

Background: The ability of arrestin-3 to facilitate activation of JNK1 and JNK2 has never been reported.
Results: Arrestin-3 binds JNK1α1 and JNK2α2 and promotes their phosphorylation by MKK4 and MKK7 in vitro and in intact cells.
Conclusion: Arrestin-3 promotes the activation of ubiquitous JNK1 and JNK2 isoforms.
Significance: Arrestin-3 scaffolds MKK4/7-JNK1/2/3 signaling modules and facilitates activation of ubiquitous JNK isoforms.
Keywords: Arrestin, Jun N-terminal Kinase (JNK), MAP Kinases (MAPKs), Protein Phosphorylation, Scaffold Proteins, Signal Transduction
Abstract
Non-visual arrestins scaffold mitogen-activated protein kinase (MAPK) cascades. The c-Jun N-terminal kinases (JNKs) are members of MAPK family. Arrestin-3 has been shown to enhance the activation of JNK3, which is expressed mainly in neurons, heart, and testes, in contrast to ubiquitous JNK1 and JNK2. Although all JNKs are activated by MKK4 and MKK7, both of which bind arrestin-3, the ability of arrestin-3 to facilitate the activation of JNK1 and JNK2 has never been reported. Using purified proteins we found that arrestin-3 directly binds JNK1α1 and JNK2α2, interacting with the latter comparably to JNK3α2. Phosphorylation of purified JNK1α1 and JNK2α2 by MKK4 or MKK7 is increased by arrestin-3. Endogenous arrestin-3 interacted with endogenous JNK1/2 in different cell types. Arrestin-3 also enhanced phosphorylation of endogenous JNK1/2 in intact cells upon expression of upstream kinases ASK1, MKK4, or MKK7. We observed a biphasic effect of arrestin-3 concentrations on phosphorylation of JNK1α1 and JNK2α2 both in vitro and in vivo. Thus, arrestin-3 acts as a scaffold, facilitating JNK1α1 and JNK2α2 phosphorylation by MKK4 and MKK7 via bringing JNKs and their activators together. The data suggest that arrestin-3 modulates the activity of ubiquitous JNK1 and JNK2 in non-neuronal cells, impacting the signaling pathway that regulates their proliferation and survival.
Introduction
Arrestins were first discovered for their ability to specifically bind active phosphorylated G protein-coupled receptors (GPCRs)4 (1), acting as negative regulators of G protein activation (2). Subsequently arrestins were shown to interact with a variety of non-receptor partners, affecting numerous signaling pathways (for review, see Refs. 3–5). The ability of overexpressed arrestin-35 to promote the activation of exogenous c-Jun N-terminal kinase 3 (JNK3) in cells was demonstrated more than a decade ago (6). Numerous subsequent studies showed that this function does not depend on arrestin-3 association with GPCRs (7–12). In all these studies JNK3, which is predominantly expressed in neurons, heart, and testes (13), was either exogenously expressed in cultured cells that normally do not contain this isoform (6–9, 11, 12) or used in purified form for in vitro reconstitution experiments (10, 12). Although arrestin-3 is expressed in virtually every cell in the body (3, 14, 15), its ability to affect the activation of equally ubiquitous JNK1 and JNK2 isoforms (16–18) has never been reported.
Here, using purified proteins, we show that arrestin-3 directly binds JNK1α1 and JNK2α2, in the latter case at the levels comparable to those observed with JNK3α2. At optimal concentrations arrestin-3 increases the phosphorylation of JNK1α1 and JNK2α2 by both MKK4 and MKK7, similar to the effect of purified arrestin-3 on JNK3α2 phosphorylation by these upstream kinases (10, 12). Endogenous arrestin-3 co-immunoprecipitates with endogenous JNK1/2. Arrestin-3 also promotes the phosphorylation of endogenous JNK1/2 in cells expressing MKK4, MKK7, or their upstream activator ASK1. Importantly, the dependence of JNK phosphorylation on arrestin-3 levels is biphasic; low arrestin-3 concentrations enhance, whereas high concentrations inhibit the phosphorylation of all JNK isoforms tested both in vitro and in intact cells. Thus, arrestin-3 similarly scaffolds signaling modules involved in the activation of JNK1, JNK2, and JNK3, suggesting that arrestin-3 plays a role in the regulation of JNK signaling in the majority of cell types.
EXPERIMENTAL PROCEDURES
Materials
All restriction and DNA modifying enzymes (T4 DNA ligase, Vent® DNA polymerase, and calf intestine alkaline phosphatase) were from New England Biolabs (Ipswich, MA). Cell culture reagents and media were from Mediatech (Manassas, VA) or Invitrogen. All other chemicals were from sources recently described (10, 12).
Protein Purification
Arrestin-3, MBP-arrestin3, JNK1α1, JNK2α2, JNK3α2, active MKK4, and active MKK7 were purified as previously described (10, 12, 19–21).
His-tag Pulldown
Binding of JNK1α1 and JNK2α2 to arrestin-3 was assayed by His-tag pulldown with purified His-JNK1α1, His-JNK2α2, and JNK3α2-His immobilized on Ni-NTA resin from Qiagen according to the manufacturer's instructions. Briefly, 50 μl of purified JNK proteins (5 μg) were incubated with 25 μl of Ni-NTA resin (50% slurry) in binding buffer (50 mm Hepes-Na, pH 7.3, 150 mm NaCl) at 4 °C with gentle rotation for 2 h. Subsequently, 50 μl of protein solutions containing arrestin-3 (5 μg) were added, and the suspensions were incubated at 4 °C for 2 h. The suspension was transferred to centrifuge filters (Ultrafree, Millipore, Bedford, MA) and washed 3 times with washing buffer (50 mm Hepes-Na, pH 7.3, 150 mm NaCl, 50 mm imidazole). The proteins were eluted from resin by the addition of 100 μl of elution buffer (250 mm imidazole, 50 mm Hepes-Na, pH 7.3, and 150 mm NaCl). Eluates were analyzed by SDS-PAGE and Western blotting.
In Vitro JNK1α1/JNK2α2 Phosphorylation
The effect of arrestins on the phosphorylation of JNK1α1/JNK2α2 by MKK7 or MKK4 was analyzed by an in vitro kinase activity assay. Briefly, the assays were conducted in 10 μl containing the following final concentrations: 50 nm active MKK7 or MKK4, 1 μm JNK1α1 or JNK2α2, and 0–24 μm arrestin-3. The mixtures were incubated individually at 30 °C for 10 s. The reactions were stopped by the addition of 15 μl of Laemmli SDS sample buffer (Sigma), and 2 μl of total reaction sample was subjected to SDS-PAGE (8%) and transferred polyvinylidene difluoride (PVDF) membranes (Millipore). Phosphorylated JNK1α1 or JNK2α2 was visualized by rabbit anti-phospho-JNK antibody (Cell Signaling), and the level of JNK phosphorylation was quantified.
Cell Culture and Transient Transfection
COS-7 African green monkey, arrestin-2 knock-out mouse embryonic fibroblasts (MEFs), and Neuro2a cells were maintained in DMEM supplemented with 10% heat-inactivated FBS (Invitrogen), penicillin, and streptomycin at 37 °C in a humidified incubator with 5% CO2. The cells were plated at 80–90% confluence and transfected using FuGENE HD (Promega) according to the manufacturer's instructions. Cells were used 48 h post-transfection and serum-starved overnight before experiments.
Western Blotting and Measurement of JNK Phosphorylation in Intact Cells
COS-7 cells were incubated with phosphatase inhibitors (50 mm NaF and 10 mm sodium orthovanadate (Na3VO4)) in serum-free medium for 15 min at 37 °C, washed with cold PBS, and lysed with SDS lysis buffer containing 1% SDS, 10 mm Tris-HCl, pH 7.4, 10 mm NaF, 100 μm Na3VO4, 2 mm EDTA, 2 mm benzamidine, and 1 mm PMSF. JNK phosphorylation was assayed by Western blotting using an antibody specific for phosphorylated JNKs to detect phospho (active) JNKs (10, 12). Whole cell lysates were boiled for 5 min and centrifuged at 10,000 × g for 10 min, and the supernatants were used for Western blotting. Protein was measured using the Bio-Rad Coomassie Blue assay. The proteins were resolved on 8% SDS-PAGE and transferred to PVDF membrane (Millipore). Blots were incubated with the primary antibodies (Cell Signaling Technology, Inc.) anti-phospho-JNK, anti-JNK, anti-HA (6E2) (1:1000 to 1:5000), anti-FLAG M2 (Sigma) (1:1000), and GAPDH (Millipore) (1:500) followed by appropriate HRP-conjugated secondary antibodies. Arrestins were visualized with F4C1 mouse monoclonal antibody (1:10,000) (22). Protein bands were detected by enhanced chemiluminescence (Pierce) followed by exposure to x-ray film. To quantify phospho-JNKs, we used serial dilutions of anisomycin (1 μg/ml)-stimulated HEK-A cell lysates to ensure that all samples were in linear range. The values for these proteins are expressed in arbitrary units.
