

Abstract
Mycobacterium tuberculosis is an important human pathogen in virtually every part of the world. Here we investigate whether distinct strains of M. tuberculosis infect different human populations and whether associations between host and pathogen populations are stable despite global traffic and the convergence of diverse strains of the pathogen in cosmopolitan urban centers. The recent global movement and transmission history of 100 M. tuberculosis isolates was inferred from a molecular epidemiologic study of tuberculosis that spans 12 years. Genetic relationships among these isolates were deduced from the distribution of large genomic deletions, which were identified by DNA microarray and confirmed by PCR and sequence analysis. Phylogenetic analysis of these deletions indicates that they are unique event polymorphisms and that horizontal gene transfer is extremely rare in M. tuberculosis. In conjunction with the epidemiological data, phylogenies reveal three large phylogeographic regions. A host's region of origin is predictive of the strain of tuberculosis he or she carries, and this association remains strong even when transmission takes place in a cosmopolitan urban center outside of the region of origin. Approximate dating of the time since divergence of East Asian and Philippine clades of M. tuberculosis suggests that these lineages diverged centuries ago. Thus, associations between host and pathogen populations appear to be highly stable.
Mycobacterium tuberculosis is a global pathogen, killing 1.9 million people each year and infecting ≈2 billion people worldwide (1, 2). Although it is primarily a scourge of the developing world, tuberculosis affects virtually every nation and every ethnicity (2). In view of this ubiquity, an important question in understanding the epidemiology and basic disease biology of the current pandemic is whether there are geographically or ethnically defined human populations within which transmission of M. tuberculosis is relatively common but between which transmission is far more limited. Do global traffic and “small world” effects result in a single, panmictic population of M. tuberculosis, or is the pathogen population somehow subdivided, with genetically distinct varieties carried in distinct populations of human hosts?
The answer to this basic question may have direct implications for the development and administration of tuberculosis vaccines. Genetic variability in the pathogen population, as in Plasmodium and Streptococcus pneumoniae, can render vaccines ineffective against certain strains or in certain geographic regions. There is substantial geographic variability in the efficacy of the world's current tuberculosis vaccine (3, 4), and the reasons for this variation are not yet well understood. Although it is possible that genetic differences among strains of M. tuberculosis play a role in the variable efficacy of bacillus Calmette–Guérin, actual associations between distinct strains of M. tuberculosis and their human host populations have not been demonstrated, and the significance of such genetic structure for vaccine design and administration has not been addressed.
The population genetic structure of such an important pathogen may also have implications for the study of human genetic variation. Work in this area, from classic studies (5) to a number of recent discoveries (6), has shown that certain human polymorphisms, some of which are responsible for genetic diseases, are also associated with resistance to common infectious diseases. An equally important but as yet incipient area of research will elucidate how the genetic variation of pathogen populations (7–11) is related to genetic structure among their human hosts (12–14). An initial step in this undertaking is to determine the nature and stability of associations between host and pathogen populations.
Cosmopolitan centers of immigration draw strains of M. tuberculosis from diverse locations around the globe. Furthermore, because of the natural history of M. tuberculosis, with which individuals may remain latently infected for decades before they develop active disease, the M. tuberculosis population in cosmopolitan centers consists of strains contracted not only in a diversity of locations but also over an extended period. Hence, such centers are concentrations of geographically and temporally disparate samples of M. tuberculosis as well as potential crossroads in global transmission. They therefore represent a promising setting in which to investigate whether M. tuberculosis shows stable associations with certain human host populations or is instead relatively panmictic. The population of M. tuberculosis in San Francisco has been the subject of a comprehensive, 12-year program of conventional and molecular epidemiology (15). This long-term dataset allows us to study strains of M. tuberculosis that arrived in San Francisco over a period of >10 years and to infer whether each case of tuberculosis is the result of recent transmission or reactivation of an earlier infection. In reactivated cases, the host's personal history provides information on the isolate's geographic provenance.
In addition to long-term data on the geographic origin and recency of transmission of a large number of tuberculosis cases, investigation of the stability of association between host and pathogen populations calls for a genetic marker with appropriate properties. The mobile insertion sequence (IS)6110 and other genetic markers associated with repetitive DNA (16–19) have proven invaluable for tracing chains of recent transmission. Furthermore, when analyzed with distance metrics and clustering algorithms, these markers suggest larger family groups, revealing population genetic structure (20, 21). However, the genotypes generated by these markers tend to change relatively rapidly, and identical patterns can occur as a consequence of convergence. Consequently, phylogenies constructed according to these markers are ambiguous if strains are not closely related, making the detection of longer-term associations between host and pathogen populations more difficult.
