


Abstract
Type I restriction enzymes (REases) are large pentameric proteins with separate restriction (R), methylation (M) and DNA sequence-recognition (S) subunits. They were the first REases to be discovered and purified, but unlike the enormously useful Type II REases, they have yet to find a place in the enzymatic toolbox of molecular biologists. Type I enzymes have been difficult to characterize, but this is changing as genome analysis reveals their genes, and methylome analysis reveals their recognition sequences. Several Type I REases have been studied in detail and what has been learned about them invites greater attention. In this article, we discuss aspects of the biochemistry, biology and regulation of Type I REases, and of the mechanisms that bacteriophages and plasmids have evolved to evade them. Type I REases have a remarkable ability to change sequence specificity by domain shuffling and rearrangements. We summarize the classic experiments and observations that led to this discovery, and we discuss how this ability depends on the modular organizations of the enzymes and of their S subunits. Finally, we describe examples of Type II restriction–modification systems that have features in common with Type I enzymes, with emphasis on the varied Type IIG enzymes.
TYPE I RESTRICTION ENZYMES
Introduction
In the early 1960s, Werner Arber and Daisy Dussoix (1,2) provided evidence that degradation and methylation of DNA lay behind a phenomenon called ‘host-controlled variation in bacterial viruses’, reported a decade earlier (3–5) and reviewed by Luria (6). ‘Variation’ referred to the observation that one cycle of growth of bacterial viruses (also called (bacterio)phages) on certain hosts affected the ability of the progeny phage to grow on other bacterial hosts, by either restricting or enlarging their host range. Unlike mutation, this change was readily reversed, and one cycle of growth in the previous host returned the virus to its original form. ‘Host-controlled variation’ came to be known by the more familiar terms, ‘restriction’ (R) and ‘modification’ (M). It was learned that modification of the cell’s DNA by methylation protected the DNA, whereas the absence of modification on the phage DNA rendered it sensitive to restriction by endonucleases. Restriction–modification (R–M) systems of all types were investigated in the same way, initially, by measuring the efficiency of plating (eop) of phage on alternate bacterial hosts (2,7–10); see references (6,11–15) for early reviews. The genes responsible for restriction and modification were found on bacterial chromosomes, on ‘resistance transfer factor’ plasmids and on the chromosomes of certain temperate phage themselves (11,12,14–16). Their products, restriction endonucleases (REases) and modification methyltransferases (MTases) began to be purified in the late 1960s and have been studied intensively ever since.
Differences in subunit composition, co-factor requirements and DNA-cleavage properties led to the early division of REases (14,17–19) into Type I (Escherichia coli EcoKI, EcoBI) and Type II (EcoRI, HindII). Subsequently, Type III enzymes (EcoP1I, EcoP15I) and the Type IV modification-dependent REases (Mcr and Mrr) were also recognized to be distinct classes (18–20). See Restriction Enzyme dataBASE (REBASE) (http://rebase.neb.com) (21) and accompanying papers in this journal. Sequencing and biochemistry have since led to several subdivisions within the Type I and the Type II classes, and boundaries are beginning to blur (20,22).
In the late 1970s, a further difference between Type I and Type II REases was demonstrated by the Linn and Yuan laboratories. Using purified EcoBI and EcoKI, respectively, they showed that Type I enzymes translocated DNA powered by ATP hydrolysis. They reported the formation of DNA loops visible by electron microscopy (EM) and considered reaction intermediates. Concomitant with a large conformational change, these enzymes appeared to translocate the DNA while remaining attached to their recognition sites (23,24).
Viruses and other mobile genetic elements (MGE) have evolved multiple strategies to evade REases, including acquiring modification from their host; synthesizing their own MTases, or hyper-modifying their DNA by incorporating unusual bases; synthesizing antirestriction proteins that mimic DNA; and avoiding specific recognition target sequences (25–27). These processes enable phages to become resistant to the REases they encounter, and once this occurs, those REases no longer protect the host from that particular phage. The solution to this decline in protection over time is for cells to change REase specificity periodically. To do this successfully, however, the specificity of the MTase must change simultaneously in exactly the same way or the new REase will restrict the improperly modified cellular DNA, and the cell will die. Unlike conventional Type II R–M systems that generally cannot do this, Type I R–M systems have evolved an efficient way to change both REase and MTase specificities harmoniously, by using a common DNA sequence-recognition (S) subunit for both restriction and modification.
Genes, families and distribution
Type I R–M systems are encoded by three genes, termed hsd for host specificity determinant: hsdR encodes the restriction (R) subunit, hsdM the modification (M) subunit and hsdS the recognition (S for specificity) subunit. The Type I enzymes studied in most detail are EcoKI, from the workhorse of molecular biology, E. coli K12, and the plasmid-encoded EcoR124. The EcoKI genes are located near those for McrBC and Mrr in a cluster called the ‘immigration control region’, a highly divergent locus with alternative DNA segments containing Type I R–M systems, and linked to serB in most E. coli isolates between the yjiS and yjiA genes (28–32). Related Type I enzymes in enteric bacteria often localize to this same region, with few exceptions (e.g. EcoR124).
The current division of Type I enzymes into five families, A-E, is based on complementation tests, antibody cross-reactivity and amino acid (aa) sequence; see accompanying paper and reviews (33–37). The enzymes from E. coli K12 and E. coli B are the founder members of Type IA, EcoAI of E. coli 15T− is the prototype of Type IB, plasmid-encoded EcoR124 is the prototype of Type IC, and Salmonella StySBLI and Klebsiella KpnBI represent Type ID and IE, respectively. Further families undoubtedly exist.
Genome sequencing over the last 15 years, coupled with bioinformatics analysis, reveals that Type I R–M systems are present in approximately one-half of all bacteria and archaea. REBASE maintains a comprehensive list of R–M components of all kinds, both characterized (i.e. biochemically confirmed), putative (unconfirmed) and decayed (disrupted aa sequence). At recent count, of 2145 sequenced bacterial and archaeal genomes in REBASE, 1140 (53%) have at least one each of the hsdR, M and S genes needed for a Type I system. In all, 835 genomes (39%) appear to have no hsd genes at all, and the remaining 170 genomes (8%) have some hsd genes, but perhaps not all. Disruptions and scrambled gene organizations are common.
Among the 1140 genomes that include Type I R–M systems are ∼2100 putative hsdR genes, 2200 putative hsdM genes and 2600 putative hsdS genes—or roughly two of each per organism. The multiplicity of systems varies considerably though. Most genomes have only one Type I system, but two, three or more is common, with as many as eight in some bacteria (e.g. Desulfococcus oleovorans). Some species of Mycoplasma have only one or two hsdR and hsdM genes but multiple hsdS genes: e.g. 10 in Mycoplasma pneumoniae, 13 in Mycoplasma suis and a record 22 in Mycoplasma haemofelis. In these organisms, different hsdS genes probably cycle on and off to effect a continuously changing defense against invaders (38,39). The individual target recognition domains (TRDs) of S subunits can shuffle into different combinations by genetic rearrangements, and so the number of different R–M specificities that these microbes can conjure up is potentially far larger than the number of hsdS genes they possess. Nearly 500 different specificities, and perhaps twice that number, could emerge from 22 hsdS genes, a remarkable defensive repertoire.
Enzyme activities
Type I R–M enzymes are pentameric proteins of composition 2R+2M+S. They require ATP, Mg2+ and S-adenosylmethionine (SAM) for activity and display both REase and MTase activities. A trimer of 2M+S acts solely as an MTase (35,40,41). Characteristically, Type I enzymes recognize bipartite DNA sequences comprising two half-sequences separated by a gap—for example, AACNNNNNNGTGC (AAC N6 GTGC) where N = any base (Table 1). This stems from the repetitive organization of the S subunit that comprises two separate TRDs, one for recognizing each half-sequence. Recombination between TRDs generates new sequence specificities and is a powerful driver of Type I R–M system diversification.
Table 1.
Characterized wild-type Type I R–M enzymes, arranged chronologically
| Enzyme | Family | Recognition sequence | Me-interval | Reference |
|---|---|---|---|---|
| EcoBI | IA | TGA N8 TGCT | 8 | (42–44) |
| EcoKI | IA | AAC N6 GTGC | 8 | (45) |
| EcoAI | IB | GAG N7 GTCA | 9 | (46,47) |
| StyLTIII | IA | GAG N6 RTAYG | 8 | (48) |
| StySPI | IA | AAC N6 GTRC | 8 | (48) |
| EcoDI | IA | TTA N7 GTCY | 8 | (49) |
| EcoDXXI | IC | TCA N7 RTTC | 8 | (50) |
| EcoR124I | IC | GAA N6 RTCG | 7 | (51) |
| EcoEI | IB | GAG N7 ATGC | 9 | (52) |
| CfrAI | IB | GCA N8 GTGG | 9 | (53) |
| EcoprrI | IC | CCA N7 RTGC | 8 | (54) |
| StySKI | IB | CGAT N7 GTTA | 9 | (55) |
| StySBLI | ID | CGA N6 TACC | 6 | (56) |
| NgoAVa | IC | GCA N8 TGC | 8 | (57) |
| Eco377I | GGA N8 ATGC | 9 | (58) | |
| Eco585Ib | GCC N6 TGCG | ? | (58) | |
| Eco646I | CCA N7 CTTC | ? | (58) | |
| Eco777I | GGA N6 TATC | ? | (58) | |
| KpnBI | IE | CAAA N6 RTCA | 7 | (59) |
| KpnAI | ID | GAA N6 TGCC | 6 | (60) |
| StySEAI | ACA N6 TYCA | 6 | (60) | |
| StySGI | TAAC N7 RTCG | 9 | (60) | |
| Eco394I | GAC N5 RTAAY | 7 | (61) | |
| Eco826I | GCA N6 CTGA | 7 | (61) | |
| Eco851I | GTCA N6 TGAY | 6 | (61) | |
| Eco912I | CAC N5 TGGC | 6 | (61) | |
| CsaII | CCAC N6 CTC | 8 | (62) | |
| VbrI | AGHA N7 TGAC | 7 | (62) | |
| VbrII | CTAG N6 RTAA | 8 | (62) | |
| CjeFII | CAAY N6ACT | 9 | (62) | |
| CjeFIV | TAAY N5 TGC | 6 | (62) | |
| BceSVI | TAAG N7 TGG | 8 | (62) | |
| EcoGIV | CCAC N8 TGAY | 9 | (63) | |
| MpuII | GA N7 TAY | 7 | (64) | |
| SauMW2I | CCAY N5 TTAA | ? | (65) | |
| SauMW2II | CCAY N6 TGT | ? | (66) | |
| SauN315I | ATCN5CCT | 9 | (66) | |
| SauN315II | CCAY N6 GTA | 8 | (66) |
aThe S subunit of NgoAV is truncated, resulting in symmetric specificity.
bNo adenine occurs in the first haf-sequence of Eco585I implying that cytosine becomes methylated instead.
Column 1: The name of the prototype enzyme; isoschizomers are not listed. Bold type signifies that the system is well-characterized. Column 2: Complementation group to which the enzyme belongs, if known. Column 3: Only one strand of the recognition sequence is shown, oriented 5′–3′. For the well-characterized enzymes, the sequence is shown in the orientation for which the first (5′) half is specified by the N-TRD of the S subunit, and the second (3′) half is specified by the C-TRD. For the remaining systems, the TRD assignment is not known, and the orientation shown is arbitrary. The numeral in the recognition sequence indicates the number of non-specific bases between the two half-sequences; ‘AAC N6 GTGC’, for example, means AACNNNNNNGTGC. Bases in bold type indicate the positions of methylation if known, generally one A in the 5′ half-sequence and another that is the complement of the T in the 3′ half-site. Column 4: Number of base pairs between the two methylated bases. Enzymes belonging to the same family have similar spacing. Column 5: Publication reporting the specificity.
REase and translocase activities
The R subunit is essential for REase activity. It contains an N-terminal endonuclease domain fused to a so-called ‘motor’ domain found in many other DNA and RNA processing enzymes with translocation or helicase activity (66–74). The R subunit contacts the M2S MTase core via the M subunits. If neither half of the recognition sequence is methylated, the R subunits translocate the flanking DNA and cleave it a variable distance away (75), approximately midway between neighboring recognition sites (76), while the enzyme remains attached with the site. This behavior generates DNA loops visible by EM (23,24) and atomic force microscopy (77,78). Mutations in the endonuclease domain can prevent DNA cleavage without affecting DNA translocation function (66).
Single-molecule studies coupled with improved biochemical and biophysical methods have illuminated the DNA translocation properties of EcoKI and EcoR124 and revealed hitherto unsuspected details about protein dimerization and DNA looping (78). Dimerization appears to be favored when the DNA molecule contains two recognition sites, while DNA looping can occur in the absence of ATP hydrolysis. This way of bringing distant DNA regions together using complex protein assemblies may be a common phenomenon. Magnetic tweezers experiments have provided details of the biophysical aspects of translocation, including the rate of translocation of DNA by the molecular motors—the R subunits—as well as their processivity and ATP dependence (79–83). These studies showed that the two motors could work independently, and that the enzyme tracked along the helical pitch of the DNA on torsionally constrained molecules. Apparently, translocation could stop and restart by disassembly and reassembly. This finding might be of interest to those working on the much larger complexes that contain the related eukaryotic DNA helicases and chromatin-remodeling proteins of the SNF2 superfamily (67). Further studies confirmed the model that ATP hydrolysis is coupled to bidirectional DNA translocation, which would explain the DNA loops seen in the EM by Bob Yuan and colleagues 25 years earlier (24,84,85). The R subunits disengage transiently roughly every other 500 bp using ATP, but translocation progresses ∼2 kb. Once translocation is blocked, either by collision with another molecule or the presence of supercoiled DNA, the DNA is cleaved. Also, there is in vitro evidence for cleavage at a replication fork (86). Though the enzyme remains attached to the DNA, it can be displaced by other translocating enzymes such as the major recombination complex of E. coli, RecBCD (84).
MTase activity
Methylation of the recognition sequence is catalyzed by both the M2S trimer (M.EcoKI) and the R2M2S pentamer (EcoKI), and it requires the co-factor SAM. Usually, methylation converts one adenine in each half-sequence to N6-methyladenine (m6A) (Table 1). Like other MTases (87–90), M.EcoKI flips the target base out of the DNA helix to carry out methyl transfer (91). M.EcoKI and related Type IA enzymes are maintenance MTases, with a preference for hemimethylated substrates in which only one of the two half-sequences is methylated (92). However, both the small Ral protein of phage λ and mutations in the N-terminus of the M subunit can turn M.EcoKI into a de novo MTase (93,94), the same activity as found for the Type IB enzyme EcoAI (46).
Enzyme structure
Atomic structures of Type I R–M enzymes have been difficult to obtain. Crystal structures of individual subunits have been solved, but not of complexes. S subunit structures were solved in 2005 (pdb:1YF2 and 1YDX) (95,96) and 2011 (pdb: 3OKG) (97), and M subunit structures in 2005 and 2007 (pdb:2AR0 and 2OKC; New York Center for Structural Genomics, and Joint Center for Structural Genomics, unpublished). These structures did not include DNA, but models of the M.EcoKI trimer with DNA (Figure 1) and with a DNA-mimic protein, Ocr, were reported in 2009 (40) based on EM single particle reconstructions (pdb:2Y7H and 2Y7C).
Figure 1.
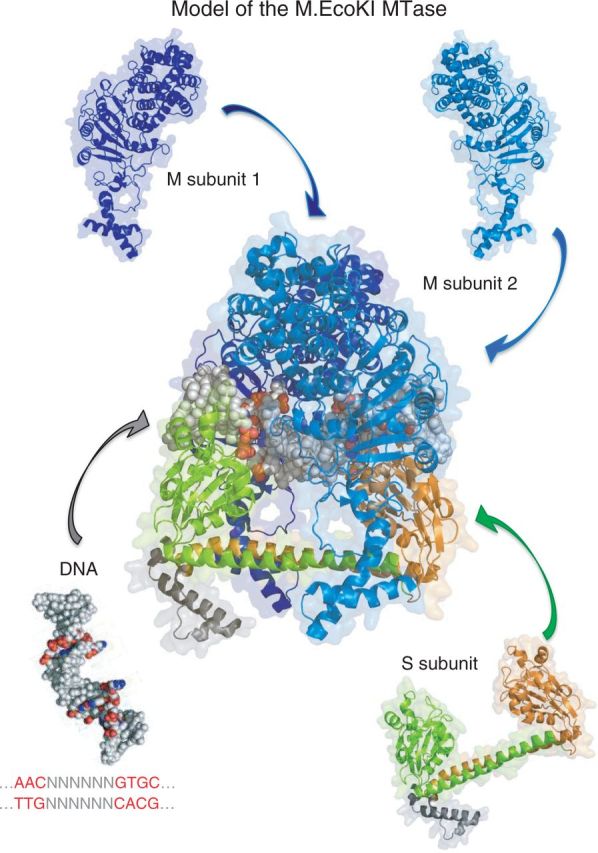
Model of the M.EcoKI MTase (pdb file 2Y7H). The S subunit is composed of two TRDs in inverted orientations. Each TRD comprises a globular DNA-binding domain and an alpha helical dimerization domain. The N-TRD (green) in this protein is specific for the sequence AAC (the 5′-half-sequence), and the C-terminal domain (orange) is specific for GCAC (the 3′ half-sequence). Zipper-like association of the helices separates the globular domains by a fixed distance and reverses the orientation of the C-TRD, resulting in the composite recognition sequence that is bipartite: AAC N6 GTGC. Each TRD also associates with one M subunit (identical, but shown here in different shades of blue for clarity) to form an M2S trimer. Neither S nor M subunits bind to DNA alone, but the trimer binds specifically at the recognition sequence and catalyzes methylation of one adenine in each half-sequence. Because the TRDs are inverted, the two M subunits have opposite orientations. Consequently, both strands of the recognition sequence become methylated, the ‘top’ strand of the 5′ half-sequence (Am6AC) and the ‘bottom’ strand of the 3′ half-sequence (GCm6AC). This enables the DNA of the host cell to be distinguished from infecting DNA during DNA replication.