Immunoprecipitation
For immunoprecipitation, COS-7 cells (on 60-mm plates) were co-transfected with arrestin-3 and FLAG-JNK1/2 and lysed in 0.75 ml of lysis buffer (50 mm Tris, 2 mm EDTA, 250 mm NaCl, 10% glycerol, 0.5% Nonidet P-40, 20 mm NaF, 1 mm Na3VO4, 10 mm N-ethylmaleimide, 2 mm benzamidine, and 1 mm PMSF) for 30–60 min at 4 °C. After centrifugation, supernatants were precleared with 35 μl of protein G-agarose (50% slurry, Millipore). Then 600 μl of supernatant was incubated with rabbit anti-FLAG antibodies overnight followed by the addition of 25 μl of protein G-agarose beads for 2 h. The beads were washed 3 times with 1 ml of lysis buffer, and the proteins were eluted with 50 μl of SDS sample buffer, boiled for 5 min, and analyzed by Western blotting as described above.
Arrestin-2 KO MEFs that only express arrestin-3 and Neuro-2A cells expressing both non-visual arrestins were used for immunoprecipitation to check for interaction between endogenous arrestin-3 and JNK1/2. Cells (on two 100-mm plates) were washed with cold PBS and lysed in 1.5 ml of lysis buffer for 60 min at 4 °C. After centrifugation at 10,000 × g for 10 min, supernatants were precleared with 35 μl of protein G-agarose. Then, protein concentration was measured using Bio-Rad Coomassie Blue assay. For immunoprecipitation, cell lysates (1 mg of total protein) were incubated with either 1 μg of arrestin-3 antibody (Santa Cruz Biotechnology, catalog #sc-13140) or 1 μg of mouse IgG (as negative control) or 3 μl of F431 (pan-arrestin rabbit polyclonal antibody (23)) or 10 μg of rabbit IgG as negative control overnight followed by the addition of 25 μl of protein G-agarose beads for 2 h. The beads were washed 3 times with 1 ml of lysis buffer, and the proteins were eluted with 50 μl of SDS sample buffer, boiled for 5 min, and analyzed by Western blotting with rabbit anti-arrestin-3 #182 antibody (24, 25), rabbit anti-JNK antibody (Cell Signaling Technology, catalog # 92520), or mouse anti-JNK antibody (Santa Cruz Biotechnology catalog #sc-7345).
Quantification and Statistical Analysis
The bands on the x-ray film were quantified using Quantity One software (Bio-Rad). Two-way analysis of covariance (StatView software, SAS Institute) was used for statistical analysis of upstream kinase-arrestin-3 co-expression, with upstream kinase (ASK1, MKK7, MKK4) concentration treated as a co-variate and the presence or absence of arrestin-3 as a factor. Significant interaction of the co-variate and the arrestin-3 factor is indicative of the change in the slope (homogeneity of slopes test). One-way ANOVA was used to analyze the effect of arrestin-3 expression of JNK activation, as the effect was clearly non-linear. ANOVA analysis was followed by Bonferroni/Dunn post hoc comparison of means, with corrections for multiple comparisons. In all cases, p < 0.05 was considered significant.
RESULTS
Arrestin-3 Binds JNK1α1/JNK2α2 in Vitro and in Intact Cells
The binding of arrestin-3 to JNK3 was inferred from co-immunoprecipitation of these two co-expressed proteins from intact cells (6, 8, 9, 11) as well as from the ability of arrestin-3 to remove JNK3 from the nucleus (26, 27). Only recently was this interaction unambiguously demonstrated using two purified proteins in vitro in the absence of any potential helpers and/or intermediaries (10, 12). However, among the three isoforms of JNK (JNK1, JNK2, and JNK3), JNK1 and JNK2 are as ubiquitous as arrestin-3, whereas JNK3 is expressed in a limited number of cell types (3, 13). Therefore, we tested whether arrestin-3 interacts with JNK1 and JNK2. To obtain a definitive answer, we used purified proteins in vitro. JNK1 and JNK2 each has four different splice variants (17). In these experiments we used a representative splice variant of each, JNK1α1 (one of the shorter 46-kDa forms of JNK1) and JNK2α2 (one of the longer 54-kDa forms of JNK2). To assess the direct interaction in vitro, we immobilized His-tagged JNK isoforms on Ni-NTA resin using binding buffer as a negative control. Specific eluates from Ni-NTA resin (with buffer containing 250 mm imidazole) were analyzed by SDS-PAGE followed by Coomassie Blue staining to determine the amount of each JNK isoform bound to the Ni-NTA column (Fig. 1A). To determine the amount of arrestin-3 retained by each JNK isoform, eluates were subjected to Western blotting with F4C1 anti-arrestin antibody (22, 28). We found that both JNK1α1 and JNK2α2 directly bind arrestin-3 (Fig. 1B). Quantitatively, the amounts of arrestin-3 retained by JNK2α2 were similar to that of JNK3α2, which was previously shown to bind arrestin-3 directly (10, 12), whereas arrestin-3 binding to JNK1α1 was somewhat lower (Fig. 1B). Binding was specific, as arrestin-3 was not retained by Ni-NTA resin (Fig. 1B). Coomassie Blue staining of JNK proteins eluted from the resin revealed that equivalent amounts of each JNK isoform were present in each binding assay (Fig. 1A). Thus, arrestin-3 directly and specifically binds JNK1α1 and JNK2α2, similarly to JNK3α2.
FIGURE 1.
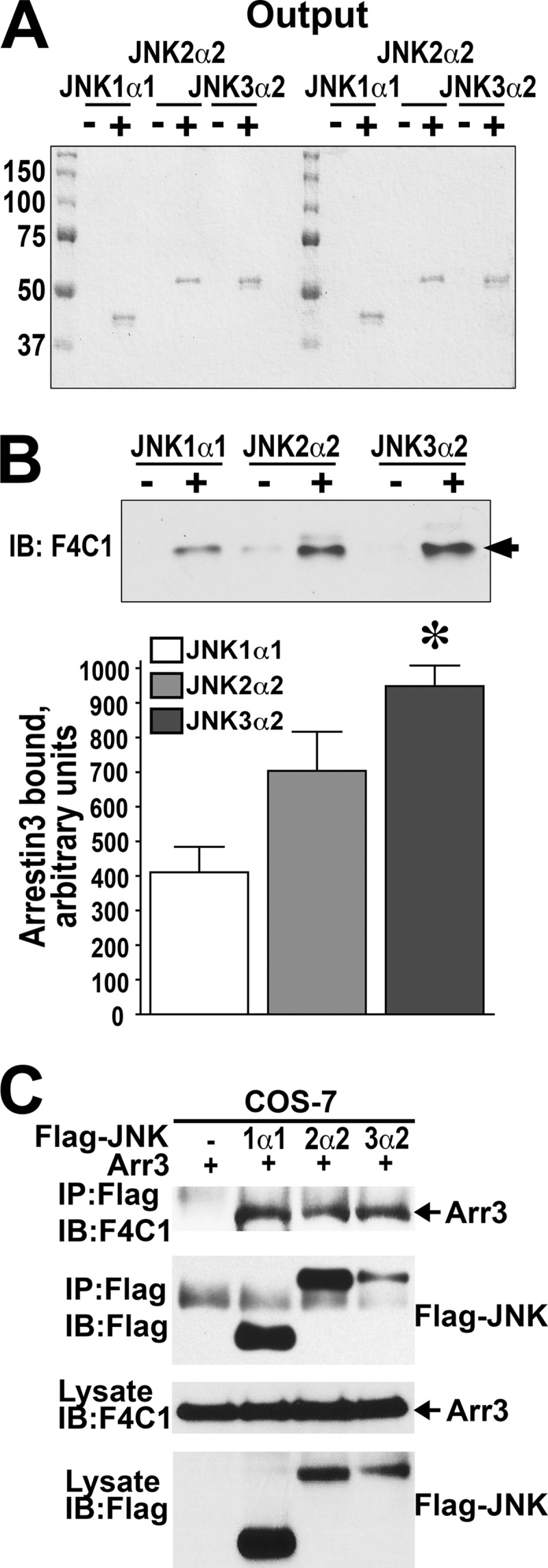
Arrestin-3 binds JNK1α1 and JNK2α2 isoforms in vitro and in cells. A, Coomassie Blue-stained gel of the eluted bait proteins (JNK1α1, JNK2α2, and JNK3α2). B, the amount of arrestin-3 retained was analyzed by Western blot (IB). Representative gels are shown. Bar graph, quantification of the Western blot data. The intensity of the arrestin-3 bands specifically bound to indicated JNK isoforms in three independent experiments was quantified and statistically analyzed. The means ± S.E. are shown. All JNK isoforms showed significant specific binding (p < 0.001 as compared with control without JNK, repeated measure ANOVA); JNK isoforms showed significantly different efficacy in arrestin-3 binding (one-way ANOVA F(2,6) = 9.975 p = 0.0124); *, −p < 0.05 compared to JNK1 according to Bonferroni/Dunn post hoc test with correction for multiple comparisons. C, results of a representative immunoprecipitation (IP) experiment in COS-7 cells expressing arrestin-3 without (−) or with FLAG-JNK1α1, FLAG-JNK2α2, or FLAG-JNK3α2 using anti-FLAG antibody. Arrows point to arrestin-3 bands.