In contrast, synonymous single-nucleotide polymorphisms (sSNPs) and genomic deletions are more likely to behave as unique event polymorphisms (UEPs), meaning that each mutation is unique and irreversible. Over 200 sSNPs were recently identified by genomic sequence comparison and used to construct a phylogeny for 306 M. tuberculosis isolates and 126 isolates from other species in the M. tuberculosis complex (22). The unambiguous identification of eight distinct clades of mycobacteria suggests that most sSNPs are indeed behaving as UEPs. Brosch et al. (23) analyzed 20 polymorphic genomic deletions identified by differential hybridization arrays, in silico comparison of Mycobacterium bovis and M. tuberculosis, and pulsed-field gel electrophoresis techniques. Eleven of these deletions were not flanked by repetitive DNA, and these genetic markers appeared to behave as UEPs, allowing Brosch et al. to construct a rooted phylogeny for the M. tuberculosis complex.
In the present study, the genomes of 100 distinct strains of M. tuberculosis isolated from patients in San Francisco were examined with DNA microarrays to comprehensively identify large-sequence polymorphisms. Roughly half of the isolates are from probable cases of reactivation of M. tuberculosis contracted earlier, providing a sample of the pathogen from 17 different countries on four continents. Each of the remaining isolates was involved in a chain of transmission within the city, but no two were involved in the same chain of transmission. DNA array hybridization and sequence analysis unambiguously identified 68 genomic deletions to the base pair, and the presence or absence of each of these deletions was determined in all 100 isolates. Initial phylogenetic analysis of this dataset showed that large genomic deletions generally behave as UEPs and can therefore be used to construct an unambiguous phylogeny for the 100 isolates. This phylogeny, in conjunction with the basic epidemiological data on the likely geographic origin of each isolate and the window of time in which it was contracted, allows us to investigate the relationship between host and pathogen population structures, delineating human populations that carry distinct genotypes of the pathogen.
Materials and Methods
Bacterial Isolates. From 1991 to 1999, 1,802 M. tuberculosis isolates from the city of San Francisco were genotyped for an IS6110 restriction fragment length polymorphism (RFLP) band pattern (16); in the event that an isolate had fewer than six bands, it was genotyped for Pro-Glu polymorphic GC-rich repetitive sequence RFLP pattern (24). We refer to isolates with identical genotypes as members of the same “strain.” From the 1802 genotyped isolates, we selected 50 that were linked by genotype to two or more tuberculosis cases in San Francisco. We refer to these isolates and the cases they were from as “clustered,” because they were implicated in chains of transmission. Note, however, that no two clustered isolates in our sample were implicated in the same chain of transmission. The other 50 selected isolates had genotypes that were unique in the San Francisco collection. We refer to these isolates and the cases they were from as “unique” or “nonclustered.”
Detection and Confirmation of Putative Deletions. Sequences that were present in H37Rv but absent from one of the 100 clinical isolates were detected by hybridization of genomic DNA to a M. tuberculosis DNA microarray. All putative deletions were confirmed and mapped to the base pair by sequencing. Details of experimental procedure are provided in ref. 25.
Construction of Phylogenies. Each deletion, defined by the address of its first and last base pair in the H37Rv genome, was considered a two state, irreversible mutation. Parsimonious trees were found by using heuristic searches in paup (26). The strict consensus tree was then used for maximum parsimony reconstruction of ancestral characters (27, 28). In subsequent tree constructions, phage-associated deletions (198a and 149) were excluded. Patient place of birth was not a character in tree construction but was traced as an unordered character on the maximum parsimony phylogeny. In comparisons of IS6110 RFLP band patterns, two bands were identified as matched if their sizes differed by <2.5% of the larger band (20). For comparison between band patterns A and B, a Jaccard distance, d, was calculated, where d = 100–100 (number of matched bands)/(number of bands in A + number of bands in B – number of matched bands). These distances were used in neighbor joining (29), with the maximum parsimony phylogeny based on deletions enforced as a constraint.
Clock Calibration. To minimize the number of undetected changes in IS6110 RFLP band patterns, we compared only isolates from the same terminal clade of the phylogeny shown in Fig. 4. These clades are (16, 44), (2, 9, 10, 15, 33, 43, 65, 68, 72, 74), (4, 14, 23, 57, 86), (7, 56, 70), (28, 58, 84), (47, 55, 67, 69, 95, 97), (46, 79), (76, 83), (18, 22, 35, 40, 81), (38, 60), (51, 96), and (61, 62, 99). The number of deletions per band change was then estimated as
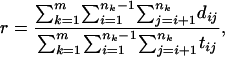 |
[1] |
where m is the number of terminal clades in the phylogeny, nk is the number of isolates in clade k, dij is the number of deletions by which isolates i and j differ, and tij is the number of bands by which the IS6110 fingerprints of isolates i and j differ.
Fig. 4.

Host place of birth mapped onto the maximum parsimony phylogeny for both reactivated and recently transmitted cases. As in Fig. 3, isolates for which deletions were not phylogenetically informative are not shown.
