

Abstract
Tyrosine hydroxylase (TH) catalyzes the rate limiting step in the synthesis of catecholamine neutrotransmitters, and a reduction of TH activity is associated with several neurological diseases. Human TH is regulated, among other mechanisms, by Ser19-phosphorylation-dependent interaction with 14-3-3 proteins. The N-terminal sequence (residues 1-43), which corresponds to an extension to the TH regulatory domain, also interacts with negatively charged membranes. By using X-ray crystallography together with molecular dynamics simulations and structural bioinformatics analysis, we have probed the conformations of the Ser19-phosphorylated N-terminal peptide (THp-(1-43)), bound to 14-3-3γ, free in solution and bound to a phospholipid bilayer, and of the unphosphorylated peptide TH-(1-43) both free and bilayer-bound. As seen in the crystal structure of THp-(1-43) complexed with 14-3-3γ, the region surrounding pSer19 adopts an extended conformation in the bound state, whereas THp-(1-43) adopts a bent conformation when free in solution, with higher content of secondary structure and higher number of internal hydrogen bonds. TH-(1-43) in solution presents the highest mobility and least defined structure of all forms studied, and it shows an energetically more favorable interaction with membranes relative to THp-(1-43). Cationic residues, notably Arg15 and Arg16, which are the recognition sites of the kinases phosphorylating at Ser19, are also contributing to the interaction with the membrane. Our results reveal the structural flexibility of this region of TH, in accordance with the functional versatility and conformational adaptation to different partners. Furthermore, this structural information has potential relevance for the development of therapeutics for neurodegenerative disorders, through modulation of TH-partner interactions.
INTRODUCTION
Tyrosine hydroxylase (TH) catalyzes the hydroxylation of L-tyrosine (L-Tyr) to L-3,4-dihydroxyphenylalanine (L-Dopa), using the cofactor tetrahydrobiopterin (BH4) and dioxygen as additional substrate. L-Dopa is the precursor of the catecholamines dopamine, noradrenaline and adrenaline, which act as neurotransmitters in the central nervous system and as hormones in the neuroendocrine system, where noradrenaline and adrenaline secreted by the chromaffin cells from the medulla of the adrenal gland initiate the fight-or-flight response. A decrease in TH activity is associated with many neuropsychiatric and neurodegenerative diseases such as Dopa-responsive dystonia, Parkinson’s and Alzheimer’s disease and others. The enzyme is thus strictly regulated both at the transcriptional and posttranscriptional levels 1; 2; 3.
Short term changes in TH activity and regulation of the synthesis of dopamine appear to be associated with post-transcriptional modifications in a sequence stretch located at the N-terminal of the protein, adjacent to the regulatory ACT domain. This sequence includes four phosphorylation sites (Thr8, Ser19, Ser31 and Ser40 in human TH isoform 1 (hTH1)) which are phosphorylated by different kinases, with considerable specificity for each site 2; 3. Thus, these phosphorylation events involve the N-terminal sequence, but little structural information is available on the conformational changes associated with each specific modification. Phosphorylation of Ser40 by cAMP-dependent protein kinase (PKA) is the most studied event, both in vitro and in vivo 2; 3. This phosphorylation activates the enzyme, and also counteracts the feedback inhibition of TH by catecholamines, an inhibition that is competitive with respect to the cofactor BH4. Hydrogen/deuterium exchange 4 and fluorescence spectroscopy 5 have revealed a more open conformation for Ser40-phosphorylated TH, with a higher exposure of regions Ile35-Leu41 and Ile42-Ala71 4. The first region includes the recognition site for PKA (residues Arg37 and Arg38), and at the same time it plays a key role in determining the inhibition by dopamine 6; 7. Phosphorylation at Ser19 by p38 regulated/activated kinase (PRAK) and by Ca++/calmodulin-dependent protein kinase II (CaMKII) activates and stabilizes TH through a mechanism that requires the binding to 14-3- 3 proteins 2; 3; 8. Conformational changes related to Ser19 phosphorylation are not so well understood, but time-resolved tryptophan fluorescence and circular dichroism (CD) measurements revealed that Ser19 phosphorylation and binding to 14-3-3 reduced the solvent exposure of residues 14 and 34 and also reduced the sensitivity of the N-terminal part of TH (first 33 residues) towards proteolysis 9. However, neither phosphorylation nor the binding to 14-3-3 proteins appeared to significantly change the secondary structure of the regulatory domain 9. Finally, the effect of phosphorylation of Thr8 and Ser31 by other kinases such as ERK and Cdk5 is much less understood but their influence in the regulation of TH activity and dopamine synthesis is starting to be elucidated 10; 11.
TH is essentially soluble and cytoplasmic, but a fraction is also found as membrane-bound in the brain, notably at nerve endings and synaptic vesicles 12; 13; 14; 15. Little is known about the significance of the interaction of TH with membranes, but it has been speculated that it could contribute to modulate the distribution of TH in the cytoplasm and the membrane fractions and coordinate dopamine synthesis and packaging in the vesicles 14; 16. Recent studies have shown that the association of TH with synaptic vesicles might occur through the formation of a complex with the vesicular monoamine transporter-2 (VMAT2) and aromatic amino acid decarboxylase (AADC) 17. However, a direct interaction of TH with the membrane has been demonstrated both with chromaffin granule membranes 18 and with synthetic phospholipidic bilayers 19, and it might be hypothesized that this direct association has a cytotoxic effect, as found for other protein:membrane interactions involved in neurodegeneration 20; 21. With respect to protein motifs involved in the interaction of the enzyme with the membrane, it has been shown that truncated forms of the recombinant hTH1 lacking up to residues 33–49 do not bind to membranes 19. Moreover, the peptide TH-(1-43), corresponding to the N-terminal sequence of TH (residues 1-43), binds to negatively charged membranes 22 and, as seen by surface plasmon resonance (SPR), the unphosphorylated TH-(1-43) peptide showed higher affinity for negatively charged membranes than the Ser19-phosphorylated THp-(1-43) peptide 22. However, little is known about the structural determinants and conformational changes associated with membrane binding. On the other hand, THp-(1-43) showed, as expected, a much higher affinity for 14-3-3γ (Kd = 0.5 µM) than the unphosphorylated counterpart (Kd = 1550 µM) 22. The structure-energetics relationships for the complex formation between 14-3-3 proteins and a number of phosphopeptides are relatively well understood from crystal structure analyses of several complexes 23; 24; 25; 26. The phosphorylated 14-3-3-interacting proteins and peptides customarily present three binding motifs; RSXpSXP (mode I), RX(Y/F)XpSXP (mode II), and carboxyl-terminal (pS/pT)X1–2-COOH (mode III), where X is not Pro 27. The TH sequence that binds to 14-3-3 proteins does not conform to these consensus motifs.
The structural analysis of full-length TH has been unattainable so far and only the catalytic and oligomerization domains have been crystallized 28. Thus, there is no structural information on neither the regulatory ACT domain nor its N-terminal extension which includes residues 1-43. While the regulatory ACT domain is characteristic of the four mammalian aromatic amino acid hydroxylases, an N-terminal extension of similar length is present in neuronal tryptophan hydroxylase isoform 2 (TPH2) 29; 30, but not in phenylalanine hydroxylase (PAH) and TPH1. The TPH2 extension does not show sequence similarity with its TH counterpart, yet it has a similar 14-3-3-interacting phosphorylatable site at Ser19 29; 31.
In this work we have studied the conformation of the unphosphorylated TH-(1-43) and Ser19-phosphorylated THp-(1-43) N-terminal peptides in three states, i.e. bound to 14-3-3γ, unbound in solution, and bound to negatively charged bilayers. Through integration of several experimental (X-ray crystallography, circular dichroism (CD) and surface plasmon resonance (SPR)) and computational methods (molecular dynamics (MD) simulations and energetic decomposition analysis by MM/PBSA) we investigated the conformational change effected by phosphorylation at Ser19 and the structural determinants for interaction of THp-(1-43) with 14-3-3γ and of both TH-(1-43) and THp-(1-43) with negatively charged bilayers. These detailed analyses reveal the conformational versatility of this N-terminal regulatory region in TH and provide a structural frame for attempts to develop drugs that stabilize/disrupt interactions with 14-3-3 proteins and membranes.
MATERIALS AND METHODS
Peptides and phospholipids
The peptides TH-(1-43) (MPTPDATTPQAKGFRRAVSELDAKQAEAIMSPRFIGRRQSLIE) and THp-(1-43) (MPTPDATTPQAKGFRRAVS(PO3)2−ELDAKQAEAIMSPRFIGRRQSLIE) were synthesized by CPC Scientific (San Jose, CA) at approximately 90% purity, as seen by mass spectroscopy, and used without further purification. Phosphatidylcholine (PC) from egg yolk lecithin (99% PC) and palmitoyl-oleyl-phosphatidylserine (POPS) were purchased from Avanti Polar Lipids, Inc.
Expression and purification of 14-3-3γ
Human 14-3-3γ was expressed using pGEX-expression vector in BL-21-CodonPlus® as glutathione S-transferase fusion proteins, and further purified on glutathione-Sepharose 4B (GE Healthcare) essentially as reported 22. The fusion protein was cleaved by thrombin (4 units/ml) while attached to the column, at 4 °C for 1 h. The dimeric 14-3-3γ protein (about 56-kDa dimer) was further purified by gel filtration in HiLoad 16/60 Superdex 200 (Amersham Biosciences) in 50 mM sodium phosphate, 150 mM NaCl, pH 7.4, 1 mM dithiothreitol and 1 mM EDTA and further exchanged to 10 mM Tris, 200 mM NaCl, pH 8 (crystallization buffer, see below).
Crystallization, data collection and X-ray structure determination
The purified 14-3-3γ protein was concentrated up to 30 mg/ml (approx. 1.07 mM subunit) using Amicon® Ultra centrifugal filter units, MWCO 30 kDa (Millipore), and the synthetic THp-(1-43) peptide was prepared in 10 mM Tris, 200 mM NaCl, pH 8, to a concentration of 20 mg/ml (approx. 4 mM). 14-3-3γ and THp-(1-43) were mixed at a molar ratio of 1:3 (14-3-3γ:THp-(1-43) complex) to a concentration of 0.6 mM 14-3-3γ subunit. After mixing and incubating on ice for 30 min, the sample was ultracentrifuged at approximately 200,000 g for 10 min. The supernatant was dispensed in a clean tube and used for crystallization.
Crystals were grown by sitting drop vapour diffusion using 96-well plates (Innovaplate SD-2, Innovadyne Technologies, Inc.) with the help of a dispensing robot (Innovadyne Screenmaker, Innovadyne Technologies, Inc.) and incubated at 10 °C. The 14-3-3γ:THp-(1-43) complex was mixed in a 1:1 ratio with buffer containing 0.1 M K thiocyanate, with 30% PEG 2K MME as precipitant. Small rectangular crystals (~60×40×20 µm) grew after 7 days, and they were harvested with a nylon cryoloop and soaked briefly in a solution consisting of 25% glycerol added to the crystallization buffer. The crystals were subsequently frozen directly into liquid nitrogen. X-ray diffraction data were collected at 100 K at the GM/CA-CAT beamline of the Advanced Photon Source (APS, at Argonne) by using a 10-µm beam collimator. Data processing was performed by using the XDS software package 32; the structure was solved using the dimeric 14-3-3γ (PDB code: 2B05) as a model for molecular replacement and refined with the programs Coot 33 and Refmac 5 from the CCP4 program suite 34.