To test whether arrestin-3 interacts with JNK1α1 or JNK2α2 in the cellular environment, we co-transfected COS-7 cells with pcDNA3-based vectors encoding arrestin-3 and FLAG-JNK1α1 or FLAG-JNK2α2. JNK isoforms were immunoprecipitated from cell lysates with FLAG-specific antibody, and co-immunoprecipitated arrestin-3 was detected by Western blot with monoclonal pan-arrestin antibody F4C1 (22). Comparable amounts of arrestin-3 co-immunoprecipitated with FLAG-JNK1α1 and FLAG-JNK2α2, similar to that co-immunoprecipitated with somewhat lower amount of FLAG-JNK3α2 (Fig. 1C). Thus, arrestin-3 interacts with JNK1α1, JNK2α2, and JNK3α2 in intact cells, which suggests that arrestin-3 may play a role in the regulation of JNK1 and JNK2 activity.
Endogenous Arrestin-3 Binds Endogenous JNK1 and JNK2 in Intact Cells
The experiments above demonstrating interactions of arrestin-3 with JNK1/2 in cells have been performed with overexpressed JNK proteins. The interaction of endogenous arrestin-3 with any JNK isoform has never been reported. Therefore, we have examined the endogenous arrestin-3-JNK1/2 interaction by immunoprecipitation. We took advantage of availability of arrestin-2 knock-out MEFs (29) expressing only arrestyin-3 (Fig. 2A), and we also performed experiments in Neuro2a cells of neuronal origin that express both non-visual subtypes but have slightly higher level of arrestin-3 than MEFs (Fig. 2B). We used two different arrestin antibodies to immunoprecipitate arerestin-3 from both cell types. As shown in Fig. 2, we were able to immunoprecipitate arrestin-3 from arrestin-2 knock-out MEFs (Fig. 2A) and Neuro2a cells (Fig. 2B), and found that endogenous JNK1/2 co-immunoprecipitates from both cells with both antibodies (Fig. 2, A and B). Thus, endogenous arrestin-3 interacts with endogenous JNK1/2 in living cells. Interestingly, only the band corresponding to a longer (p54) isoforms of JNK1/2 was detected co-immunoprecipitating with arrestin-3. This may reflect higher affinity of these isoforms for arrestin-3 and is consistent with the in vitro data demonstrating higher binding of longer JNK2α2 than of shorter JNK1α1 isoform to arrestin-3 (Fig. 1, A and B).
FIGURE 2.

Endogenous arrestin-3 interacts with endogenous JNK1/2 in non-neuronal and neuronal cells. A, immunoprecipitation of endogenous arrestin-3 from arrestin-2 knock-out MEFs using rabbit polyclonal arrestin antibody (ab) F431 (upper panels) or mouse monoclonal arrestin-3 antibody (lower panels). Respective rabbit or mouse IgGs were used as controls. Co-immunoprecipitated JNK isoforms were visualized using mouse or rabbit JNK antibodies. Black arrows point to arrestin-3 bands, black arrowheads point to JNK bands, and white arrows point to IgG bands. IB, immunoblot. B, immunoprecipitation experiments performed in Neuro2a cells. All conditions and labels are the same as in panel A. Rabbit arrestin antibody immunoprecipitated arrestin-3 much more efficiently than mouse arrestin-3 antibody, as seen in the images in the left in A and in B. Note that the amount of arrestin-3 standard loaded on each gel was the same (Arr3 Std), but the exposure times differed. Thus, the amount of co-precipitated JNK1/2 was also higher when rabbit arrestin antibody was used.
Arrestin-3 Facilitates ASK1-, MKK7-, and MKK4-mediated Phosphorylation of JNK1/2 in Intact Cells
Arrestin-3 was previously shown to bind JNK3 and upstream kinases ASK1, MKK4, and MKK7 and scaffold ASK1-MKK4/7-JNK3 signaling cascades to promote JNK3 activation (6–9, 11, 12). Our finding that arrestin-3 binds JNK1 and JNK2 (Figs. 1 and 2) suggested that it might be involved in the regulation of these ubiquitous JNK isoforms. Therefore, we tested whether arrestin-3 facilitates the phosphorylation of JNK1α1 and JNK2α2 in cells expressing ASK1, MKK4, or MKK7. To this end increasing amounts of HA-ASK1 were co-transfected into COS-7 cells with constant levels of either empty pcDNA3 or pcDNA3-arrestin-3. COS-7 cells endogenously express eight different isoforms of JNK1 and JNK2 (17). The level of the phosphorylation of endogenous JNK1/2 isoforms was determined by Western blotting of cell lysates with an antibody that specifically recognizes JNK double-phosphorylated at Thr and Tyr in the activation loop. Cells transfected with empty pcDNA3 vector were used as a control for the basal JNK activity. We quantified and statistically analyzed the level of phosphorylation of endogenous JNK isoforms. The phosphorylation levels of p46 upper (p46H), lower (p46L), and p54 JNK isoforms progressively increased with the amount of transfected HA-ASK1 (p < 0.0001) (Fig. 3A). The elevated concentration of arrestin-3 significantly altered phosphorylation of the upper band in p46 and p54 JNK groups (Fig. 3A). Co-expression of arrestin-3 with HA-ASK1 significantly increased the slope of the JNK1/2 activation curves for both p46H and p54 (p < 0.0001 for ASK1 x arrestin3 interaction) (Fig. 3B). At higher levels of ASK1 expression, the magnitude of arrestin-3-induced increase of p46H JNK1/2 activation reached ∼2-fold, as compared with HA-ASK1 alone, whereas the magnitude of p54 activation was smaller (Fig. 3, A and B). Thus, arrestin-3 facilitates ASK1-dependent phosphorylation of several isoforms of endogenous JNK1/2.
FIGURE 3.

Arrestin-3 promotes JNK1/2 activation induced by the expression of ASK1. A, representative Western blot (IB) showing phosphorylation of endogenous JNK1/2 isoforms with or without arrestin-3 in COS-7 cells expressing varying amounts of ASK1. The upper p-JNK blot is the same as the lower blot exposed for a longer time to visualize p54 isoforms. B, quantification of phosphorylation of JNK p46H and p54 isoforms with or without arrestin-3. Analysis of covariance with Arr3 as factor and ASK1 concentration as co-variate showed a significant effect of ASK1 concentration on the level of p46H and p54 phosphorylation (p < 0.0001). Significant Arr3 × ASK1 interaction (p < 0.0001) indicated a difference in slopes due to co-expression of arrestin-3. *, −p < 0.05; **, −p < 0.01 to −Arr3; Student's t test for individual points.