Time to Most Recent Common Ancestor (TMRCA). Unbiased estimation of the TMRCA based on deletions requires that the reference isolate (in our case, H37Rv, the sequence used to create the microarray) is an outgroup to the isolates whose most recent common ancestor is dated. H37Rv belongs to the same clade as most American isolates. Therefore, when estimating a TMRCA, we considered only the East Asian clade (see EA in Fig. 4) and the Philippine clade (see P in Fig. 4), for which H37Rv is in fact an outgroup. The TMRCA was estimated by the frequentist method of Tang et al. (30) and by maximum likelihood. In the frequentist method, isolates were divided into the predefined clades EA and P. In the likelihood model, a rate matrix without zero entries was constructed by assuming that back-mutation occurs at a rate 10–9 times the rate of deletion. A molecular clock was enforced, and likelihood maximization was performed by the method implemented in paup (26).
Results and Discussion
Phylogenetic Analysis Reveals Two Categories of Deletions and Indicates That Horizontal Gene Transfer Is Rare. Microarray analysis of genomic DNA from each of the 100 isolates, followed by PCR confirmation of all putatively deleted sequences (see Materials and Methods), revealed a total of 68 distinct genomic regions that were present in the fully sequenced M. tuberculosis strain H37Rv but absent from one or more of the 100 clinical isolates. Although it has long been thought that horizontal gene transfer is very rare in M. tuberculosis, population genetic investigation of this supposition has begun only very recently (31). If horizontal transfer is indeed very infrequent, then genomic deletions are effectively irreversible, and deletions distributed around the genome all participate in a single coalescent genealogy. In this case, application of maximum parsimony to our dataset of deletions should reveal an unambiguous phylogeny in which each deletion occurs only once and is not reversed. By contrast, if horizontal transfer occurs, then deletions are not irreversible and different sites in the genome participate in distinct genealogies. In this case, different deletions will suggest different parsimonious phylogenies.
Initially, the application of maximum parsimony to our dataset of 68 deletions in 100 isolates did not yield a single, unambiguous phylogeny, but instead suggested contradictions among deletions. We therefore constructed the strict consensus tree of multiple heuristic searches and used maximum parsimony reconstruction of ancestral characters to trace each deletion on the polytomous consensus tree (see Materials and Methods). This tracing revealed that the two deletions comprising mycobacteriophage DNA behaved differently from the other deletions. As illustrated in Fig. 1, phage-associated deletions exhibit a distribution on the consensus phylogeny that can only be explained by invoking multiple independent genetic events. This pattern is consistent with the mobility of phage DNA, and we do not know whether the independent genetic events represent phage-specific excisions, insertions, or a combination of the two. By contrast, all deletions not associated with phage are monophyletic on the consensus tree; their distributions among isolates can be explained without invoking multiple independent events.
Fig. 1.

The distribution of a phage-associated deletion (deletion 149) on the strict consensus tree. Lineages in which deletion 149 was observed or reconstructed by maximum parsimony reconstruction are gray. The other phage-associated deletion (deletion 198a) also exhibited more than one occurrence on the consensus tree. By contrast, deletions that were not associated with phage occurred only once. For example, deletions 145, 178, and 182 exhibited the same pattern of occurrence, appearing in lineages shown in black.
Excluding the two phage-associated deletions, we repeated the heuristic search for parsimonious phylogenies. This yielded two equally parsimonious trees. In both trees, 65 of 66 deletions behaved as UEPs. In one of the two maximum parsimony trees, deletion 236 exhibited two independent occurrences; in the other, deletion 147c exhibited two independent occurrences. Sequencing of the region around deletion 147c revealed that it is flanked by copies of IS6110, which is known to mediate deletion by homologous recombination (32). This deletion was therefore deemed more likely to exhibit independent occurrences, and we accepted as the most likely phylogeny the tree in which deletion 236 is a UEP and 147c is not.
In summary, when we set aside three deletions associated with mobile DNA, all deletions behave as UEPs, allowing maximum parsimony to converge on a single tree in which there is no homoplasy, i.e., no two deletions exhibit contradiction. This is noteworthy for two reasons. First, it offers confidence that the resulting phylogeny reflects the actual evolutionary history of the isolates. Second, it is consistent with the long-held but little tested belief that horizontal gene exchange is extremely rare in M. tuberculosis.
In principle, there are two plausible explanations for the complete absence of homoplasy in the phylogeny. First, perhaps M. tuberculosis participates in horizontal gene transfer extremely rarely. Second, perhaps horizontal gene transfer occurs, but is invisible in our sample simply because the population is so strongly structured that genetic exchange never takes place between individuals who are distinct at our markers. However, as will be clear when we consider the association between geographic and phylogenetic structure, there is no evidence of genetic exchange even within apparently sympatric populations of bacteria, suggesting that biological properties of M. tuberculosis do in fact result in low rates of horizontal transfer. Nonetheless, we cannot rule out the possibility that extremely fine-scale geographic structure invisible in the current study contributes to the apparent absence of horizontal transfer.