Preparation of liposomes
Liposomes were prepared as large unilamellar vesicles, made of either PC or POPS as described 22. Briefly, the chloroform-dissolved phospholipids were dried under nitrogen stream and subjected to vacuum for 2 h. The lipids were resuspended in 10 mM Na-Hepes, 150 mM NaCl, pH 7.4, and left in an incubator overnight in the dark at 37 °C and 150 rpm. The hydrated phospholipid solutions were then frozen and thawed seven times, followed by extrusion (11 times) with the Avanti Mini-Extruder using a membrane of 0.1 µm pore size. The hydrodynamic diameter was measured by dynamic light scattering using a Zetasizer Nano-Instrument (Malvern), and estimated to be 92.4 ± 0.6 and 115.4 ± 2.5 nm for liposomes made of PC and POPS, respectively.
Surface plasmon resonance (SPR)
SPR analyses to monitor the interaction of TH-(1-43) and THp-(1-43) with liposomes composed of either PC or POPS were carried out using the Biacore 3000 biosensor (Biacore AB) and an L1 sensor chip at 25 °C. The conditions for deposition of the liposomes, application of the peptides to the captured liposomes, and regeneration of the sensor chip surface was performed essentially as reported 22. The values for non-specific binding measured in the reference cell (with liposomes consisting of PC) were subtracted. The BIA evaluation program, version 3.2 (Biacore AB) was used for analysis of the sensorgrams. The binding isotherms for maximal response units (RU) versus concentration of peptide were found to be hyperbolic and were processed by nonlinear least-squares analysis. The concentration of peptide providing half-maximal binding (S0.5) is thus a measure of the inverse of the apparent association or partition constant (1/Ka(app)). The S0.5 values provide comparative and operational measurements of affinity.
Circular dichroism
CD experiments were performed in the far-UV spectral range (180–260 nm) with a Jasco J-810 spectropolarimeter equipped with a Peltier element for temperature control, at 25 °C, using a data pitch of 0.1 nm, a bandwidth of 2 nm and a scan speed of 50 nm/min. Sample solutions were loaded into a 1 mm quartz cuvette (Hellma) and CD spectra of TH-(1-43) and THp-(1-43) (both 50 µM) either in water or in 10 mM Na-Hepes, 150 mM NaCl, pH 7.4, were acquired. Spectra in the presence of liposomes made of POPS (0.6 mM liposome phospholipid) were taken in the same buffer with 40 µM peptide. Baseline spectra of buffer or liposomes without peptide were obtained and subtracted using the software provided by the instrument manufacturers, and data were smoothed using running average with a sampling proportion of 0.05. The mean residue ellipticity was determined using the formula [θ]MRW = MRW × θ / (10 × c ×, where MRW is the mean residue weight (g/mol), θ is the ellipticity (deg), 10 is a scaling factor, c is the protein concentration (g/ml) and l is the path length of the cuvette (cm). MRW is calculated from MRW = M / (N − 1), where M is the molecular mass (g/mol), and N is the number of amino acids in the chain. Three parallels were averaged after subtraction, calculation of mean residue ellipticity and estimation of secondary structure by using the CDNN software 35.
MD simulations
14-3-3γ with bound THp-(1-43) peptide
MD simulation was initiated using coordinates generated from the 3D X-ray structure including data up to 3.1 Å resolution. The final model of the 14-3-3γ:THp-(1-43) complex included residues Phe14-Ala26/Ala11-Asp22 of the THp fragment (System A in Table 1), and the model was built as follows: Two 14-3-3γ dimers with the THp fragments bound to each of the monomers were aligned using Accelrys Discovery Studio 36 and one of them subsequently deleted due to a very high degree of similarity between the two dimer conformations. The longest peptide fragments in the alignment were kept and overlapping residues removed to give a single peptide bound to each monomer. These peptide fragments were included in the simulation structure and capped with ACE and NME groups, as is standard in AMBER. In consequence, the system consisted of a 14-3-3γ dimer with a 13-residue (Phe14-Ala26) and a 12-residue fragment (Ala11-Asp22) bound to each monomer, respectively (Table 1). The complex was solvated by an octahedral-shaped water box and subjected to the following minimization and simulation steps: i) Minimization for 10000 steps; ii) 5 ps gradual heating from 0 to 100 K (NVT) with restraints on 14-3-3γ and peptide fragments; iii) 100 ps gradual heating from 100 to 300 K (NPT) with restraints on 14-3-3γ and peptide fragments; iv) 100 ps equilibration (NPT) with restraints on peptide fragments; v) 1 ns equilibration (NPT) without restraints; vi) 100 ns simulation (NPT) without restraints.
Table 1.
Summary of the different simulation systems involving TH peptide variants.
| System | Peptide:protein | Number of POPS lipids |
Number of sodium ions |
Number of TIP3P waters |
Simulation time |
|---|---|---|---|---|---|
| A | 14-3-3γdimer: THp fragmentsa | - | - | 21124 | 100 ns |
| B | TH-(1-43) | - | - | 3159 | 500 ns |
| C | THp-(1-43) | - | - | 2773 | 500 ns |
| D | TH-(1-43) | 128 | 128 | 13290 | 500 ns |
| E | THp-(1-43) | 128 | 128 | 13296 | 500 ns |
THp-(14-26) and THp-(11-22), each bound to a 14-3-3γmonomer
Pure POPS membrane
The start configuration for the simulation of the pure POPS bilayer was the equilibrated end structure from an MD study by Mukhopadhyay et al. 37, obtained as a PDB file by correspondence with co-author Luca Monticelli. The system had been simulated for 40 ns and consisted of a united atom representation of 128 POPS molecules, 128 neutralizing sodium ions and 5391 water molecules. Following the addition of aliphatic hydrogens to obtain an all-atom model, six of the POPS conformations were used for charge derivation in Gaussian03 38. The resulting electrostatic potential surfaces were fitted to atomic point charges by virtue of a RESP procedure 39, and the average charges calculated from the six conformations were applied in all the simulations involving POPS. Antechamber 40 assigned GAFF atom types 41 to the membrane lipids. Subsequent minimization and equilibration of the pure membrane system were performed as previously described 42. The area per lipid was kept constant at 53.54 Å2, close to the average value of 55 +/− 1 Å2 derived by Mukhopadhyay et al. 37.
TH peptides in water and with the POPS membrane
The peptides TH-(1-43) and THp-(1-43) were built as random coils with the protein builder in Accelrys Discovery Studio 36 and simulated for 100 ns in explicit octahedral-shaped water boxes. The final peptide conformations from these simulations were subjected to another 500 ns of aqueous simulation at 300 K using the NPT ensemble (Systems B and C in Table 1), and the same peptide conformations were inserted into the aqueous phase of the pure POPS bilayer system (see above). To fully accommodate the peptides, the water phase had to be extended through addition of extra water molecules in Packmol 43. The resulting membrane/peptide simulation systems are described in Table 1 (Systems D and E). Both were subjected to the same minimization and simulation procedure: i) Minimization for 10000 steps without restraints; ii) 5 ps gradual heating from 0 to 100 K (NVT) with restraints on membrane and peptide; iii) 100 ps gradual heating from 100 to 300 K (NPT) with restraints on membrane and peptide; iv) 1 ns equilibration of water (NPT) with restraints on membrane and peptide; v) 500 ns simulation at constant volume (NVT) without restraints.
All simulations were performed with AMBER12 44. Periodic boundary conditions were applied for all systems and enabled the particle mesh Ewald method 45; 46 to sum the electrostatic interactions by fourth-order B-spline interpolation and with a grid spacing of < 1 Å. Van der Waals forces were truncated by a 10 Å cut-off. By means of the SHAKE algorithm 47 the lengths of the covalent bonds involving hydrogen were constrained, which allowed for time steps of 2 fs. Pressure was regulated anisotropically for the constant pressure portion of the membrane simulations, whilst isotropic scaling was applied for the peptides in pure water and for the simulation of the 14-3-3γ:THp peptide complex. In both cases, the target pressure was maintained at 1 bar. The Langevin thermostat 48 controlled the temperature. Force constants for the positional restraints referred to in previous sections differed depending on the type of calculation, with 500 kcal/mol Å2 used for minimization and 10 kcal/mol Å2 applied in the relevant simulation steps. All the peptides and 14-3-3γ were modelled by the ff99SB force field 49, apart from the phosphoserine group in THp-(1-43) and in the THp-(1-43) fragments in the complex, which required specific parameters developed by Homeyer et al 50. Water molecules were described by the TIP3P model 51.
Trajectory analysis
Several types of MD trajectory analysis were performed, including B factor, hydrogen bonding and secondary structure analysis. A brief description of each of them is provided in the following.
In the context of MD simulations, B factors are defined to be directly proportional to the squared root mean square fluctuations (RMSF) of the atomic positions:
| (Eqn.1) |
As such, they represent how much atoms move in the course of the simulation and thereby give a measure of flexibility. B factors were derived according to Equation 1 using ptraj 44.
Hydrogen bond counts and occupancies were calculated by the cpptraj facility 44. For an interaction to be classified as a hydrogen bond, the distance between the non-hydrogen atoms had to be <3.5 Å and the donor-hydrogen-acceptor angle >20 degrees.
Secondary structure content was estimated by DSSP 52 as implemented in the ptraj secstruct program 44. The proportions of each peptide that adopted α-helix (including 3–10 helices), β-sheet (both parallel and antiparallel) and turn were averaged over the time frame of analysis.
MM/PBSA per residue energy decomposition
Free energy of binding (ΔGbinding) quantifies how favourable or unfavourable it is for a ligand to go from the solvated to the receptor-bound state and can be broken down into the accompanying changes in enthalpic (ΔH) and entropic (ΔS) energy (Equation 2). T denotes the temperature. The more negative the free energy, the more favourable is the binding between ligand and receptor relative to their existence free in solution.
| (Eqn.2) |
In the molecular mechanics Poisson-Boltzmann surface area (MM/PBSA) approach 53 a combination of several methods is applied to obtain values for ΔH and ΔS and thereby ΔGbinding, according to the following relationship:
| (Eqn.3) |
where ΔEMM is the molecular mechanics energy, ΔGPB is the Poisson-Boltzmann energy and ΔGnon-polar is the non-polar solvation energy. The two latter terms can collectively be referred to as ΔGPBSA. ΔEMM encompasses terms for internal, van der Waals and Coloumbic energies and can be calculated using a molecular mechanics force field. Representative conformational ensembles of the ligand, receptor and complex to be subjected to MM/PBSA analysis are often generated in explicit solvent MD simulations. Water and ions are stripped from the snapshots and replaced by an implicit continuum model before the calculations. We applied the single trajectory protocol, in which all the structures are derived from the complex trajectory 53; 54; 55; 56; 57.
