

Abstract
DM (myotonic dystrophy) is a dominantly inherited genetic disorder that is the most common cause of muscular dystrophy in adults affecting 1 in 8500 individuals worldwide. Different microsatellite expansions in two loci cause different forms of the disease that share similar features: DM1 (DM type 1) is caused by a tri- (CTG) nucleotide expansion within the DMPK (dystrophia myotonica protein kinase) 3′-untranslated region and DM2 (DM type 2) is caused by a tetra- (CCTG) nucleotide expansion within intron 1 of the ZNF9 (zinc finger 9) gene. The pathogenic mechanism of this disease involves the RNA transcribed from the expanded allele containing long tracts of (CUG)n or (CCUG)n. The RNA results in a toxic effect through two RNA-binding proteins: MBNL1 (muscleblind-like 1) and CUGBP1 (CUG-binding protein 1). In DM1, MBNL1 is sequestered on CUG repeat-containing RNA resulting in its loss-of-function, while CUGBP1 is up-regulated through a signalling pathway. The downstream effects include disrupted regulation of alternative splicing, mRNA translation and mRNA stability, which contribute to the multiple features of DM1. This review will focus on the RNA gain-of-function disease mechanism, the important roles of MBNL1 and CUGBP1 in DM1, and the relevance to other RNA dominant disorders.
Keywords: CUG repeat, CUG-binding protein 1 (CUGBP1), dystrophia myotonica protein kinase (DMPK), muscleblind-like 1 (MBNL1), myotonic dystrophy type 1 (DM1), RNA gain-of-function
Introduction
DM (myotonic dystrophy) is a multisystemic disorder and the second most common form of muscular dystrophy. Major features of the disease include myotonia (hyperexcitability of skeletal muscle), muscle wasting, insulin resistance, cardiac conduction defects, cataracts, cognitive dysfunction and mental retardation in the most severe congenital form of the disease. DM is among the class of diseases called microsatellite expansion disorders in which a tandem nucleotide repeat within a specific gene, typically containing a variable number of repeats within the population, expands to a pathogenic range. More than 20 microsatellite expansion disorders have been described [1]. In addition to DM, this class of diseases includes Huntington’s disease, SCA (spinocerebellar ataxia) and fragile X syndrome. There are two types of DM caused by microsatellite expansions in different genes. DM1 (DM type 1) is the more common of two forms in the United States. Our understanding of the disease has increased enormously over the past decade. This review discusses the important findings regarding the pathogenic mechanisms of DM1.
Evidence of RNA gain-of-function
In 1992, the DM1 mutation was found to be expanded (up to >4000) CTG repeats located within the 3′-UTR (3′-untranslated region) of the DMPK (dystrophia myotonica protein kinase) gene in exon 15 [2]. Unaffected individuals contain 5–38 CTG repeats, while symptoms occur in individuals with as few as 50 repeats. Larger repeat size correlates with increased severity of symptoms and decreased age of onset [3]. Several hypotheses have been proposed to explain the pathogenesis of this multisystemic disorder. One is that the repeats inhibit DMPK mRNA or protein production, resulting in DMPK haploinsufficiency. This was supported by studies demonstrating decreased expression of DMPK mRNA and protein in DM1 muscle [4]. Surprisingly, Dmpk-knockout mice displayed only mild myopathy [5] and cardiac conduction defects in older animals [6]. More importantly, the mice did not exhibit myotonia, one of the characteristic symptoms of DM1. Therefore the multisystemic features of DM1 cannot be explained solely by DMPK haploinsufficiency. To date, no DMPK loss-of-function mutation has been identified in DM1 patients.
Although DMPK haploinsufficiency is not the predominant cause of the disease, the CTG repeats might influence expression of adjacent genes. A nuclease resistant region indicating condensed chromatin structure was found downstream of the CTG repeat expansion [7]. A homoeodomain-encoding gene named SIX5 is located in this region and its mRNA level is indeed decreased in DM1 patients [8]. However, Six5-knockout mice develop only cataracts and no muscle pathology was observed [9]. Therefore decreased SIX5 expression is not the primary cause of DM1.