The first crystal structure of a Type I R subunit (EcoR124) was published in 2009 [pdb: 2W00; (98,99)]. The catalytic site for DNA-cleavage in these proteins belongs to the ‘PD-(D/E)XK’ endonuclease superfamily, the same kind found in many Type II restriction endonucleases [(100–104); Figure 2]. The motor domain belongs to the RecA-like ATP-dependent family, a clade of the Additional Strand Conserved E family (107), of which the well-known AAA+ motor proteins are another clade (108). The location of the nuclease domain opposite the translocase domain suggests a coupling between DNA translocation and cleavage. The crystal structure of an N-terminal fragment of a putative Type I R subunit from Vibrio sp. has also been reported [pdb:3H1T; (109,110)]. It contains three globular domains with a nucleolytic core and the ATPase site close to the putative site for DNA-translocation.
Figure 2.
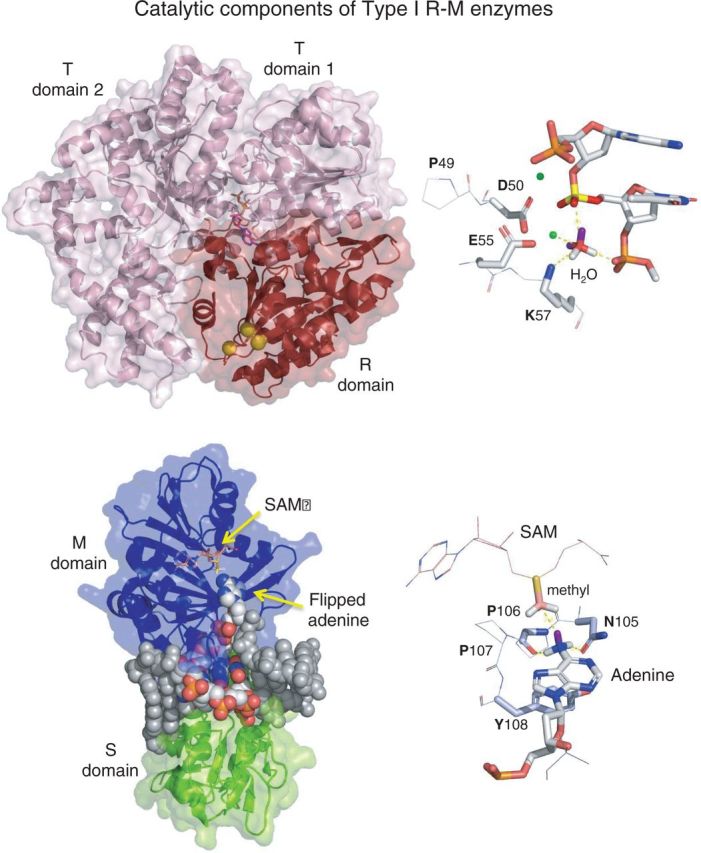
Catalytic components of Type I R–M enzymes. Upper figure, left: The R/T subunit of EcoR124I (pdb: 2W00). The R domain (red), responsible for DNA cleavage, comprises the N-terminal ∼260 aa. It contains the common PD-D/EXK endonuclease catalytic site, composed of D151, E165 and K167 (yellow spheres). Upper figure, right. The PD-D/EXK catalytic site of the Type II REase, MvaI (pdb:2OAA). D50 and E55 coordinate divalent metal ions (in this case two Ca2+ ions, shown as green spheres at reduced scale for clarity). The hydrolytic water molecule is oriented by interaction with a metal ion, the general base K57, and a phosphate oxygen from the adjacent base. These interactions position a lone pair electron orbital (purple sticks) of the water molecule for in-line nucleophilic attack on the phosphorus atom (bright yellow), initiating the DNA cleavage reaction. Catalysis occurs in the presence of Mg2+, but not in the presence of Ca2+; hence, this structure represents the pre-cleavage complex. Lower figure, left: The monomeric γ-MTase, M.TaqI, with bound DNA (pdb: 1G38). SAM was absent in this complex, which represents the pre-methylation complex. SAM has been added here through modeling by structural alignment with pdb:2ADM. Lower figure, right: The catalytic NPPY of M.TaqI is composed of N105, P106, P107 and Y108. When the target adenine is flipped into the catalytic site, the hydrogens of the 6-amino group form hydrogen bonds with the side chain amide carbonyl of N104 and the main chain carbonyl of P105. These lie below the plane of the base and likely induce the nitrogen to switch from the planar sp2 orbital configuration it normally possesses, to the tetrahedral sp3 configuration (105). In this latter configuration, the lone pair orbital of nitrogen (purple stick) is appropriately positioned for in-line nucleophilic attack on the carbon thiol (pink) of SAM, initiating the DNA methylation reaction (105,106).
Together, these structures provided a framework for how the complete pentameric enzyme assembles, and how the motor domains might translocate the DNA. Recently, the complete structure of both the EcoKI and EcoR124I enzymes was elucidated by EM single particle reconstruction studies.
CONTROL OF R–M ACTIVITY
Restriction alleviation
Type I R–M systems are suppressed following DNA damage by ultraviolet irradiation, 2-aminopurine and nalidixic acid, a phenomenon termed ‘restriction alleviation’ (RA) (111). Following the damage event, a cell population that normally restricts phage to an eop of ∼10−5, say, instead restricts to only ∼10−1. This mechanism is presumed to protect the cell while the damage is being repaired or during homologous recombination (112). During this period, unmodified sites can be created in the host chromosome, and RA prevents these from being restricted. Restriction (but not modification) is also suppressed following transfer of R–M genes to a new host; that is, expression of the new genes does not lead to cleavage of the unmodified host chromosome. Investigation of the dynamics of appearance of restriction activity in the transfer recipient revealed a long delay after entry of the EcoKI hsdR, M and S genes. This delay was genetically dependent on a host function, at first called HsdC (113,114). Further genetic investigation demonstrated that the delay mechanism was mediated by the ClpX chaperone and its proteolytic form, the ClpXP complex (115).
The two sorts of RA were unified in further experiments, which studied stability, by western blot, of components of the Type IA and Type IB R–M systems (115,116). Degradation occurs specifically when the R subunit is assembled into a translocation-competent complex. Mutations in any of the R subunit DEAD box motifs, which impair ATPase activity and translocation both in vivo and in vitro (117,118), relieve sensitivity of the R subunit to ClpXP (119). Mutations in the endonuclease motif, in contrast, do not relieve sensitivity to degradation (119).
The EcoR124I Type IC R–M system is insensitive to ClpXP-mediated control but also displays RA (120,121). RA could result from disassembly of the pentameric restriction complex (during endogenous RA when the complex will be attempting to translocate on the host chromosome) or inefficent assembly (during establishment of RM in a new host) for this plasmid-borne system. Still unresolved is the mechanism by which these enzymes distinguish between an invading unmodified DNA and an unmodified new or old host. A recent article showed that the R subunit of EcoKI (but not those of representative Type IB and IC enzymes) can be phosphorylated on threonine. This could play a role in localization, disassembly or proteolysis (122).
Antirestriction and antirestriction-modification
Antirestriction (antiR) and antirestriction–modification (antiR–M) systems have evolved in many MGEs such as phage, plasmids and transposons. Their activities increase the probability that the MGE will survive the cell’s R–M systems and initiate a successful infection. Phage T4 has the most numerous antiR and antiR–M mechanisms (26,27,123–125). Early work on antiR–M was carried out by the groups of Studier at NIH, Kruger in Berlin, Bickle in Basel (126–131) and by the groups of Belogurov and Zavil’gel’skii in Russia; reviewed in (22,26,27,132–134). The Wilkins laboratory in the UK solved the riddle of control of antiR encoded by the self-transmissible IncI plasmid: the ardA gene was transiently induced during conjugation by transcription from a single-stranded promoter (135–138). The evolutionary struggle between antiR–M and R–M has been likened to an ‘arms race’ with new mechanisms and counter-mechanisms developing continuously (22,26,27,132–134,138–140). We describe examples of these later in the text, including alteration of the methylation preference of M.EcoKI and the synthesis of antiR–M proteins: Ocr from phage T7 (141), ArdA from transposon Tn916 of Enterococcus faecalis (142), ArdB (143) from a pathogenicity island of E. coli CFT073, and KlcA [an ArdB homolog (144)] from plasmid pBP136 of Bordetella pertussis (145).
De novo methylation
M.EcoKI and related Type IA modification enzymes are ‘maintenance’ MTases that exhibit a strong preference for hemi-methylated recognition sites. This enables the MTase to be highly active on replicating DNA, but very slow to modify foreign unmodified DNA when it enters. This property can be foiled by antirestriction activities. Interaction with the phage lambda Ral protein converts M.EcoKI to a ‘de novo’ methyltransferase (93,146). Induction of ral (restriction alleviation) under the control of an inducible promoter enhanced survival of unmodified phage lambda 800-fold within 1 min. This fast reaction suggested direct interaction of the Ral protein with EcoKI perhaps inducing a conformational change to enhance methylation over restriction (93). Mutations in the hsdM gene can achieve the same result, thus effectively converting EcoKI from a maintenance to a de novo MTase in the absence of Ral (94). A model (147) was proposed to explain the differences in activities between wild-type and mutant enzymes based on biochemical and structural data (148).
AntiR and antiR–M proteins
The Ocr, ArdA and ArdB proteins vary in their occurrence in nature (149–151). Ocr seems to be confined to phage, particularly T7 and its relatives (150–153). In T7, it is encoded by gene 0.3, the first gene to enter the E. coli host on infection. It is synthesized in large amounts for the first 2 min of infection, during which insertion of further T7 DNA halts (154–156). Sufficient Ocr protein is produced in this brief period to completely inhibit the host’s Type I enzymes, following which Ocr synthesis stops and the rest of the phage genome enters the cell in safety. (In vitro, Ocr also binds to E. coli RNA polymerase (157); whether this is relevant in vivo has not been investigated.)
ArdA and ArdB proteins are generally encoded by conjugative plasmids and transposons, and they are also often the first genes to enter the recipient cell (149). As the entering DNA is single stranded and resistant to restriction, an unusual promoter is formed: the DNA of plasmid ColIb-P9 forms a double-stranded hairpin to allow transcription of ardA (135–137). Production of ArdA or ArdB leads to rapid inhibition of the host’s Type I enzymes. This novel transcription method may well be more common and deserves further research. Importantly, both ardA and ardB genes are widespread (as determined by genomic sequencing projects) and frequently accompany antibiotic-resistance genes. Their presence might have a considerable impact on the rate of spread of resistance in bacterial populations.
The structure of Ocr
Ocr is a striking example of DNA mimicry by a protein. It is a homodimer of two 116-aa subunits (127) that interface by a complementary fit of hydrophobic surfaces [(141,158); pdb:1S7Z and 2Y7C; Figure 3]. The dimer is banana shaped with a length of ∼7.5 nm and a general width of 2 nm thickening to 2.5 nm at the dimer interface (141,159,160). Each monomer contains several well-packed alpha helices, a long loop and flexible N- and C-termini. The narrowness of the structure means that it has a minimal hydrophobic core, which contains a considerable number of aromatic aa, perhaps akin to the aromatic core of another DNA mimic, Qnr (161). Despite this small core, Ocr is stable to heat and chemical denaturation (160). Arrayed on the surface of each monomer are 34 negatively charged aa and only 6 positive aa (∼12 of each would be expected for a typical globular protein of this size), although not all of these are required for activity (162–164). The negative surface charges have roughly the same spacing as the phosphate groups on 24 bp of B-form DNA with a bend in the center, which explains its affinity for Type I enzymes (165).
Figure 3.
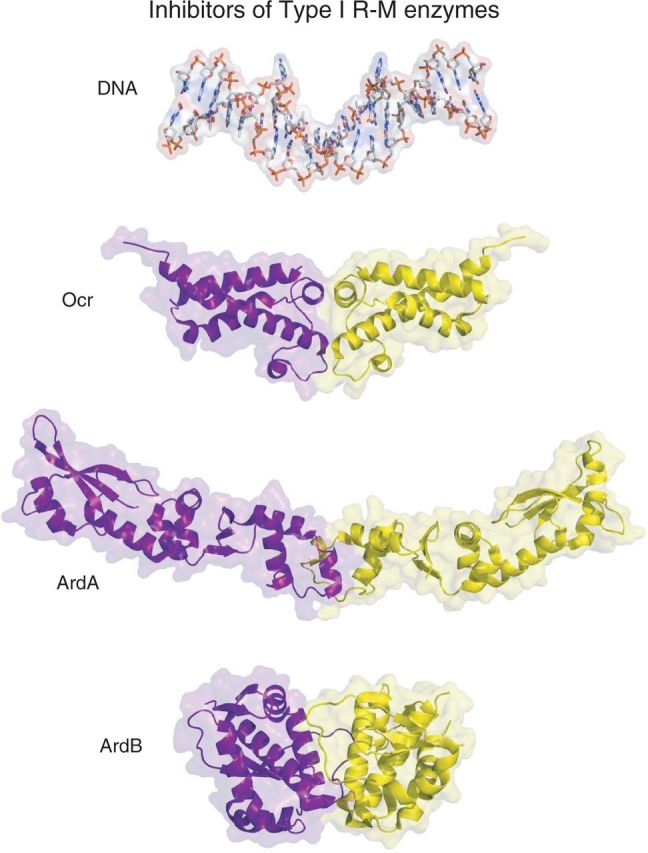
Protein inhibitors of Type I R–M enzymes. Top panel: DNA model (hydrogen atoms omitted) from pdb:2Y7H displayed on the same scale as the proteins for structural comparisons. Panel b: Ocr (pdb:1S7Z and 2Y7C) from bacteriophage T7; panel c: ArdA (pdb:2W82) from Tn916 of E. faecalis (117); panel d: ArdB (pdb:2WJ9) from a pathogenicity island of E. coli CFT073. All three proteins are homodimeric. Their subunits are identical, but are displayed here in different colors.
The structure of ArdA
ArdA also mimics B-form DNA in geometry and electrostatics (166). The crystal structure shows ArdA to be a homodimer, but it can exist as both monomer and dimer in solution [(142); pdb:2W82; Figure 3]. The ArdA encoded by Tn916 has 166 aa per subunit; the dimerization interface is smaller than in Ocr. The dimer is a highly elongated bent cylinder ∼15 nm long and 2 nm in diameter along its entire length. The narrowness of the protein again means that it has a minimal hydrophobic core, but unlike Ocr, ArdA is not resistant to denaturation (142). The fold of ArdA is completely different to that of Ocr with each ArdA monomer comprising three small loosely packed domains. The domain folds have been found in other protein structures and comprise a mix of alpha helices, beta strands and loops. The surface of each monomer is covered with numerous carboxyl groups such that the dimer mimics ∼42 bp of bent B-form DNA. The domain organization and low stability of ArdA imply structural flexibility and suggest that the protein might mold itself to the contorted, S-shaped, DNA-binding groove of Type I enzymes (41), with different domains interacting with the different R, M and S subunits (40).