Exactly how low are the rates of horizontal transfer in M. tuberculosis? Statistical tests of congruence among gene trees (33) and analysis of sequences from sequential isolates (34) have suggested that Helicobacter pylori has high rates of horizontal transfer; Neisseria meningitidis, Streptococcus pneumoniae, and Staphylococcus aureus have somewhat lower rates of horizontal transfer; and Escherichia coli and Hemopholis influenzae have still lower rates of transfer. Comparison of our own results with microarray investigations of the genomes of several of these pathogens provides at least a relative estimate of the rate of horizontal transfer in M. tuberculosis. In a microarray investigation of the genomes of 36 strains of S. aureus (9), most of the 18 large genomic regions that were present in one strain but absent from others were widely distributed among strains of divergent lineages. Microarray-based genomic comparisons of 26 pathogenic E. coli (35) strains allowed hierarchical clustering, but did not reveal a unique tree without homoplasy. Similarly, in the close relative of E. coli, Salmonella enterica, at least five major horizontal transfer events were detected in a phylogeny of 20 isolates (36). In contrast with these results, in our set of 100 M. tuberculosis isolates, only three of 68 deletions appear in divergent lineages; two of these three are mycobacteriophage, and one is likely to represent a parallel deletion due to recombination between nearby copies of IS6110. Thus, M. tuberculosis appears to have substantially lower rates of horizontal transfer than most other pathogenic bacteria.
Geotemporal Sampling of M. tuberculosis. Because of the comprehensive nature of the San Francisco epidemiological database, we can be fairly confident that cases of tuberculosis that cannot be linked to any other case by conventional contact tracing or DNA-fingerprint clustering are not due to recent transmission in the city of San Francisco. These nonclustered cases are far more likely to be caused by reactivation of an infection acquired outside of San Francisco. In such cases, the patient's place of birth provides information on the likely geographic origin of the isolate, and the patient's dates of birth and immigration to the U.S. indicate the temporal interval in which the infection was acquired (Fig. 2a). Consider, for example, the nonclustered patient represented by the top bar in Fig. 2a (indicated by an arrow). We may reasonably infer that this patient's M. tuberculosis infection was contracted between February 10, 1962 (the patient's date of birth) and July 15, 1983 (the patient's date of arrival in the US). The most likely geographic origin of the infection is Cambodia, the patient's place of birth. Taking the right side of each bar as the most recent possible date that each patient's infection could have taken place, we note that for each of the fifteen countries sampled, at least one isolate was contracted more than three years ago. For most regions (Mexico and Central America, China, South-East Asia, and the Philippines) we have sampled at least one isolate that was contracted >20 years ago.
Fig. 2.

(a) The temporal intervals in which nonclustered isolates were contracted by their patients. Each nonclustered patient's place of birth and, thus, the strain's likely place of origin is indicated by a color corresponding to the points on the world map in b. Each horizontal bar represents a single, nonclustered patient. The bar spans the time interval over which the M. tuberculosis isolate is likely to have been contracted by the patient, starting from date of birth and ending at immigration to the U.S. The point to the right of each bar indicates the date of diagnosis of active tuberculosis in the U.S. For the five nonclustered patients born in the U.S., the bar spans the interval from date of birth to date of diagnosis. (b) The places of birth of the patients whose isolates were studied. Each point represents a patient, and each color corresponds to a country; both clustered and nonclustered cases are indicated on the map.
Inference of Phylogeographic Structure of M. tuberculosis. If M. tuberculosis exhibits genetic differentiation between geographic regions and this population genetic structure is stable over the time interval sampled by the isolates in this study, then a nonclustered patient's place of birth should be predictive of the genotype of that patient's M. tuberculosis isolate. In short, if stable phylogeographic regions exist, then each case of reactivated tuberculosis should be caused by an isolate belonging to a phylogeographic clade corresponding to the patient's place of birth.
To determine whether this is in fact the case, we mapped each nonclustered patient's place of birth onto the maximum parsimony phylogeny of nonclustered patients' isolates. When we divide places of birth into four larger regions, an association between patient place of birth and isolate phylogenetic clade is apparent (Fig. 3a). One clade is dominated by isolates from East Asia, a separate clade is dominated by isolates from the Philippines, and in two other clades isolates from North America predominate. This suggests that in the time frame sampled by the isolates in our phylogeny, there is far less transmission of tuberculosis between these continental regions than there is within them. (Approximate estimation of this time frame will be discussed below.)
Fig. 3.

