M, Franklyn JA, H. Medical Research Council National Survey of, S. Development, and T. Data Collection Childhood weight gain and thyroid autoimmunity at age 60-64 years: the 1946 British birth cohort study. The Journal of clinical endocrinology and metabolism. 2013;98:1435–1442. doi: 10.1210/jc.2012-3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hersoug LG, Linneberg A. The link between the epidemics of obesity and allergic diseases: does obesity induce decreased immune tolerance? Allergy. 2007;62:1205–1213. doi: 10.1111/j.1398-9995.2007.01506.x. [DOI] [PubMed] [Google Scholar]
- 53.Duntas LH. Environmental factors and thyroid autoimmunity. Annales d'endocrinologie. 2011;72:108–113. doi: 10.1016/j.ando.2011.03.019. [DOI] [PubMed] [Google Scholar]
- 54.Gerriets VA, Rathmell JC. Metabolic pathways in T cell fate and function. Trends Immunol. 2012;33:168–173. doi: 10.1016/j.it.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marelli-Berg FM, Fu H, Mauro C. Molecular mechanisms of metabolic reprogramming in proliferating cells: implications for T-cell-mediated immunity. Immunology. 2012;136:363–369. doi: 10.1111/j.1365-2567.2012.03583.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Piccio L, Stark JL, Cross AH. Chronic calorie restriction attenuates experimental autoimmune encephalomyelitis. Journal of leukocyte biology. 2008;84:940–948. doi: 10.1189/jlb.0208133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu Y, Yu Y, Matarese G, La Cava A. Cutting edge: fasting-induced hypoleptinemia expands functional regulatory T cells in systemic lupus erythematosus. Journal of immunology. 2012;188:2070–2073. doi: 10.4049/jimmunol.1102835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Procaccini C, De Rosa V, Galgani M, Carbone F, Cassano S, Greco D, Qian K, Auvinen P, Cali G, Stallone G, Formisano L, La Cava A, Matarese G. Leptin-induced mTOR activation defines a specific molecular and transcriptional signature controlling CD4+ effector T cell responses. Journal of immunology. 2012;189:2941–2953. doi: 10.4049/jimmunol.1200935. [DOI] [PubMed] [Google Scholar]
- 60.De Rosa V, Procaccini C, Cali G, Pirozzi G, Fontana S, Zappacosta S, La Cava A, Matarese G. A key role of leptin in the control of regulatory T cell proliferation. Immunity. 2007;26:241–255. doi: 10.1016/j.immuni.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 61.Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, Elstrom RL, June CH, Thompson CB. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16:769–777. doi: 10.1016/s1074-7613(02)00323-0. [DOI] [PubMed] [Google Scholar]
- 62.Dallman MF, Strack AM, Akana SF, Bradbury MJ, Hanson ES, Scribner KA, Smith M. Feast and famine: critical role of glucocorticoids with insulin in daily energy flow. Frontiers in neuroendocrinology. 1993;14:303–347. doi: 10.1006/frne.1993.1010. [DOI] [PubMed] [Google Scholar]
- 63.Makimura H, Mizuno TM, Isoda F, Beasley J, Silverstein JH, Mobbs CV. Role of glucocorticoids in mediating effects of fasting and diabetes on hypothalamic gene expression. BMC physiology. 2003;3:5. doi: 10.1186/1472-6793-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Franchimont D. Overview of the actions of glucocorticoids on the immune response: a good model to characterize new pathways of immunosuppression for new treatment strategies. Ann N Y Acad Sci. 2004;1024:124–137. doi: 10.1196/annals.1321.009. [DOI] [PubMed] [Google Scholar]
- 65.Gruver AL, Ventevogel MS, Sempowski GD. Leptin receptor is expressed in thymus medulla and leptin protects against thymic remodeling during endotoxemia-induced thymus involution. J Endocrinol. 2009;203:75–85. doi: 10.1677/JOE-09-0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fantuzzi G. Leptin: nourishment for the immune system. Eur J Immunol. 2006;36:3101–3104. doi: 10.1002/eji.200636770. [DOI] [PubMed] [Google Scholar]
- 67.Tanaka M, Suganami T, Kim-Saijo M, Toda C, Tsuiji M, Ochi K, Kamei Y, Minokoshi Y, Ogawa Y. Role of central leptin signaling in the starvation-induced alteration of B-cell development. J Neurosci. 2011;31:8373–8380. doi: 10.1523/JNEUROSCI.6562-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.