In the MM/PBSA part of this study our objectives were to identify residues important for binding of TH peptides to 14-3-3γ and to the POPS membrane and to evaluate differences between TH-(1-43) and THp-(1-43) with regards to membrane binding. For this reason and because of the high computational costs related to entropy calculations, we chose to focus on the enthalpic contribution and disregard the entropic term (TΔS) in Equation 3:
| (Eqn.4) |
The single quotation mark in ΔG’ serves the purpose of differentiating the enthalpic free energy from the binding free energy (Equation 3). The non-polar portion of the solvation free energy was estimated by linearly relating it to the solvent accessible surface area (SASA):
| (Eqn.5) |
A solvent probe of radius 1.4 Å was applied in order to derive SASA values. In accordance with the Parse parameter set 58, the regression coefficient γ and offset parameter β were set to 0.00542 and 0.92 kcal/mol, respectively. The electrostatic contribution to the solvation free energy (ΔGPB) was calculated by solving the Poisson-Boltzmann equation with a maximum of 1000 iterations per snapshot. The internal dielectric constant was 1 whilst the external counterpart was 80 in the calculations. A grid spacing of 0.5 Å was used, along with a ratio of 4.0 between the longest dimension of the grid and that of the solute. Ionic strength was set to 150 mM, which approximately corresponds to physiological salt concentrations. Analyses were performed using the MMPBSA.py program incorporated into AmberTools 1.5 44. The script allows for the binding enthalpy ΔG’ (Equation 4) to be decomposed into the contributions made by each residue in the system, in our case individual residues in the TH peptides and 14-3-3γ, and individual POPS lipids. Prior to analysis, radii in the topology files were converted to the Parse variant 58. 1000 conformations extracted evenly from the last 100 ns from each of the peptide:membrane simulations (Systems D and E in Table 1) were subjected to MM/PBSA per residue decomposition analysis. In order to reduce the computational cost, only the peptide-interacting monolayer (i.e. 64 lipid molecules) comprised the receptor in the calculations. In terms of the 14-3-3γ:peptide system (System A in Table 1), 1000 frames were taken with equally spaced time steps from the last 50 ns of simulation. The 14-3-3γ:THp complex was divided into two ligand:receptor pairs for the MM/PBSA analysis, with each 14-3-3γ monomer representing the receptor and the bound peptide fragment regarded as the ligand in each case. The sets of snapshots represent satisfactorily equilibrated systems, as demonstrated by stable RMSd values (Supplementary Figures S1 and S2). Secondly, the time windows between frames (100 ps and 50 ps, respectively) are large enough to ensure that the snapshots are independent. The number of frames chosen was based on an attempt to reasonably balance statistical sample size and computational demand.
RESULTS AND DISCUSSION
The conformation of THp-(1-43) bound to 14-3-3γ
In order to identify the structural determinants for interaction of Ser19-phosphorylated TH with 14-3-3γ the crystal structure of 14-3-3γ with THp-(1-43) was determined (PDB code 4J6S; crystallographic statistics in Table 2). The structure was solved by molecular replacement (see Materials and Methods), and the crystallographic details of the complex are in Table 2. The final model included 4 chains in the asymmetric unit (A–D) (Figure 1A). The dimers were almost identical (RMSD for Cα atoms of chains A,B (dimer 1) and chains C,D (dimer 2) = 0.481 Å). The structure of 14-3-3γ in the present complex is similar to previously solved structures of peptides bound to 14-3-3γ, which adopt an extended conformation, such as PDB 2B05 (RMSD for Cα atoms for chains A,B in both structures = 0.827 Å) and PDB 3UZD (RMSD = 1.504 Å) 59. The structure superimposed with PDB 2B05 is shown in Figure 1B. The present structure of 14-3-3γ extends to residue Asp239, i.e. 5 residues longer than the other available structures, and reveals the start of the C-terminal stretch, which seems to adopt a random coil configuration (Figure 1B). This stretch has been found to have an important regulatory role and folds into the phosphopeptide binding site in the absence of ligand, while it is released in the ligand-bound state 60; 61.The phosphorylated TH peptide with satisfactory resolution included in the model (PDB 4J6S) consisted of residues RRAVpSELDA. This sequence does not totally concur with the consensus binding modes of 14-3-3 proteins 24; 25; 27. The positioning and conformation of the observed fragment of THp-(1-43) is nevertheless similar to that of the model peptide in PDB 2B05 (RAIpSLP), which belongs to the canonical mode I motif (Figure 1B; see also below).
Table 2.
Crystal and X-ray data collection and crystallographic refinement statistics for the 14-3-3γ:THp-(1-43) complex which resulted in structural information for a dimeric 14-3-3γ and THp-(14-26) and THp-(11-22) fragments, each bound to a 14-3-3γ monomer.
| Crystal data | |
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 84.19, 115.12, 136.90 |
| α=β=γ (°) | 90.0 |
| Data collection | |
| Processing software | XDS |
| Wavelength (Å) | 1.0331 |
| Resolution (Å) | 40.0-3.08 (3.16-3.08) |
| Rmerge (%) | 11.1 (49.2) |
| I / σI | 11.6 (3.3) |
| Completeness (%) | 87.1 (90.0) |
| No. of unique reflections | 22015 |
| Redundancy | 5.1 (5.1) |
| Refinement | |
| Processing software | Refmac 5 |
| Resolution (Å) | 3.08(3.16-3.08) |
| Reflections | 20915(1565) |
| Rwork / Rfree (%) | 21.0(28.1)/25.4(36.4) |
| No. atoms | 7970 |
| 14-3-3gamma protein | 7690 |
| THp peptide fragment | 280 |
| Average B overall (Å2) | 69.7 |
| 14-3-3gamma protein | 69.0 |
| THp peptide fragment | 86.2 |
| R.m.s.d. bond length (Å) | 0.009 |
| R.m.s.d. bond angle (°) | 1.192 |
| PDB entry | 4J6S |
Figure 1. The crystal structure of 14-3-3γ.
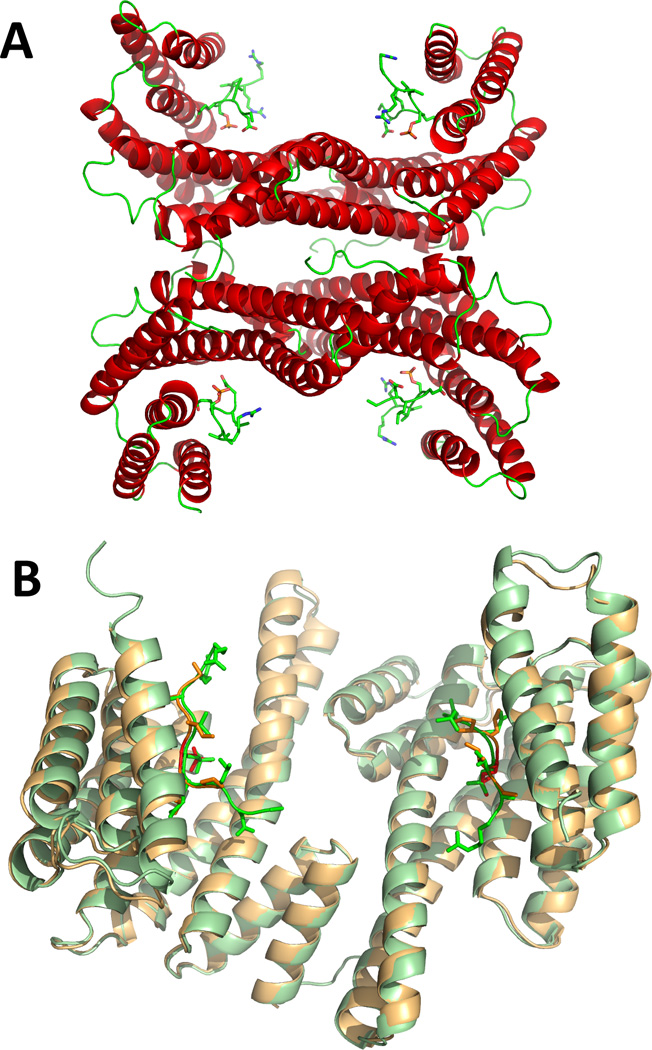
A) The two 14-3-3γ dimers (chains A–D) in the asymmetric unit (from this work; PDB 4J6S), as ribbons coloured by secondary structure, complexed to the TH peptides, represented by sticks coloured by atom type. B) A dimer of 14-3-3γ (chains A and B in PDB 4J6S), as pale green ribbons, with the TH peptides as bright green sticks, superimposed with the equivalent dimeric structure of 14-3-3γ from PDB 2B05, as pale orange ribbons, with the phosphopeptide (RAIpSLP) represented by red sticks.
For the MD simulation we used a template where, by implementing weaker portions of the electron density for modeling, the THp fragment residues were extended to 12 and 13 residues in the two monomers, respectively. More specifically, the final model consisted of a 14-3-3γ dimer with a THp-(1-43) peptide fragment bound to each monomer, one containing 13 residues (Phe14-Ala26) and the other one composed of 12 residues (Ala11-Asp22) (System A in Table 1). The simulated system was divided into two receptor:ligand pairs prior to MM/PBSA analysis, each unit consisting of a 14-3-3γ monomer with bound phosphorylated TH peptide fragment. A representative snapshot of the 14-3-3γ:THp system, along with close-ups of the most important hydrogen bond interactions taking place between the THp fragments and 14-3-3γ are depicted in Figure 2 (panels A–C).
Figure 2. Representative structure and interactions from the 14-3-3γ:THp peptide complex simulation.

Panel A depicts the 14-3-3γ dimer as a grey surface with THp-(Ala11-Asp22) bound to the left monomer and THp-(Phe14-Ala26) bound to the right monomer. The peptides are coloured as continuous gradients going from blue (N-terminal) to red (C-terminal). In addition, the Ser19 residue in each THp fragment is highlighted in the form of a stick model. Panels B and C (THp-(Ala11-Asp22) and THp-(Phe14-Ala26), respectively) are close-ups revealing the most important hydrogen bond interactions (dashed lines) taking place in the panel A complex structure. 14-3-3γ residues are shown as ball-and-stick representations and THp residues as sticks. Water has been removed for clarity.
MM/PBSA decomposition analyses were performed in order to obtain more detailed information on the interactions between the THp peptide fragments and 14-3-3γ. Table 3 presents the total binding enthalpy (ΔG’) decomposed into its component energy terms given in Equation 4. For simplicity, the polar energy terms were summed into a total polar contribution. ΔEelec, the electrostatic term calculated from the force field, makes a very favourable contribution whilst ΔGPB makes an equally unfavourable contribution to the binding enthalpy. Their opposing effects on the enthalpic energy change and comparable sizes render the sum of them more informative than the large individual contributions.
Table 3.