The RNA gain-of-function hypothesis proposes that the mutant RNA transcribed from the expanded allele is sufficient to induce symptoms of the disease. This was suggested by several observations: (i) loss of function of DMPK or surrounding genes did not reproduce major features of DM1 [5,9], (ii) the expanded CTG repeats are transcribed into CUG repeats that accumulate in discrete nuclear foci [10], (iii) expression of only the DMPK 3′-UTR with 200 CTG repeats is sufficient to inhibit myogenesis [11]. Direct experimental support of the RNA gain-of-function hypothesis was provided by a mouse model (HSALR) expressing 250 CTG repeats in the 3′-UTR of the human skeletal α-actin gene. These mice develop myotonia and exhibit muscle histology similar to that of DM1, including increased central nuclei and ring fibres [12]. This demonstrates that CUG repeats alone, regardless of the gene context, are sufficient to induce pathogenic features of DM1. Additional mouse models for DM1 further support the RNA gain-of-function model. Seznec et al. [13] generated transgenic mice that carry a large genomic fragment containing either the normal (20 CTGs, DM20) or mutant (>300 CTGs, DM300) allele of DMPK as well as two flanking genes. DM300 mice developed DM1-like phenotype, while DM20 mice were normal, suggesting that the length of the repeats is responsible for the phenotype [13]. Two inducible mouse models expressing 960 CUG repeats in the DMPK 3′-UTR specifically in heart or muscle also recapitulate DM1 symptoms [14,15].
In 1998, a second type of DM not caused by the DM1 mutation was identified and termed DM2 (DM type 2) [16]. DM2 patients have symptoms similar to DM1, with the exception that no congenital form of DM2 has been reported. DM2 results from CCTG repeats within intron 1 of the ZNF9 (zinc finger 9) gene [17]. The fact that two repeat sequences located in entirely different genes can cause such similar disease features implies a common pathogenic mechanism by RNA gain-of-function. In summary, it is now clear that the mutant RNA is the predominant cause of DM1 pathogenesis, although loss of DMPK and SIX5 may also play a minor role.
One explanation of how repeat-containing RNA can cause disease symptoms is through interaction with RNA-binding proteins. A CUG expansion of more than 11 repeats tends to fold into a hairpin-like secondary structure in which C-G base pairs are interrupted by U–U mismatches [18] (Figure 1). It was proposed that specific proteins are sequestered on the double-stranded hairpins, resulting in depletion below a functional threshold. Two important proteins were identified by their ability to bind to CUG repeats: MBNL1 (muscleblind-like 1) and CUGBP1 (CUG-binding protein 1), which will be the focus of the following sections.
Figure 1. Expanded CUG repeats in DM1 result in MBNL1 loss-of-function and CUGBP1 gain-of-function.
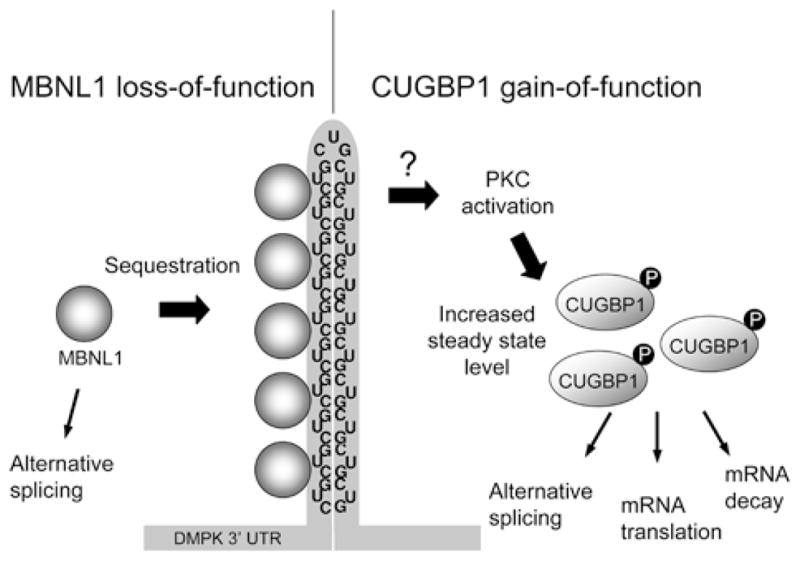
MBNL1 is sequestered to the double-stranded hairpin structure formed by CUG repeats, depleting it from the nucleoplasm. The CUG repeats stimulate PKC activation through an unknown mechanism, which induces CUGBP1 hyperphosphorylation and stabilization. The downstream effects include disruption of alternative splicing, mRNA translation and mRNA decay.