All JNKs are phosphorylated by two upstream MAP kinase kinases (MAPKK), MKK4 and MKK7 (30, 31), which are activated by several MAPKKKs, including ASK1 (18, 32). Recently, using purified proteins we demonstrated that arrestin-3 directly binds MKK4 and MKK7 and promotes JNK3α2 phosphorylation by both MKK4 and MKK7 in vitro as well as in intact cells (10, 12). Therefore, we next tested the ability of arrestin-3 to promote JNK1/2 phosphorylation by MKK7. To this end we co-expressed increasing amounts of MKK7 without or with arrestin-3 in COS-7 cells and monitored the phosphorylation of endogenous JNK isoforms. Increasing amounts of MKK7 resulted in a dose-dependent increase of JNK1/2 phosphorylation in the absence of arrestin-3 (Fig. 4, A and B). Similar to its effect on ASK1-dependent JNK1/2 phosphorylation, arrestin-3 markedly increased the rate of phosphorylation of JNK1/2 p54 and slower migrating p46H JNK isoforms, with the arrestin-3-dependent difference reaching ∼1.7-fold for p46H (quantified in Fig. 4B). The phosphorylation of p46H was elevated by arrestin-3 across a wide range of MKK7 concentrations, as evidenced by a significant effect of arrestin-3 factor (p = 0.0057). Co-expression of arrestin-3 also increased the slope of the MKK7 effect on p46H JNK1/2 phosphorylation, as reflected in significant arrestin-3 × MKK7 interaction (p = 0.0335). The level of p54 phosphorylation was lower than that of p46H, and the effect of arrestin-3 was evident in the increase of the slope (p < 0.0001).
FIGURE 4.

Arrestin-3 enhances MKK7-dependent phosphorylation of endogenous JNK1/2 in intact cells. A, a representative Western blot (IB) showing phosphorylation of endogenous JNK1/2 isoforms with or without arrestin-3 in COS-7 cells expressing varying amounts of MKK7. B, quantification of phosphorylation of JNK p46H and p54 isoforms with or without arrestin-3. Analysis of covariance with Arr3 as factor and MKK7 concentration as co-variate showed a significant effect of MKK7 concentration on the level of p46H and p54 phosphorylation (p < 0.0001). The presence of Arr3 significantly affected the level of p46H, but not p54, phosphorylation across MKK7 concentrations (F(1,38) = 8.592; p = 0.0057). Significant Arr3 × MKK7 interaction (p = 0.0335 for p46H and p < 0.0001 for p54) indicated a difference in slopes due to co-expression of arrestin-3. *, −p < 0.05 to −Arr3; Student's t test for individual points.
Previous reports showed that scaffold proteins in the JNK pathway have distinct capabilities to interact with upstream MAPKKs, MKK4, and MKK7. JIP-1 (JNK interacting protein-1) and JIP-2 specifically bind MKK7 but cannot bind MKK4 (33, 34), whereas JIP-3 can associate with either of these two MAPKKs (34–36). To investigate whether arrestin-3 facilitates MKK4-mediated JNK1/2 activation, increasing amounts of MKK4 were co-transfected into COS-7 cells with empty vector or a constant amount of arrestin-3 followed by Western blotting of lysates with phospho-JNK antibody (Fig. 5). In contrast to MKK7 (Fig. 4), exogenous expression of MKK4 per se did not result in an increase of JNK1/2 phosphorylation even at the highest level (Fig. 5). Without arrestin-3, progressive enhancement of phosphorylation by increasing amounts of MKK4 was not visible on Western blot even for p46H isoform, which responded the strongest to overexpression of ASK1 and MKK7 (Figs. 3 and 4). However, the addition of arrestin-3 strongly increased the slope of the curve for p46H, as evidenced by highly significant MKK4-arrestin-3 interaction (Fig. 5, A and B). Moreover, MKK4-dependent increase in p54 phosphorylation was only detectable in cells co-expressing arrestin-3 (Fig. 5A). Collectively, the data in Figs. 3–5 show that arrestin-3 scaffolds ASK1-MKK4/7-JNK1/2 signaling modules, similar to the reported scaffolding of ASK1-MKK4/7-JNK3α2 modules (6, 8, 9, 11).
FIGURE 5.

Arrestin-3 enhances MKK4-dependent phosphorylation of endogenous JNK1/2 in intact cells. A, representative Western blot (IB) showing phosphorylation of endogenous JNK1/2 isoforms with or without arrestin-3 in COS-7 cells expressing varying amounts of MKK4. Note that there is no progression in the level of p46H phosphorylation with increased MKK4 concentration without arrestin-3 co-expression. P54 isoforms were poorly activated by the expression of MKK4, in contrast to ASK1 and MKK7 (Figs. 3 and 4), although some increase in the presence of arrestin-3 is visible. B, quantification of phosphorylation of JNK p46H isoforms with or without arrestin-3. Analysis of covariance with Arr3 as factor and MKK4 concentration as co-variate showed significant Arr3 X MKK4 interaction (p < 0.0001), indicating a difference in slopes due to co-expression of arrestin-3. *, −p < 0.05; **, −p < 0.01; *, −p < 0.001 −arrestin-3; Student's t test for individual points.
Biphasic Effect of Arrestin-3 on JNK1α1 or JNK2α2 Activation by MKK4 and MKK7 in Vitro
To directly test whether arrestin-3 acts as a scaffold facilitating the phosphorylation of JNK1α1 or JNK2α2 by MKK4 or MKK7, using purified proteins we assembled four signaling modules, MKK4-JNK1α1, MKK7-JNK1α1, MKK4-JNK2α2, and MKK7-JNK2α2, in the presence and absence of purified arrestin-3 (Fig. 6). Constant concentrations of MKK4 or MKK7 (50 nm) as well as their substrates JNK1α1 or JNK2α2 (1 μm) were incubated with varying concentration of arrestin-3 (0.2–24 μm), and the extent of JNK1α1 or JNK2α2 phosphorylation was determined by Western blot with phospho-JNK antibody (Fig. 6). We found that low concentrations of arrestin-3 enhanced the phosphorylation of both JNK1α1 and JNK2α2 by MKK4, whereas high concentrations of arrestin-3 markedly attenuated the ability of MKK4 to phosphorylate JNK1α1 or JNK2α2 (Fig. 6, A–C). The experiments testing the phosphorylation of JNK1α1 and JNK2α2 by MKK7 yielded similar results; arrestin-3 biphasically affected the phosphorylation of JNK1α1 and JNK2α2 by MKK7 (Fig. 6, D–F). Thus, qualitatively arrestin-3 plays the same role in the phosphorylation of JNK1α1 and JNK2α2 by MKK4 and MKK7. In contrast to our observations with JNK3α2, where arrestin-3 concentrations optimal for its phosphorylation by MKK4 and MKK7 were very different (0.6 and 5–8 μm, respectively (10, 12)), optimal arrestin-3 concentrations for scaffolding MKK4-JNK1α1, MKK4-JNK2α2, MKK7-JNK1α1, and MKK7-JNK2α2 modules were all in the range of 5–10 μm (Fig. 6). We previously found that in case of arrestin-3 scaffolding of MKK7-JNK3α2 module, when all possible complexes are taken into account by a thermodynamically valid model, the optimal scaffold concentration can exceed that of either kinase (12). The presence of arrestin-3 had a stronger effect on the phosphorylation of JNK1α1 by both MKK4 and MKK7, increasing it by 1.7–1.8-fold at optimal concentrations, than on the phosphorylation of JNK2α2, which was enhanced by ∼20–45% (Fig. 6). An arrestin-dependent increase in JNK1/2 phosphorylation in intact cells (Figs. 3–5) tends to be similar to that observed in vitro with JNK1α1 (Fig. 6) or even greater. Because we cannot determine whether endogenous phospho-JNK bands in cells represented splice variants of JNK1 or JNK2, it is possible that JNK1 isoforms were mostly affected. However, we more often observed an arrestin-3-dependent increase in the phosphorylation of shorter (p46) isoforms of JNK1/2 (Figs. 3–5) that were represented in the in vitro assay by JNK1α1, suggesting that longer (p54) splice variants, like JNK2α2, may be affected less in cells. Because in vitro we only tested two purified forms of JNK1/2 of eight existing, it is also conceivable that some of the more sensitive isoforms that are revealed by in-cell assays (Figs. 3–5) were not tested in the experiments with purified proteins (Fig. 6). Importantly, the phosphorylation of JNK1α1 or JNK2α2 by MKK4 as well as MKK7 showed a biphasic dependence on arrestin-3 concentration (Fig. 6), suggesting that arrestin-3 acts as a scaffold bringing MKK4 or MKK7 to their substrates JNK1α1 or JNK2α2 via direct binding of both kinases in each cascade (Fig. 6). This is similar to the earlier finding that arrestin-3 acts as a scaffold tethering the two kinases in MKK4/7-JNK3α2 signaling modules (10, 12).