(a) Phylogeny of M. tuberculosis isolates from cases that were nonclustered and, therefore, very likely to be caused by reemergence of latent infection. Patient place of birth, the likely location of origin of the M. tuberculosis isolate, is mapped onto the maximum parsimony tree based on deletions. Four large geographic regions are indicated by colors: blue corresponds to East Asia; pink to the Philippines; black to the Americas; and orange to Africa, Europe, and the Middle East. Here and in b and Fig. 4, country of origin is indicated by the abbreviations at the top of the tree: CB, Cambodia; CH, China; CO, Colombia; ET, Ethiopia; HK, Hong Kong; IR, Iran; MC, Macau; MX, Mexico; RP, Philippines; RQ, Puerto Rico; US, United States; VM, Vietnam; YO, Yugoslavia. Isolates for which deletions are not phylogenetically informative (i.e., they do not reveal membership in any clade with more than one nonclustered member) are not shown. (b) Phylogeny of M. tuberculosis isolates from cases that were clustered and, therefore, very likely to be from recent transmission in the city of San Francisco. As in a, patient place of birth is mapped onto the maximum parsimony tree based on deletions. Note that genetic groups of M. tuberculosis remain associated with patients from certain regions, despite the occurrence of infection in San Francisco.
When we focus on a finer geographic scale, turning our attention to the individual nations in which patients were born, there is less evidence of phylogeographic structure (see taxon labels in Fig. 3a). Although the isolates from Hong Kong and Macau are grouped together, isolates from Vietnam appear to be as closely related to isolates from China as they are to isolates from Cambodia or to other isolates from Vietnam. In principle, the absence of population genetic structure at a finer geographic scale could be due either to real panmixis within the larger, more clearly delineated regions or to our inability to resolve the close genetic relationships within each of the larger groups in the phylogeny. It could be, for instance, that the Vietnamese isolates are closely related to each other but that this relationship is not apparent because we are using a genetic marker that changes relatively slowly and, therefore, does not resolve close relationships in the phylogeny. Below, we discuss the use of a marker with a higher rate of change to determine whether the panmixis within continental regions is real or only apparent.
Association Between Host and Pathogen Populations Is Stable in a Cosmopolitan Urban Setting. Among nonclustered cases, patient place of birth is indicative of the place tuberculosis was contracted. By contrast, cases that are “clustered,” linked by DNA fingerprinting to local outbreaks, are very likely to be due to recent transmission, indicating that tuberculosis was contracted in San Francisco, irrespective of the host's place of birth. Does the urban convergence of diverse host and parasite populations erode the phylogeographic pattern found among hosts who contracted their tuberculosis abroad, or do the associations between host and parasite populations remain stable even when distinct host populations live in proximity? As is shown in Fig. 3b, when patient place of birth is mapped onto the maximum parsimony tree for clustered isolates, the phylogeographic regions defined among nonclustered isolates once again occupy separate regions of the tree. Thus, hosts who contract tuberculosis in San Francisco tend to contract a genotype associated with the phylogeographic region in which they were born. An “East Asian” strain, for example, is very likely to be found not only in a host who contracts tuberculosis in China, but also in a Chinese individual who contracts tuberculosis in San Francisco. This population genetic structure would be entirely unsurprising if our clustered isolates were members of the same outbreak. However, no two isolates in our sample belong to the same cluster, and they represent outbreaks that occurred over a 12-year period (1990–2002).
To observe the combined effects of geographic differentiation and stable associations between host and pathogen populations, we map host place of birth onto the maximum parsimony phylogeny for both reactivated and recently transmitted cases (Fig. 4). The clear segregation of distinct regions of host origination into separate regions of the tree suggests that over the time frame sampled by our phylogeny, distinct families of M. tuberculosis are associated with both large geographical regions and emigrant host populations who are from those regions. As in the phylogeny of nonclustered isolates alone, there is no clear indication of differentiation of M. tuberculosis within the large continental regions. Although two Vietnamese isolates (76 and 83) are closely related, other Vietnamese isolates are more closely related to Chinese isolates. Similarly, the Mexican isolates are not clearly separated from other American groups.
Population Genetic Structure on a Finer Scale. To determine whether the apparent absence of population genetic structure on the geographic scale of national boundaries was due to relatively fluid movement of M. tuberculosis within the larger phylogeographic regions or rather to a limitation in the resolution of our genetic marker, we considered genetic relationships defined by a second, more labile marker. As discussed above, IS6110 fingerprints are ideal for tracing the close relationships present in outbreaks. In addition, although they are not UEPs, fingerprint comparisons can be converted to distance metrics to provide approximate measures of more distant relationships as well (20, 21). It is this intermediate range of genetic distances that interests us, and we resort to this marker to resolve relationships when the deletion genotypes change too slowly to reveal differences. For example, we would like to use IS6110 to determine whether isolate 10 (Mexico) is more closely related to 15 (Mexico) than it is to 72 (Cuba) (Fig. 4).
By enforcing the phylogeny based on deletions as a constraint, we resolved genetic relationships by applying the neighbor-joining algorithm to a matrix of Jaccard distances based on IS6110 fingerprints (see Materials and Methods). The resolved tree (Fig. 5, which is published as supporting information on the PNAS web site) does not suggest genetic structure within the broad regions defined above (East Asia, Philippines, Americas, Africa, and Europe). Distinct clades are not composed of isolates from any one intracontinental region, such as South East Asia (Vietnamese and Cambodian), South China Sea (Hong Kong and Macau), or Mexico. This suggests that the movement of M. tuberculosis within the broad boundaries defined above is more fluid than it is between them.