Binding enthalpy from the simulated dimeric 14-3-3γ:THp peptide complex simulation decomposed into its component terms
| Energy terms | Monomeric componenta | |
|---|---|---|
| THp-(Phe14-Ala26):14-3-3γ | THp-(Ala11-Asp22):14-3-3γ | |
| ΔEvdW | −69.5 ± 8.8 | −49.8 ± 6.4 |
| ΔEelec + ΔGPB | 21.6 ± 10.6 | 17.7 ± 10.4 |
| ΔGnon-polar | −10.7 ± 0.9 | −9.0 ± 0.5 |
| ΔG’ | −58.6 ± 8.6 | −41.1 ± 7.2 |
The simulated dimeric 14-3-3γ:THp peptide complex was divided into its component 14-3-3γ monomer:peptide fragment pairs before both were subjected to MM/PBSA analysis.
The non-polar desolvation energy (ΔGnon-polar) is not decomposable for MM/PBSA using the MMPBSA.py implementation of AmberTools 1.5 44. It is therefore not included in the ΔGdecomp energies featured in Figure 3, which shows the energies from the MM/PBSA per residue decomposition for the THp peptide fragments (panels A and B) and for the 14-3-3γ monomers (panels C and D). Considering that the ΔGnon-polar values are small compared to the other energy terms in Table 3, and result from the contribution of many residues, it is fair to assume that they are negligible for individual residues (Figure 3). The two analyses on the 14- 3-3γ monomers (Figure 3C and D) are in good agreement with each other and paint the same picture of the interaction with the THp peptide fragments, both in terms of identifying the main contributing residues and with regard to the energy values themselves. The few differences observed might be a consequence of the difference in fragment sequence coverage. Although the two simulated fragments are partially overlapping sequences from the same phosphorylated TH peptide, they still constitute two different peptides. Both THp-(14- 26) and THp-(11-22) displayed negative ΔG’ values in the MM/PBSA analysis (Table 3), implying that the enthalpy changes related to binding of the TH peptide fragments to 14-3-3γ contribute favourably to the free energy of binding. It is important to remember, though, that the entropy term is omitted from the analysis. As evidenced by the large and negative ΔEvdW values (Table 3) and by the non-polar contributions in Figure 3, van der Waals interactions contribute significantly to the favourable enthalpies of binding. Yet, the per residue decomposition analyses also disclose that Arg132, Arg57 and Tyr133 play an important role and identify these residues as making the most favourable contributions to the total binding enthalpy (Figure 3C and D). These residues are conserved in the 14-3-3 protein family and are involved in phosphopeptide ligand binding 62; 63. Our results are thus consistent with a peptide-binding groove with partly hydrophobic and partly basic properties 23; 25.
Figure 3. MM/PBSA per residue decomposition energies for the crystallographic 14-3-3γ:THp peptide complex after simulation.

The calculations were performed for the two TH peptide fragments (Panels A and B) and for the corresponding monomers of 14-3-3γ (Panels C and D), as calculated from 1000 frames representing the last 50 ns of the crystallographic 14-3-3γ:THp peptide complex simulation (System A in Table 1). ΔGdecomp corresponds to the binding enthalpy (ΔG’ in Equation 4 and Table 3) minus the non-decomposable, but most likely negligible, non-polar contribution to the solvation energy (ΔGnon-polar in Equation 4 and Table 3). Panels A and C are sorted by amino acid sequence. Panels B and D are sorted in ascending order based on the decomposition energies for THp-(14-26) and 14-3-3γ monomer 1, respectively. In terms of 14-3-3γ, only the residues making contributions greater than ±0.5 kcal/mol are shown.
The occupancies for hydrogen bonds involving the three main contributors to a favourable enthalpy in Figure 3 and the Ser19 phosphate oxygens agree with the crucial role of the phosphate group in binding the THp fragments to 14-3-3γ (Table 4). Hydrogen bond occupancy corresponds to the percentage of analyzed simulation time in which the relevant hydrogen bond existed. To distinguish the participating atoms from each other, different atom names were given to them in Table 4. Very stable interactions were maintained between the Ser19 phosphate in the TH fragments and Arg132, Arg57 and Tyr133, explaining the strong favourable contributions made by those 14-3-3γ residues in the energy decomposition analysis (Figure 3). These results are in accordance with the fact that the Arg132-Arg57-Tyr133 triad corresponds to the conserved phosphopeptide binding motif in 14-3-3 proteins 23; 24; 25.
Table 4.
Occupancies for hydrogen bond interactions between the three residues in 14-3-3γ that contributed most to the negative total enthalpic energy and the phosphate oxygens of Ser19 in the phosphorylated THp peptide fragments.
| 14-3-3γ residuea | Ser19 oxygenb | Hydrogen bond occupancy (%)c | |
|---|---|---|---|
| THp-(Phe14-Ala26) | THp-(Ala11-Asp22) | ||
| Arg132 (HH22) | O2P | 100.0 | 93.0 |
| Arg132 | (HH12) O1P | 100.0 | 92.9 |
| Arg132 (HH12) | O2P | 40.7 | 44.1 |
| Arg132 (HH22) | O1P | 24.7 | 16.1 |
| Arg57 (HH12) | O3P | 100.0 | 89.0 |
| Arg57 (HH22) | O1P | 100.0 | 89.4 |
| Arg57 (HH12) | O1P | 61.3 | 62.7 |
| Tyr133 (HH) | O2P | 100.0 | 93.0 |
Given in parentheses are hydrogens in the residue; HH12 and HH22 are amine hydrogens in the arginine side chain, whilst HH refers to the hydroxyl hydrogen in tyrosine
O1P, O2P and O3P correspond to the phosphate oxygens in THp-(Ser19)
Percentage of the last 50 ns of 14-3-3γ:THp simulation time in which the interaction partners formed a hydrogen bond interaction (characterized by a distance < 3.5 Å between the non-hydrogen atoms and a donor-hydrogen-acceptor angle > 120 degrees).
On the other hand, the sequence of the neighbouring residues of Ser19 in TH does not comply with sequences considered to be optimal for 14-3-3 binding, i.e. RSXpSXP (mode 1), and RX(Y/F)XpSXP (mode 2) 23; 25. It was therefore of interest to conduct a detailed analysis of the non-canonical intermolecular interactions involving THp residues in the vicinity of Ser19. The occupancies for hydrogen bonds formed during the last 50 ns of simulation between residues of 14-3-3γ and those surrounding Ser19 in both THp peptide fragments were calculated (Table 5). Considerable interaction was observed between the side chains of Asn229 and Asn178 in 14-3-3γ and the backbone of the pSer-1 and the pSer+1 residues, respectively (Table 5 and Figure 2). These interactions agree with those previously reported for the corresponding residues in several 14-3-3 isoforms bound to mode 1 or mode 2 phosphopeptides 23; 25. TH has an alanine in the pSer-2 position, as opposed to Ser (mode 1) or Tyr/Phe (mode 2). It is worth noting, though, that despite the difference in residue size, the Ala17 side chain in both of our simulated peptide fragments was directed towards the same hydrophobic pocket in which the pSer-2 Tyr resides in mode-2 phosphopeptide/14-3-3ζ complexes 25. The pSer-4 Arg in mode-2 phosphopeptides complexed to 14-3-3ζ folds back into the phosphopeptide binding site and forms favourable salt bridges with the Ser19 phosphate and the Glu180 side chain (corresponds to Glu185 in γ) 25. Instead, we see that Arg15 interacts favourably with Asp228 while Glu185 forms hydrogen bonds with Arg16, creating a well packed high affinity complex.
Table 5.
Occupancies for hydrogen bond interactions between residues surrounding Ser19 in both phosphorylated TH peptide fragments and 14-3-3γ residues.
| THp residuea | 14-3-3γ residuea | Hydrogen bond occupancy (%)b | |
|---|---|---|---|
| THp-(Phe14-Ala26) | THp-(Ala11-Asp22) | ||
| Arg15 (HH12) | Asp228 (OD1) | 33.4 | 70.6 |
| Arg15 (HH12) | Asp228 (OD2) | 32.4 | 55.4 |
| Arg15 (HH22) | Asp228 (OD2) | 29.3 | 74.4 |
| Arg15 (HH22) | Asp228 (OD1) | 28.7 | 66.2 |
| Arg16 (HH21) | Glu185 (OE2) | 29.6 | 49.6 |
| Arg16 (HE) | Glu185 (OE1) | 26.1 | 35.7 |
| Val18 (H) | Asn229 (OD1) | 95.7 | 71.8 |
| Val18 (O) | Asn229 (HD21) | 56.9 | 91.2 |
| Glu20 (H) | Asn178 (OD1) | 80.6 | 59.1 |
Given in parentheses are names that were given to the different interacting atoms in the simulation: HH12, HH21, HH22 and HE are different guanidinium hydrogens in the arginine side chain; OD1/OD2 in aspartate and OE1/OE2 in glutamate are the side chain carboxyl oxygens; HD21 and OD1 in asparagine denote one of the amine hydrogens and the carbonyl oxygen in the side chain; O and H refer to the backbone amide oxygen and amide hydrogen in the specified residue
Percentage of the last 50 ns of 14-3-3γ:THp simulation time in which the interaction partners formed a hydrogen bond interaction (characterized by a distance of < 3.5 Å between the non-hydrogen atoms and a donor-hydrogen-acceptor angle > 120 degrees). Only interactions with occupancies above 25 % in both monomer:peptide pairs are shown
The conformation of unbound TH-(1-43) and effect of phosphorylation at Ser19
In order to investigate the structural propensities of the TH N-terminal sequence in its unphosphorylated (TH-(1-43)) and phosphorylated (THp-(1-43)) states, we subjected the peptide sequences to MD simulation, using the configurations of the minimized structures as the starting structures for the simulations performed in water (Systems B and C in Table 1). Given in Figure 4A are the B factors for the α-carbon atoms in TH-(1-43) and THp-(1-43), computed from the last 100 ns of both the water and membrane simulations (Systems B–E in Table 1). Very high values for most of the TH-(1-43) atoms imply that the unphosphorylated peptide was highly flexible in the aqueous simulation. Moreover, it can be inferred from the overall greater values in Figure 4A that TH-(1-43) was considerably more flexible in water than THp-(1-43). The B factor analysis is consistent with the propensity for formation of internal hydrogen bonds in the two peptides. As an example, only two residues (Arg16 and Glu20) in TH-(1-43) engaged in sidechain-sidechain hydrogen bond interactions with occupancies >25% in the last 100 ns of aqueous simulation. This reflects the high flexibility of the peptide, which precludes the visualization of a representative structure of TH-(1-43) from the simulation in water. Comparatively, quite a few stable sidechain-sidechain interactions involving several residues were formed in THp-(1-43), and a representative conformation from the aqueous simulation is presented in Figure 4B. All these interactions involved arginines, and the phosphate at Ser19 participated in stable hydrogen bonding with the Arg37 sidechain. This is in agreement with the B factor analysis, since the highlighted residues in Figure 4B also gave the lowest B factors in THp-(1-43) (Figure 4A).