FIGURE 6.

Arrestin-3 facilitates phosphorylation of purified JNK1α1 and JNK2α2 by MKK4 and MKK7. Representative blots show JNK1α1 (A and D) or JNK2α2 (B and E) phosphorylation in the presence of MKK4 (A and B) or MKK7 (D and E) and increasing amounts of arrestin-3. Quantification of phosphorylation data for JNK1α1 and JNK2α2 in the presence of MKK4 (C) or MKK7 (F) yielded bell-shaped curves as functions of arrestin-3 concentration. Means ± S.D. of three independent experiments are shown. IB, immunoblot. ANOVA analysis with arrestin-3 as the main factor demonstrated significance of arrestin-3 concentration in the presence of MKK4 for both JNK1α1 and JNK2α2 (p < 0.001) and in the presence of MKK7 for both JNK isoforms (p < 0.001). *, −p < 0.001; **, −p < 0.01; *, −p < 0.05 to maximal values (at 5 or 10 μm arrestin-3, respectively) according to Bonferroni/Dunn post-hoc test with correction for multiple comparisons.
Arrestin-3 Modulates ASK1- and MKK7-stimulated JNK1/2 Phosphorylation in Cells in a Concentration-dependent Manner
Our results suggested that arrestin-3 functions as a scaffold for the MKK4/7-JNK1/2 modules in vitro. Scaffolds that bring the two proteins close to each other have two defining hallmarks: they directly bind both components, and they enhance signaling at low concentrations but attenuate it when the amounts of scaffold molecules are too high, demonstrating a bell-shaped concentration dependence (37–39). The inhibitory effect of high concentrations is attributable to the sequestration of individual components, which results in the formation of non-productive incomplete complexes (38, 39). This prediction of mathematical modeling has been recently experimentally validated by the analysis of arrestin-3-mediated scaffolding of MKK4-JNK3α2 and MKK7-JNK3α2 signaling modules reconstructed from pure proteins (10, 12). Because this is clearly the case with JNK1/2 phosphorylation in vitro (Fig. 6), we tested the effect of arrestin-3 expression levels on ASK1-dependent phosphorylation of endogenous JNK1/2 in COS7 cells (Fig. 7, A and B). We found that ASK1 overexpression increases the phosphorylation of JNK1/2 in the absence of arrestin-3. As could be expected, arrestin-3 expression at low to medium levels gradually increased the phosphorylation of several faster migrating (p46) endogenous JNK isoforms, whereas higher levels of arrestin-3 markedly reduced it (Fig. 7A). The quantification of phosphorylated p46 JNK confirmed the biphasic effect of arrestin-3 concentration on ASK1-dependent JNK1/2 phosphorylation in the cellular context, demonstrating statistical significance of an increase in JNK phosphorylation at low arrestin-3 levels and a decrease at high arrestin-3 levels (overall effect of arrestin-3 concentration by ANOVA, p < 0.0001) (Fig. 7B). Next, we used the same approach to test the concentration dependence of arrestin-3 effect on JNK1/2 phosphorylation by MKK7. To this end, FLAG-MKK7 was co-expressed in COS7 cells with increasing amounts of arrestin-3, and the resulting JNK1/2 phosphorylation was measured by Western blot (Fig. 7, C and D). MKK7 expression per se increased the phosphorylation level of endogenous JNK1/2, particularly of the slower-migrating p46 isoforms. Arrestin-3 co-expression further increased MKK7-mediated JNK1/2 phosphorylation within certain range, whereas at the highest level of arrestin-3 the inhibitory effect was observed (Fig. 7C). As in case of ASK1-induced JNK phosphorylation, both an increase at low and a decrease at high levels of arrestin-3 were statistically significant (overall arrestin-3 effect significant at p < 0.0001) (Fig. 7D). These results are consistent with in vitro biphasic effect of arrestin-3 (Fig. 6), indicating that in intact cells arrestin-3 also acts as a scaffold tethering the kinases. Thus, the level of arrestin-3 expression, like that of most MAPK scaffolds, can positively and negatively regulate JNK1/2 activity in cells. The productive signaling unit can only be assembled when the relative concentrations of kinases and arrestin-3 are optimal, as was predicted by mathematical modeling (38, 39) and recently demonstrated experimentally for MKK4/7-JNK3 signaling modules (10, 12).
FIGURE 7.

Arrestin-3 enhances ASK1- and MKK7-dependent phosphorylation of endogenous JNK1/2 in intact cells. A and C, representative Western blots (IB) showing phosphorylation of endogenous JNK1/2 isoforms in cells expressing ASK1 (A) or MKK7 (C) in the presence of increasing concentrations of arrestin-3. B and D, quantification of the levels of p46H JNK isoform phosphorylation. One-way ANOVA analysis with arrestin-3 concentration as factor yielded a significant effect of arrestin-3 on JNK phosphorylation both in the presence of ASK1 and MKK7 (p < 0.0001). *, −p < 0.001; **, −p < 0.01; *, −p < 0.05 to the value at 0 μg of arrestin-3; b, −p < 0.01; c, −p < 0.001 to the maximal value (at 1 or 4 μg of arrestin-3 DNA) according to the Bonferroni/Dunn post-hoc test.
DISCUSSION
The kinases of the JNK family play a major role in multiple stress responses, including those to UV irradiation, heat shock, oxidative stress, protein synthesis inhibition, and exposure to inflammatory cytokines (40, 41). JNK signaling is also involved in the normal physiological processes of cell proliferation, apoptosis, differentiation, and migration (16). JNKs, like other MAP kinases, are activated by MAPKKs, which in their turn are activated by MAPKKKs (40, 41). In most cases, the three kinases constituting a signaling module associate with scaffold proteins, which increase the efficiency of signaling by bringing two or more components of the cascade together (41, 42). Several scaffold proteins were described for the JNK signaling pathway. Among the four mammalian arrestin subtypes (3), arrestin-3 is the only one capable of scaffolding the signaling module that leads to the activation of JNK3α2 (6–9). The first report suggested that in response to GPCR activation arrestin-3 binds JNK3α2 and ASK1, whereas the recruitment of MKK4, which is necessary to make a complete signaling module, was believed to be indirect (6). Subsequent work showed that arrestin-3 promotes JNK3α2 phosphorylation independently of GPCRs (7–9, 11), in agreement with the finding that conformationally biased arrestin mutants, both the pre-activated 3A form with enhanced propensity to bind GPCRs (43–47) and the one “frozen” in basal-like conformation with impaired ability to bind receptors (11, 48, 49), interact with JNK3α2 similarly to WT arrestin-3 (26, 49). Arrestin-3 was also shown to co-immunoprecipitate with MKK4 essentially as efficiently as with ASK1 and JNK3α2 (8, 9, 11). The construction of mutants deficient in either GPCR binding or the ability to promote JNK activation showed that these functions do not correlate (11). Most importantly, the creation of a mutant arrestin-3 that binds all three kinases in ASK1-MKK4-JNK3α2 module but does not facilitate JNK3α2 activation showed that binding per se does not necessarily result in productive scaffolding (11). Work with purified proteins showed that arrestin-3 interactions with JNK3α2, MKK4 (10), and MKK7 (12) are direct and do not require any intermediaries, which could not have been definitively demonstrated by co-immunoprecipitation from cells. These studies also showed that the dependence of JNK3α2 phosphorylation by MKK4 (10) or MKK7 (12) on arrestin-3 concentration is biphasic: arrestin-3 facilitates these reactions at lower concentrations but inhibits them at higher concentrations. This was consistent with the predicted behavior of scaffolds that act by bringing the components together (38, 39).
JNK3 is encoded by one of the three genes in this family, the other two encoding JNK1 and JNK2. In mammals JNK3 has two splice variants and shows limited expression in very few cell types, mostly in neurons, heart, and testes (13), whereas JNK1 and JNK2, each having four splice variants, are ubiquitously expressed (16, 17). The comparison of JNK1−/−JNK2−/− and WT MEFs showed that large amounts of 46-kDa JNK1 and 54-kDa JNK2 isoforms and small amounts of 54-kDa JNK1 and 46-kDa JNK2 isoforms are expressed in these cells (50).