The structure of ArdB
The structures of two members of the ArdB family have been solved by crystallography [ArdB from E. coli CFT073 (143); pdb:2WJ9; Figure 3] and nuclear magnetic resonance spectroscopy [KlcA from B.pertussis (145); pdb:2KMG]. The proteins are close homologs with >30% aa sequence identity. Klc genes form part of the kor operon involved in the regulatory network of these promiscuous plasmids (144). The ArdB structures are different from those of Ocr and ArdA, being small, rather normal-looking, globular proteins although with a novel polypeptide fold. They are neither elongated nor possess significant charged patches so are unlikely to cause antiR via DNA mimicry. KlcA is a monomer in solution, but the ArdB structure is a dimer with two intermolecular disulphide bridges and a well-packed hydrophobic interface. As the cysteine residues are not conserved, dimer-formation may not be required for activity and may not even arise in the reducing environment of the bacterial cytoplasm. The novel fold of ArdB shows two alpha helices projecting out from the protein with a smaller third helix nearby completing a domain. This domain is then ‘cupped’ or held in the palm of a hand by another domain comprised of helices, beta strands and loops. The absence of conserved charged patches or a narrow structure to allow association with a DNA-binding site suggests a different mechanism of inhibition from ArdA and Ocr, although it is possible that the ArdB structures could be different when in the cytoplasm.
Effect on restriction and modification
The effectiveness of Ocr, ArdA and ArdB in inhibiting restriction and modification by Type I enzymes has usually been investigated in vivo by comparing the eop of phage on restriction-proficient hosts with or without antiR–M genes (139,141,142,145,158,167). Active antiR enhances the number of recovered phage, and these in turn can then be tested for modification by their eop on restriction-proficient and -deficient strains. Quantitative comparisons in these experiments are difficult due to varying experimental details, but the in vivo titration assay, in which an inducible promoter and antiR–M expression is controlled by inducer concentration, recently introduced by the group of Zavil’gel’skii, suggests a good way forward for quantitative comparison of antiR–M systems (139,158,167).
The eop assays show that Ocr effectively blocks both restriction and modification by all Type I families. This is a direct consequence of the extremely strong binding of Ocr to the DNA-binding groove in the MTase core (170). ArdA and ArdB block restriction in all Type I families but are much less effective at blocking modification (139,141,145,158,167). This minimal anti-M activity is due to the binding of ArdA to the MTase core being of similar or weaker strength than DNA binding to the core. Although the binding is weak, it is sufficient to prevent restriction. ArdB also shows little or no anti-M effect in vivo, and no interaction has been observed in vitro between ArdB and the MTase core. Furthermore, although ArdB causes antiR in vivo, no effect could be demonstrated in vitro on restriction. Therefore, the mechanism of antiR used by ArdB is indirect rather than the binding mechanisms used by Ocr and ArdA. It would be useful to define an active core for ArdB activity by constructing truncated variants of the protein.
Our understanding of antiR–M is still in its infancy. Aside from the three systems described earlier in the text, few others have been studied beyond their initial discovery. Given their synergistic role with R–M systems in regulating horizontal gene transfer and the ‘resistome’ (171,172), this deficiency in our knowledge needs to be addressed.
EcoprrI, an apoptosis enzyme
An especially elaborate example of defense and counter-defense concerns EcoprrI. The prrABD genes encode a conventional Type IC R–M system (54). The prrC gene, embedded in this operon, encodes a latent anticodon nuclease (ACN) that sacrifices the host on phage infection. PrrC is normally sequestered within the Type I enzyme complex, but it is released to act when phage T4, which is completely resistant to Type I REases, infects the host. Lethal damage is inflicted on the infected cell and expression of phage proteins is inhibited, by cutting the anticodon loop of tRNAlys. T4 counters the DNA/RNA restriction activity of EcoprrABCD with three dedicated T4 proteins (173), T4 polynucleotide kinase and RNA ligase that repair the cleaved tRNA, and Stp (174) that inhibits the Type I activity but liberates the ACN in the process [see (175) for review].
Interestingly, ACN appear to be involved in stress responses in both prokaryotic and eukaryotic cells: e.g. DNA repair defects sensitize yeast to ACN activity (176), and angiogenin induces tRNA cleavage in mouse cells (177). Such an ‘RNA-based innate immune system that distinguishes self from non-self’ (178) may also be considered programmed cell death after infection (179).
TYPE I DNA SEQUENCE SPECIFICITY
Characterization
Few Type I R–M enzymes were known to exist before the advent of genome sequencing, and these were confined to E. coli and close bacterial relatives. Even today, the number of characterized Type I enzymes is small compared with Type II enzymes (Table 1). This disparity reflects the difficulty of identifying Type I enzymes and determining their recognition sequences. R–M systems of all types were studied in the same way at first, by their effects on plating efficiencies of phages. As the enzymology of restriction and modification began to be explored in the 1970s, DNA-cleavage assays of bacterial cell extracts largely replaced phage-plating assays. This led to the discovery of many new Type II REases, as these cleave DNA at fixed positions and produce discreet DNA fragment patterns, but it had little effect on the discovery of Type I enzymes (19). Type I REases cleave DNA at variable positions with respect to their recognition sites, and so they do not produce signature fragment patterns. Consequently, they cannot be identified easily in cell extracts, and their recognition sites are far more difficult to map.
By 1980, only two Type I enzymes had been discovered—EcoBI (42–44) and EcoKI (45)—compared with 56 different Type II REases (180). Eight more Type I specificities were discovered in the 1980s [versus over 100 new Type II REases (181)] and another two in the 1990s (Table 1)—together with some remarkable variant specificities that had arisen by chance or by design (Table 2). Fourteen more Type I specificities were discovered early in the following decade, due mainly to work in Junichi Ryu’s laboratory based on the restriction of sets of sequenced plasmids during transformation (58–61), and 11 more have been added very recently from work elsewhere (62,63,65,192). The original systems are well-characterized for the most part, but little is known yet about the newer ones. Characterizing Type I enzymes would likely have remained difficult but for a development that is poised to revolutionize this field. Next-generation sequencing of genomic DNA provides a rapid way of determining sequence specificity based on the sites of methylation rather than the sites of cleavage (61,62).
Table 2.
Derivative Type I R-M systems with altered specificities
| Enzyme | Recognition sequence | S subunit organization | Reference |
|---|---|---|---|
| StySB | GAG N6 RTAYG | [GAG]∼[CRTAY] | |
| StySP | AAC N6 GTRC | [AAC]∼[GYAC] | |
| StySQ | AAC N6 RTAYG | [AAC]∼[CRTAY] | (48,180–182) |
| StySJ | GAG N6 GTRC | [GAG]∼[GYAC] | (185) |
| EcoAI | GAG N7 GTCA | [GAG]∼[TGAC] | |
| StySKI | CGAT N7 GTTA | [CGAT]∼[TAAC] | |
| SKI/AI | CGAT N7 GTCA | [CGAT]∼[TGAC] | (55) |
| EcoR124I | GAA N6 RTCG | [GAA]∼[CGAY] | |
| EcoDXXI | TCA N7 RTTC | [TCA]∼∼[GAAY] | |
| EcoDR2 | TCA N6 RTCG | [TCA]∼[CGAY] | (186) |
| EcoRD2 | GAA N6 RTTC | [GAA]∼[GAAY] | (186) |
| EcoDR3 | TCA N7 RTCG | [TCA]∼∼[CGAY] | (186) |
| EcoRD3 | GAA N7 RTTC | [GAA]∼∼[GAAY] | (186) |
| EcoR124I | GAA N6 RTCG | [GAA]∼[CGAY] | |
| EcoR124II | GAA N7 RTCG | [GAA]∼∼[CGAY] | (51,187) |
| EcoR124I | GAA N6 RTCG | [GAA]∼[CGAY] | |
| EcoR124IΔ50 | GAA N7 TTC | 2[GAA] | (188) |
| EcoDXXI | TCA N7 RTTC | [TCA]∼∼[GAAY] | |
| EcoDXXIΔC | TCA N8 TGA | 2[TCA] | (189) |
| EcoDXXIΔN | GAAYN5 RTTC | 2[GAAY] | (190) |
| EcoAI | GAG N7 GTCA | [GAG]∼[TGAC] | |
| EcoAI cp380 | GAG N7 GTCA | [TGAC]∼[GAG] | (191) |
Column 1: Enzymes in regular type are parental; these are also listed in Table 1. Enzymes in bold are derivatives. Column 2: The recognition sequence is printed in the orientation for which the 5′ half-sequence is specified by the N-TRD, and the 3′ half-sequence is specified by the C-TRD. Numerals indicate the number of non-specific bases between the two half-sites. Bold type indicates the bases known or inferred to be methylated. Column 3: Inferred compositions of the S subunits. The specificities of the individual TRDs are given in the order in which they occur in the S subunit, e.g. ‘[GAG]∼[CRTAY]’, indicates that the N-TRD recognizes GAG, and the C-TRD recognizes CRTAY. ‘∼’ indicates that the TRDs are joined into a single protein chain. ‘∼∼’ indicates that this linkage contains additional aa that increase their separation. S subunits that are homodimers of a single TRD are depicted as ‘2[GAA]’, for example, meaning that it comprises two molecules of a GAA-specific TRD. Because the orientation of the second TRD in S subunits is inverted with respect to the first TRD, the 3′ half-sequence is always the ‘complement’ of the specificity of that TRD. Column 4: Publication reporting the derivative.
Single-Molecule Real-Time (SMRT) DNA-sequencing instruments of the kind manufactured by Pacific Biosciences not only distinguish the bases from one another but also distinguish their modification states, differentiating m6A from adenine, and m4C, hm5C and to some extent m5C, from cytosine (64). As modification always takes place within the recognition sequence in all types of R–M systems, identifying the sequence contexts in genomic DNA that contain modified bases reveals the methylation profile of the organism—its ‘methylome’—and the specificities of all of its active Mtases (193). Bioinformatics analysis of the genome reveals the MTases that are present and their likely characteristics, and then it becomes a matter of correctly matching the MTases to the DNA sequences methylated.
Methylation data have begun to emerge from SMRT sequencing projects and are growing rapidly (62,63,168,169,192). New R–M specificities of various types have already been uncovered, and REBASE has added a new section to track the accumulating information (http://rebase.neb.com/rebase/rebase.charts.html>Specialized Information> PacBio). Because multiple R–M systems as well as solitary MTases are often present in each genome, sub-cloning and methylome re-analysis will be needed in many instances to confirm which specificity belongs to which system. In addition, for Type I enzymes, there is the added task of establishing which TRD of the two present in each S subunit recognizes which half of its recognition sequence. Amino acid sequence comparisons combined with experiments of the kinds described later in the text suggest ways in which these latter assignments can be made. Structural similarities between crystallized Type I S and M subunits (40,95,96), γ-class adenine MTases (105), and Type IIG RM enzymes (194,195), coupled with our growing understanding of how these proteins recognize DNA specifically (196), make it plausible that by the end of this decade, we will be able to ‘decode’ the specificities of many Type I enzymes by simply inspecting the aa sequences of their S subunits.
Type I S subunits
The S subunits of Type I enzymes are responsible for DNA sequence recognition. They have a duplicated organization comprising two TRDs in tandem (Figure 4). Each TRD specifies one half of the bi-partite recognition sequence. The TRDs consist of a globular specificity domain (S) and an alpha-helical, ‘dimerization’ domain (D). The TRDs occur as direct repeats in the linear aa sequence, but they assume inverted orientations in the folded protein as the helices associate (‘dimerize’) in antiparallel to make a coiled-coil ‘leucine-zipper’ (Figure 5). In simplest form, the N-terminal TRD (N-TRD) of the S subunit has the composition S1-D1, the C-terminal TRD (C-TRD) has the composition S2-D2, and the entire S subunit has the composition S1-D1∼S2-D2. In some Type I families, circular permutation of this organization alters the positions of these domains.
Figure 4.
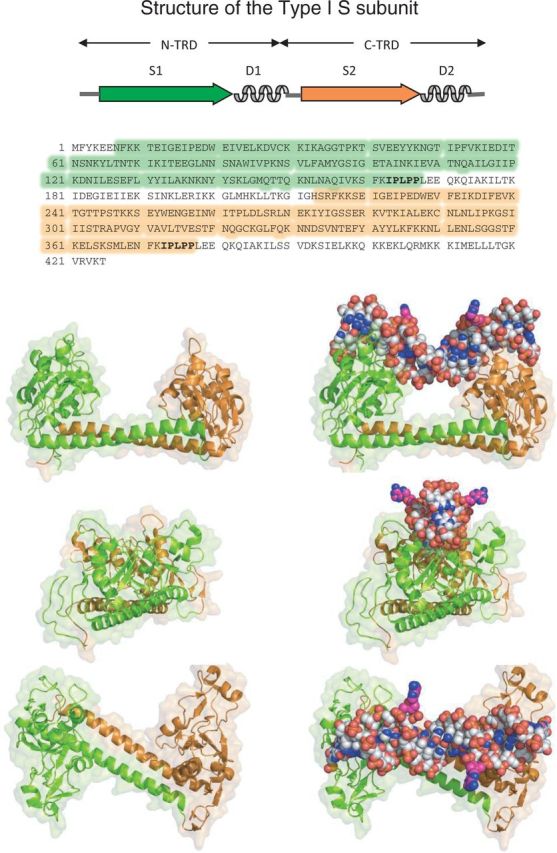
Structure of the Type I S subunit (pdb:1YF2). The recognition sequence of this protein, S-MjaXI, from Methanocaldococcus (formerly Methanococcus) jannaschii is not known. It is closely related to the EcoKI-family (Type IA) of enzymes depicted in Figure 1. The upper diagram shows the domain organization of the protein; arrows represent DNA-binding domains, and curly lines represent dimerization alpha helices. The aa sequence of the protein is shown below, with the domains in corresponding colors. Below this are three views of the structure, from three perpendicular directions, ‘sideways’, ‘end-on’, and ‘above’. The panels on the left depict the protein; those on the right depict the protein with modeled DNA positioned approximately as it is bound. The DNA was taken from pdb:2Y7H and transferred by structural alignment of the S subunits.
Figure 5.

Dimerization helices of Type I S subunits. Upper diagram: aa sequence alignment of the dimerization helices of S-MjaXI (pdb:1YF2; D1 and D2 in Figure 3) show that they are similar but not identical (top). The proline-rich motif that precedes each helix (IPLPP) is a hallmark of Type I S subunits and likely plays a structural role establishing the correct architectural relationship between the S and D domains. The helices interact in opposite orientations to form an antiparallel coiled-coil (bottom). Adjacent pairs of aa (red and blue) form the dimerization interface and occur with the 4-3-4-3 … spacing characteristic of leucine zippers. Middle diagram: Exposed dimerization surfaces of two helices. Side chains of red aa point ‘up’ toward the bound DNA, and those of blue aa point ‘down’. To form the coiled coil, D1 must be rotated 180 degrees around the vertical axis and docked against the surface of D2 shown. Lower diagram: Within the coiled coil, red aa from one helix interdigitate with red aa from the other helix, forming the ‘upper’ surface of the coiled coil, the surface closest to the bound DNA (left). And blue aa from one helix inter-digitate with blue aa from the other helix to form the ‘lower’ surface on the other side (red). Within the coiled-coiled, red side chains from one helix stack on blue side chains from the other helix in an alternating pattern. At this interface, the helices have complimentary topologies such that a ridge or bump in one is accommodated by a valley or depression in the other, resulting in a close hydrophobic fit.
Specificity changes
TRD exchanges
The structure of S subunits enables the specificities of Type I R-M systems to change spontaneously and robustly. The first observation of such a change was reported in 1976. Using phage transduction to transfer chromosomal DNA from a strain of Salmonella that expressed the Type I SP system (now StySPI) to a derivative that expressed the SB system (now StyLTIII), a transductant was recovered that expressed a novel specificity, ‘SQ’ (182). With considerable insight, the authors attributed this new system to a genetic cross-over in the middle of the parental hsdS genes.
Heteroduplex analysis of the hsdS genes showed the same organization as other members of the EcoKI family (197), consisting of an N-terminal variable region (=S1), a central conserved region (=D1), a second variable region (=S2) and another conserved region (=D2) at the C-terminus. The SQ hsdS gene was a hybrid consisting of the N-TRD of the SP gene joined to the C-TRD of the SB gene (183,184). Independently, the recognition sequence of StySQ was determined and also found to be a hybrid comprising the 5′ half-site of SP and the 3′ half-site of SB (Table 2) (48,198).
These experiments suggested strongly that specificity was determined by the variable regions of the S subunits, and that each specified one-half of the recognition sequence independently of the other. The finding that discrete protein domains recognized discrete DNA sequences was a revelation at the time and marked a significant advance in our understanding of how proteins interact with DNA. To complete matters, the reciprocal hybrid was intentionally created by genetic recombination and was shown to have the reciprocal specificity. In this system, ‘StySJ’, the N-TRD came from SB, and the C-TRD from SP, and as predicted, its specificity combined the 5′ half-site of SB with the 3′ half-site of SP (Table 2) (185). The nucleotide sequences of the parental and hybrid S genes confirmed their structures and showed that crossing-over had occurred in a 130-nt stretch of near sequence identity that we now know encodes the first of the two dimerization helices (184,185).