Figure 4. B factors for the α-carbons of the N-terminal fragments of TH and representative structure and interactions from the aqueous THp-(1-43) simulation.

B factors (Panel A) were calculated from the aqueous simulations for TH-(1-43) (black line) and THp-(1-43) (red line) and from the peptide/membrane simulations for TH-(1-43) (blue line) and THp-(1-43) (green line), and are represented as a function of residue number. Panel B: Representative structure of THp-(1-43) which shows the intrapeptide sidechain-sidechain hydrogen bonds with occupancies >25% (dashed lines) in the aqueous simulation. The peptide is coloured as a continuous gradient going from blue (N-terminal) to red (C-terminal). The last 100 ns of each simulation were used for both analyses.
We also calculated the total number of intramolecular hydrogen bonds in each of the two peptides averaged over the last 100 ns of aqueous simulation, obtaining 24.0 ± 3.8 for TH-(1-43) and 37.3 ± 4.1 for THp-(1-43). In support of the B factor analysis, it indicates that internal hydrogen bond interactions are much more prominent in the phosphorylated peptide, as revealed by the substantially higher bond count. Intrapeptide hydrogen bonds stabilize the structure. At the same time, the difference correlates well with the secondary structure content estimated for the two peptides (Table 6). For the aqueous simulations, more secondary structure is found in THp-(1-43). Furthermore, a count of the backbone-backbone NH-CO hydrogen bonds formed between amino acids separated by two, three or four residues provides values in accordance with the differences in flexibility and helix formation between the two peptides. Using a cut-off of 50% occupancy within the last 100 ns of simulation, the number of such interactions was found to be 12 in THp-(1-43) compared to 4 in TH-(1-43).
Table 6.
Content of secondary structure in the unphosphorylated TH-(1–43) and Ser19-phosphorylated THp-(1–43) peptides, calculated from the simulations both in water and with the POPS bilayer.
| Interaction partner |
Alpha helix (%)a | Beta sheet (%)a | Turn (%)a | |||
|---|---|---|---|---|---|---|
| TH-(1–43) | THp-(1–43) | TH-(1–43) | THp-(1–43) | TH-(1–43) | THp-(1–43) | |
| POPS bilayer | 11.7 ± 5.7 | 8.0 ± 5.3 | 4.2 ± 1.2 | 0.3 ± 1.1 | 26.8 ± 6.0 | 22.0 ± 6.3 |
| Water | 8.9 ± 5.5 | 23.7 ± 5.8 | 0.2 ± 1.0 | 1.8 ± 2.2 | 19.1 ± 6.8 | 21.8 ± 5.9 |
The proportion of the peptide adopting the specified type of secondary structure, averaged over the last 100 ns of simulation
Together, these analyses suggest that phosphorylation of Ser19 induces a structuration resulting in a more stable, compact and less flexible peptide with more secondary structure content. However, these results do not seem to be supported by previous studies, including CD analysis, which indicated very similar structures for TH-(1-43) and THp-(1-43) in 20 mM citric acid-phosphate, pH 7.4, with a similar α-helical content of about 15–16% 22. For some peptides the CD spectra and estimated secondary structure content are also dependent on the ionic strength and type of buffer in the peptide solutions (see e.g. 64). We therefore performed CD analyses of the peptides at 2.5-fold higher concentration than in our previous study, both in water and in 10 mM Hepes, 150 mM NaCl, pH 7.4. As previously 22, we obtained very similar spectra for both peptides, which appear to adopt a higher structuration in water than in buffer, with a clearly higher negative molar ellipticity at 222 nm, corresponding to higher helical content (Figure S3). At the same time, the estimated secondary structure contents were only slightly higher in water than in buffer. For instance, the α-helical and random coil content estimated for THp-(1-43) by CDNN 35 were about 15% and 49% in water versus 13% and 51% in buffer, respectively. In fact, these values were not so different from the estimates for TH-(1-43). Similar values were obtained with other programs (data not shown), and significant differences were not proven for the two peptides. For loosely folded peptides and proteins, many possible conformations exist, as is expected from the Boltzmann distribution. CD will detect an average of these distributions across the sample volume and time-resolution of the experiment, while MD simulations follow one of many possibilities that are close in energy and conformational space, probably catching the dissimilarity of TH-(1-43) and THp-(1-43) with respect to conformational propensity.
A structuration upon phosphorylation is in agreement with structural analyses on the effect of phosphorylation of PAH at Ser16, which was found to cause a localized conformational change in the N-terminal segment 66. The region surrounding Ser16 adopted a bent and thereby more compact configuration, a reorientation postulated to primarily result from favourable electrostatic interaction between the introduced phosphate group and Arg13. Phosphorylation was also accompanied by an increase in α-helix content. Moreover, Stultz et al. 67 deduced from fluorescence resonance energy transfer measurements and MD simulations that the peptide TH-(24-33) becomes more compact upon phosphorylation of Ser31 due to formation of a salt bridge between the phosphate and Arg33.
The binding of TH-(1-43) and THp-(1-43) to the POPS membrane
Previous studies have shown that full-length TH 19 and both TH-(1-43) and THp-(1-43) 22 interact selectively with membranes of zwitterionic and negatively charged lipids. The unphosphorylated peptide showed 5–8-fold higher affinity than the phosphorylated peptide 22, as measured by the S0.5 (concentration of peptide for half-maximal binding obtained from binding isotherms). In the present study and in order to investigate the conformational determinants for the peptide-membrane interaction by MD simulations, we selected a bilayer made of POPS (see Materials and Methods section). Membrane binding of TH-(1-43) and THp-(1-43) was previously tested by Halskau et al. using liposomes made of phospholipid mixtures, which can favour protein interaction relative to bilayers composed of a unique negatively charged phospholipid 68. In this study we therefore confirmed by SPR analyses that the peptides also interacted with liposomes made of pure POPS. The peptides showed only slight binding to liposomes made of PC, and the response units (RU) with these liposomes (at the reference channel) were subtracted from the RU in the channel with POPS to compensate for non-specific binding. The sensorgrams for TH-(1-43) binding to POPS liposomes at increasing peptide concentration are shown in Figure 5A. The binding was concentration dependent (Figure 5B), and although S0.5 was found to be similar for both peptides (53.3 ± 9.2 µM and 48.2 ± 7.4 µM for TH-(1-43) and THp-(1-43), respectively), the unphosphorylated peptide certainly showed a larger extent of membrane binding.
Figure 5. Binding of TH-(1-43) and THp-(1-43) to POPS liposomes by surface plasmon resonance and circular dichroism.

A) Representative sensorgrams for the interaction of TH-(1-43) with POPS liposomes on a L1 sensor chip at increasing concentrations of peptide. B) Peptide concentration dependency of TH-(1-43) (black) and THp-(1-43) (red) when bound to POPS liposomes. RUs were measured as a function of peptide concentration. S0.5 values were extracted from the hyperbolic, single-rectangular, two-parameter curve fitting and resulted in S0.5 of 53.3 ± 9.2 µM and 48.2 ± 7.4 µM for TH-(1-43) and THp-(1-43), respectively. C) Far-UV CD spectra of 40 µM TH-(1-43) (black lines) or THp-(1-43) (red lines) without (solid lines) or with (dotted lines) POPS liposomes (0.6 mM phospholipid). Samples were prepared in 10 mM Na-Hepes, 150 mM NaCl, pH 7.4, and spectra were recorded at 25 °C.
We then performed simulations of TH-(1-43) and THp-(1-43) with the POPS bilayer model (Systems D and E in Table 1). Representative structures from these simulations, together with close-ups of the most important hydrogen bond interactions in each case are depicted in Figure 6A–D. As determined by the B factors for the α-carbon atoms, both peptides were much more structurally stable and a lot less flexible in the presence of the lipid bilayer than was the case in the aqueous simulations (Figure 4A). The membrane-interacting peptides showed rather similar B factor profiles apart from the last couple of residues in the C-terminal region, where the THp-(1-43) residues proved more flexible. Contrary to the situation in water, TH-(1-43) shows a trend towards higher content than THp-(1-43) of all three types of secondary structure in the membrane simulations (Table 6). However, the estimations are hampered by large standard deviations. The findings appear to be in accordance with experimental results obtained by CD for the interaction of the peptides with negatively charged phospholipid bicelles and with the increased propensity of TH-(1-43) for acquiring secondary structure in response to increasing concentrations of trifluoroethanol used as a membrane-mimicking solvent 22. Furthermore, the higher stabilization of the membrane-bound peptides with respect to their conformations in water is also in agreement with earlier results showing that TH is stabilized against thermal denaturation in the membrane-bound state 19. Here, we performed CD analyses of the peptides in the presence of liposomes made of POPS. Bicelles are superior to liposomes with regard to use in spectroscopic techniques due to their smaller volume, which causes less noise in CD measurements 22. However, bicelles cannot be prepared with a sole composition of phospholipid, and CD spectra could not be taken in the presence of high liposome content. With a liposome content corresponding to 0.6 mM phospholipid, similar CD spectra were obtained for both peptides, with increased ellipticity at 222 nm compared to the spectra obtained without liposomes (Figure 5C). This may reflect their stabilization at the membrane. The liposomes are not stable in water and measurements can only be performed in buffer.
Figure 6. Representative structures and interactions from the peptide/membrane simulations.