Here we tested the ability of arrestin-3 to bind and activate ubiquitous JNK1 and JNK2. Using purified proteins we demonstrated direct binding of arrestin-3 to JNK1α1 and JNK2α2 (Fig. 1). The results showed that arrestin-3 directly binds JNK2α2 essentially as well as JNK3α2, which we used as a positive control, and also binds JNK1α1, albeit less efficiently (Fig. 1). Arrestin-3 associates with these JNKs in cells, as revealed by co-immunoprecipitation of FLAG-JNK1α1 and FLAG-JNK2α2 with arrestin-3 (Fig. 1). Moreover, endogenous arrestin-3 interacts with endogenous JNK1 and JNK2 isoforms in cells (Fig. 2). We showed that arrestin-3 enhances the phosphorylation of endogenous JNK1 and JNK2 in response to ASK1, MKK4, and MKK7 in intact cells (Figs. 3–5). Furthermore, we reconstructed signaling modules containing MKK4 or MKK7 and JNK1α1 or JNK2α2, and tested the effect of arrestin-3 on JNK phosphorylation by either upstream kinase under these controlled conditions (Fig. 6). Collectively, these data suggest that arrestin-3 acts as a molecular scaffold that regulates JNK signaling in a broad range of cells and tissues, not only in neurons and a few additional cell types expressing JNK3. Because JNK1/2 isoforms are critically involved in numerous physiological and pathological processes (16, 40, 41, 50) and arrestin-3 is expressed as ubiquitously as JNK1/2 (3), this finding has broad biological implications. For example, JNK3 isoform, due to its preferential expression in the nervous system, has been shown to be involved in neuronal death (13, 51–53). In contrast, JNK1/2 plays a wide role in cell death caused by a variety of toxic stimuli in different cell types. Thus, JNK1 and JNK2 isoforms play a critical role in apoptosis-induced UV irradiation or by genotoxic drugs and cytochrome c release from mitochondria during this process (50, 55). Furthermore, JNK1/2 has been shown to be involved in the regulation of apoptosis induced via a TNFα- and p53-dependent pathway (56–63). The role of arrestin-3-dependent JNK1/2 activation in these pathways has never been examined, because arrestin-3 was believed to promote the activation of exclusively JNK3 isoform. However, the role of arrestin-3 in the control of activity of JNK1/2 isoforms described here suggests that arrestin-3 can be involved in the regulation of cell death and survival across a broad range of cell types.
Scaffold proteins are known to play two functional roles. First, scaffolds serve as platforms to bring enzyme and substrate together, as was shown for Ste5, MP1 (MAPK partner 1), JIP-1 (JNK interacting protein-1), JSAP-1 (JNK/SAPK-activating protein1), and KSR (kinase suppressor of Ras) (64–68). Second, some scaffolding proteins also have catalytic roles, activating different components of the signaling pathway in addition to bringing them to each other (42). This mechanism was most convincingly demonstrated for yeast scaffold Ste5 (64, 65). Mathematical modeling showed that when the scaffold tethers the components, bringing them to each other, the scaffold concentration affects the signaling biphasically, increasing it at lower, then reducing it at higher concentrations, when the excess of scaffold over the kinases increases the probability of formation of incomplete complexes, containing only one kinase (38, 39). Previously we found that arrestin-3 promotes the activation of JNK3α2 by MKK4 or MKK7, demonstrating biphasic concentration dependence, so that JNK3α2 phosphorylation by either MKK reaches maximum at optimal concentration of arrestin-3 (10, 12). Using purified proteins in the same paradigm we found that the phosphorylation of JNK1α1 or JNK2α2 by either MKK4 or MKK7 also shows biphasic dependence on arrestin-3 concentrations (Fig. 6). An alternative explanation of the findings would be that high concentration of arrestin-3 promotes its oligomerization (69, 70) and that oligomers are unable to serve as scaffold leading to reduced efficacy of arrestin-3-dependent JNK activation. However, two lines of evidence argue against this explanation. First, based on dimerization constant of arrestin-3 in the absence of IP6, which was determined to be ∼100 μm (54),6 the fraction of arrestin-3 oligomers at the concentrations used in our in vitro experiments (20 μm or lower) is expected to be very low. Thus, the data indicate that arrestin-3 functions as a scaffold for MKK4/7-JNK1/2 signaling modules, similar to the case of JNK3α2 phosphorylation by MKK4 (10) and MKK7 (12).
The biphasic dependence of signaling on scaffold concentration was predicted by mathematical modeling more than a decade ago (38) and confirmed by assembling the signaling complex using purified proteins in vitro (10, 12). However, the enhancement of kinase activity by scaffold proteins and the inhibitory effect of excessive levels of scaffold at the same time have never been reported in the JNK signaling pathway in the cellular context. Our data showed that arrestin-3 expression in intact cells increased the phosphorylation of JNK1/2 by ASK1 or MKK7 within the limited range, whereas higher levels of arrestin-3 suppressed the JNK1/2 phosphorylation (Fig. 7). The dose-response analysis of arrestin-3 in the activation of endogenous JNK1/2 by ASK1 or MKK7 revealed that different expression levels of arrestin-3 can negatively or positively regulate ASK1/MKK7-induced JNK1/2 activation in cellular environment.
In conclusion, here we present the first evidence that arrestin-3 regulates the activation by ASK1, MKK4, and MKK7 of all three types of JNKs (JNK1, JNK2, and JNK3) expressed in a wide variety of cell types. This regulation can be positive or negative, depending on the level of arrestin-3 expression. Further experimentation is necessary to reveal the physiological role of arrestin-3-dependent activation of different JNK isoforms in cell biology, particularly cell death and survival, as well as for devising ways of targeting this process for therapeutic purposes.
This work was supported, in whole or in part, by National Institutes of Health Grants NS065868 and DA030103 (to E. V. G.), GM077561, GM081756, and EY011500 (to V. V. G.), and GM059802 (to K. N. D.). This work was also supported by the Welch Foundation (F-1390) (to K. N. D.).
We use the systematic names of arrestin proteins: arrestin-1 (historic names S-antigen, 48-kDa protein, visual or rod arrestin), arrestin-2 (β-arrestin or β-arrestin1), arrestin-3 (β-arrestin2 or hTHY-ARRX), and arrestin-4 (cone or X-arrestin; for unclear reasons its gene is called “arrestin 3” in the HUGO database).
Q. Chen, Y. Zhuo, D. J. Francis, S. A. Vishnivetskiy, S. M. Hanson, X. Zhan, E. K. Brooks, T. I. Iverson, C. Altenbach, W. L. Hubbell, C. S. Klug, and V. V. Gurevich, manuscript in preparation.
- GPCR
- G protein-coupled receptor
- Ni-NTA
- nickel-nitrilotriacetic acid
- MEF
- mouse embryonic fibroblast
- ANOVA
- analysis of variance.