StySB and StySP belong to the Type IA family, but domain shuffling is not confined to enzymes of this group. The S subunits of other Type I families also have a tandemly repeated organization of alternating specificity and dimerization domains and are similarly suited to exchange these by recombination. A hybrid S subunit constructed in vitro by joining the N-TRD of StySKI to the C-TRD of EcoAI—both members of the Type IB family—displayed a hybrid specificity and enabled the specificities of the individual domains to be assigned (55). Likewise, reciprocal hybrids between the Type IC enzymes EcoR124I and EcoDXXI were constructed and shown to possess the expected hybrid specificities (Table 2) (186). There are no reports of hybrids between members of different Type I families, but there are indications that this occurs in nature, such as the N-TRDs of StyLTIII and EcoEI (55).
Gap length changes
Enzymes of the Type IC family also change specificity by unequal crossing-over at a short sequence-repeat in D1, the first of two helices that connect the DNA-binding domains. The length of these helices determines the separation between the two specificity domains (Figure 4), and this in turn determines the gap in the recognition sequence—or more precisely, the separation between the two bases that becomes methylated. Studies in several laboratories showed that EcoR124I and a variant, EcoR124II, recognize the same DNA sequence half-sites, but differ in how far they are apart: EcoR124I is specific for a gap of 6 bp (GAA N6 RTCG), and EcoR124II is specific for a gap of 7 bp (GAA N7 RTCG) (187,199–201). This change was found to depend on a 12 bp sequence in conserved domain D1 that was repeated twice in EcoR124I but three times in EcoR124II (51,187,201). The repeat codes for Thr-Ala-Glu-Leu (TAEL)—just over one alpha helical turn—and the extra repeat in EcoR124II moves the two S domains 0.34 nm further apart and rotates them by 36 degrees, similar to the offset between adjacent bp in duplex DNA (187). Two other enzymes, EcoDXXI and EcoPrrI, also contain three TAEL repeats in D1, and this appears to be the optimum number based on aa sequence alignments (190). During the construction of hybrids between EcoR124I and EcoDXXI, the number of TAEL repeats was also changed, and the hybrid recognition sequences were found to change in precisely the manner predicted (Table 2) (186).
The crystal structures of putative S subunits show that the alpha helices encoded by the dimerization domains interconnect to form an antiparallel coiled coil (95–97). Amino acid side chains down their lengths interlock like tines of a zipper and form a hydrophobic core that holds the two helices together (Figure 5). Addition or loss of a turn in one helix but not in the other, due to a change in the number of TAEL repeats, must change the register between of the aa in the zipper because, if it did not, the gap would not change. How this perturbation is accommodated is not clear. Introducing a compensatory change in the other helix to restore the original register would be worth investigating.
Homodimeric S subunits
In the changes described earlier in the text, the order of the specificity domains remained the same, one N-TRD being swapped for another N-TRD, and so on. Experiments in the 1990’s showed that the order makes little biological difference because, as they adopt inverse orientations in the folded protein, the S subunit is rotationally symmetric. This is due to the antiparallel nature of the coiled coil: it both separates the TRDs and inverts their orientations. In hindsight, a structure with rotational symmetry was only to be expected: the S subunit must orient the two M subunits to methylate different strands of the recognition sequence so that both can become methylated. As the strands of DNA have opposite orientations, the M subunits must also have opposite orientations, and because they derive their orientations from their association with the S domains, these must have opposite orientations as well. That this was indeed the case emerged from experiments with EcoR124I.
A mutant of EcoR124I was isolated, HsdS(Δ50), that lacked ∼160 aa from the C-terminus of the S subunit, including most of the C-TRD. This mutant was active, and it was found to have a new specificity, GAA N7 TTC, that was a palindrome of the sequence recognized by the N-TRD (Table 2) (188). The fact that this sequence was bipartite, like regular Type I enzymes, indicated that a second molecule of the N-TRD was substituting for the missing C-TRD, and that the two TRDs were therefore interchangeable. It also indicated that the TRDs have inverted orientations, and that they need not be joined in a continuous polypeptide chain to function together. Perhaps more surprising is that the helices forming the coiled coil can also substitute for each other. D1 forms an antiparallel zipper with D2 in the normal ‘heterodimeric’ S subunit. In HsdS(Δ50), D1 evidently zippers with itself, and does so in a conservative way that preserves the parental 7-nt separation between methylated adenines. Considerable sequence similarity exists between the dimerization domains of Type IC S subunits, perhaps explaining why one helix can satisfactorily replace the other.
A comparable truncation arose by insertion of transposon Tn5 into the C-TRD of the EcoDXXI S subunit, and likewise resulted in a homodimer specific for a palindrome of the N-TRD specificity (Table 2) (189). The separation between the methylated bases—8 nt in this case due to the presence of a third TAEL repeat—was also preserved. To test whether the C-TRD could substitute for the N-TRD in the reverse scenario, the N-terminus of the EcoDXXI S subunit was intentionally deleted. This mutant behaved in the same way as the other homodimers and recognized an interrupted palindrome of the C-TRD specificity (190). When the N- and C-terminal-truncated S genes were co-expressed to create a mixture, wild-type EcoDXXI activity was regenerated, confirming further that the two TRDs need not be joined into a single protein chain to work together. That they are joined in all of the characterized Type I systems and most of the putative ones, too, suggests that fusion nevertheless confers a selective advantage. Fusion fixes the TRD stoichiometry to 1:1 and limits the combinatorial pairing possibilities when multiple TRDs are present in the same cell.
One natural Type I system has been found that has a palindromic specificity: NgoAV (Table 1). The S gene of this system is fragmented into several open reading frames (ORFs). The first ORF encodes a complete N-TRD similar in size and composition to the EcoR124I(Δ50) and EcoDXXI truncations described earlier in the text, and it evidently behaves in the same way. The C-terminal domain is incomplete and inactive (57). EcoR124I, EcoDXXI and NgoAV all belong to the Type IC family. No equivalent homodimers have been reported for enzymes from other Type I families. The D1 and D2 helical domains of Type IA S subunits have little sequence similarity, suggesting they might not be able to substitute for each other as can those of the Type IC S subunits.
Circular permutation of S subunits
Within Type I families, the S subunits have a consistent organization, but between families, they are circular permutations of one another. For Type IA S subunits, the domain order is S1-D1-S2-D2, whereas for Type IB S subunits, it is D2-S1-D1-S2. The Type IC S subunits are in-between, beginning and ending within D2. These forms differ in where, within a circle of alternating specificity and dimerization domains, the N- and C-termini occur. That these forms are structurally equivalent was demonstrated by circularly permuting the EcoAI S subunit, a Type IB family member (191). In one permutation, most of the N-terminal conserved domain was moved to the C-terminus, mimicking the organization of Type IC S subunits. This construct, cp91, was active in both methylation and restriction. From the equivalent position in the central conserved region, a second permutation was constructed, cp380, in which the entire N-terminal half of the protein was switched to the C-terminus, reversing the order of the TRDs. This construct, a Type IC mimic in reverse, was active in only methylation but, tellingly, it displayed the same EcoAI specificity as its parent, confirming the inverted orientations of the two domains that make up the S subunit. Two other permutations, one a mimic of Type IA S subunits, the other a Type IB mimic but reversed, proved to be inactive (191). Evidently, the permuted forms are not necessarily functionally equivalent. The S domain order seems not to matter internally, but the positions of the N- and C-termini do matter, perhaps affecting proper folding or interaction with the M and R subunits or resistance to proteases (191).
Indications that S domains switch order in nature can be inferred from aa sequence comparisons. The first specificity domain of StySKI (S1), for example, specifies CGAT. A similar domain occupies the second position (S2) in EcoR124I where it specifies a similar sequence CGAY (55). Other possible examples can be found among the uncharacterized systems in GenBank and REBASE (202).
Recognition sequence orientations
Most of the natural and derivative Type I specificities are asymmetric, reflecting the ‘heterodimeric’ composition of the S subunit. Because of this asymmetry, Type I recognition sequences can be written in two ways depending on which strand is referenced. EcoKI, for example, can be written as ‘AAC N6 GTGC’ or as ‘GCAC N6 GTT’. Ideally, these sequences should all be oriented in the same, biologically meaningful, way where the 5′ half-site is specified by the N-TRD of the S subunit, and the 3′-half site is specified by the C-TRD. However, deciding which strand should be used for the sequence in any instance requires first knowing the specificities of those TRDs; without this information, the orientation is arbitrary. Nonetheless, because the TRDs have inverted orientations in the S subunit, the specificity of the C-TRD is not the sequence of the second half-site of the recognition sequence, but rather its complement. This becomes important when comparing the specificities of TRDs that occupy different positions, such as the N-TRD of StySKI and the C-TRD of EcoR124I, referred to earlier in the text (55).
RELATIVES OF TYPE I SYSTEMS
Type I systems are large complex R–M enzymes, which at first encounter can be puzzling. Why three subunits? Why is the S subunit internally duplicated while the other subunits are not? Why is the stoichiometry 1S:2M:2R? This organization begins to make sense when Type I enzymes are viewed from the perspective of variations on a theme, or more accurately variations on two themes, one being the inviolable necessity to protectively modify both DNA strands of the recognition sequence, and the other being the benefit derived from coupling R and M activities to the same sequence-specificity determinant. Type I enzymes represent only one of many variations on these themes, albeit a prominent one.
Classes of MTases
Three major classes of amino-MTases occur in prokaryotes, termed alpha, beta and gamma (203,204). Members of all three classes catalyze the transfer of a methyl group from SAM to the exocyclic amino group of adenine, forming m6A. Many members of the α and β classes methylate the exocyclic amino group of cytosine instead of adenine, to form m4C. MTases of the γ class methylate mainly adenine, and only rarely [e.g. the unusual M.NgoMXV and its homologs (205,206)] methylate cytosine instead. A further major class of MTases occurs in both prokaryotes and eukaryotes—the m5C-MTases. These exclusively methylate carbon-5 of the cytosine ring to form 5-methylcytosine (m5C), and they are catalytically distinct from the amino-MTases. Bioinformatics analysis suggests that additional kinds of amino-MTases might exist, representing minor classes (204). The m4C-specific MTase M.MwoI, for example, has been proposed to be a member of the delta class based on differences with M.SfiI (207), which is a β-class m4C-MTase, and M.TvoORF1413P has been proposed to be member of the zeta group (208).
Gamma-class MTases
Gamma-class MTases are the most diverse of the three major amino-MTase classes. They comprise a 250–450-aa catalytic domain and a sequence-specificity domain that is either fused to form the C-terminus of a single protein (M∼S) or is present as a separate subunit (M+S). A curious mechanistic difference distinguishes these groups in that the γ-MTases extract the base to be methylated from one DNA strand, whereas the α− and β-MTases extract it from the other strand. This difference can be seen by comparing the DNA co-crystal structure of the γ-MTase M.TaqI (pdb:1G38) (105), for example, with that of the α-MTase T4Dam (pdb:1YFL) (209). Type I enzymes belong to the γ-class, and a consistent strand-specificity is evident in their recognition sequences. Invariably, the base that becomes methylated in the 5′ half-sequence is located in the ‘top’ strand, and the base that becomes methylated in the 3′ half-sequence is located in the ‘bottom’ strand.
In aa sequence alignments of γ-MTases, two motifs stand out: motif I, which forms the SAM binding site, and motif IV, which is typically Asn-Pro-Pro-aromatic [i.e. NPP(Y/F/W)] and forms the catalytic site for methyl transfer. The four catalytic aa are located in a slot in the surface of the protein into which the target base flips before methyl transfer. The aromatic aa stacks with the flipped base, compensating for the loss of DNA base stacking (210), and the exocyclic amino group forms two hydrogen bonds with the protein, compensating for the loss of the Watson–Crick base-pairing hydrogen bonds (105,106) (Figure 2). Methyl transfer occurs directly, without formation of a covalent protein–DNA intermediate such as occurs in the m5C-MTases.
Gamma-class MTases form the M2S core of Type I R–M systems, and also the MTase components of many Type II R–M systems. Consequently, they represent an evolutionary bridge between these two types of R–M systems (Figure 6). We discuss later in the text some of the varied organizations in which the γ-MTases occur.
Figure 6.

Organizations of gamma-class modification MTases. γ-MTases catalyze transfer of a methyl group from SAM to the exocyclic amino group of adenine (or in rare cases, cytosine) at specific target sequences in duplex DNA. Their organizations vary depending on whether they methylate both DNA strands, or only one, and whether they cleave the DNA instead if the sequence is completely unmodified. At minimum, γ-MTases comprise an M component for base flipping and methyl transfer (blue oblongs), and an S component for sequence-recognition (green and orange ovals). These can be separate subunits or discrete domains. S components can function individually, or as inverted pairs that associate through antiparallel dimerization helices. The members of such pairs can be identical, in which case the structure is homodimeric, or they can differ, in which case the structure is functionally heterodimeric albeit usually connected into a single protein chain. Type I and Type IIG R–M enzymes are γ-MTases that also restrict DNA. For Type I enzymes, a supplementary R/T subunit (large red ovals) catalyzes endonuclease and DNA-translocase activities. For Type IIG enzymes, an R domain (red triangle) is permanently present at the N-terminus. The latter combine with S components in configurations that mirror those of the monofunctional γ-MTases. The figure is not exhaustive; it depicts only the commonest variants of γ-MTases but reveals a close evolutionary connection between Type I R–M systems and certain Type II R–M systems. An example of each organization is given in parentheses.
Monomeric γ-MTases
M.TaqI is a typical γ-MTase. It is a simple monomer of ∼420 aa comprising an N-terminal catalytic domain (245 aa) connected to a C-terminal specificity domain (165 aa) by a 10-aa linker (Figure 2). M.TaqI is specific for the sequence TCGA. As this sequence is symmetric, M.TaqI can bind to it in either orientation and methylate both of the DNA strands, first one and then the other. This enables M.TaqI to serve as the sole modification component of the Type II TaqI R–M system. M.SalI, M.PstI, M.HincII and many other γ-MTases function in the same way in their respective Type II systems (Table 3). With the recent, and unpublished, exception of MmeI, M.TaqI is the only γ-MTase whose crystal structure has been solved bound to DNA (105,211,212).
Table 3.
Varied organizations of γ-MTases found in Type II R–M systems
| Enzyme | Recognition sequence | Enzyme composition |
|---|---|---|
| M.TaqI | TCGA | γM∼[TCGA] |
| M.SalI | GTCGAC | γM∼[GTCGAC] |
| M.HincII | GTYRAC | γM∼[GTYRAC] |
| M.PstI | CTGCAG | γM∼[CTGCAG] |
| M.Eco57I | CTGAAG | γM∼[CTNNAG] |
| M1.Eco31I | GGTCTC | γM∼[GAGACC] |
| M.Esp3I | CGTCTC | γM∼[GAGACG]∼m5C-MTase |
| M.BseRI | GAGGAG | m4C-MTase∼γM∼[GAGGAG] |
| M.XmnI | GAA N4 TTC | 2γM∼[GAA] |
| M.DrdI | GAC N6 GTC | 2γM∼[GAC] |
| M.XcmI | CCA N9 TGG | 2γM∼[CCA] |
| M.BsaBI | GAT N4 ATC | 2γM + 2[GAT] |
| M.PshAI | GAC N4 GTC | 2γM + 2[GAC] |
| M.AhdI | GAC N5 GTC | 2γM + 2[GAC] |
| M.BstXI | CCA N6 TGG | 2γM + [CCA]∼[CCA] |
Differences occur in the number of catalytic domains/subunits (γM), the number of specificity domains/subunits (S) and in their linkage: whether they are separate proteins (+) or joined into a continuous protein chain (∼) Column 1: MTases are differentiated from other components of R–M systems by the prefix ‘M.’. Column 2: Numerals within the sequence indicate the number of non-specific bases between the two half-sequences. Bold type indicates the bases known or inferred to be methylated. ‘A’ signifies that the adenine shown is methylated, and ‘T’ signifies that the complementary adenine on the other strand is methylated. Most of these enzymes methylate both DNA strands. Column 3: Domain/subunit composition of the MTase. ‘+’ signifies the components are separate subunits, ‘∼’ signifies they are covalently joined. The specificity of the TRD is given in square brackets. ‘[GAG]’ means that the TRD recognizes GAG and methylates the A. Some enzymes comprise two MTases fused together; in these cases, the presence of the other MTase is indicated by ‘m4C-MTase’ or ‘m5C-MTase’.