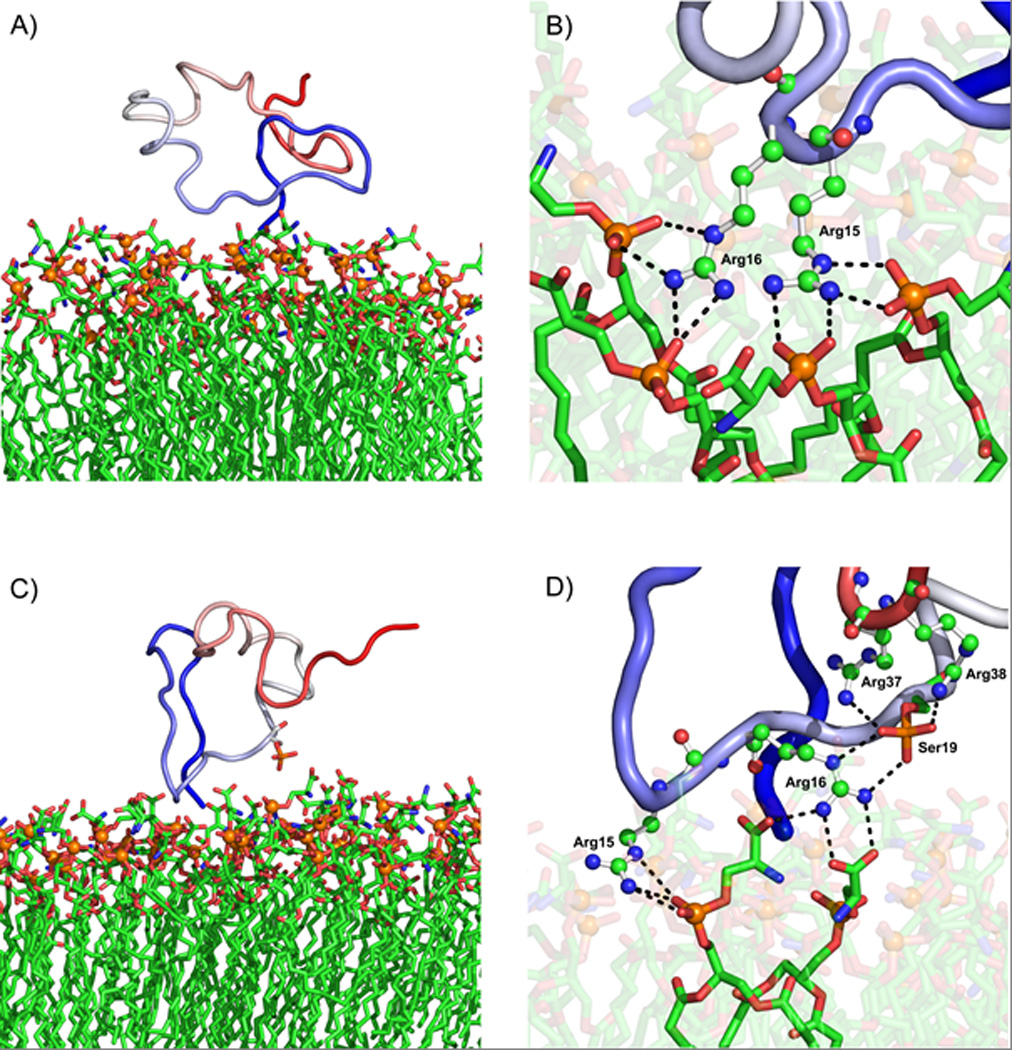
TH-(1-43)/POPS is presented in panels A and B, and THp-(1-43)/POPS is presented in panels C and D. Panels A and C show the peptide – coloured as a continuous gradient going from blue (N-terminal) to red (C-terminal) – together with most of the peptideinteracting leaflet of the POPS bilayer, the orange spheres representing the head group phosphorus atoms. Panels B and D provide close-ups of the most important hydrogen bond interactions (dashed lines) in each case, where the relevant TH residues are displayed as ball-and-stick models and the POPS lipids as sticks. Water and ions have been removed for clarity.
To get a more detailed picture of the peptide-membrane interactions, MM/PBSA decomposition analyses were carried out. The total enthalpic energy changes (ΔG’) for each system and the contributions from the energy terms in Equation 4 are presented in Table 7. In addition, the ΔΔ values for each term, calculated as the energy differences between the TH-(1-43)/POPS and the THp-(1-43)/POPS systems, are given. Figure 7 shows the decomposed energies of the two peptides from the MM/PBSA analysis on a per residue basis. As in the analysis of the 14-3-3γ:THp simulations (Figure 3), ΔGdecomp refers to binding enthalpy (ΔG’), but without inclusion of the non-decomposable ΔGnon-polar term. Favourable binding enthalpies were observed for both peptides upon interaction with the POPS bilayer model, as shown by the negative ΔG’ values in Table 7. However, the binding enthalpy is approximately 40 kcal/mol smaller for the THp-(1-43)/POPS system, indicating that binding of TH-(1-43) to the POPS membrane is much more enthalpically favourable than is the case for the Ser19-phosphorylated counterpart. Differences between the two peptides with regards to van der Waals interactions (ΔEvdW) and non-polar solvation energy (ΔGnon-polar) are modest. Most of the difference in overall binding enthalpy arises from the polar terms (ΔEelec + ΔGPB). In contrast to the TH-(1-43)/POPS system, the polar terms involved in THp-(1-43)/POPS interaction contribute unfavourably to the binding enthalpy. The phosphorylated Ser19 residue accounts for nearly the whole deviation in decomposition energies for the two peptides (Figure 7). Excluding Ser19, only small differences between the peptides are seen in the per residue decomposition energies. Accordingly, it seems that the introduction of the phosphate group causes repulsion between the peptide and the membrane. Not surprisingly, the positively charged arginines and lysines were the main contributors to the favourable enthalpy of binding to the POPS membrane observed for both peptides, with Arg15 and Arg16 being the most important (Figures 6 and 7). Negatively charged aspartates and glutamates, together with the phosphorylated Ser19 in THp-(1-43), contributed unfavourably to the total binding enthalpy (Figure 7), implying that the decomposition results are reasonable from an electrostatic point of view.
Table 7.
Binding enthalpy for the peptide/membrane simulations decomposed into its component terms.
| Energy termsa | Systemb | Difference ΔΔ (kcal/mol) |
|
|---|---|---|---|
| TH-(1–43)/POPS | THp-(1–43)/POPS | ||
| ΔEvdW | −26.1 ± 5.5 | −21.8 ± 7.4 | −4.3 |
| ΔEelec + ΔGPB | −21.8 ± 11.7 | 12.6 ± 13.8 | −34.4 |
| ΔGnon-polar | −5.2 ± 0.6 | −4.3 ± 0.9 | −0.9 |
| ΔG’ | −53.1 ± 10.8 | −13.5 ± 9.8 | −39.6 |
ΔG’ refers to the change in total enthalpy as given in Equation 4
The peptide-interacting leaflet of the POPS bilayer constituted the receptor in these calculations
Figure 7. MM/PBSA per residue decomposition energies for TH-(1-43) and THp-(1-43) from the peptide/membrane simulations.
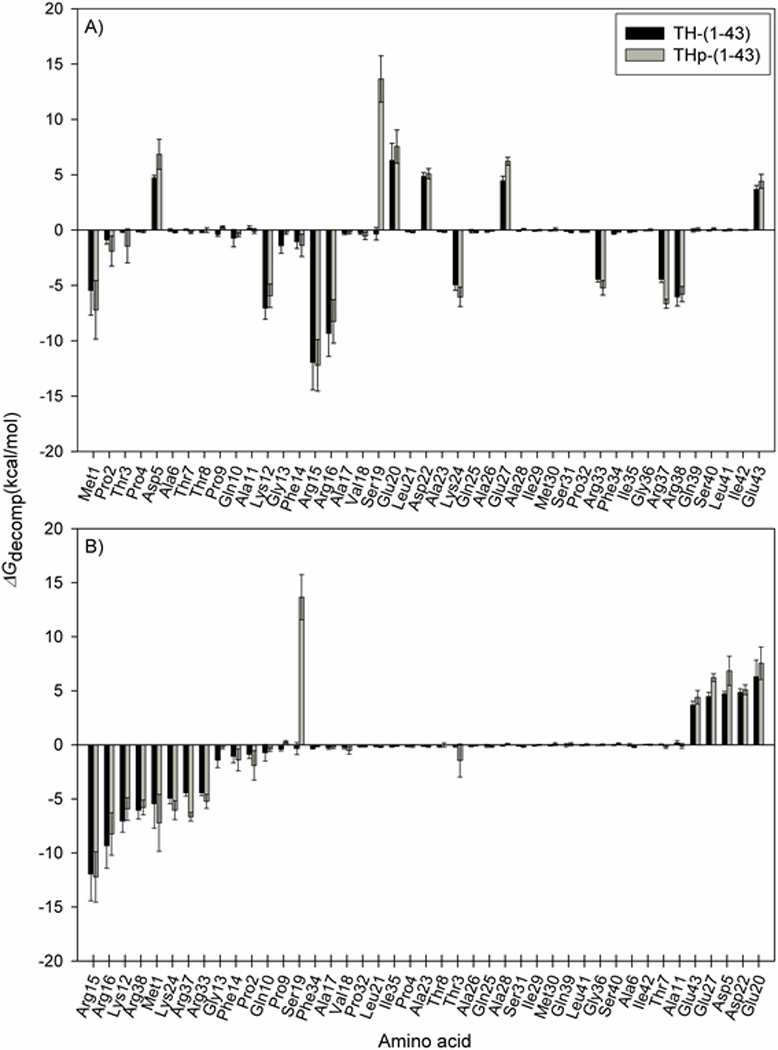
The energies are computed using 1000 frames extracted from the last 100 ns of each peptide/membrane simulation (systems D and E in Table 1). ΔGdecomp = ΔG’ – ΔGnon-polar. The black bars represent the energies derived for each residue in TH-(1-43), and the grey bars are the THp-(1-43) equivalents. A) Sorted by amino acid sequence; B) Sorted in ascending order according to the TH-(1-43) decomposition energies.
The importance of Arg15 and Arg16 in the interaction with the membrane becomes more apparent in Figure 6B and D, where extensive interaction between POPS lipids and the two arginines in both peptides is shown. However, Arg16 in THp-(1-43) is partly diverted away from the bilayer through favourable interaction with the Ser19 phosphate (panel D) rather than fulfilling its hydrogen bonding potential by binding to the membrane only (panel B). This intramolecular interaction might also contribute to the observed difference in ΔG’ between TH-(1-43)/POPS and THp-(1-43)/POPS (Table 7).
Structural comparison of the TH N-terminal peptides in different states
We further measured the distances between the terminal α-carbons of each peptide fragment in the 14-3-3γ dimer:THp fragment complex (System A, Table 1) as a function of simulation time and averaged over the last 50 ns. The resulting values were compared with the distances between the corresponding α-carbons averaged over the last 100 ns of both the aqueous and membrane-containing simulations of THp-(1-43) and TH-(1-43) (Systems B–E) (Table 8). These estimations provide a crude measurement of how extended the peptides were in each state. The distance between the α-carbons of Phe14 and Ala26 was 8 Å greater when bound to 14-3-3γ than in water. Following the same trend, the separation between the Ala11 and Asp22 α-carbons was 13.1 Å greater when bound to 14-3-3γ than in water. In conclusion, this indicates that the region surrounding the Ser19 phosphate group in THp-(1-43) stretches out and accommodates a more extended conformation upon shifting from “free in solution” to the 14-3-3γ-bound state. The membrane-interacting version of the phosphorylated TH peptide gives distances that are intermediate compared to the 14-3-3γ-bound and solvated conformations. Positively charged residues in THp-(1-43), that in water are available for intramolecular interactions with negatively charged residues (Figure 4), interact favourably with the POPS bilayer (Figs 6D and 7). With regards to TH-(1-43), the results are ambiguous and appear to be more random (Table 8). Also, the standard deviations related to the distances derived from the aqueous simulation are three times higher than for THp-(1-43), in line with the greater flexibility of the unphosphorylated peptide discussed earlier (Figure 4A). These observations suggest that the conformational sampling of the region around Ser19 is less dependent on the Ser19 residue than is the case in THp-(1-43).
Table 8.