MBNL1
MBNL1 was identified in a screen for proteins that specifically bound to long expansions of CUG repeats [19]. The Drosophila homologue of MBNL1 is essential for muscle terminal differentiation [20], suggestive of an ideal candidate for the protein sequestration model. MBNL1 co-localizes to CUG-containing nuclear foci in DM1 cells and is depleted from the nucleoplasm [21,22]. To determine the role of MBNL1 in DM1, Kanadia et al. [23] generated knockout mice (Mbnl1ΔE3/ΔE3) with targeted deletion of Mbnl1 exon 3 where an RNA-binding motif is located. Eliminating the CUG-binding isoforms of Mbnl1 mimics the molecular environment in DM1, where these isoforms are depleted from the nucleoplasm. These mice develop several characteristic features of DM1, including myotonia and cataracts [23]. In addition, AAV (adeno-associated virus)-mediated overexpression of mouse Mbnl1 in skeletal muscle of HSALR mice is sufficient to reverse the myotonia [24]. These data demonstrate that MBNL1 depletion indeed contributes to DM1 pathogenesis, but how? The answer lies in the role of MBNL1 as a regulator of alternative splicing.
An important molecular feature of DM1 is the mis-regulation of alternative splicing. More than two dozen splicing events are mis-regulated in DM1 [25] and some events correlate directly with DM1 symptoms. For example, the myotonia seen in DM1 is due to the abnormal splicing of the skeletal muscle-specific ClC-1 (chloride channel 1). Increased inclusion of exons containing premature stop codons result in down-regulation of ClC-1 mRNA and protein [26,27], which is sufficient to cause myotonia. A direct link between ClC-1 and myotonia was established by a study in which modified DNA oligonucleotides (morpholinos) were introduced into skeletal muscle of HSALR mice to reverse the splicing of ClC-1 and rescue myotonia [28]. Another abnormal splicing event seen in DM1 is increased skipping of the IR (insulin receptor) exon 11. The spliced isoform lacking exon 11 has lower signalling capacity [29] and increased expression correlates with insulin resistance in DM1 individuals [30]. There is also increased inclusion of cTNT (cardiac troponin T) exon 5 in DM1 [31], which possibly contributes to the cardiac conduction defects.
Mbnl1ΔE3/ΔE3 mice display the same splicing defects observed in DM1, strongly suggesting a role of MBNL1 in alternative splicing regulation [23]. Ho et al. [32] showed that knockdown of MBNLl in cultured cells results in disrupted splicing of both cTNT and IR. Furthermore, overexpression of MBNL1 in DM1 myoblasts is sufficient to rescue the IR splicing defect [33]. AAV-mediated delivery of Mbnl1 to HSALR skeletal muscle restored normal ClC-1 splicing and reversed myotonia, further demonstrating direct links between MBNL1, ClC-1 splicing and myotonia [24]. MBNL1 promotes skipping of cTNT exon 5 by binding directly to the upstream intron [32], which blocks recruitment of the essential splicing factor U2AF65 [34]. Intriguingly, the MBNL1-binding site on this intron forms a hairpin structure containing mismatches similar to the expanded CUG repeats [35,36]. This implies that MBNL1 is sequestered to the repeat RNA because the secondary structure resembles its natural binding site. An interesting observation is that all of these mis-spliced events are developmentally regulated and the predominant transcript switches from the adult isoform to the embryonic isoform in DM1. MBNL1 localization switches from cytoplasmic to predominantly nuclear during normal skeletal muscle development [22], hence it is proposed that MBNL1 sequestration reverses a subset of splicing targets to their embryonic isoform, contributing to multiple symptoms of DM1.
Although the MBNL1 sequestration model (Figure 1) is well established, it is not the only mechanism of DM1 pathogenesis. One example is that Mbnl1ΔE3/ΔE3 mice do not display muscle wasting, suggesting that some aspects of the disease may not result from loss of MBNL1 function [23]. Also, Ho et al. [37] showed that while MBNL1 can be sequestered to both CUG- and CAG-containing foci with similar affinity, only CUG repeats induced abnormal splicing of cTNT and IR minigenes. Thus MBNL1 sequestration alone may not be sufficient to induce all the splicing changes in this disease.
CUGBP1
Another protein involved in DM1 is CUGBP1, which was originally identified by its ability to bind short single-stranded CUG repeats [38]. CUGBP1 does not bind to double-stranded CUG repeats or co-localize with RNA nuclear foci [39,40], thus it is not a sequestered factor like MBNL1. Philips et al. [31] reported that CUGBP1 binds to CUG sequences within splicing enhancers located downstream of the cTNT exon 5 and increases inclusion of this alternative exon in DM1. Subsequent studies showed that CUGBP1 promotes the embryonic splice form of both IR [30] and ClC-1 [26], which are predominant in DM1. CUGBP1 induces skipping of IR exon 11 by binding to both an exonic splicing enhancer on exon 11 and another intronic splicing enhancer on the upstream intron [30,41]. Interestingly, in these three examples (cTNT, IR and ClC-1), when CUGBP1 induces inclusion of an exon, MBNL1 increases exclusion of the same exon and vice versa. Kalsotra et al. [42] performed a large screen to identify alternative splicing events regulated during mouse heart development and identified CUGBP1 and MBNL1 as regulators of more than half of the splicing transitions tested. Many of these developmentally regulated splicing events are modulated exclusively by CUGBP1 or MBNL1, but all events regulated by both proteins exhibit antagonistic responses [42]. In addition, while MBNL1 nuclear levels increase during development, CUGBP1 nuclear levels decrease [22,42], supporting the hypothesis that the level and localization of these two proteins control a fetal to adult splicing transition, which is reversed in DM1 tissues (Figure 2).