REFERENCES
- 1. Gurevich V. V., Gurevich E. V. (2004) The molecular acrobatics of arrestin activation. Trends Pharmacol. Sci. 25, 105–111 [DOI] [PubMed] [Google Scholar]
- 2. Carman C. V., Benovic J. L. (1998) G-protein-coupled receptors. Turn-ons and turn-offs. Curr. Opin. Neurobiol. 8, 335–344 [DOI] [PubMed] [Google Scholar]
- 3. Gurevich E. V., Gurevich V. V. (2006) Arrestins are ubiquitous regulators of cellular signaling pathways. Genome Biology 7, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. DeWire S. M., Ahn S., Lefkowitz R. J., Shenoy S. K. (2007) β-Arrestins and cell signaling. Annu. Rev. Physiol. 69, 483–510 [DOI] [PubMed] [Google Scholar]
- 5. Gurevich V. V., Gurevich E. V. (2006) The structural basis of arrestin-mediated regulation of G protein-coupled receptors. Pharmacol. Ther. 110, 465–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McDonald P. H., Chow C. W., Miller W. E., Laporte S. A., Field M. E., Lin F. T., Davis R. J., Lefkowitz R. J. (2000) β-arrestin 2. A receptor-regulated MAPK scaffold for the activation of JNK3. Science 290, 1574–1577 [DOI] [PubMed] [Google Scholar]
- 7. Miller W. E., McDonald P. H., Cai S. F., Field M. E., Davis R. J., Lefkowitz R. J. (2001) Identification of a motif in the carboxyl terminus of β-arrestin2 responsible for activation of JNK3. J. Biol. Chem. 276, 27770–27777 [DOI] [PubMed] [Google Scholar]
- 8. Song X., Coffa S., Fu H., Gurevich V. V. (2009) How does arrestin assemble MAPKs into a signaling complex? J. Biol. Chem. 284, 685–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Seo J., Tsakem E. L., Breitman M., Gurevich V. V. (2011) Identification of arrestin-3-specific residues necessary for JNK3 activation. J. Biol. Chem. 286, 27894–27901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhan X., Kaoud T. S., Dalby K. N., Gurevich V. V. (2011) Non-visual arrestins function as simple scaffolds assembling MKK4-JNK3α2 signaling complex. Biochemistry 50, 10520–10529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Breitman M., Kook S., Gimenez L. E., Lizama B. N., Palazzo M. C., Gurevich E. V., Gurevich V. V. (2012) Silent scaffolds. Inhibition of c-Jun N-terminal kinase 3 activity in the cell by a dominant-negative arrestin-3 mutant. J. Biol. Chem. 287, 19653–19664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhan X., Kaoud T. S., Kook S., Dalby K. N., Gurevich V. V. (2013) JNK3 enzyme binding to arrestin-3 differentially affects the recruitment of upstream mitogen-activated protein (MAP) kinase kinases. J. Biol. Chem. 288, 28535–28547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang D. D., Kuan C. Y., Whitmarsh A. J., Rincón M., Zheng T. S., Davis R. J., Rakic P., Flavell R. A. (1997) Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 389, 865–870 [DOI] [PubMed] [Google Scholar]
- 14. Attramadal H., Arriza J. L., Aoki C., Dawson T. M., Codina J., Kwatra M. M., Snyder S. H., Caron M. G., Lefkowitz R. J. (1992) β-Arrestin2, a novel member of the arrestin/β-arrestin gene family. J. Biol. Chem. 267, 17882–17890 [PubMed] [Google Scholar]
- 15. Sterne-Marr R., Gurevich V. V., Goldsmith P., Bodine R. C., Sanders C., Donoso L. A., Benovic J. L. (1993) Polypeptide variants of β-arrestin and arrestin3. J. Biol. Chem. 268, 15640–15648 [PubMed] [Google Scholar]
- 16. Davis R. J. (2000) Signal transduction by the JNK group of MAP kinases. Cell 103, 239–252 [DOI] [PubMed] [Google Scholar]
- 17. Gupta S., Barrett T., Whitmarsh A. J., Cavanagh J., Sluss H. K., Dérijard B., Davis R. J. (1996) Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 15, 2760–2770 [PMC free article] [PubMed] [Google Scholar]
- 18. Johnson G. L. (2011) Defining MAPK interactomes. ACS Chem. Biol. 6, 18–20 [DOI] [PubMed] [Google Scholar]
- 19. Madsen J. A., Kaoud T. S., Dalby K. N., Brodbelt J. S. (2011) 193-nm photodissociation of singly and multiply charged peptide anions for acidic proteome characterization. Proteomics 11, 1329–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yan C., Kaoud T., Lee S., Dalby K. N., Ren P. (2011) Understanding the specificity of a docking interaction between JNK1 and the scaffolding protein JIP1. J. Phys. Chem. B 115, 1491–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kaoud T. S., Park H., Mitra S., Yan C., Tseng C. C., Shi Y., Jose J., Taliaferro J. M., Lee K., Ren P., Hong J., Dalby K. N. (2012) Manipulating JNK signaling with (−)-zuonin A. ACS Chem. Biol. 7, 1873–1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Donoso L. A., Gregerson D. S., Smith L., Robertson S., Knospe V., Vrabec T., Kalsow C. M. (1990) S-antigen. Preparation and characterization of site-specific monoclonal antibodies. Curr. Eye. Res. 9, 343–355 [DOI] [PubMed] [Google Scholar]
- 23. Song X., Vishnivetskiy S. A., Seo J., Chen J., Gurevich E. V., Gurevich V. V. (2011) Arrestin-1 expression in rods. Balancing functional performance and photoreceptor health. Neuroscience 174, 37–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gurevich E. V., Benovic J. L., Gurevich V. V. (2002) Arrestin2 and arrestin3 are differentially expressed in the rat brain during postnatal development. Neuroscience 109, 421–436 [DOI] [PubMed] [Google Scholar]
- 25. Gurevich E. V., Benovic J. L., Gurevich V. V. (2004) Arrestin2 expression selectively increases during neural differentiation. J. Neurochem. 91, 1404–1416 [DOI] [PubMed] [Google Scholar]
- 26. Song X., Raman D., Gurevich E. V., Vishnivetskiy S. A., Gurevich V. V. (2006) Visual and both non-visual arrestins in their “inactive” conformation bind JNK3 and Mdm2 and relocalize them from the nucleus to the cytoplasm. J. Biol. Chem. 281, 21491–21499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Scott M. G., Le Rouzic E., Périanin A., Pierotti V., Enslen H., Benichou S., Marullo S., Benmerah A. (2002) Differential nucleocytoplasmic shuttling of β-arrestins. Characterization of a leucine-rich nuclear export signal in β-arrestin2. J. Biol. Chem. 277, 37693–37701 [DOI] [PubMed] [Google Scholar]
- 28. Wacker W. B., Donoso L. A., Kalsow C. M., Yankeelov J. A., Jr., Organisciak D. T. (1977) Experimental allergic uveitis. Isolation, characterization, and localization of a soluble uveitopathogenic antigen from bovine retina. J. Immunol. 119, 1949–1958 [PubMed] [Google Scholar]
- 29. Kohout T. A., Lin F. S., Perry S. J., Conner D. A., Lefkowitz R. J. (2001) β-Arrestin 1 and 2 differentially regulate heptahelical receptor signaling and trafficking. Proc. Natl. Acad. Sci. U.S.A. 98, 1601–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lawler S., Fleming Y., Goedert M., Cohen P. (1998) Synergistic activation of SAPK1/JNK1 by two MAP kinase kinases in vitro. Curr. Biol. 8, 1387–1390 [DOI] [PubMed] [Google Scholar]
- 31. Haeusgen W., Herdegen T., Waetzig V. (2011) The bottleneck of JNK signaling. Molecular and functional characteristics of MKK4 and MKK7. Eur. J. Cell Biol. 90, 536–544 [DOI] [PubMed] [Google Scholar]
- 32. Keshet Y., Seger R. (2010) The MAP kinase signaling cascades. A system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 661, 3–38 [DOI] [PubMed] [Google Scholar]
- 33. Yasuda J., Whitmarsh A. J., Cavanagh J., Sharma M., Davis R. J. (1999) The JIP group of mitogen-activated protein kinase scaffold proteins. Mol. Cell. Biol. 19, 7245–7254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Engström W., Ward A., Moorwood K. (2010) The role of scaffold proteins in JNK signalling. Cell Prolif. 43, 56–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Matsuura H., Nishitoh H., Takeda K., Matsuzawa A., Amagasa T., Ito M., Yoshioka K., Ichijo H. (2002) Phosphorylation-dependent scaffolding role of JSAP1/JIP3 in the ASK1-JNK signaling pathway. A new mode of regulation of the MAP kinase cascade. J. Biol. Chem. 277, 40703–40709 [DOI] [PubMed] [Google Scholar]