Comparison of the distances between the terminal amino acids of the THp-(14–26) and THp-(11–22) fragments from the 14-3-3γ:THp simulation and the corresponding distances taken from the simulations of THp-(1–43) and TH-(1–43) in water and in the presence of the POPS bilayer.
| System | Distance (Å) | |
|---|---|---|
| Phe14-Ala26 | Ala11-Asp22 | |
| THp fragments (in the 14-3-3γ:THp complex)a | 22.9 ± 2.1 | 26.9 ± 1.6 |
| THp-(1–43)/waterb | 14.9 ± 0.9 | 13.8 ± 1.2 |
| TH-(1–43)/waterb | 12.3 ± 2.7 | 18.9 ± 3.6 |
| THp-(1–43)/POPSb | 20.9 ± 0.8 | 15.3 ± 0.3 |
| TH-(1–43)/POPSb | 9.8 ± 0.6 | 21.3 ± 0.8 |
Averaged over the last 50 ns of simulation
Averaged over the last 100 ns of simulation
Finally, we consider it probable that the binding mode displayed by the truncated THp-(1-43) peptide in its interaction with 14-3-3γ is the same as for this sequence in fulllength TH. There is no structural information available for the 1-43 N-terminal segment in TH, which is an extension of the regulatory ACT domain equivalent to the extended N-terminus in TPH2 29; 30. This N-terminal segment is associated with increased instability of TPH2 compared to TPH1, and phosphorylation of Ser19, which is also the determinant for binding to 14-3-3 in TPH2, increases its intracellular stability 29; 69. Furthermore, the N-terminal sequences in TH and TPH2 are predicted to be highly disordered using PONDR-FIT and DisProt 70; 71. Intrinsically disordered segments are most likely separated from the rest of the folded cores of the proteins but become structured by post-translational modification and/or binding to a partner 72. Consequently, it is highly probable that the rearrangements observed in the TH peptides upon phosphorylation and further by binding to 14-3-3 reflect the changes to the segment when connected to the enzyme. Nevertheless, other regions of TH might interact with 14-3-3 at areas remote from the phosphopeptide binding site, as observed for serotonin N-acetyltransferase bound to 14-3-3ζ 7373.
CONCLUSIONS
As expected, the crystal structure of phosphorylated TH fragments complexed to 14-3-3γ, complemented by MD simulations and subsequent MM/PBSA analysis, indicate that the phosphate at Ser19 forms strong and stable interactions with the conserved phosphopeptide binding motif of 14-3-3γ. The region surrounding Ser19 adopted a more extended conformation when bound to 14-3-3γ than was the case in water, and established similar interactions with the protein as found for other peptides with canonical mode 1 and mode 2 14-3-3-binding motifs. Our aqueous simulations suggest that phosphorylation of Ser19 induces a localized conformational change in the N-terminal segment of TH, resulting in a more compact and rigid configuration and an apparent increase in α-helix content. Both TH-(1-43) and THp-(1-43) bind to anionic POPS membranes, as indicated by SPR experiments and favourable binding enthalpies calculated for the initial stage of the peptide-membrane interactions using MM/PBSA. Binding seems to be mediated mainly by positively charged arginines and lysines. Electrostatic repulsion between the Ser19 phosphate and the negatively charged serine lipid head groups of the membrane gives a less favourable enthalpy of binding for THp-(1-43). Interaction with the POPS bilayer model appears to stabilize the structure of both peptides, notably TH-(1-43). Our results provide further insights on the conformational versatility of the N-terminal region (residues 1-43) of TH, and on the influence of phosphorylation at Ser19 as a post-translational modification that regulates the interaction of the enzyme with 14-3-3 and membranes, with concomitant changes in function and localization.
Supplementary Material
Acknowledgements
The authors would like to thank The Norwegian Research Council, The Kristian Gerhard Jebsen Foundation, the Western Norway Health Authorities, and the Meltzer fund for funding. This work was also supported by NIH Roadmap grant GM073197 (to MM and RCS). We thank the Amber community for support and advice. Notur and uniComputing are acknowledged for providing resources for computational studies.
Abbreviations
- CaMKII
Ca++/calmodulin-dependent protein kinase II
- CD
circular dichroism
- PC
phosphatidylcholine
- PKA
cAMP dependent protein kinase
- POPS
palmitoyl-oleoyl-phosphatidylserine
- PRAK
p38 regulated/activated kinase
- RU
response units
- SPR
surface plasmon resonance
- TH
tyrosine hydroxylase
- TH-(1-43)
truncated human hTH1 containing N-terminal residues 1-43
- THp-(1-43)
Ser19-phosphorylated human TH-(1-43).
References
- 1.Aumann T, Horne M. Activity-dependent regulation of the dopamine phenotype in substantia nigra neurons. Journal of neurochemistry. 2012;121:497–515. doi: 10.1111/j.1471-4159.2012.07703.x. [DOI] [PubMed] [Google Scholar]
- 2.Daubner SC, Le T, Wang S. Tyrosine hydroxylase and regulation of dopamine synthesis. Arch. Biochem. Biophys. 2011;508:1–12. doi: 10.1016/j.abb.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dunkley PR, Bobrovskaya L, Graham ME, von Nagy-Felsobuki EI, Dickson PW. Tyrosine hydroxylase phosphorylation: regulation and consequences. J Neurochem. 2004;91:1025–1043. doi: 10.1111/j.1471-4159.2004.02797.x. [DOI] [PubMed] [Google Scholar]
- 4.Wang S, Sura GR, Dangott LJ, Fitzpatrick PF. Identification by hydrogen/deuterium exchange of structural changes in tyrosine hydroxylase associated with regulation. Biochemistry. 2009;48:4972–4979. doi: 10.1021/bi9004254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang S, Lasagna M, Daubner SC, Reinhart GD, Fitzpatrick PF. Fluorescence spectroscopy as a probe of the effect of phosphorylation at serine 40 of tyrosine hydroxylase on the conformation of its regulatory domain. Biochemistry. 2011;50:2364–2370. doi: 10.1021/bi101844p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakashima A, Mori K, Suzuki T, Kurita H, Otani M, Nagatsu T, Ota A. Dopamine inhibition of human tyrosine hydroxylase type 1 is controlled by the specific portion in the N-terminus of the enzyme. J Neurochem. 1999;72:2145–2153. doi: 10.1046/j.1471-4159.1999.0722145.x. [DOI] [PubMed] [Google Scholar]
- 7.Nakashima A, Hayashi N, Mori K, Kaneko YS, Nagatsu T, Ota A. Positive charge intrinsic to Arg(37)-Arg(38) is critical for dopamine inhibition of the catalytic activity of human tyrosine hydroxylase type 1. FEBS Lett. 2000;465:59–63. doi: 10.1016/s0014-5793(99)01704-4. [DOI] [PubMed] [Google Scholar]
- 8.Ichimura T, Isobe T, Okuyama T, Takahashi N, Araki K, Kuwano R, Takahashi Y. Molecular cloning of cDNA coding for brain-specific 14-3-3 protein, a protein kinase-dependent activator of tyrosine and tryptophan hydroxylases. Proc Natl Acad Sci U S A. 1988;85:7084–7088. doi: 10.1073/pnas.85.19.7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Obsilova V, Nedbalkova E, Silhan J, Boura E, Herman P, Vecer J, Sulc M, Teisinger J, Dyda F, Obsil T. The 14-3-3 protein affects the conformation of the regulatory domain of human tyrosine hydroxylase. Biochemistry. 2008;47:1768–1777. doi: 10.1021/bi7019468. [DOI] [PubMed] [Google Scholar]
- 10.Gordon SL, Bobrovskaya L, Dunkley PR, Dickson PW. Differential regulation of human tyrosine hydroxylase isoforms 1 and 2 in situ: Isoform 2 is not phosphorylated at Ser35. Biochim Biophys Acta. 2009;1793:1860–1867. doi: 10.1016/j.bbamcr.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 11.Salvatore MF, Pruett BS. Dichotomy of tyrosine hydroxylase and dopamine regulation between somatodendritic and terminal field areas of nigrostriatal and mesoaccumbens pathways. PLoS ONE. 2012;7:e29867. doi: 10.1371/journal.pone.0029867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuczenski RT, Mandell AJ. Regulatory properties of soluble and particulate rat brain tyrosine hydroxylase. J Biol Chem. 1972;247:3114–3122. [PubMed] [Google Scholar]
- 13.McGeer EG, McGeer PL, Wada JA. Distribution of tyrosine hydroxylase in human and animal brain. J Neurochem. 1971;18:1647–1658. doi: 10.1111/j.1471-4159.1971.tb03738.x. [DOI] [PubMed] [Google Scholar]
- 14.Chen R, Wei J, Fowler SC, Wu JY. Demonstration of functional coupling between dopamine synthesis and its packaging into synaptic vesicles. J Biomed Sci. 2003;10:774–781. doi: 10.1159/000073965. [DOI] [PubMed] [Google Scholar]
- 15.Tsudzuki T, Tsujita M. Isoosmotic isolation of rat brain synaptic vesicles, some of which contain tyrosine hydroxylase. J Biochem. 2004;136:239–243. doi: 10.1093/jb/mvh113. [DOI] [PubMed] [Google Scholar]
- 16.Morita K, Hamano S, Oka M. Ionic factors affecting the association of tyrosine hydroxylase with chromaffin granules in the adrenal medullary cell. Neurochem. Int. 1994;25:403–411. doi: 10.1016/0197-0186(94)90015-9. [DOI] [PubMed] [Google Scholar]
- 17.Cartier EA, Parra LA, Baust TB, Quiroz M, Salazar G, Faundez V, Egana L, Torres GE. A biochemical and functional protein complex involving dopamine synthesis and transport into synaptic vesicles. J Biol Chem. 2010;285:1957–1966. doi: 10.1074/jbc.M109.054510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morita K, Teraoka K, Oka M. Interaction of cytoplasmic tyrosine hydroxylase with chromaffin granule. In vitro studies on association of soluble enzyme with granule membranes and alteration in enzyme activity. J. Biol. Chem. 1987;262:5654–5658. [PubMed] [Google Scholar]
- 19.Thórólfsson M, Døskeland AP, Muga A, Martínez A. The binding of tyrosine hydroxylase to negatively charged lipid bilayers involves the N-terminal region of the enzyme. FEBS Lett. 2002;519:221–226. doi: 10.1016/s0014-5793(02)02745-x. [DOI] [PubMed] [Google Scholar]
- 20.Bucciantini M, Cecchi C. Biological membranes as protein aggregation matrices and targets of amyloid toxicity. Methods in molecular biology. 2010;648:231–243. doi: 10.1007/978-1-60761-756-3_15. [DOI] [PubMed] [Google Scholar]
- 21.Anderluh G, Lakey JH. Disparate proteins use similar architectures to damage membranes. Trends Biochem Sci. 2008;33:482–490. doi: 10.1016/j.tibs.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 22.Halskau O, Ying M, Baumann A, Kleppe R, Rodriguez-Larrea D, Almas B, Haavik J, Martinez A. Three-way Interaction between 14-3-3 Proteins, the N-terminal Region of Tyrosine Hydroxylase, and Negatively Charged Membranes. J Biol Chem. 2009;284:32758–32769. doi: 10.1074/jbc.M109.027706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang X, Lee WH, Sobott F, Papagrigoriou E, Robinson CV, Grossmann JG, Sundstrom M, Doyle DA, Elkins JM. Structural basis for protein-protein interactions in the 14-3-3 protein family. Proc Natl Acad Sci U S A. 2006;103:17237–17242. doi: 10.1073/pnas.0605779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, Gamblin SJ, Smerdon SJ, Cantley LC. The structural basis for 14-3-3 : phosphopeptide binding specificity. Cell. 1997;91:961–971. doi: 10.1016/s0092-8674(00)80487-0. [DOI] [PubMed] [Google Scholar]