DoubleMTases
Many MTases are specific for sequences that are not symmetric. These enzymes can bind to their recognition sequence in one orientation only, and consequently they can methylate only one DNA strand. Such MTases can nevertheless participate in R–M systems by partnering with a complementary MTase that methylates the other strand. Such MTase partnerships comprise the modification components of most Type IIS R–M systems. For example, in the Eco31I/BsaI R–M systems, the modification component consists of a γ-MTase partnered with a separate m5C-MTase. In the related Esp3I/BsmBI R-M systems, the modification component is a fusion of a γ-MTase and an m5C-MTase (213,214). In the BssSI and BseRI systems, the modification components are fusions of γ-MTases and m4C-MTases (Table 3).
Homodimeric γ-MTases
An alternative way in which γ-MTases with asymmetric specificities participate in R–M systems is through dimerization (215). Instead of partnering with a complementary MTase, these pair with a second molecule in opposite orientation, achieving similar results. These homodimeric MTases act only in this paired state, and their recognition sequences are interrupted palindromes—inverted repeats of the asymmetric specificity separated by a gap. The modification enzymes of most of the Type II R–M systems with bipartite recognition sequences are γ-MTases that likely act in this way, including M.XmnI, M.DrdI and M.XcmI (Table 3; Figure 6). No proteins of this kind have been crystallized, and so their structures remain unproven, but their C-terminal specificity domains resemble TRDs of Type I S subunits, including motifs suggestive of coiled-coil dimerization helices.
‘Type 1½’ MTases
The relationship between Type II R–M systems and Type I MTases is even closer for enzymes such as M.BsaBI, M.PshAI and M.AhdI (216). These consist of separate M and S subunits rather than composite M∼S chains, and they are close natural equivalents of the EcoR124IΔ and EcoDXXIΔ mutants described earlier (Tables 2 and 3; Figure 6). M.AhdI has the composition 2M+2S (216). Small-angle neutron scattering shows that its structure is similar to that modeled for Type I MTases (217). Dubbed ‘Type 1½’ MTases, these enzymes are intermediate between typical Type I and typical Type II γ-MTases, and they likely represent the ancestral form from which contemporary Type I enzymes evolved by fusion of previously separate TRDs. Finally, the modification component of the BstXI system is not just closely related to Type I MTases, it appears to be one. M.BstXI comprises separate M and S subunits, but unlike the S subunits of Type 1½ MTases, which are single TRDs, the BstXI S subunit has a duplicated organization with two TRDs. These TRDs are similar in aa sequence but not identical, and evidently, they recognize the same half-sequence, CCA, resulting in a recognition sequence that is symmetric overall (Table 3).
Type IIG R–M enzymes
Type IIG enzymes [also termed Type IIC; (20)] are γ-MTases with N-terminal DNA-cleavage domains (Table 4; Figure 6). They are bi-functional R and M enzymes that cleave at fixed distances from their recognition sequences, one or two turns of the helix away. When first discovered, they were so distinct from other Type II REases that they were assigned to a new class, ‘Type IV’ (218). Subsequent agreement to classify all REases that cut at fixed positions as ‘Type II’, regardless of phylogeny, led to their being re-designated ‘Type IIG’, and ‘Type IV’ was assigned to the modification-dependent enzymes instead (20). Type IIG enzymes are widespread in bacteria and archaea. They are less common than Type I enzymes but more diverse and occurring in several distinct organizations (Table 4). Type IIG enzymes are present in 833 of 2145 sequenced genomes in REBASE, or roughly 40%. In all, 1779 systems are distributed among these genomes with an average multiplicity of two per genome. Most genomes have only one or two Type IIG systems but some have many, and strains of Borrelia burgdorferi, the agent of tick-borne Lyme disease, have up to an astonishing 20!
Table 4.
Varied organizations of Type IIG R-M enzymes
| Enzyme | Recognition sequence and cleavage positions | System organization |
|---|---|---|
| RM.BpuSI | GGGAC 10/14 | R∼γM∼[GGGAC] & M1 & M2 |
| RM.BseRI | GAGGAG 10/8 | R∼γM∼[GAGGAG] & M1∼M2 |
| RM.Eco57I | CTGAAG 16/14 | R∼γM∼[CTGAAG] & M |
| RM.BpmI | CTGGAG 16/14 | R∼γM∼[CTGGAG] & M |
| RM.Tth111II | CAARCA 11/9 | 1-2R∼γM∼[CAARCA] |
| RM.MmeI | TCCRAC 20/18 | 1-2R∼γM∼[TCCRAC] |
| RM.NmeAIII | GCCGAG 20/18 | 1-2R∼γM∼[GCCGAG] |
| RM.AquIV | GRGGAAG 20/18 | 1-2R∼γM∼[GRGGAAG] |
| RM.BaeI | 10/15 AC N4 GTAYC 12/7 | 2R∼γM + S1∼S2 |
| RM.BcgI | 10/12 CGA N6 TGC 12/10 | 2R∼γM + S1∼S2 |
| RM.CspCI | 10/12 CCAC N5 TTG 12/10 | 2R∼γM + [CCAC]∼[CAA] |
| RM.BsaXI | 9/12 AC N5 CTCC 10/7 | 2R∼γM + S1∼S2 |
| RM.NgoAVIII | 10/12 TCA N5 GTC 13/11 | 2R∼γM + [TCA]∼[GAC] |
| RM.HaeIV | 7/13 GAY N5 RTC 14/9 | 2R∼γM∼[GAY] |
| RM.AloI | 7/12 GGA N6 GTTC 12/7 | R∼γM∼[GGA]∼[GAAC] |
| RM.PpiI | 8/13 GAG N5 GTTC 12/7 | R∼γM∼[GAG]∼[GAAC] |
| RM.TstI | 7/12 GGA N6 GTG 13/8 | R∼γM∼[GGA]∼[CAC] |
Differences occur in the number of catalytic domains/subunits (RM), the number of specificity domains/subunits (S) and in the linkages between them. Frequently, these mirror the different forms of γ-MTases listed in Table 3. Column 1: Type IIG enzymes are γ-MTases with N-terminal DNA-cleavage domains. They are differentiated from other system components by the prefix ‘RM.’. For brevity, this prefix is often omitted. Column 2: Numerals inside the sequences indicate the number of non-specific bases between the half-sequences. Numerals outside indicate the positions of cleavage. GGGAC 10/14, for example, signifies that cleavage takes place to the right of the sequence shown, 10 bases further down on that strand and 14 bases further down on the other strand. Column 3: Organization of the system, and domain/subunit composition of the R-M enzyme. ‘∼’ indicates the components are covalently joined; ‘+’ indicates they are separate subunits. ‘&’ indicates that additional MTases (M, M1, M2) are components of the system and provide protective modification. Stoichiometry, known or inferred, is indicated by the numeral preceding the protein. MmeI-like enzymes are monomers but likely act as homodimers/homotetramers; this bimodal behavior is indicated by ‘1–2’. Where known, the specificities of the TRDs are given in square brackets and are shown in the order in which they occur within the protein. When the order is not known, the TRDs are designated S1 and S2.
The DNA-cleavage domains of Type IIG enzymes comprise approximately the N-terminal 200 aa and belong primarily to the ‘PD-(D/E)XK’ family of endonucleases, the same as found in Type I R subunits, and the commonest among the Type IIP REases. The Asp and Glu residues of this motif, in combination with a phosphate oxygen and water molecules, coordinate one or two divalent metal ions, typically magnesium or manganese. These ions are essential for catalysis and are thought to counter the build up of negative charge on the phosphorus during the transition state. They might also induce a positive charge on the phosphorus atom before catalysis. The Lys acts as a general base by deprotonating a structured water molecule, producing the hydroxide ion that attacks the phosphorus (Figure 2). A single catalytic site is present in Type IIG R domains, and cleavage of duplex DNA likely involves transient dimerization between neighboring enzyme molecules. The crystal structure of only one Type IIG enzyme has been published: BpuSI (GGGAC 10/14; see footnote to Table 4 for convention) (194). The cleavage domain of BpuSI resembles the well-characterized C-terminal cleavage domain of the Type IIS enzyme, R.FokI, and it cleaves DNA with the same staggered geometry, producing 4-base 5′-overhangs (219,220). BpuSI is unusual in this regard, as most Type IIG enzymes create 2-base 3′-overhangs, indicating that their catalytic sites cleave across the minor groove of DNA rather than across the major groove. BpuSI was crystallized without DNA, and comparison with M.TaqI indicates that it must undergo significant structural rearrangements to bind to its recognition sequence and effect catalysis (194).
Monomeric Type IIG enzymes: Eco57I
Eco57I (CTGAAG 16/14) was the first member of this new class to be characterized (218). Its large size (997 aa; 117 kDa) is typical for these enzymes and reflects their composite nature of three joined domains, R, M and S (Figure 6). The recognition sequence of Eco57I is asymmetric but continuous (Table 4). The enzyme methylates only one strand and cleaves on only one side, indicating that it binds to the sequence as a monomer in one orientation only. Requisite methylation of the other strand is accomplished by a separate MTase, M.Eco57I, also a γ-MTase (218). This latter enzyme has a degenerate specificity (CTNNAG) enabling it to methylate both strands of the sequence, the same A in the top strand methylated by Eco57I, and the sole A in the bottom strand (Table 4). The need for an accompanying MTase confines the evolutionary diversification that Eco57I can undergo to just the central 2 bp. Related enzymes with specificity differences at these positions are known, but they are few in number.
The crystallized BpuSI also acts as a monomer and methylates the single A in the top strand. Two additional MTases accompany this enzyme: a separate γ-MTase that methylates the same adenine, and an m5C-MTase that methylates a cytosine in the bottom strand. In the BseRI Type IIG system, the accompanying modification enzyme is a fusion of an m4C-MTase joined to a γ-MTase (Table 4) (221). In all of these monomeric Type IIG systems, the accompanying MTases methylate both DNA strands, hinting that the intrinsic methylation activity of the R–M enzymes themselves might play a role other than protective modification.
Multimeric Type IIG enzymes: BcgI
BcgI (10/12 CGA N6 TGC 12/10) was discovered shortly after Eco57I and has the novel property of cleaving on both sides of its recognition sequence, releasing a small fragment containing the recognition sequence at every site it cuts (222). BcgI comprises two proteins, an RM subunit and an S subunit (Figure 6), and it represents a trimmed-down fixed-cleaving version of Type I enzymes (223–225). More enzymes of this kind, also termed Type IIB (226), have been discovered and characterized including BaeI, CspCI, BsaXI, SdeOSI and NgoAVIII (227), but they are far less common than Type I enzymes (Table 4). The gap between the two half-sites of their recognition sequences varies, as also does the ‘reach’ to the cleavage site. However, if the methylated bases are taken as the reference points rather than the boundaries of the half-sites, then bi-lateral symmetry emerges reflecting the fact that the same RM subunit catalyzes the reactions on both sides.
Both subunits of BcgI-like enzymes (Type IIB) are required for cleavage activity and for methylation activity. The subunit stoichiometry is 2RM:1S (223), mirroring the 2M:1S stoichiometry of Type I MTases. The S subunits, though generally smaller than Type I S subunits, appear to have a similar organization of tandemly repeated specificity and dimerization domains. Unlike the monomeric Type IIG enzymes described in the previous section, BcgI-like enzymes protectively modify both strands of their recognition sequence without the need for accompanying MTases. The cleavage domain of the RM subunits contains only one catalytic site, and double-strand cleavage likely requires transient dimerization between neighboring enzyme molecules. Because cleavage takes place on both sides of the recognition sequence, multiple molecules are involved at the same time (228–231).
Cleavage geometry
As the RM subunits of Type IIB enzymes methylate the half-sequence to which they are bound, it seems logical that they would cleave on the same side, too. If so, the RM subunit would be cleaving on the 5′ side of the methylated base, upstream of the recognition site. Type IIG enzymes that bind continuous, rather than bipartite, recognition sequences invariably cleave on the 3′ side of the methylated base, downstream, in the opposite direction. The RM subunit of BcgI is similar in size and predicted structure to the corresponding RM domain of MmeI (described later in the text), suggesting that these two enzymes cleave and methylate with similar geometries. Nevertheless, BcgI cleaves 12/14 upstream, whereas MmeI cleaves 21/19 downstream (Table 4). How to reconcile this considerable catalytic discrepancy is not clear. One possibility is for the BcgI subunits to cleave on the other side of the recognition sequence, in which case they would be cleaving 21/19 downstream, exactly like MmeI. Another possibility is for MmeI to act as a dimer or tetramer and to cleave 12/14 upstream, but not of the sequence to which it is bound, but instead to a flanking, inverted sequence, that lies side-by-side.
Single-chain Type IIG enzymes: AloI
A number of single-chain Type IIG enzymes also cleave on both sides of bipartite recognition sequences. HaeIV, for example, cleaves on both sides of a recognition sequence that is symmetric, suggesting that it acts as a homodimer—the Type IIG equivalent of MTases like M.DrdI (Table 4) (232). In other enzymes, such as AloI and CjeI, the two subunits of a BcgI-like ancestor are fused into a single protein that possesses two TRDs at the C-terminus (233–235). These enzymes are ∼1250 aa in length, and both TRDs appear to be functional, as shown by domain-swap experiments (236). Yet, how they act is a mystery because the normal stoichiometry of 1RM:1TRD such as occurs in BcgI and all other Type II G enzymes, is reduced to 1RM:2TRD in CjeI and AloI (Figure 6). This reduction deprives one of the two TRDs of an RM domain with which to methylate or to cleave (226).
Bimodal Type IIG enzymes: MmeI
MmeI (TCCRAC 20/18) was the first Type IIG enzyme to be purified (237), but it was not well-characterized until many years later (238). MmeI belongs to a large family of closely related enzymes whose specificities have diversified extensively (196). ‘By-eye-oinformatics’ analysis correlating TRDs with sequences recognized, in combination with mutagenesis experiments, have led to the identification of most of the ‘contact’ amino acids responsible for sequence-recognition (196). Remarkably, when these aa are judiciously changed the specificities of the enzymes also change, predictably and robustly (196). A model of MmeI bound to DNA has been reported (239), and the X-ray crystal structures of MmeI bound to DNA, and of NmeAIII without DNA, have recently been solved [(195); Scott Callahan and Aneel Aggarwal, personal communication]. The DNA-bound TRD of MmeI superimposes on M.TaqI and the crystallized Type I S subunits (95,96), enabling the identities of some of the contact residues to be transferred. This might allow Type I R–M enzyme specificities to be changed rationally, base-by-base, in the same way they can now be changed in MmeI-family enzymes (196). Eventually, it might also allow the specificities of some of the thousands of the putative S subunits in REBASE to be assigned by simple inspection of their aa sequences.
MmeI-family enzymes purify as monomers, but it seems likely that in vivo they act as homodimers in somewhat the same way as Type III R–M enzymes are thought to do (240,241). MmeI-enzymes protectively modify their DNA without the need for accompanying MTases, and because their specificities are generally asymmetric, they methylate only one strand (238). This has led to the suggestion they be termed ‘Type IIL’ enzymes, for Lone-strand modification (242). It would be astonishing if the MmeI-family enzymes did rely on modification of only one strand for protection, however, because then they would have no way to distinguish unmodified, infecting DNA from unmodified, but newly replicated, host DNA. A priori, it seems likely that they rely instead on pairs of recognition sequences in opposite, head-to-head, orientations. One member of such pairs will always be modified in newly replicated DNA, whereas neither will be modified in infecting DNA.
Type I R–M enzymes faced this same problem during evolution and solved it by binding to two inverted half-sequences approximately one DNA helix turn apart. Things are somewhat different for the MmeI enzymes because the opposing sites are not a fixed distance apart, implying that protein–protein interaction must depend on sliding or DNA looping (243,244). MmeI-enzymes are predicted to have alpha-helical domains at their C-termini (239) that could act as dimerization zippers to interconnect adjacent enzyme molecules. Such helices occur at the C-termini of γ-MTases that act as dimers such as M.DrdI and M.AhdI, but they do not occur at the C-termini of γ-MTases such as M.TaqI that act as monomers and have no need for them (Figure 6). It seems plausible, then, that MmeI-type enzymes bind to their recognition sequences as monomers, but cleave DNA only after assembling into homodimers, the overall organizations of which are similar to Type I MTases. If so, what we refer to as the ‘recognition sequence’ of MmeI (i.e. TCCRAC) is the equivalent of a Type I half-sequence, and its complete binding site is the indefinite symmetric sequence: TCCRAC Ni GTYGGA (where i can be any number of base pairs between ∼50 and 5000).
‘Type ISP’ enzymes
The recently christened ‘Type ISP’ enzymes combine features of both Type I and MmeI-type enzymes. They are long single-polypeptide chain fusions of, in order, an N-terminal Mrr-like endonuclease domain (typically, PD-QXK instead of PD-EXK), a DNA-translocase domain, a γ-MTase and a C-terminal TRD (245–247) (Figure 6). Type ISP enzymes recognize continuous, asymmetric sequences, or perhaps more accurately ‘half-sequences’, and they behave essentially like ‘half-Type I R-M enzymes’ showing ATP-dependent DNA translocation and a requirement for two target sites in head-to-head orientation for cleavage. Cleavage requires two molecules of the enzyme, and it occurs at a random location between the two sites (248). Translocation produces loops of DNA, but only a single recognition site, and a single copy of the enzyme, is needed for this to take place (249).