Figure 2. The levels of MBNL1 and CUGBP1 in the nucleus control a subset of developmentally regulated splicing events that are reversed in DM1.
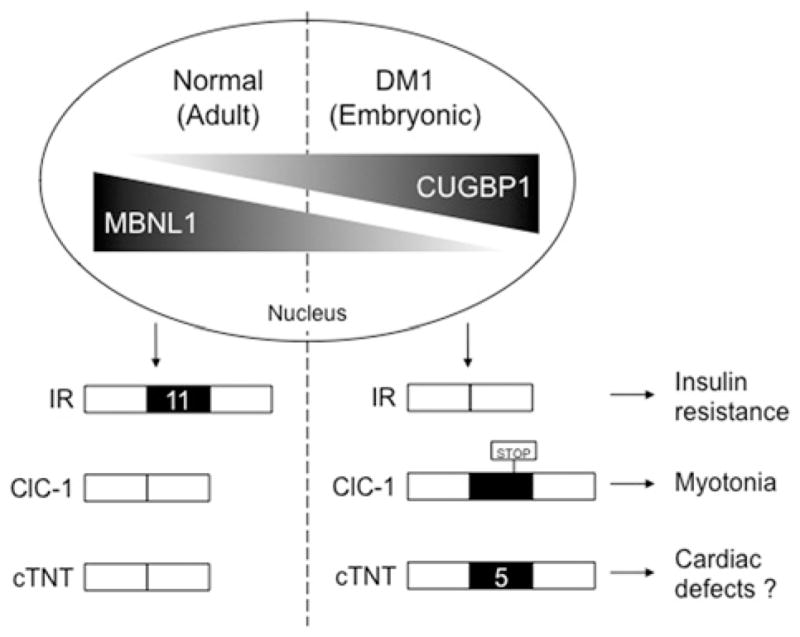
In the embryonic stage, MBNL1 nuclear levels are low and CUGBP1 levels are high. During development, MBNL1 nuclear levels increase while CUGBP1 levels decrease, inducing an embryonic-to-adult transition of downstream splice targets (including IR exon 11, ClC-1 exons containing stop codons and cTNT exon 5). In DM1, MBNL1 is sequestered to CUG repeats, resulting in a decrease of functional MBNL1, while CUGBP1 levels are increased due to phosphorylation and stabilization. This simulates the embryonic condition and enhances expression of embryonic isoforms in adults, resulting in multiple disease symptoms.
CUGBP1 protein steady-state levels are increased in DM1 muscle [31] and heart tissue [43]. But does up-regulation of CUGBP1 contribute to DM1 pathogenesis? Two different transgenic mice overexpressing CUGBP1 in muscle were generated and both displayed muscle abnormalities similar to DM1 [44,45]. One mouse model exhibited splicing defects, providing further evidence that CUGBP1 contributes to mis-regulated splicing in DM1 [44]. The other model revealed another role of CUGBP1 as a regulator of translation. CUGBP1 interacts with the mRNA transcript of MEF2A (myocyte enhancer factor 2A), a transcription factor involved in myogenesis, and increases its translation [45]. MEF2A levels are increased in CUGBP1 transgenic mice and DM1 skeletal muscle tissues, which is proposed to contribute to delayed myogenesis [45]. Another known translational target of CUGBP1 is p21, which could cause impaired cell cycle withdrawal in DM1 muscle cells [46].
Besides regulating splicing and translation, CUGBP1 also plays a role in mRNA decay. Moraes et al. [47] reported that CUGBP1 binds to AREs (AU-rich elements) in c-fos and TNFα (tumour necrosis factor α), followed by recruitment of the polyA-specific ribonuclease, which deadenylates and destabilizes the transcripts. Furthermore, TNF mRNA is stabilized in cells overexpressing the DMPK 3′-UTR containing expanded CUG repeats [48]. In addition to binding to AREs, CUGBP1 mediates mRNA decay of short-lived transcripts by interaction with GU-rich elements [49]. These studies reveal putative targets of CUGBP1 that may also contribute to DM1.