Longevity of the Observed Associations. Fig. 4 shows associations between pathogen and host populations delineated by three broad geographic regions. To obtain a rough estimate of the time frame over which M. tuberculosis carried by distinct human populations has been separated, we estimated the TMRCA of East Asian and Philippine clades of M. tuberculosis. Calculation of times from phylogenetic branch-lengths measured in numbers of deletions requires an estimate of the number of deletions per year. Direct measurement of mutation events per year has not been attempted for deletions and probably would not be feasible, but it has been performed for IS6110 RFLP band patterns (37–41). We therefore calibrated the rate of deletions against the rate of change in IS6110 fingerprints. We must note, however, that for two important reasons the rate estimate obtained in this way is quite rough. First, the rate of change of IS6110 fingerprints is itself uncertain. The instantaneous probability of RFLP band loss or gain is likely to be a nonlinear function of the number of IS6110 in the genome and may depend on the infection's state of activity (37–41). We therefore considered a fairly wide range of values, assuming that a single band change in IS6110 fingerprint occurs, on average, once every 5–20 years.
The second reason calibration with IS6110 fingerprints provides only a rough estimate of the rate of deletion is that some changes in RFLP band patterns may be invisible to us because they occur and are reversed in the time period spanned by our sequence comparisons. This problem of “multiple hits” causes us to underestimate the actual time between deletions, because many transposition events go undetected. To minimize the bias introduced by the problem of multiple hits, we compared only the most closely related pairs of sequences in Fig. 4. Specifically, we confined sequence comparisons to the terminal clades in the phylogeny (see Materials and Methods). Within these clades, we performed all pairwise comparisons, counting the numbers of deletions and band changes (gains or losses) in each comparison. Ninety-four sequence comparisons in 12 terminal clades yielded an estimate of 13 changes in IS6110 genotype per deletion. However, even among our comparisons between relatively closely related isolates, undetected fingerprint changes are likely to have occurred, which is evident in the frequency of band differences between the fingerprints of isolates that share exactly the same deletion genotype (Fig. 6, which is published as supporting information on the PNAS web site). With a mean of 8 (and as many as 17) band differences separating isolates with identical deletion genotypes, it is certain that many differences have gone undetected. Therefore, our estimate of the average time between deletions is likely to be biased downward.
The TMRCA of the East Asian and Philippine clades shown in Fig. 4 was estimated by two different methods (see Materials and Methods). A maximum likelihood approach with enforcement of a molecular clock placed the TMRCA at 3.94 deletions. A frequentist estimator yielded a similar result, placing the TMRCA at 3.7 deletions. If we adopt our lower limit of 5 years per change in IS6110 fingerprint and the estimate of 13 band changes per deletion, which is also likely to be biased downward, the TMRCA estimates suggest that East Asian and Philippine lineages of M. tuberculosis have been separated for at least 240 years. Slower rates of fingerprint change (one event every 10–20 years) would place the TMRCA of East Asian and Philippine clades at 500–1,000 years. Performing the same calculations to obtain the maximum likelihood depths of the East Asian and Philippine clades separately, we find that the most recent common ancestor of the East Asian clade occurred at least 130 years ago and perhaps as long ago as 500 years. Similarly, the TMRCA of the Philippine clade occurred at least 100 years ago, and perhaps as many as 400 years ago.
In summary, our TMRCA estimates suggest that East Asian and Philippine human populations carry M. tuberculosis belonging to distinct lineages that have been separated for 240–1,000 years. Furthermore, in view of the finding that the last common ancestor of East Asian strains occurred well over a century ago, we can infer that the strong association between host place of birth and parasite genotype is not due to the movement of individual M. tuberculosis lineages through distinct human populations over the last few decades. The strong associations between host and parasite populations we see today are likely to be well over 1, and perhaps as many as 10, centuries old.
Concluding Remarks
Because this study focused on a single urban center, the geographic diversity of our sample of M. tuberculosis isolates was dictated largely by the composition of the local immigrant population. Consequently, we have sampled certain geographic regions, such as East Asia, far more thoroughly than others, such as Africa and the Indian subcontinent. Within the bounds of this limitation, however, this study suggests that M. tuberculosis is organized into several large, genetically differentiated populations, which are stably associated with host populations that can be delineated according to place of origin.
In our view, the most important factors in the maintenance of stable associations between host and pathogen populations are likely to be epidemiological and sociological. M. tuberculosis is not a highly contagious pathogen, and transmission generally requires extensive contact. In combination with sociological patterns of assortment, this low transmissibility might generate the kind of stable association between host and pathogen populations that we observe. This explanation is consistent with previous observations of very limited transmission from ethnically defined, foreign-born patient populations to U.S.-born individuals (42, 43). However, the apparent longevity of the associations revealed by this study is surprising. Such stability of population structure increases the likelihood of adaptation by specific M. tuberculosis lineages to the genetic, cultural, or environmental characteristics of particular populations of hosts. Although this study does not provide direct evidence for the adaptation of different clades of M. tuberculosis to different host populations, it does suggest that associations between host and parasite populations are sufficiently stable for such adaptation to evolve.