CONCLUSION
Type I R–M enzymes and their relatives represent a huge group of sequence-specific DNA-binding proteins with varied specificities, oligomeric organizations and catalytic properties. Traditionally, they have been difficult to characterize, but genome sequencing, bioinformatics and, very recently, methylome analysis by next-generation SMRT sequencing have thrown open a wide door for identifying them and determining their target recognition sequences. As more of these enzymes are analyzed, a detailed understanding of how they recognize DNA will emerge. This could allow new specificities to be engineered at will, and new biochemical functions to be delivered to DNA sequences of choice. Type I enzymes and their relatives offer numerous opportunities for protein design and fabrication. Although they have been studied for >50 years, their potential as molecular tools and as sources of nano-scale biochemical components is only now becoming apparent.
FUNDING
Funding for open access charge: New England Biolabs.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Werner Arber, Rich Roberts and Joe Bertani for reprints from their archives, and encouragement. They also thank the anonymous reviewers of a previous manuscript for care taken with the review process, greatly improving the present document.
REFERENCES
- 1.Dussoix D, Arber W. Host specificity of DNA produced by Escherichia coli. II. Control over acceptance of DNA from infecting phage lambda. J. Mol. Biol. 1962;5:37–49. doi: 10.1016/s0022-2836(62)80059-x. [DOI] [PubMed] [Google Scholar]
- 2.Arber W, Dussoix D. Host specificity of DNA produced by Escherichia coli. I. Host controlled modification of bacteriophage lambda. J. Mol. Biol. 1962;5:18–36. doi: 10.1016/s0022-2836(62)80058-8. [DOI] [PubMed] [Google Scholar]
- 3.Luria SE, Human ML. A nonhereditary, host-induced variation of bacterial viruses. J. Bacteriol. 1952;64:557–569. doi: 10.1128/jb.64.4.557-569.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson ES, Felix A. Variation in Vi-phage II of Salmonella typhi. Nature. 1952;170:492–494. doi: 10.1038/170492b0. [DOI] [PubMed] [Google Scholar]
- 5.Bertani G, Weigle JJ. Host controlled variation in bacterial viruses. J. Bacteriol. 1953;65:113–121. doi: 10.1128/jb.65.2.113-121.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luria SE. Host-induced modifications of viruses. Cold Spring Harb. Symp. Quant. Biol. 1953;18:237–244. doi: 10.1101/sqb.1953.018.01.034. [DOI] [PubMed] [Google Scholar]
- 7.Arber W, Hattman S, Dussoix D. On the host-controlled modification of bacteriophage lambda. Virology. 1963;21:30–35. doi: 10.1016/0042-6822(63)90300-3. [DOI] [PubMed] [Google Scholar]
- 8.Glover W, Schell J, Symonds N, Stacey KA. The control of host-induced modification by phage P1. Genet. Res. 1963;4:480–482. doi: 10.1016/0006-291x(63)90193-1. [DOI] [PubMed] [Google Scholar]
- 9.Lederberg S. Genetics of host-controlled restriction and modification of deoxyribonucleic acid in Escherichia coli. J. Bacteriol. 1966;91:1029–1036. doi: 10.1128/jb.91.3.1029-1036.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franklin NC, Dove WF. Genetic evidence for restriction targets in the DNA of phages lambda and phi 80. Genet. Res. 1969;14:151–157. doi: 10.1017/s0016672300001981. [DOI] [PubMed] [Google Scholar]
- 11.Arber W. Host-controlled modification of bacteriophage. Annu. Rev. Microbiol. 1965;19:365–378. doi: 10.1146/annurev.mi.19.100165.002053. [DOI] [PubMed] [Google Scholar]
- 12.Arber W, Linn S. DNA modification and restriction. Annu. Rev. Biochem. 1969;38:467–500. doi: 10.1146/annurev.bi.38.070169.002343. [DOI] [PubMed] [Google Scholar]
- 13.Arber W. Host-Controlled Cariation. NY: Cold Spring Harbor Laboratory; 1971. [Google Scholar]
- 14.Boyer HW. DNA restriction and modification mechanisms in bacteria. Annu. Rev. Microbiol. 1971;25:153–176. doi: 10.1146/annurev.mi.25.100171.001101. [DOI] [PubMed] [Google Scholar]
- 15.Meselson M, Yuan R, Heywood J. Restriction and modification of DNA. Annu. Rev. Biochem. 1972;41:447–466. doi: 10.1146/annurev.bi.41.070172.002311. [DOI] [PubMed] [Google Scholar]
- 16.Arber W. Host specificity of DNA produced by Escherichia coli. V. The role of methionine in the production of host specificity. J. Mol. Biol. 1965;11:247–256. doi: 10.1016/s0022-2836(65)80055-9. [DOI] [PubMed] [Google Scholar]
- 17.Smith HO, Nathans D. Letter: A suggested nomenclature for bacterial host modification and restriction systems and their enzymes. J. Mol. Biol. 1973;81:419–423. doi: 10.1016/0022-2836(73)90152-6. [DOI] [PubMed] [Google Scholar]
- 18.Nathans D, Smith HO. Restriction endonucleases in the analysis and restructuring of dna molecules. Annu. Rev. Biochem. 1975;44:273–293. doi: 10.1146/annurev.bi.44.070175.001421. [DOI] [PubMed] [Google Scholar]
- 19.Roberts RJ. Restriction endonucleases. CRC Crit Rev. Biochem. 1976;4:123–164. doi: 10.3109/10409237609105456. [DOI] [PubMed] [Google Scholar]
- 20.Roberts RJ, Belfort M, Bestor T, Bhagwat AS, Bickle TA, Bitinaite J, Blumenthal RM, Degtyarev SK, Dryden DT, Dybvig K, et al. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003;31:1805–1812. doi: 10.1093/nar/gkg274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE–a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2010;38:D234–D236. doi: 10.1093/nar/gkp874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tock MR, Dryden DT. The biology of restriction and anti-restriction. Curr. Opin. Microbiol. 2005;8:466–472. doi: 10.1016/j.mib.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 23.Rosamond J, Endlich B, Linn S. Electron microscopic studies of the mechanism of action of the restriction endonuclease of Escherichia coli B. J. Mol. Biol. 1979;129:619–635. doi: 10.1016/0022-2836(79)90472-8. [DOI] [PubMed] [Google Scholar]
- 24.Yuan R, Hamilton DL, Burckhardt J. DNA translocation by the restriction enzyme from E. coli K. Cell. 1980;20:237–244. doi: 10.1016/0092-8674(80)90251-2. [DOI] [PubMed] [Google Scholar]
- 25.Warren RA. Modified bases in bacteriophage DNAs. Annu. Rev. Microbiol. 1980;34:137–158. doi: 10.1146/annurev.mi.34.100180.001033. [DOI] [PubMed] [Google Scholar]
- 26.Kruger DH, Bickle TA. Bacteriophage survival: multiple mechanisms for avoiding the deoxyribonucleic acid restriction systems of their hosts. Microbiol. Rev. 1983;47:345–360. doi: 10.1128/mr.47.3.345-360.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bickle TA, Kruger DH. Biology of DNA restriction. Microbiol. Rev. 1993;57:434–450. doi: 10.1128/mr.57.2.434-450.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arber W, Wauters-Willems D. Host specificity of DNA produced by Escherichia coli. XII. The two restriction and modification systems of strain 15T- Mol. Gen. Genet. 1970;108:203–217. doi: 10.1007/BF00283350. [DOI] [PubMed] [Google Scholar]
- 29.Bullas LR, Colson C, Neufeld B. Deoxyribonucleic acid restriction and modification systems in Salmonella: chromosomally located systems of different serotypes. J. Bacteriol. 1980;141:275–292. doi: 10.1128/jb.141.1.275-292.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryu J, Rajadas PT, Bullas LR. Complementation and hybridization evidence for additional families of type I DNA restriction and modification genes in Salmonella serotypes. J. Bacteriol. 1988;170:5785–5788. doi: 10.1128/jb.170.12.5785-5788.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barcus VA, Titheradge AJ, Murray NE. The diversity of alleles at the hsd locus in natural populations of Escherichia coli. Genetics. 1995;140:1187–1197. doi: 10.1093/genetics/140.4.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sibley MH, Raleigh EA. Cassette-like variation of restriction enzyme genes in Escherichia coli C and relatives. Nucleic Acids Res. 2004;32:522–534. doi: 10.1093/nar/gkh194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murray NE. Type I restriction systems: sophisticated molecular machines (a legacy of Bertani and Weigle) Microbiol. Mol. Biol. Rev. 2000;64:412–434. doi: 10.1128/mmbr.64.2.412-434.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murray NE. 2001 Fred Griffith review lecture. Immigration control of DNA in bacteria: self versus non-self. Microbiology. 2002;148:3–20. doi: 10.1099/00221287-148-1-3. [DOI] [PubMed] [Google Scholar]
- 35.Dryden DT, Murray NE, Rao DN. Nucleoside triphosphate-dependent restriction enzymes. Nucleic Acids Res. 2001;29:3728–3741. doi: 10.1093/nar/29.18.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loenen WA. Tracking EcoKI and DNA fifty years on: a golden story full of surprises. Nucleic Acids Res. 2003;31:7059–7069. doi: 10.1093/nar/gkg944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bourniquel AA, Bickle TA. Complex restriction enzymes: NTP-driven molecular motors. Biochimie. 2002;84:1047–1059. doi: 10.1016/s0300-9084(02)00020-2. [DOI] [PubMed] [Google Scholar]
- 38.Sitaraman R, Dybvig K. The hsd loci of Mycoplasma pulmonis: organization, rearrangements and expression of genes. Mol. Microbiol. 1997;26:109–120. doi: 10.1046/j.1365-2958.1997.5571938.x. [DOI] [PubMed] [Google Scholar]
- 39.Dybvig K, Sitaraman R, French CT. A family of phase-variable restriction enzymes with differing specificities generated by high-frequency gene rearrangements. Proc. Natl Acad. Sci. U. S. A. 1998;95:13923–13928. doi: 10.1073/pnas.95.23.13923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kennaway CK, Obarska-Kosinska A, White JH, Tuszynska I, Cooper LP, Bujnicki JM, Trinick J, Dryden DT. The structure of M.EcoKI Type I DNA methyltransferase with a DNA mimic antirestriction protein. Nucleic Acids Res. 2009;37:762–770. doi: 10.1093/nar/gkn988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kennaway CK, Taylor JE, Song CF, Potrzebowski W, Nicholson W, White JH, Swiderska A, Obarska-Kosinska A, Callow P, Cooper LP, et al. Structure and operation of the DNA-translocating type I DNA restriction enzymes. Genes Dev. 2012;26:92–104. doi: 10.1101/gad.179085.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ravetch JV, Horiuchi K, Zinder ND. Nucleotide sequence of the recognition site for the restriction-modification enzyme of Escherichia coli B. Proc. Natl Acad. Sci. USA. 1978;75:2266–2270. doi: 10.1073/pnas.75.5.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lautenberger JA, Edgell MH, Hutchison CA, III, Godson GN. The DNA sequence on bacteriophage G4 recognized by the Escherichia coli B restriction enzyme. J. Mol. Biol. 1979;131:871–875. doi: 10.1016/0022-2836(79)90206-7. [DOI] [PubMed] [Google Scholar]
- 44.Sommer R, Schaller H. Nucleotide sequence of the recognition site of the B-specific restriction modification system in E. coli. Mol. Gen. Genet. 1979;168:331–335. doi: 10.1007/BF00271504. [DOI] [PubMed] [Google Scholar]
- 45.Kan NC, Lautenberger JA, Edgell MH, Hutchison CA., III The nucleotide sequence recognized by the Escherichia coli K12 restriction and modification enzymes. J. Mol. Biol. 1979;130:191–209. doi: 10.1016/0022-2836(79)90426-1. [DOI] [PubMed] [Google Scholar]
- 46.Suri B, Shepherd JC, Bickle TA. The EcoA restriction and modification system of Escherichia coli 15T-: enzyme structure and DNA recognition sequence. EMBO J. 1984;3:575–579. doi: 10.1002/j.1460-2075.1984.tb01850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kroger M, Hobom G. The nucleotide recognized by the Escherichia coli A restriction and modification enzyme. Nucleic Acids Res. 1984;12:887–899. doi: 10.1093/nar/12.2.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nagaraja V, Shepherd JC, Pripfl T, Bickle TA. Two type I restriction enzymes from Salmonella species. Purification and DNA recognition sequences. J. Mol. Biol. 1985;182:579–587. doi: 10.1016/0022-2836(85)90243-8. [DOI] [PubMed] [Google Scholar]
- 49.Nagaraja V, Stieger M, Nager C, Hadi SM, Bickle TA. The nucleotide sequence recognised by the Escherichia coli D type I restriction and modification enzyme. Nucleic Acids Res. 1985;13:389–399. doi: 10.1093/nar/13.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Piekarowicz A, Goguen JD. The DNA sequence recognized by the EcoDXX1 restriction endonuclease. Eur. J. Biochem. 1986;154:295–298. doi: 10.1111/j.1432-1033.1986.tb09396.x. [DOI] [PubMed] [Google Scholar]
- 51.Price C, Shepherd JC, Bickle TA. DNA recognition by a new family of type I restriction enzymes: a unique relationship between two different DNA specificities. EMBO J. 1987;6:1493–1497. doi: 10.1002/j.1460-2075.1987.tb02391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cowan GM, Gann AA, Murray NE. Conservation of complex DNA recognition domains between families of restriction enzymes. Cell. 1989;56:103–109. doi: 10.1016/0092-8674(89)90988-4. [DOI] [PubMed] [Google Scholar]
- 53.Kannan P, Cowan GM, Daniel AS, Gann AA, Murray NE. Conservation of organization in the specificity polypeptides of two families of type I restriction enzymes. J. Mol. Biol. 1989;209:335–344. doi: 10.1016/0022-2836(89)90001-6. [DOI] [PubMed] [Google Scholar]
- 54.Tyndall C, Meister J, Bickle TA. The Escherichia coli prr region encodes a functional type IC DNA restriction system closely integrated with an anticodon nuclease gene. J. Mol. Biol. 1994;237:266–274. doi: 10.1006/jmbi.1994.1230. [DOI] [PubMed] [Google Scholar]
- 55.Thorpe PH, Ternent D, Murray NE. The specificity of sty SKI, a type I restriction enzyme, implies a structure with rotational symmetry. Nucleic Acids Res. 1997;25:1694–1700. doi: 10.1093/nar/25.9.1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Titheradge AJ, King J, Ryu J, Murray NE. Families of restriction enzymes: an analysis prompted by molecular and genetic data for type ID restriction and modification systems. Nucleic Acids Res. 2001;29:4195–4205. doi: 10.1093/nar/29.20.4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Piekarowicz A, Klyz A, Kwiatek A, Stein DC. Analysis of type I restriction modification systems in the Neisseriaceae: genetic organization and properties of the gene products. Mol. Microbiol. 2001;41:1199–1210. doi: 10.1046/j.1365-2958.2001.02587.x. [DOI] [PubMed] [Google Scholar]