Several recent DM1 mouse models, all with repeats located within the DMPK 3′-UTR, exhibit increased CUGBP1 levels. Orengo et al. [14] generated inducible bitransgenic mice with muscle-specific expression of 960 CUG repeats that display many features of DM1 including RNA foci, myotonia, splicing defects, and importantly, increased CUGBP1 and muscle wasting. A heart specific mouse model expressing the same construct developed DM1-like cardiac conduction and contraction defects, and displayed elevated CUGBP1 levels within only 6 h after induction of the repeats [15]. Furthermore, CUGBP1 was up-regulated specifically in the nuclei in which RNA foci were detected [15]. This provides evidence that elevated CUGBP1 is a primary response to the mutant DMPK transcript. Mahadevan et al. [50] generated transgenic mice expressing the DMPK 3′-UTR containing only five CUGs as part of a GFP (green fluorescent protein) transcript. Surprisingly, these mice develop DM1-like phenotype and CUGBP1 is up-regulated in skeletal muscle; however, there was no formation of RNA foci and no obvious redistribution of nuclear MBNL1, although depletion of MBNL1 on ‘micro-foci’ cannot be ruled out [50].
How does expression of the DM1 repeat mutation result in increased CUGBP1 protein steady-state level? Kuyumcu-Martinez et al. [51] found that this is due to PKC (protein kinase C)-mediated hyperphosphorylation. Expression of expanded CUG repeats in the DMPK 3′-UTR induces CUGBP1 phosphorylation and is dependent on PKC activation. Activation of PKC increases CUGBP1 protein stability and PKC directly phosphorylates CUGBP1 in vitro. Moreover, PKC is activated in heart tissues from DM1 patients and a DM1 mouse model [51]. Thus it is hypothesized that the mutant RNA activates the PKC signalling pathway through an unknown mechanism, which induces CUGBP1 hyperphosphorylation and stabilization (Figure 1).
Relevance to other RNA gain-of-function disorders
RNA toxicity caused by expression of non-coding repeat expansions is now known to be operative in other diseases. Huntington’s disease-like 2 is caused by expanded CTG repeats located in the junctophilin-3 gene. The repeats can be in the coding or UTR depending on the alternative splicing pattern [52]. The CTG repeats form RNA foci that include MBNL1, similar to what is observed in DM1 [53]. MBNL1 is also found within nuclear inclusions in neurons of patients with FXTAS (fragile X associated tremor/ataxia syndrome) [54]. FXTAS is another RNA dominant disorder caused by CGG repeats in the FMR1 (fragile X mental retardation 1) gene 5′-UTR. This raises the possibility that sequestration of MBNL1 is a common pathogenic mechanism. Using a Drosophila model, Sofola et al. [55] further identified CUGBP1 as a suppressor of neurodegeneration induced by 90 CGG repeats in the FMR1 5′-UTR. The role of CUGBP1 in FXTAS is not yet understood. Most forms of SCA are caused by CAG expansions within coding regions, leading to a polyglutamine gain-of-function; however, several forms of SCA are caused by non-coding repeat expansions, including SCA8, SCA10 and SCA12 [25]. Interestingly, evidence from a Drosophila model indicates that, while CAG repeats of the SCA3 gene encode toxic protein, the RNA itself still contributes to the neurodegeneration phenotype [56].
Conclusions
DM1 is one of the best characterized RNA dominant diseases. The pathogenic mechanisms involve protein sequestration, stimulation of signalling pathways, disruption of alternative splicing, mRNA translation and possibly mRNA stability. Understanding the mechanisms of this disease not only promotes the development of potential therapies but also provides insight for other diseases that share a common pathogenic mechanism.
Acknowledgments
Funding
Work is our laboratory is supported by the National Institutes of Health and the Muscular Dystrophy Association.