Supplementary Material
Acknowledgments
We thank Dmitri Petrov and Mark Tanaka for helpful discussions and two anonymous reviewers for extremely valuable suggestions. This work was supported by National Institute of Allergy and Infectious Disease Grant AI 34238 (to P.M.S.) and National Institutes of Health Grants GM 28016 and GM 28424 (to M.W.F.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: sSNP, synonymous single-nucleotide polymorphism; UEP, unique event polymorphism; IS, insertion sequence; RFLP, restriction fragment length polymorphism; TMRCA, time to the most recent common ancestor.
See Commentary on page 4721.
References
- 1.Navin, T. R., McNabb, S. J. & Crawford, J. T. (2002) Emerg. Infect. Dis. 8, 1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dye, C., Scheele, S., Dolin, P., Pathania, V. & Raviglione, M. C. (1999) J. Am. Med. Assoc. 282, 677–686. [DOI] [PubMed] [Google Scholar]
- 3.Colditz, G. A., Brewer, T. F., Berkey, C. S., Wilson, M. E., Burdick, E., Fineberg, H. V. & Mosteller, F. (1994) J. Am. Med. Assoc. 271, 698–702. [PubMed] [Google Scholar]
- 4.McMurray, D. N. (2003) Int. J. Parasitol. 33, 547–554. [DOI] [PubMed] [Google Scholar]
- 5.Allison, A. C. (1964) Cold Spring Harbor Symp. Quant. Biol. 29, 137–149. [DOI] [PubMed] [Google Scholar]
- 6.Hill, A. V. (2001) Annu. Rev. Genomics Hum. Genet. 2, 373–400. [DOI] [PubMed] [Google Scholar]
- 7.Tibayrenc, M. & Ayala, F. J. (2002) Trends Parasitol. 18, 405–410. [DOI] [PubMed] [Google Scholar]
- 8.Salama, N., Guillemin, K., McDaniel, T. K., Sherlock, G., Tompkins, L. & Falkow, S. (2000) Proc. Natl. Acad. Sci. USA 97, 14668–14673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fitzgerald, J. R., Sturdevant, D. E., Mackie, S. M., Gill, S. R. & Musser, J. M. (2001) Proc. Natl. Acad. Sci. USA 98, 8821–8826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kato-Maeda, M., Rhee, J. T., Gingeras, T. R., Salamon, H., Drenkow, J., Smittipat, N. & Small, P. M. (2001) Genome Res. 11, 547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reid, S. D., Green, N. M., Buss, J. K., Lei, B. & Musser, J. M. (2001) Proc. Natl. Acad. Sci. USA 98, 7552–7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cavalli-Sforza, L. L. & Feldman, M. W. (2003) Nat. Genet. 33, Suppl., 266–275. [DOI] [PubMed] [Google Scholar]
- 13.Falush, D., Wirth, T., Linz, B., Pritchard, J. K., Stephens, M., Kidd, M., Blaser, M. J., Graham, D. Y., Vacher, S., Perez-Perez, G. I., et al. (2003) Science 299, 1582–1585. [DOI] [PubMed] [Google Scholar]
- 14.Rosenberg, N. A., Pritchard, J. K., Weber, J. L., Cann, H. M., Kidd, K. K., Zhivotovsky, L. A. & Feldman, M. W. (2002) Science 298, 2381–2385. [DOI] [PubMed] [Google Scholar]
- 15.Small, P. M., Hopewell, P. C., Singh, S. P., Paz, A., Parsonnet, J., Ruston, D. C., Schecter, G. F., Daley, C. L. & Schoolnik, G. K. (1994) N. Engl. J. Med. 330, 1703–1709. [DOI] [PubMed] [Google Scholar]
- 16.van Embden, J. D., Cave, M. D., Crawford, J. T., Dale, J. W., Eisenach, K. D., Gicquel, B., Hermans, P., Martin, C., McAdam, R., Shinnick, T. M., et al. (1993) J. Clin. Microbiol. 31, 406–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ross, B. C., Raios, K., Jackson, K. & Dwyer, B. (1992) J. Clin. Microbiol. 30, 942–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Groenen, P. M., Bunschoten, A. E., van Soolingen, D. & van Embden, J. D. (1993) Mol. Microbiol. 10, 1057–1065. [DOI] [PubMed] [Google Scholar]
- 19.McNabb, S. J., Braden, C. R. & Navin, T. R. (2002) Emerg. Infect. Dis. 8, 1314–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cowan, L. S. & Crawford, J. T. (2002) Emerg. Infect. Dis. 8, 1294–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sola, C., Filliol, I., Legrand, E., Mokrousov, I. & Rastogi, N. (2001) J. Mol. Evol. 53, 680–689. [DOI] [PubMed] [Google Scholar]
- 22.Gutacker, M. M., Smoot, J. C., Migliaccio, C. A., Ricklefs, S. M., Hua, S., Cousins, D. V., Graviss, E. A., Shashkina, E., Kreiswirth, B. N. & Musser, J. M. (2002) Genetics 162, 1533–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brosch, R., Gordon, S. V., Marmiesse, M., Brodin, P., Buchrieser, C., Eiglmeier, K., Garnier, T., Gutierrez, C., Hewinson, G., Kremer, K., et al. (2002) Proc. Natl. Acad. Sci. USA 99, 3684–3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chaves, F., Yang, Z., el Hajj, H., Alonso, M., Burman, W. J., Eisenach, K. D., Dronda, F., Bates, J. H. & Cave, M. D. (1996) J. Clin. Microbiol. 34, 1118–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsolaki, A. G., Hirsh, A. E., DeRiemer, K., Enciso, J. A., Wong, M. Z., Hannan, M., Goguet de la Salmoniere, Y.-O. L., Aman, K., Kato-Maeda, M. & Small, P. M. (2004) Proc. Natl. Acad. Sci. USA 101, 4865–4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swofford, D. L. (2002) paup, Phylogenetic Analysis Using Parsimoney (and Other Methods) (Sinauer Associates, Sunderland, MA), Version 4.