- 58.Kasarjian JK, Iida M, Ryu J. New restriction enzymes discovered from Escherichia coli clinical strains using a plasmid transformation method. Nucleic Acids Res. 2003;31:e22. doi: 10.1093/nar/gng022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chin V, Valinluck V, Magaki S, Ryu J. KpnBI is the prototype of a new family (IE) of bacterial type I restriction-modification system. Nucleic Acids Res. 2004;32:e138. doi: 10.1093/nar/gnh134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kasarjian JK, Hidaka M, Horiuchi T, Iida M, Ryu J. The recognition and modification sites for the bacterial type I restriction systems KpnAI, StySEAI, StySENI and StySGI. Nucleic Acids Res. 2004;32:e82. doi: 10.1093/nar/gnh079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kasarjian JK, Kodama Y, Iida M, Matsuda K, Ryu J. Four new type I restriction enzymes identified in Escherichia coli clinical isolates. Nucleic Acids Res. 2005;33:e114. doi: 10.1093/nar/gni114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Murray IA, Clark TA, Morgan RD, Boitano M, Anton BP, Luong K, Fomenkov A, Turner SW, Korlach J, Roberts RJ. The methylomes of six bacteria. Nucleic Acids Res. 2012;40:11450–11462. doi: 10.1093/nar/gks891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fang G, Munera D, Friedman DI, Mandlik A, Chao MC, Banerjee O, Feng Z, Losic B, Mahajan MC, Jabado OJ, et al. Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat. Biotechnol. 2012;30:1232–1239. doi: 10.1038/nbt.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Clark TA, Murray IA, Morgan RD, Kislyuk AO, Spittle KE, Boitano M, Fomenkov A, Roberts RJ, Korlach J. Characterization of DNA methyltransferase specificities using single-molecule, real-time DNA sequencing. Nucleic Acids Res. 2012;40:e29. doi: 10.1093/nar/gkr1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Roberts GA, Houston PJ, White JH, Chen K, Stephanou AS, Cooper LP, Dryden DT, Lindsay JA. Impact of target site distribution for Type I restriction enzymes on the evolution of methicillin-resistant Staphylococcus aureus (MRSA) populations. Nucleic Acids Res. 2013;141:7472–7484. doi: 10.1093/nar/gkt535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Davies GP, Martin I, Sturrock SS, Cronshaw A, Murray NE, Dryden DT. On the structure and operation of type I DNA restriction enzymes. J. Mol. Biol. 1999;290:565–579. doi: 10.1006/jmbi.1999.2908. [DOI] [PubMed] [Google Scholar]
- 67.Fairman-Williams ME, Guenther UP, Jankowsky E. SF1 and SF2 helicases: family matters. Curr. Opin. Struct. Biol. 2010;20:313–324. doi: 10.1016/j.sbi.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gorbalenya AE, Koonin EV. Endonuclease (R) subunits of type-I and type-III restriction-modification enzymes contain a helicase-like domain. FEBS Lett. 1991;291:277–281. doi: 10.1016/0014-5793(91)81301-n. [DOI] [PubMed] [Google Scholar]
- 69.Gorbalenya AE, Koonin EV, Donchenko AP, Blinov VM. A conserved NTP-motif in putative helicases. Nature. 1988;333:22. doi: 10.1038/333022a0. [DOI] [PubMed] [Google Scholar]
- 70.Hall MC, Matson SW. Helicase motifs: the engine that powers DNA unwinding. Mol. Microbiol. 1999;34:867–877. doi: 10.1046/j.1365-2958.1999.01659.x. [DOI] [PubMed] [Google Scholar]
- 71.Singleton MR, Dillingham MS, Wigley DB. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 72.Szczelkun MD, Friedhoff P, Seidel R. Maintaining a sense of direction during long-range communication on DNA. Biochem. Soc. Trans. 2010;38:404–409. doi: 10.1042/BST0380404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tuteja N, Tuteja R. Prokaryotic and eukaryotic DNA helicases. Essential molecular motor proteins for cellular machinery. Eur. J. Biochem. 2004;271:1835–1848. doi: 10.1111/j.1432-1033.2004.04093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Umate P, Tuteja N, Tuteja R. Genome-wide comprehensive analysis of human helicases. Commun. Integr. Biol. 2011;4:118–137. doi: 10.4161/cib.4.1.13844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Horiuchi K, Zinder ND. Cleavage of bacteriophage fl DNA by the restriction enzyme of Escherichia coli B. Proc. Natl Acad. Sci. U. S. A. 1972;69:3220–3224. doi: 10.1073/pnas.69.11.3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Studier FW, Bandyopadhyay PK. Model for how type I restriction enzymes select cleavage sites in DNA. Proc. Natl Acad. Sci. U. S. A. 1988;85:4677–4681. doi: 10.1073/pnas.85.13.4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.vanNoort J, van der Heijden T, Dutta CF, Firman K, Dekker C. Initiation of translocation by Type I restriction-modification enzymes is associated with a short DNA extrusion. Nucleic Acids Res. 2004;32:6540–6547. doi: 10.1093/nar/gkh999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Neaves KJ, Cooper LP, White JH, Carnally SM, Dryden DT, Edwardson JM, Henderson RM. Atomic force microscopy of the EcoKI Type I DNA restriction enzyme bound to DNA shows enzyme dimerization and DNA looping. Nucleic Acids Res. 2009;37:2053–2063. doi: 10.1093/nar/gkp042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Seidel R, van NJ, van der Scheer C, Bloom JG, Dekker NH, Dutta CF, Blundell A, Robinson T, Firman K, Dekker C. Real-time observation of DNA translocation by the type I restriction modification enzyme EcoR124I. Nat. Struct. Mol. Biol. 2004;11:838–843. doi: 10.1038/nsmb816. [DOI] [PubMed] [Google Scholar]
- 80.Seidel R, Bloom JG, van NJ, Dutta CF, Dekker NH, Firman K, Szczelkun MD, Dekker C. Dynamics of initiation, termination and reinitiation of DNA translocation by the motor protein EcoR124I. EMBO J. 2005;24:4188–4197. doi: 10.1038/sj.emboj.7600881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stanley LK, Seidel R, van der Scheer C, Dekker NH, Szczelkun MD, Dekker C. When a helicase is not a helicase: dsDNA tracking by the motor protein EcoR124I. EMBO J. 2006;25:2230–2239. doi: 10.1038/sj.emboj.7601104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Seidel R, Dekker C. Single-molecule studies of nucleic acid motors. Curr. Opin. Struct. Biol. 2007;17:80–86. doi: 10.1016/j.sbi.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 83.Seidel R, Bloom JG, Dekker C, Szczelkun MD. Motor step size and ATP coupling efficiency of the dsDNA translocase EcoR124I. EMBO J. 2008;27:1388–1398. doi: 10.1038/emboj.2008.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bianco PR, Hurley EM. The type I restriction endonuclease EcoR124I, couples ATP hydrolysis to bidirectional DNA translocation. J. Mol. Biol. 2005;352:837–859. doi: 10.1016/j.jmb.2005.07.055. [DOI] [PubMed] [Google Scholar]
- 85.Bianco PR, Xu C, Chi M. Type I restriction endonucleases are true catalytic enzymes. Nucleic Acids Res. 2009;37:3377–3390. doi: 10.1093/nar/gkp195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ishikawa K, Handa N, Kobayashi I. Cleavage of a model DNA replication fork by a Type I restriction endonuclease. Nucleic Acids Res. 2009;37:3531–3544. doi: 10.1093/nar/gkp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Klimasauskas S, Kumar S, Roberts RJ, Cheng X. HhaI methyltransferase flips its target base out of the DNA helix. Cell. 1994;76:357–369. doi: 10.1016/0092-8674(94)90342-5. [DOI] [PubMed] [Google Scholar]
- 88.Roberts RJ, Cheng X. Base flipping. Annu. Rev. Biochem. 1998;67:181–198. doi: 10.1146/annurev.biochem.67.1.181. [DOI] [PubMed] [Google Scholar]
- 89.Cheng X, Roberts RJ. AdoMet-dependent methylation, DNA methyltransferases and base flipping. Nucleic Acids Res. 2001;29:3784–3795. doi: 10.1093/nar/29.18.3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cheng X, Blumenthal RM. Cytosines do it, thymines do it, even pseudouridines do it–base flipping by an enzyme that acts on RNA. Structure. 2002;10:127–129. doi: 10.1016/s0969-2126(02)00710-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Su TJ, Tock MR, Egelhaaf SU, Poon WC, Dryden DT. DNA bending by M.EcoKI methyltransferase is coupled to nucleotide flipping. Nucleic Acids Res. 2005;33:3235–3244. doi: 10.1093/nar/gki618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dryden DT, Cooper LP, Murray NE. Purification and characterization of the methyltransferase from the type 1 restriction and modification system of Escherichia coli K12. J. Biol. Chem. 1993;268:13228–13236. [PubMed] [Google Scholar]
- 93.Loenen WA, Murray NE. Modification enhancement by the restriction alleviation protein (Ral) of bacteriophage lambda. J. Mol. Biol. 1986;190:11–22. doi: 10.1016/0022-2836(86)90071-9. [DOI] [PubMed] [Google Scholar]
- 94.Kelleher JE, Daniel AS, Murray NE. Mutations that confer de novo activity upon a maintenance methyltransferase. J. Mol. Biol. 1991;221:431–440. doi: 10.1016/0022-2836(91)80064-2. [DOI] [PubMed] [Google Scholar]
- 95.Calisto BM, Pich OQ, Pinol J, Fita I, Querol E, Carpena X. Crystal structure of a putative type I restriction-modification S subunit from Mycoplasma genitalium. J. Mol. Biol. 2005;351:749–762. doi: 10.1016/j.jmb.2005.06.050. [DOI] [PubMed] [Google Scholar]
- 96.Kim JS, DeGiovanni A, Jancarik J, Adams PD, Yokota H, Kim R, Kim SH. Crystal structure of DNA sequence specificity subunit of a type I restriction-modification enzyme and its functional implications. Proc. Natl Acad. Sci. USA. 2005;102:3248–3253. doi: 10.1073/pnas.0409851102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gao P, Tang Q, An X, Yan X, Liang D. Structure of HsdS subunit from Thermoanaerobacter tengcongensis sheds lights on mechanism of dynamic opening and closing of type I methyltransferase. PLoS One. 2011;6:e17346. doi: 10.1371/journal.pone.0017346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lapkouski M, Panjikar S, Kuta SI, Csefalvay E. Purification, crystallization and preliminary X-ray analysis of the HsdR subunit of the EcoR124I endonuclease from Escherichia coli. Acta Crystallogr. Sect. F. Struct. Biol. Cryst. Commun. 2007;63:582–585. doi: 10.1107/S174430910702622X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lapkouski M, Panjikar S, Janscak P, Smatanova IK, Carey J, Ettrich R, Csefalvay E. Structure of the motor subunit of type I restriction-modification complex EcoR124I. Nat. Struct. Mol. Biol. 2009;16:94–95. doi: 10.1038/nsmb.1523. [DOI] [PubMed] [Google Scholar]
- 100.Bujnicki JM, Rychlewski L. Grouping together highly diverged PD-(D/E)XK nucleases and identification of novel superfamily members using structure-guided alignment of sequence profiles. J. Mol. Microbiol. Biotechnol. 2001;3:69–72. [PubMed] [Google Scholar]
- 101.Niv MY, Ripoll DR, Vila JA, Liwo A, Vanamee ES, Aggarwal AK, Weinstein H, Scheraga HA. Topology of Type II REases revisited; structural classes and the common conserved core. Nucleic Acids Res. 2007;35:2227–2237. doi: 10.1093/nar/gkm045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Orlowski J, Bujnicki JM. Structural and evolutionary classification of Type II restriction enzymes based on theoretical and experimental analyses. Nucleic Acids Res. 2008;36:3552–3569. doi: 10.1093/nar/gkn175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Laganeckas M, Margelevicius M, Venclovas C. Identification of new homologs of PD-(D/E)XK nucleases by support vector machines trained on data derived from profile-profile alignments. Nucleic Acids Res. 2011;39:1187–1196. doi: 10.1093/nar/gkq958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Steczkiewicz K, Muszewska A, Knizewski L, Rychlewski L, Ginalski K. Sequence, structure and functional diversity of PD-(D/E)XK phosphodiesterase superfamily. Nucleic Acids Res. 2012;40:7016–7045. doi: 10.1093/nar/gks382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Goedecke K, Pignot M, Goody RS, Scheidig AJ, Weinhold E. Structure of the N6-adenine DNA methyltransferase M.TaqI in complex with DNA and a cofactor analog. Nat. Struct. Biol. 2001;8:121–125. doi: 10.1038/84104. [DOI] [PubMed] [Google Scholar]
- 106.Blumenthal RM, Cheng X. A Taq attack displaces bases. Nat. Struct. Biol. 2001;8:101–103. doi: 10.1038/84072. [DOI] [PubMed] [Google Scholar]
- 107.Iyer LM, Leipe DD, Koonin EV, Aravind L. Evolutionary history and higher order classification of AAA+ ATPases. J. Struct. Biol. 2004;146:11–31. doi: 10.1016/j.jsb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 108.Bustamante C, Cheng W, Mejia YX. Revisiting the central dogma one molecule at a time. Cell. 2011;144:480–497. doi: 10.1016/j.cell.2011.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Uyen NT, Nishi K, Park SY, Choi JW, Lee HJ, Kim JS. Crystallization and preliminary X-ray diffraction analysis of the HsdR subunit of a putative type I restriction enzyme from Vibrio vulnificus YJ016. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2008;64:926–928. doi: 10.1107/S1744309108027516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Uyen NT, Park SY, Choi JW, Lee HJ, Nishi K, Kim JS. The fragment structure of a putative HsdR subunit of a type I restriction enzyme from Vibrio vulnificus YJ016: implications for DNA restriction and translocation activity. Nucleic Acids Res. 2009;37:6960–6969. doi: 10.1093/nar/gkp603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Thoms B, Wackernagel W. Genetic control of damage-inducible restriction alleviation in Escherichia coli K12: an SOS function not repressed by lexA. Mol. Gen. Genet. 1984;197:297–303. doi: 10.1007/BF00330977. [DOI] [PubMed] [Google Scholar]
- 112.Blakely GW, Murray NE. Control of the endonuclease activity of type I restriction-modification systems is required to maintain chromosome integrity following homologous recombination. Mol. Microbiol. 2006;60:883–893. doi: 10.1111/j.1365-2958.2006.05144.x. [DOI] [PubMed] [Google Scholar]
- 113.Prakash-Cheng A, Ryu J. Delayed expression of in vivo restriction activity following conjugal transfer of Escherichia coli hsdK (restriction-modification) genes. J. Bacteriol. 1993;175:4905–4906. doi: 10.1128/jb.175.15.4905-4906.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Prakash-Cheng A, Chung SS, Ryu J. The expression and regulation of hsdK genes after conjugative transfer. Mol. Gen. Genet. 1993;241:491–496. doi: 10.1007/BF00279890. [DOI] [PubMed] [Google Scholar]
- 115.Makovets S, Titheradge AJ, Murray NE. ClpX and ClpP are essential for the efficient acquisition of genes specifying type IA and IB restriction systems. Mol. Microbiol. 1998;28:25–35. doi: 10.1046/j.1365-2958.1998.00767.x. [DOI] [PubMed] [Google Scholar]
- 116.Makovets S, Doronina VA, Murray NE. Regulation of endonuclease activity by proteolysis prevents breakage of unmodified bacterial chromosomes by type I restriction enzymes. Proc. Natl Acad. Sci. USA. 1999;96:9757–9762. doi: 10.1073/pnas.96.17.9757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Davies GP, Kemp P, Molineux IJ, Murray NE. The DNA translocation and ATPase activities of restriction-deficient mutants of Eco KI. J. Mol. Biol. 1999;292:787–796. doi: 10.1006/jmbi.1999.3081. [DOI] [PubMed] [Google Scholar]
- 118.Garcia LR, Molineux IJ. Translocation and specific cleavage of bacteriophage T7 DNA in vivo by EcoKI. Proc. Natl Acad. Sci. USA. 1999;96:12430–12435. doi: 10.1073/pnas.96.22.12430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Doronina VA, Murray NE. The proteolytic control of restriction activity in Escherichia coli K-12. Mol. Microbiol. 2001;39:416–428. doi: 10.1046/j.1365-2958.2001.02232.x. [DOI] [PubMed] [Google Scholar]
- 120.Kulik EM, Bickle TA. Regulation of the activity of the type IC EcoR124I restriction enzyme. J. Mol. Biol. 1996;264:891–906. doi: 10.1006/jmbi.1996.0685. [DOI] [PubMed] [Google Scholar]
- 121.Makovets S, Powell LM, Titheradge AJ, Blakely GW, Murray NE. Is modification sufficient to protect a bacterial chromosome from a resident restriction endonuclease? Mol. Microbiol. 2004;51:135–147. doi: 10.1046/j.1365-2958.2003.03801.x. [DOI] [PubMed] [Google Scholar]
- 122.Cajthamlova K, Sisakova E, Weiser J, Weiserova M. Phosphorylation of Type IA restriction–modification complex enzyme EcoKI on the HsdR subunit. FEMS Microbiol. Lett. 2007;270:171–177. doi: 10.1111/j.1574-6968.2007.00663.x. [DOI] [PubMed] [Google Scholar]