Abbreviations used
- AAV
adeno-associated virus
- ARE
AU-rich element
- ClC-1
chloride channel 1
- cTNT
cardiac troponin T
- CUGBP1
CUG-binding protein 1
- DM
myotonic dystrophy
- DM1
DM type 1
- DMPK
dystrophia myotonica protein kinase
- FMR1
fragile X mental retardation 1
- FXTAS
fragile X associated tremor/ataxia syndrome
- IR
insulin receptor
- MBNL1
muscleblind-like 1
- MEF2A
myocyte enhancer factor 2A
- PKC
protein kinase C
- SCA
spinocerebellar ataxia
- TNF
tumour necrosis factor
- UTR
untranslated region
References
- 1.Brouwer JR, Willemsen R, Oostra BA. Microsatellite repeat instability and neurological disease. BioEssays. 2009;31:71–83. doi: 10.1002/bies.080122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- 3.Harper P. Myotonic Dystrophy. W.B, Saunders; London: 2001. [Google Scholar]
- 4.Fu YH, Friedman DL, Richards S, Pearlman JA, Gibbs RA, Pizzuti A, Ashizawa T, Perryman MB, Scarlato G, Fenwick RG, Caskey CT. Decreased expression of myotonin-protein kinase messenger RNA and protein in adult form of myotonic dystrophy. Science. 1993;260:235–238. doi: 10.1126/science.8469976. [DOI] [PubMed] [Google Scholar]
- 5.Jansen G, Groenen P, Bachner D, Jap PHK, Coerwinkel M, Oerlemans F, vandenBroek W, Gohlsch B, Pette D, Plomp JJ, et al. Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat Genet. 1996;13:316–324. doi: 10.1038/ng0796-316. [DOI] [PubMed] [Google Scholar]
- 6.Berul CI, Maguire CT, Aronovitz MJ, Greenwood J, Miller C, Gehrmann J, Housman D, Mendelsohn ME, Reddy S. DMPK dosage alterations result in atrioventricular conduction abnormalities in a mouse myotonic dystrophy model. J Clin Invest. 1999;103:R1–R7. doi: 10.1172/JCI5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Otten AD, Tapscott SJ. Triplet repeat expansion in myotonic dystrophy alters the adjacent chromatin structure. Proc Natl Acad Sci USA. 1995;92:5465–5469. doi: 10.1073/pnas.92.12.5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thornton CA, Wymer JP, Simmons Z, McClain C, Moxley RT. Expansion of the myotonic dystrophy CTG repeat reduces expression of the flanking DMAHP gene. Nat Genet. 1997;16:407–409. doi: 10.1038/ng0897-407. [DOI] [PubMed] [Google Scholar]
- 9.Klesert TR, Cho DH, Clark JI, Maylie J, Adelman J, Snider L, Yuen EC, Soriano P, Tapscott SJ. Mice deficient in Six5 develop cataracts: implications for myotonic dystrophy. Nat Genet. 2000;25:105–109. doi: 10.1038/75490. [DOI] [PubMed] [Google Scholar]
- 10.Davis BM, McCurrach ME, Taneja KL, Singer RH, Housman DE. Expansion of a CUG trinucleotide repeat in the 3′ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc Natl Acad Sci USA. 1997;94:7388–7393. doi: 10.1073/pnas.94.14.7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amack JD, Paguio AP, Mahadevan MS. Cis and trans effects of the myotonic dystrophy (DM) mutation in a cell culture model. Hum Mol Genet. 1999;8:1975–1984. doi: 10.1093/hmg/8.11.1975. [DOI] [PubMed] [Google Scholar]
- 12.Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, Krym M, Thornton CA. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000;289:1769–1772. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- 13.Seznec H, Agbulut O, Sergeant N, Savouret C, Ghestem A, Tabti N, Willer JC, Ourth L, Duros C, Brisson E, et al. Mice transgenic for the human myotonic dystrophy region with expanded CTG repeats display muscular and brain abnormalities. Hum Mol Genet. 2001;10:2717–2726. doi: 10.1093/hmg/10.23.2717. [DOI] [PubMed] [Google Scholar]
- 14.Orengo JP, Chambon P, Metzger D, Mosier DR, Snipes GJ, Cooper TA. Expanded CTG repeats within the DMPK 3′ UTR causes severe skeletal muscle wasting in an inducible mouse model for myotonic dystrophy. Proc Natl Acad Sci USA. 2008;105:2646–2651. doi: 10.1073/pnas.0708519105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang GS, Kearney DL, De Biasi M, Taffet G, Cooper TA. Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J Clin Invest. 2007;117:2802–2811. doi: 10.1172/JCI32308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ranum LPW, Rasmussen PF, Benzow KA, Koob MD, Day JW. Genetic mapping of a second myotonic dystrophy locus. Nat Genet. 1998;19:196–198. doi: 10.1038/570. [DOI] [PubMed] [Google Scholar]
- 17.Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LPW. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 18.Napierala M, Krzyzosiak WJ. CUG repeats present in myotonin kinase RNA form metastable “slippery” hairpins. J Biol Chem. 1997;272:31079–31085. doi: 10.1074/jbc.272.49.31079. [DOI] [PubMed] [Google Scholar]