- 27.Maddison, D. R. & Maddison, W. P. (2000) MacClade 4: Analysis of Phylogeny and Character Evolution (Sinauer Associates, Sunderland, Massachusetts).
- 28.Maddison, W. P. (1989) Cladistics 5, 365–377. [DOI] [PubMed] [Google Scholar]
- 29.Saitou, N. & Nei, M. (1987) Mol. Biol. Evol. 4, 406–425. [DOI] [PubMed] [Google Scholar]
- 30.Tang, H., Siegmund, D. O., Shen, P., Oefner, P. J. & Feldman, M. W. (2002) Genetics 161, 447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Supply, P., Warren, R. M., Banuls, A. L., Lesjean, S., Van Der Spuy, G. D., Lewis, L. A., Tibayrenc, M., Van Helden, P. D. & Locht, C. (2003) Mol. Microbiol. 47, 529–538. [DOI] [PubMed] [Google Scholar]
- 32.Fang, Z., Doig, C., Kenna, D. T., Smittipat, N., Palittapongarnpim, P., Watt, B. & Forbes, K. J. (1999) J. Bacteriol. 181, 1014–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feil, E. J., Holmes, E. C., Bessen, D. E., Chan, M. S., Day, N. P., Enright, M. C., Goldstein, R., Hood, D. W., Kalia, A., Moore, C. E., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Falush, D., Kraft, C., Taylor, N. S., Correa, P., Fox, J. G., Achtman, M. & Suerbaum, S. (2001) Proc. Natl. Acad. Sci. USA 98, 15056–15061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anjum, M. F., Lucchini, S., Thompson, A., Hinton, J. C. & Woodward, M. J. (2003) Infect. Immun. 71, 4674–4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Porwollik, S., Wong, R. M. & McClelland, M. (2002) Proc. Natl. Acad. Sci. USA 99, 8956–8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Warren, R. M., van der Spuy, G. D., Richardson, M., Beyers, N., Borgdorff, M. W., Behr, M. A. & van Helden, P. D. (2002) J. Clin. Microbiol. 40, 1705–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lillebaek, T., Dirksen, A., Vynnycky, E., Baess, I., Thomsen, V. O. & Andersen, A. B. (2003) J. Infect. Dis. 188, 1032–1039. [DOI] [PubMed] [Google Scholar]
- 39.de Boer, A. S., Borgdorff, M. W., de Haas, P. E., Nagelkerke, N. J., van Embden, J. D. & van Soolingen, D. (1999) J. Infect. Dis. 180, 1238–1244. [DOI] [PubMed] [Google Scholar]
- 40.Yeh, R. W., Ponce de Leon, A., Agasino, C. B., Hahn, J. A., Daley, C. L., Hopewell, P. C. & Small, P. M. (1998) J. Infect. Dis. 177, 1107–1111. [DOI] [PubMed] [Google Scholar]
- 41.Tanaka, M. M. & Rosenberg, N. A. (2001) Stat. Med. 20, 2409–2420. [DOI] [PubMed] [Google Scholar]
- 42.Jasmer, R. M., Ponce de Leon, A., Hopewell, P. C., Alarcon, R. G., Moss, A. R., Paz, E. A., Schecter, G. F. & Small, P. M. (1997) Int. J. Tuberc. Lung. Dis. 1, 536–541. [PubMed] [Google Scholar]
- 43.Chin, D. P., DeRiemer, K., Small, P. M., de Leon, A. P., Steinhart, R., Schecter, G. F., Daley, C. L., Moss, A. R., Paz, E. A., Jasmer, R. M., et al. (1998) Am. J. Respir. Crit. Care Med. 158, 1797–1803. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}