- 25.Gardino AK, Smerdon SJ, Yaffe MB. Structural determinants of 14-3-3 binding specificities and regulation of subcellular localization of 14-3-3-ligand complexes: a comparison of the X-ray crystal structures of all human 14-3-3 isoforms. Semin Cancer Biol. 2006;16:173–182. doi: 10.1016/j.semcancer.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 26.Obsil T, Obsilova V. Structural basis of 14-3-3 protein functions. Semin Cell Dev Biol. 2011 doi: 10.1016/j.semcdb.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 27.Aitken A. 14-3-3 proteins: a historic overview. Semin Cancer Biol. 2006;16:162–172. doi: 10.1016/j.semcancer.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 28.Goodwill KE, Sabatier C, Marks C, Raag R, Fitzpatrick PF, Stevens RC. Crystal structure of tyrosine hydroxylase at 2.3 A and its implications for inherited neurodegenerative diseases. Nat. Struct. Biol. 1997;4:578–585. doi: 10.1038/nsb0797-578. [DOI] [PubMed] [Google Scholar]
- 29.Murphy KL, Zhang X, Gainetdinov RR, Beaulieu JM, Caron MG. A regulatory domain in the N terminus of tryptophan hydroxylase 2 controls enzyme expression. J Biol Chem. 2008;283:13216–13224. doi: 10.1074/jbc.M706749200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Torrente MP, Gelenberg AJ, Vrana KE. Boosting serotonin in the brain: is it time to revamp the treatment of depression? Journal of psychopharmacology. 2012;26:629–635. doi: 10.1177/0269881111430744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winge I, McKinney JA, Knappskog PM, Haavik J. Characterization of wild-type and mutant forms of human tryptophan hydroxylase 2. J Neurochem. 2007;100:1648–1657. doi: 10.1111/j.1471-4159.2006.04290.x. [DOI] [PubMed] [Google Scholar]
- 32.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. Journal of Applied Crystallography. 1993;26:795–800. [Google Scholar]
- 33.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 34.CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 35.Bohm G, Muhr R, Jaenicke R. Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng. 1992;5:191–195. doi: 10.1093/protein/5.3.191. [DOI] [PubMed] [Google Scholar]
- 36.Accelrys Software Inc., D. S. M. E., Release 3.1. Discovery Studio Modeling Environment Release 3.1 edit. San Diego: Accelrys Software Inc; 2012. [Google Scholar]
- 37.Mukhopadhyay P, Monticelli L, Tieleman DP. Molecular dynamics simulation of a palmitoyl-oleoyl phosphatidylserine bilayer with Na+ counterions and NaCl. Biophys J. 2004;86:1601–1609. doi: 10.1016/S0006-3495(04)74227-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, C SM, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03 Revision B. 05 edit. Pittsburgh PA: Gaussian, Inc; 2003. [Google Scholar]
- 39.Bayly CI, Cieplak P, Cornell WD, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J Phys Chem. 1993;97:10269–10280. [Google Scholar]
- 40.Wang J, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 41.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 42.Skjevik ÅA, Haug BE, Lygre H, Teigen K. Intramolecular hydrogen bonding in articaine can be related to superior bone tissue penetration: a molecular dynamics study. Biophys Chem. 2011;154:18–25. doi: 10.1016/j.bpc.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 43.Martínez L, Andrade R, Birgin EG, Martínez JM. PACKMOL: a package for building initial configurations for molecular dynamics simulations. J Comput Chem. 2009;30:2157–2164. doi: 10.1002/jcc.21224. [DOI] [PubMed] [Google Scholar]
- 44.Case DA, Darden TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Wang B, Hayik S, Roitberg A, Seabra G, Kolossváry I, Wong KF, Paesani F, Vanicek J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh MJ, Cui G, Roe DR, Mathews DH, Seetin MG, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA. AMBER 11. San Francisco: University of California; 2010. [Google Scholar]
- 45.Darden T, York D, Pedersen L. Particle mesh Ewald: an Nlog(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 46.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 47.Ryckaert J-P, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 48.Loncharich RJ, Brooks BR, Pastor RW. Langevin dynamics of peptides: the frictional dependence of isomerization rates of N-acetylalanyl-N'-methylamide. Biopolymers. 1992;32:523–535. doi: 10.1002/bip.360320508. [DOI] [PubMed] [Google Scholar]
- 49.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Homeyer N, Horn AHC, Lanig H, Sticht H. AMBER force-field parameters for phosphorylated amino acids in different protonation states: phosphoserine, phosphothreonine, phosphotyrosine, and phosphohistidine. J Mol Model. 2006;12:281–289. doi: 10.1007/s00894-005-0028-4. [DOI] [PubMed] [Google Scholar]
- 51.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 52.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 53.Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE., III Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 54.Hou T, Wang J, Li Y, Wang W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model. 2011;51:69–82. doi: 10.1021/ci100275a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kar P, Knecht V. Origin of decrease in potency of darunavir and two related antiviral inhibitors against HIV-2 compared to HIV-1 protease. J Phys Chem B. 2012;116:2605–2614. doi: 10.1021/jp211768n. [DOI] [PubMed] [Google Scholar]
- 56.Kar P, Lipowsky R, Knecht V. Importance of polar solvation for cross-reactivity of antibody and its variants with steroids. J Phys Chem B. 2011;115:7661–7669. doi: 10.1021/jp201538t. [DOI] [PubMed] [Google Scholar]
- 57.Lafont V, Schaefer M, Stote RH, Altschuh D, Dejaegere A. Protein-protein recognition and interaction hot spots in an antigen-antibody complex: free energy decomposition identifies"efficient amino acids". Proteins. 2007;67:418–434. doi: 10.1002/prot.21259. [DOI] [PubMed] [Google Scholar]
- 58.Sitkoff D, Sharp KA, Honig B. Accurate calculation of hydration free energies using macroscopic solvent models. J Phys Chem. 1994;98:1978–1988. [Google Scholar]
- 59.Xu C, Jin J, Bian C, Lam R, Tian R, Weist R, You L, Nie J, Bochkarev A, Tempel W, Tan CS, Wasney GA, Vedadi M, Gish GD, Arrowsmith CH, Pawson T, Yang XJ, Min J. Sequence-specific recognition of a PxLPxI/L motif by an ankyrin repeat tumbler lock. Science signaling. 2012;5:ra39. doi: 10.1126/scisignal.2002979. [DOI] [PubMed] [Google Scholar]
- 60.Obsilova V, Herman P, Vecer J, Sulc M, Teisinger J, Obsil T. 14-3-3zeta C-terminal stretch changes its conformation upon ligand binding and phosphorylation at Thr232. J Biol Chem. 2004;279:4531–4540. doi: 10.1074/jbc.M306939200. [DOI] [PubMed] [Google Scholar]
- 61.Silhan J, Obsilova V, Vecer J, Herman P, Sulc M, Teisinger J, Obsil T. 14-3-3 protein C-terminal stretch occupies ligand binding groove and is displaced by phosphopeptide binding. The Journal of biological chemistry. 2004;279:49113–49119. doi: 10.1074/jbc.M408671200. [DOI] [PubMed] [Google Scholar]
- 62.Liu D, Bienkowska J, Petosa C, Collier RJ, Fu H, Liddington R. Crystal structure of the zeta isoform of the 14-3-3 protein. Nature. 1995;376:191–194. doi: 10.1038/376191a0. [DOI] [PubMed] [Google Scholar]
- 63.Xiao B, Smerdon SJ, Jones DH, Dodson GG, Soneji Y, Aitken A, Gamblin SJ. Structure of a 14-3-3 protein and implications for coordination of multiple signalling pathways. Nature. 1995;376:188–191. doi: 10.1038/376188a0. [DOI] [PubMed] [Google Scholar]
- 64.Kalita DJ, Kumar A, Kumar S. Structure-function studies of Bubalus bubalis lingual antimicrobial peptide analogs. Veterinary research communications. 2009;33:149–161. doi: 10.1007/s11259-008-9081-7. [DOI] [PubMed] [Google Scholar]
- 65.Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nature protocols. 2006;1:2876–2890. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miranda FF, Teigen K, Thorolfsson M, Svebak RM, Knappskog PM, Flatmark T, Martínez A. Phosphorylation and mutations of Ser(16) in human phenylalanine hydroxylase. Kinetic and structural effects. J. Biol. Chem. 2002;277:40937–40943. doi: 10.1074/jbc.M112197200. [DOI] [PubMed] [Google Scholar]
- 67.Stultz CM, Levin AD, Edelman ER. Phosphorylation-induced conformational changes in a mitogen-activated protein kinase substrate. Implications for tyrosine hydroxylase activation. J. Biol. Chem. 2002;277:47653–47661. doi: 10.1074/jbc.M208755200. [DOI] [PubMed] [Google Scholar]
- 68.Rodland I, Halskau O, Martinez A, Holmsen H. alpha-Lactalbumin binding and membrane integrity--effect of charge and degree of unsaturation of glycerophospholipids. Biochim Biophys Acta. 2005;1717:11–20. doi: 10.1016/j.bbamem.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 69.Winge I, McKinney JA, Ying M, D'Santos CS, Kleppe R, Knappskog PM, Haavik J. Activation and stabilization of human tryptophan hydroxylase 2 by phosphorylation and 14-3-3 binding. Biochem J. 2008;410:195–204. doi: 10.1042/BJ20071033. [DOI] [PubMed] [Google Scholar]
- 70.Xue B, Dunbrack RL, Williams RW, Dunker AK, Uversky VN. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochimica et biophysica acta. 2010;1804:996–1010. doi: 10.1016/j.bbapap.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sickmeier M, Hamilton JA, LeGall T, Vacic V, Cortese MS, Tantos A, Szabo B, Tompa P, Chen J, Uversky VN, Obradovic Z, Dunker AK. DisProt: the Database of Disordered Proteins. Nucleic Acids Res. 2007;35:D786–D793. doi: 10.1093/nar/gkl893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kovacs D, Szabo B, Pancsa R, Tompa P. Intrinsically disordered proteins undergo and assist folding transitions in the proteome. Archives of biochemistry and biophysics. 2013;531:80–89. doi: 10.1016/j.abb.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 73.Obsil T, Ghirlando R, Klein DC, Ganguly S, Dyda F. Crystal structure of the 14-3-3zeta:serotonin N-acetyltransferase complex. a role for scaffolding in enzyme regulation. Cell. 2001;105:257–267. doi: 10.1016/s0092-8674(01)00316-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.