- 36. Kelkar N., Gupta S., Dickens M., Davis R. J. (2000) Interaction of a mitogen-activated protein kinase signaling module with the neuronal protein JIP3. Mol. Cell. Biol. 20, 1030–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dhanasekaran D. N., Kashef K., Lee C. M., Xu H., Reddy E. P. (2007) Scaffold proteins of MAP-kinase modules. Oncogene 26, 3185–3202 [DOI] [PubMed] [Google Scholar]
- 38. Levchenko A., Bruck J., Sternberg P. W. (2000) Scaffold proteins may biphasically affect the levels of mitogen-activated protein kinase signaling and reduce its threshold properties. Proc. Natl. Acad. Sci. U.S.A. 97, 5818–5823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Levchenko A., Bruck J., Sternberg P. W. (2004) Regulatory modules that generate biphasic signal response in biological systems. Syst. Biol. (Stevenage) 1, 139–148 [DOI] [PubMed] [Google Scholar]
- 40. Kyriakis J. M., Avruch J. (2001) Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 81, 807–869 [DOI] [PubMed] [Google Scholar]
- 41. Widmann C., Gibson S., Jarpe M. B., Johnson G. L. (1999) Mitogen-activated protein kinase. Conservation of a three-kinase module from yeast to human. Physiol. Rev. 79, 143–180 [DOI] [PubMed] [Google Scholar]
- 42. Burack W. R., Shaw A. S. (2000) Signal transduction. Hanging on a scaffold. Curr. Opin. Cell Biol. 12, 211–216 [DOI] [PubMed] [Google Scholar]
- 43. Gurevich V. V. (1998) The selectivity of visual arrestin for light-activated phosphorhodopsin is controlled by multiple nonredundant mechanisms. J. Biol. Chem. 273, 15501–15506 [DOI] [PubMed] [Google Scholar]
- 44. Pan L., Gurevich E. V., Gurevich V. V. (2003) The nature of the arrestin x receptor complex determines the ultimate fate of the internalized receptor. J. Biol. Chem. 278, 11623–11632 [DOI] [PubMed] [Google Scholar]
- 45. Carter J. M., Gurevich V. V., Prossnitz E. R., Engen J. R. (2005) Conformational differences between arrestin2 and pre-activated mutants as revealed by hydrogen exchange mass spectrometry. J. Mol. Biol. 351, 865–878 [DOI] [PubMed] [Google Scholar]
- 46. Celver J., Vishnivetskiy S. A., Chavkin C., Gurevich V. V. (2002) Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J. Biol. Chem. 277, 9043–9048 [DOI] [PubMed] [Google Scholar]
- 47. Kovoor A., Celver J., Abdryashitov R. I., Chavkin C., Gurevich V. V. (1999) Targeted construction of phosphorylation-independent b-arrestin mutants with constitutive activity in cells. J. Biol. Chem. 274, 6831–6834 [DOI] [PubMed] [Google Scholar]
- 48. Vishnivetskiy S. A., Hirsch J. A., Velez M.-G., Gurevich Y. V., Gurevich V. V. (2002) Transition of arrestin in the active receptor-binding state requires an extended interdomain hinge. J. Biol. Chem. 277, 43961–43967 [DOI] [PubMed] [Google Scholar]
- 49. Hanson S. M., Cleghorn W. M., Francis D. J., Vishnivetskiy S. A., Raman D., Song X., Nair K. S., Slepak V. Z., Klug C. S., Gurevich V. V. (2007) Arrestin mobilizes signaling proteins to the cytoskeleton and redirects their activity. J. Mol. Biol. 368, 375–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tournier C., Hess P., Yang D. D., Xu J., Turner T. K., Nimnual A., Bar-Sagi D., Jones S. N., Flavell R. A., Davis R. J. (2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288, 870–874 [DOI] [PubMed] [Google Scholar]
- 51. Gao Y., Signore A. P., Yin W., Cao G., Yin X. M., Sun F., Luo Y., Graham S. H., Chen J. (2005) Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. J. Cereb. Blood Flow Metab. 25, 694–712 [DOI] [PubMed] [Google Scholar]
- 52. Namgung U., Xia Z. (2000) Arsenite-induced apoptosis in cortical neurons is mediated by c-Jun N-terminal protein kinase 3 and p38 mitogen-activated protein kinase. J. Neurosci. 20, 6442–6451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Okuno S., Saito A., Hayashi T., Chan P. H. (2004) The c-Jun N-terminal protein kinase signaling pathway mediates Bax activation and subsequent neuronal apoptosis through interaction with Bim after transient focal cerebral ischemia. J. Neurosci. 24, 7879–7887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hanson S. M., Vishnivetskiy S. A., Hubbell W. L., Gurevich V. V. (2008) Opposing effects of inositol hexakisphosphate on rod arrestin and arrestin2 self-association. Biochemistry 47, 1070–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zanke B. W., Boudreau K., Rubie E., Winnett E., Tibbles L. A., Zon L., Kyriakis J., Liu F. F., Woodgett J. R. (1996) The stress-activated protein kinase pathway mediates cell death following injury induced by cis-platinum, UV irradiation, or heat. Curr. Biol. 6, 606–613 [DOI] [PubMed] [Google Scholar]
- 56. Chang L., Kamata H., Solinas G., Luo J. L., Maeda S., Venuprasad K., Liu Y. C., Karin M. (2006) The E3 ubiquitin ligase itch couples JNK activation to TNFα-induced cell death by inducing c-FLIP(L) turnover. Cell 124, 601–613 [DOI] [PubMed] [Google Scholar]
- 57. De Smaele E., Zazzeroni F., Papa S., Nguyen D. U., Jin R., Jones J., Cong R., Franzoso G. (2001) Induction of gadd45β by NF-κB down-regulates pro-apoptotic JNK signalling. Nature 414, 308–313 [DOI] [PubMed] [Google Scholar]
- 58. Deng Y., Ren X., Yang L., Lin Y., Wu X. (2003) A JNK-dependent pathway is required for TNFα-induced apoptosis. Cell 115, 61–70 [DOI] [PubMed] [Google Scholar]
- 59. Dhanasekaran D. N., Reddy E. P. (2008) JNK signaling in apoptosis. Oncogene 27, 6245–6251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fogarty M. P., Downer E. J., Campbell V. (2003) A role for c-Jun N-terminal kinase 1 (JNK1), but not JNK2, in the β-amyloid-mediated stabilization of protein p53 and induction of the apoptotic cascade in cultured cortical neurons. Biochem. J. 371, 789–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kamata H., Honda S., Maeda S., Chang L., Hirata H., Karin M. (2005) Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120, 649–661 [DOI] [PubMed] [Google Scholar]
- 62. Oleinik N. V., Krupenko N. I., Krupenko S. A. (2007) Cooperation between JNK1 and JNK2 in activation of p53 apoptotic pathway. Oncogene 26, 7222–7230 [DOI] [PubMed] [Google Scholar]
- 63. Tang G., Minemoto Y., Dibling B., Purcell N. H., Li Z.-W., Karin M., Lin A. (2001) Inhibition of JNK activation through NF-κB target genes. Nature 414, 313–317 [DOI] [PubMed] [Google Scholar]
- 64. Good M., Tang G., Singleton J., Reményi A., Lim W. A. (2009) The Ste5 scaffold directs mating signaling by catalytically unlocking the Fus3 MAP kinase for activation. Cell 136, 1085–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bhattacharyya R. P., Reményi A., Good M. C., Bashor C. J., Falick A. M., Lim W. A. (2006) The Ste5 scaffold allosterically modulates signaling output of the yeast mating pathway. Science 311, 822–826 [DOI] [PubMed] [Google Scholar]
- 66. Pullikuth A., McKinnon E., Schaeffer H. J., Catling A. D. (2005) The MEK1 scaffolding protein MP1 regulates cell spreading by integrating PAK1 and Rho signals. Mol. Cell. Biol. 25, 5119–5133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Willoughby E. A., Perkins G. R., Collins M. K., Whitmarsh A. J. (2003) The JNK-interacting protein-1 scaffold protein targets MAPK phosphatase-7 to dephosphorylate JNK. J. Biol. Chem. 278, 10731–10736 [DOI] [PubMed] [Google Scholar]
- 68. Kortum R. L., Lewis R. E. (2004) The molecular scaffold KSR1 regulates the proliferative and oncogenic potential of cells.. Mol. Cell. Biol. 24, 4407–4416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Milano S. K., Kim Y. M., Stefano F. P., Benovic J. L., Brenner C. (2006) Nonvisual arrestin oligomerization and cellular localization are regulated by inositol hexakisphosphate binding. J. Biol. Chem. 281, 9812–9823 [DOI] [PubMed] [Google Scholar]
- 70. Storez H., Scott M. G., Issafras H., Burtey A., Benmerah A., Muntaner O., Piolot T., Tramier M., Coppey-Moisan M., Bouvier M., Labbé-Jullié C., Marullo S. (2005) Homo- and hetero-oligomerization of β-arrestins in living cells. J. Biol. Chem. 280, 40210–40215 [DOI] [PubMed] [Google Scholar]