- 19.Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS. Recruitment of human muscleblind proteins to (CUG)n expansions associated with myotonic dystrophy. EMBO J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Artero R, Prokop A, Paricio N, Begemann G, Pueyo I, Mlodzik M, Perez-Alonso M, Baylies MK. The muscleblind gene participates in the organization of Z-bands and epidermal attachments of Drosophila muscles and is regulated by Dmef2. Dev Biol. 1998;195:131–143. doi: 10.1006/dbio.1997.8833. [DOI] [PubMed] [Google Scholar]
- 21.Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet. 2004;13:3079–3088. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- 22.Lin XY, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, Swanson MS, Thornton CA. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet. 2006;15:2087–2097. doi: 10.1093/hmg/ddl132. [DOI] [PubMed] [Google Scholar]
- 23.Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 24.Kanadia RN, Shin J, Yuan Y, Beattie SG, Wheeler TM, Thornton CA, Swanson MS. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc Natl Acad Sci USA. 2006;103:11748–11753. doi: 10.1073/pnas.0604970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ranum LPW, Cooper TA. RNA-mediated neuromuscular disorders. Annu Rev Neurosci. 2006;29:259–277. doi: 10.1146/annurev.neuro.29.051605.113014. [DOI] [PubMed] [Google Scholar]
- 26.Charlet-B N, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell. 2002;10:45–53. doi: 10.1016/s1097-2765(02)00572-5. [DOI] [PubMed] [Google Scholar]
- 27.Mankodi A, Takahashi MP, Jiang H, Beck CL, Bowers WJ, Moxley RT, Cannon SC, Thornton CA. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell. 2002;10:35–44. doi: 10.1016/s1097-2765(02)00563-4. [DOI] [PubMed] [Google Scholar]
- 28.Wheeler TM, Lueck JD, Swanson MS, Dirksen RT, Thornton CA. Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J Clin Invest. 2007;117:3952–3957. doi: 10.1172/JCI33355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kellerer M, Lammers R, Ermel B, Tippmer S, Vogt B, Obermaierkusser B, Ullrich A, Haring HU. Distinct α-subunit structures of human insulin A and receptor B variants determine differences in tyrosine kinase activities. Biochemistry. 1992;31:4588–4596. doi: 10.1021/bi00134a008. [DOI] [PubMed] [Google Scholar]
- 30.Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet. 2001;29:40–47. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- 31.Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998;280:737–741. doi: 10.1126/science.280.5364.737. [DOI] [PubMed] [Google Scholar]
- 32.Ho TH, Charlet-B N, Poulos MG, Singh G, Swanson MS, Cooper TA. Muscleblind proteins regulate alternative splicing. EMBO J. 2004;23:3103–3112. doi: 10.1038/sj.emboj.7600300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dansithong W, Paul S, Comai L, Reddy S. MBNL1 is the primary determinant of focus formation and aberrant insulin receptor splicing in DM1. J Biol Chem. 2005;280:5773–5780. doi: 10.1074/jbc.M410781200. [DOI] [PubMed] [Google Scholar]
- 34.Warf MB, Diegel JV, von Hippel PH, Berglund JA. The protein factors MBNL1 and U2AF65 bind alternative RNA structures to regulate splicing. Proc Natl Acad Sci USA. 2009;106:9203–9208. doi: 10.1073/pnas.0900342106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Warf MB, Berglund JA. MBNL binds similar RNA structures in the CUG repeats of myotonic dystrophy and its pre-mRNA substrate cardiac troponin T. RNA. 2007;13:2238–2251. doi: 10.1261/rna.610607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yuan Y, Compton SA, Sobczak K, Stenberg MG, Thornton CA, Griffith JD, Swanson MS. Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucleic Acids Res. 2007;35:5474–5486. doi: 10.1093/nar/gkm601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ho TH, Savkur RS, Poulos MG, Mancini MA, Swanson MS, Cooper TA. Colocalization of muscleblind with RNA foci is separable from mis-regulation of alternative splicing in myotonic dystrophy. J Cell Sci. 2005;118:2923–2933. doi: 10.1242/jcs.02404. [DOI] [PubMed] [Google Scholar]
- 38.Timchenko LT, Timchenko NA, Caskey CT, Roberts R. Novel proteins with binding specificity for DNA CTG repeats and RNA CUG repeats: Implications for myotonic dystrophy. Hum Mol Genet. 1996;5:115–121. doi: 10.1093/hmg/5.1.115. [DOI] [PubMed] [Google Scholar]
- 39.Fardaei M, Larkin K, Brook JD, Hamshere MG. In vivo co-localisation of MBNL protein with DMPK expanded-repeat transcripts. Nucleic Acids Res. 2001;29:2766–2771. doi: 10.1093/nar/29.13.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mankodi A, Teng-Umnuay P, Krym M, Henderson D, Swanson M, Thornton CA. Ribonuclear inclusions in skeletal muscle in myotonic dystrophy types 1 and 2. Ann Neurol. 2003;54:760–768. doi: 10.1002/ana.10763. [DOI] [PubMed] [Google Scholar]
- 41.Sen S, Talukdar I, Webster NJG. SRp20 and CUG-BP1 modulate insulin receptor exon 11 alternative splicing. Mol Cell Biol. 2009;29:871–880. doi: 10.1128/MCB.01709-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kalsotra A, Xiao XS, Ward AJ, Castle JC, Johnson JM, Burge CB, Cooper TA. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc Natl Acad Sci USA. 2008;105:20333–20338. doi: 10.1073/pnas.0809045105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Timchenko NA, Cai ZJ, Welm AL, Reddy S, Ashizawa T, Timchenko LT. RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1. J Biol Chem. 2001;276:7820–7826. doi: 10.1074/jbc.M005960200. [DOI] [PubMed] [Google Scholar]
- 44.Ho TH, Bundman D, Armstrong DL, Cooper TA. Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy. Hum Mol Genet. 2005;14:1539–1547. doi: 10.1093/hmg/ddi162. [DOI] [PubMed] [Google Scholar]
- 45.Timchenko NA, Patel R, Iakova P, Cai ZJ, Quan L, Timchenko LT. Overexpression of CUG triplet repeat-binding protein, CUGBP1, in mice inhibits myogenesis. J Biol Chem. 2004;279:13129–13139. doi: 10.1074/jbc.M312923200. [DOI] [PubMed] [Google Scholar]
- 46.Timchenko NA, Iakova P, Cai ZJ, Smith JR, Timchenko LT. Molecular basis for impaired muscle differentiation in myotonic dystrophy. Mol Cell Biol. 2001;21:6927–6938. doi: 10.1128/MCB.21.20.6927-6938.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moraes KCM, Wilusz CJ, Wilusz J. CUG-BP binds to RNA substrates and recruits PARN deadenylase. RNA. 2006;12:1084–1091. doi: 10.1261/rna.59606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang LB, Lee JE, Wilusz J, Wilusz CJ. The RNA-binding protein CUGBP1 regulates stability of tumor necrosis factor mRNA in muscle cells: implications for myotonic dystrophy. J Biol Chem. 2008;283:22457–22463. doi: 10.1074/jbc.M802803200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vlasova IA, Tahoe NM, Fan D, Larsson O, Rattenbacher B, John JRS, Vasdewani J, Karypis G, Reilly CS, Bitterman PB, Bohjanen PR. Conserved GU-rich elements mediate mRNA decay by binding to CUG-binding protein 1. Mol Cell. 2008;29:263–270. doi: 10.1016/j.molcel.2007.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mahadevan MS, Yadava RS, Yu Q, Balijepalli S, Frenzel-McCardell DC, Bourne TD, Phillips LH. Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nat Genet. 2006;38:1066–1070. doi: 10.1038/ng1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuyumcu-Martinez NM, Wang GS, Cooper TA. Increased steady-state in levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol Cell. 2007;28:68–78. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Holmes SE, O’Hearn E, Rosenblatt A, Callahan C, Hwang HS, Ingersoll-Ashworth RG, Fleisher A, Stevanin G, Brice A, Potter NT, et al. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet. 2001;29:377–378. doi: 10.1038/ng760. [DOI] [PubMed] [Google Scholar]
- 53.Rudnicki DD, Holmes SE, Lin MW, Thornton CA, Ross CA, Margolis RL. Huntington’s disease-like 2 is associated with CUG repeat-containing RNA foci. Ann Neurol. 2007;61:272–282. doi: 10.1002/ana.21081. [DOI] [PubMed] [Google Scholar]
- 54.Iwahashi CK, Yasui DH, An HJ, Greco CM, Tassone F, Nannen K, Babineau B, Lebrilla CB, Hagerman RJ, Hagerman PJ. Protein composition of the intranuclear inclusions of FXTAS. Brain. 2006;129:256–271. doi: 10.1093/brain/awh650. [DOI] [PubMed] [Google Scholar]
- 55.Sofola OA, Jin P, Qin YL, Duan RH, Liu HJ, de Haro M, Nelson DL, Botas J. RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron. 2007;55:565–571. doi: 10.1016/j.neuron.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li LB, Yu ZM, Teng XY, Bonini NM. RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature. 2008;453:1107–1109. doi: 10.1038/nature06909. [DOI] [PMC free article] [PubMed] [Google Scholar]