- 123.Miller ES, Kutter E, Mosig G, Arisaka F, Kunisawa T, Ruger W. Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 2003;67:86–156. doi: 10.1128/MMBR.67.1.86-156.2003. table of content. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Petrov VM, Ratnayaka S, Nolan JM, Miller ES, Karam JD. Genomes of the T4–related bacteriophages as windows on microbial genome evolution. Virol. J. 2010;7:292. doi: 10.1186/1743-422X-7-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rifat D, Wright NT, Varney KM, Weber DJ, Black LW. Restriction endonuclease inhibitor IPI* of bacteriophage T4: a novel structure for a dedicated target. J. Mol. Biol. 2008;375:720–734. doi: 10.1016/j.jmb.2007.10.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Dunn JJ, Studier FW. Effect of RNAase III, cleavage on translation of bacteriophage T7 messenger RNAs. J. Mol. Biol. 1975;99:487–499. doi: 10.1016/s0022-2836(75)80140-9. [DOI] [PubMed] [Google Scholar]
- 127.Dunn JJ, Elzinga M, Mark KK, Studier FW. Amino acid sequence of the gene 0.3 protein of bacteriophage T7 and nucleotide sequence of its mRNA. J. Biol. Chem. 1981;256:2579–2585. [PubMed] [Google Scholar]
- 128.Dunn JJ, Studier FW. Nucleotide sequence from the genetic left end of bacteriophage T7 DNA to the beginning of gene 4. J. Mol. Biol. 1981;148:303–330. doi: 10.1016/0022-2836(81)90178-9. [DOI] [PubMed] [Google Scholar]
- 129.Mark KK, Studier FW. Purification of the gene 0.3 protein of bacteriophage T7, an inhibitor of the DNA restriction system of Escherichia coli. J. Biol. Chem. 1981;256:2573–2578. [PubMed] [Google Scholar]
- 130.Kruger DH, Schroeder C, Hansen S, Rosenthal HA. Active protection by bacteriophages T3 and T7 against E. coli B- and K-specific restriction of their DNA. Mol. Gen. Genet. 1977;153:99–106. doi: 10.1007/BF01036001. [DOI] [PubMed] [Google Scholar]
- 131.Kruger DH, Reuter M, Hansen S, Schroeder C. Influence of phage T3 and T7 gene functions on a type III(EcoP1) DNA restriction-modification system in vivo. Mol. Gen. Genet. 1982;185:457–461. doi: 10.1007/BF00334140. [DOI] [PubMed] [Google Scholar]
- 132.Zavil'gel'skii GB. Antirestriction [in Russian] Mol. Biol. (Mosk) 2000;34:854–862. [PubMed] [Google Scholar]
- 133.Thomas AT, Brammar WJ, Wilkins BM. Plasmid R16 ArdA protein preferentially targets restriction activity of the type I restriction-modification system EcoKI. J. Bacteriol. 2003;185:2022–2025. doi: 10.1128/JB.185.6.2022-2025.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dryden DT. DNA mimicry by proteins and the control of enzymatic activity on DNA. Trends Biotechnol. 2006;24:378–382. doi: 10.1016/j.tibtech.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 135.Bates S, Roscoe RA, Althorpe NJ, Brammar WJ, Wilkins BM. Expression of leading region genes on IncI1 plasmid ColIb-P9: genetic evidence for single-stranded DNA transcription. Microbiology. 1999;145(Pt. 10):2655–2662. doi: 10.1099/00221287-145-10-2655. [DOI] [PubMed] [Google Scholar]
- 136.Althorpe NJ, Chilley PM, Thomas AT, Brammar WJ, Wilkins BM. Transient transcriptional activation of the Incl1 plasmid anti-restriction gene (ardA) and SOS inhibition gene (psiB) early in conjugating recipient bacteria. Mol. Microbiol. 1999;31:133–142. doi: 10.1046/j.1365-2958.1999.01153.x. [DOI] [PubMed] [Google Scholar]
- 137.Nasim MT, Eperon IC, Wilkins BM, Brammar WJ. The activity of a single-stranded promoter of plasmid ColIb-P9 depends on its secondary structure. Mol. Microbiol. 2004;53:405–417. doi: 10.1111/j.1365-2958.2004.04114.x. [DOI] [PubMed] [Google Scholar]
- 138.Wilkins BM. Plasmid promiscuity: meeting the challenge of DNA immigration control. Environ. Microbiol. 2002;4:495–500. doi: 10.1046/j.1462-2920.2002.00332.x. [DOI] [PubMed] [Google Scholar]
- 139.Zavil'gel'skii GB, Rastorguev SM. Antirestriction proteins ardA and Ocr as effective inhibitors of the type I restriction-modification enzymes [in Russian] Mol. Biol. (Mosk) 2009;43:264–273. [PubMed] [Google Scholar]
- 140.Putnam CD, Tainer JA. Protein mimicry of DNA and pathway regulation. DNA Repair (Amst) 2005;4:1410–1420. doi: 10.1016/j.dnarep.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 141.Walkinshaw MD, Taylor P, Sturrock SS, Atanasiu C, Berge T, Henderson RM, Edwardson JM, Dryden DT. Structure of Ocr from bacteriophage T7, a protein that mimics B-form DNA. Mol. Cell. 2002;9:187–194. doi: 10.1016/s1097-2765(02)00435-5. [DOI] [PubMed] [Google Scholar]
- 142.Serfiotis-Mitsa D, Roberts GA, Cooper LP, White JH, Nutley M, Cooper A, Blakely GW, Dryden DT. The Orf18 gene product from conjugative transposon Tn916 is an ArdA antirestriction protein that inhibits type I DNA restriction-modification systems. J. Mol. Biol. 2008;383:970–981. doi: 10.1016/j.jmb.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 143.Oke M, Carter LG, Johnson KA, Liu H, McMahon SA, Yan X, Kerou M, Weikart ND, Kadi N, Sheikh MA, et al. The Scottish Structural Proteomics Facility: targets, methods and outputs. J. Struct. Funct. Genomics. 2010;11:167–180. doi: 10.1007/s10969-010-9090-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Larsen MH, Figurski DH. Structure, expression, and regulation of the kilC operon of promiscuous IncP alpha plasmids. J. Bacteriol. 1994;176:5022–5032. doi: 10.1128/jb.176.16.5022-5032.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Serfiotis-Mitsa D, Herbert AP, Roberts GA, Soares DC, White JH, Blakely GW, Uhrin D, Dryden DT. The structure of the KlcA and ArdB proteins reveals a novel fold and antirestriction activity against Type I DNA restriction systems in vivo but not in vitro. Nucleic Acids Res. 2010;38:1723–1737. doi: 10.1093/nar/gkp1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zabeau M, Friedman S, Van MM, Schell J. The ral gene of phage lambda. I. Identification of a non-essential gene that modulates restriction and modification in E. coli. Mol. Gen. Genet. 1980;179:63–73. doi: 10.1007/BF00268447. [DOI] [PubMed] [Google Scholar]
- 147.Winter M. Thesis/Dissertation. Edinburgh University; 1997. Investigation of de novo methylation activity in mutants of the EcoKI methyltransferase. [Google Scholar]
- 148.Dryden DT, Sturrock SS, Winter M. Structural modelling of a type I DNA methyltransferase. Nat. Struct. Biol. 1995;2:632–635. doi: 10.1038/nsb0895-632. [DOI] [PubMed] [Google Scholar]
- 149.Chilley PM, Wilkins BM. Distribution of the ardA family of antirestriction genes on conjugative plasmids. Microbiology. 1995;141(Pt. 9):2157–2164. doi: 10.1099/13500872-141-9-2157. [DOI] [PubMed] [Google Scholar]
- 150.Gefter M, Hausmann R, Gold M, Hurwitz J. The enzymatic methylation of ribonucleic acid and deoxyribonucleic acid. X. Bacteriophage T3-induced S-adenosylmethionine cleavage. J. Biol. Chem. 1966;241:1995–2006. [PubMed] [Google Scholar]
- 151.Hausmann R. Synthesis of an S-adenosylmethionine-cleaving enzyme in T3-infected Escherichia coli and its disturbance by co-infection with enzymatically incompetent bacteriophage. J. Virol. 1967;1:57–63. doi: 10.1128/jvi.1.1.57-63.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hausmann R, Messerschmid M. Inhibition of gene expression of T7-related phages by prophage P1. Mol. Gen. Genet. 1988;212:543–547. doi: 10.1007/BF00330862. [DOI] [PubMed] [Google Scholar]
- 153.Hausmann R. The T7 group. In: Calendar R, editor. The Bacteriophages. Vol. 1. New York: Plenum Publishing Corp; 1988. pp. 259–290. [Google Scholar]
- 154.Moffatt BA, Studier FW. Entry of bacteriophage T7 DNA into the cell and escape from host restriction. J. Bacteriol. 1988;170:2095–2105. doi: 10.1128/jb.170.5.2095-2105.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Garcia LR, Molineux IJ. Transcription-independent DNA translocation of bacteriophage T7 DNA into Escherichia coli. J. Bacteriol. 1996;178:6921–6929. doi: 10.1128/jb.178.23.6921-6929.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Molineux IJ. No syringes please, ejection of phage T7 DNA from the virion is enzyme driven. Mol. Microbiol. 2001;40:1–8. doi: 10.1046/j.1365-2958.2001.02357.x. [DOI] [PubMed] [Google Scholar]
- 157.Ratner D. The interaction bacterial and phage proteins with immobilized Escherichia coli RNA polymerase. J. Mol. Biol. 1974;88:373–383. doi: 10.1016/0022-2836(74)90488-4. [DOI] [PubMed] [Google Scholar]
- 158.Zavil'gel'skii GB, Kotova VI, Rastorguev SM. Antirestriction and antimodification activities of the T7 Ocr protein: effect of mutations in interface [in Russian] Mol. Biol. (Mosk) 2009;43:103–110. [PubMed] [Google Scholar]
- 159.Blackstock JJ, Egelhaaf SU, Atanasiu C, Dryden DT, Poon WC. Shape of Ocr, the gene 0.3 protein of bacteriophage T7: modeling based on light scattering experiments. Biochemistry. 2001;40:9944–9949. doi: 10.1021/bi010587+. [DOI] [PubMed] [Google Scholar]
- 160.Atanasiu C, Byron O, McMiken H, Sturrock SS, Dryden DT. Characterisation of the structure of ocr, the gene 0.3 protein of bacteriophage T7. Nucleic Acids Res. 2001;29:3059–3068. doi: 10.1093/nar/29.14.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hegde SS, Vetting MW, Roderick SL, Mitchenall LA, Maxwell A, Takiff HE, Blanchard JS. A fluoroquinolone resistance protein from Mycobacterium tuberculosis that mimics DNA. Science. 2005;308:1480–1483. doi: 10.1126/science.1110699. [DOI] [PubMed] [Google Scholar]
- 162.Stephanou AS, Roberts GA, Tock MR, Pritchard EH, Turkington R, Nutley M, Cooper A, Dryden DT. A mutational analysis of DNA mimicry by ocr, the gene 0.3 antirestriction protein of bacteriophage T7. Biochem. Biophys. Res. Commun. 2009;378:129–132. doi: 10.1016/j.bbrc.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 163.Stephanou AS, Roberts GA, Cooper LP, Clarke DJ, Thomson AR, MacKay CL, Nutley M, Cooper A, Dryden DT. Dissection of the DNA mimicry of the bacteriophage T7 Ocr protein using chemical modification. J. Mol. Biol. 2009;391:565–576. doi: 10.1016/j.jmb.2009.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Roberts GA, Stephanou AS, Kanwar N, Dawson A, Cooper LP, Chen K, Nutley M, Cooper A, Blakely GW, Dryden DT. Exploring the DNA mimicry of the Ocr protein of phage T7. Nucleic Acids Res. 2012;40:8129–8143. doi: 10.1093/nar/gks516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Su TJ, Tock MR, Egelhaaf SU, Poon WC, Dryden DT. DNA bending by M.EcoKI methyltransferase is coupled to nucleotide flipping. Nucleic Acids Res. 2005;33:3235–3244. doi: 10.1093/nar/gki618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.McMahon SA, Roberts GA, Johnson KA, Cooper LP, Liu H, White JH, Carter LG, Sanghvi B, Oke M, Walkinshaw MD, et al. Extensive DNA mimicry by the ArdA anti-restriction protein and its role in the spread of antibiotic resistance. Nucleic Acids Res. 2009;37:4887–4897. doi: 10.1093/nar/gkp478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zavil'gel'skii GV, Kotova VI, Rastorguev SM. Antimodification activity of the ArdA and Ocr proteins [in Russian] Genetika. 2011;47:159–167. [PubMed] [Google Scholar]
- 168.Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, Korlach J, Turner SW. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods. 2010;7:461–465. doi: 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Song CX, Clark TA, Lu XY, Kislyuk A, Dai Q, Turner SW, He C, Korlach J. Sensitive and specific single-molecule sequencing of 5-hydroxymethylcytosine. Nat. Methods. 2012;9:75–77. doi: 10.1038/nmeth.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Atanasiu C, Su TJ, Sturrock SS, Dryden DT. Interaction of the ocr gene 0.3 protein of bacteriophage T7 with EcoKI restriction/modification enzyme. Nucleic Acids Res. 2002;30:3936–3944. doi: 10.1093/nar/gkf518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Stern A, Sorek R. The phage-host arms race: shaping the evolution of microbes. Bioessays. 2011;33:43–51. doi: 10.1002/bies.201000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wright GD. Antibiotic resistance in the environment: a link to the clinic? Curr. Opin. Microbiol. 2010;13:589–594. doi: 10.1016/j.mib.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 173.David M, Borasio GD, Kaufmann G. Bacteriophage T4-induced anticodon-loop nuclease detected in a host strain restrictive to RNA ligase mutants. Proc. Natl Acad. Sci. USA. 1982;79:7097–7101. doi: 10.1073/pnas.79.23.7097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Penner M, Morad I, Snyder L, Kaufmann G. Phage T4-coded Stp: double-edged effector of coupled DNA and tRNA-restriction systems. J. Mol. Biol. 1995;249:857–868. doi: 10.1006/jmbi.1995.0343. [DOI] [PubMed] [Google Scholar]
- 175.Kaufmann G. Anticodon nucleases. Trends Biochem. Sci. 2000;25:70–74. doi: 10.1016/s0968-0004(99)01525-x. [DOI] [PubMed] [Google Scholar]
- 176.Klassen R, Wemhoff S, Krause J, Meinhardt F. DNA repair defects sensitize cells to anticodon nuclease yeast killer toxins. Mol. Genet. Genomics. 2011;285:185–195. doi: 10.1007/s00438-010-0597-5. [DOI] [PubMed] [Google Scholar]
- 177.Fu H, Feng J, Liu Q, Sun F, Tie Y, Zhu J, Xing R, Sun Z, Zheng X. Stress induces tRNA cleavage by angiogenin in mammalian cells. FEBS Lett. 2009;583:437–442. doi: 10.1016/j.febslet.2008.12.043. [DOI] [PubMed] [Google Scholar]
- 178.Jain R, Poulos MG, Gros J, Chakravarty AK, Shuman S. Substrate specificity and mutational analysis of Kluyveromyces lactis gamma-toxin, a eukaryal tRNA anticodon nuclease. RNA. 2011;17:1336–1343. doi: 10.1261/rna.2722711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Fukuyo M, Sasaki A, Kobayashi I. Success of a suicidal defense strategy against infection in a structured habitat. Sci. Rep. 2012;2:238. doi: 10.1038/srep00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Roberts RJ. Restriction and modification enzymes and their recognition sequences. Nucleic Acids Res. 1980;8:r63–r80. doi: 10.1093/nar/8.1.197-d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Roberts RJ. Restriction enzymes and their isoschizomers. Nucleic Acids Res. 1990;18(Suppl.):2331–2365. doi: 10.1093/nar/18.suppl.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Bullas LR, Colson C, Van PA. DNA restriction and modification systems in Salmonella. SQ, a new system derived by recombination between the SB system of Salmonella typhimurium and the SP system of Salmonella potsdam. J. Gen. Microbiol. 1976;95:166–172. doi: 10.1099/00221287-95-1-166. [DOI] [PubMed] [Google Scholar]
- 183.Fuller-Pace FV, Bullas LR, Delius H, Murray NE. Genetic recombination can generate altered restriction specificity. Proc. Natl Acad. Sci. USA. 1984;81:6095–6099. doi: 10.1073/pnas.81.19.6095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Fuller-Pace FV, Murray NE. Two DNA recognition domains of the specificity polypeptides of a family of type I restriction enzymes. Proc. Natl Acad. Sci. USA. 1986;83:9368–9372. doi: 10.1073/pnas.83.24.9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Gann AA, Campbell AJ, Collins JF, Coulson AF, Murray NE. Reassortment of DNA recognition domains and the evolution of new specificities. Mol. Microbiol. 1987;1:13–22. doi: 10.1111/j.1365-2958.1987.tb00521.x. [DOI] [PubMed] [Google Scholar]
- 186.Gubler M, Braguglia D, Meyer J, Piekarowicz A, Bickle TA. Recombination of constant and variable modules alters DNA sequence recognition by type IC restriction-modification enzymes. EMBO J. 1992;11:233–240. doi: 10.1002/j.1460-2075.1992.tb05046.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Price C, Lingner J, Bickle TA, Firman K, Glover SW. Basis for changes in DNA recognition by the EcoR124 and EcoR124/3 type I DNA restriction and modification enzymes. J. Mol. Biol. 1989;205:115–125. doi: 10.1016/0022-2836(89)90369-0. [DOI] [PubMed] [Google Scholar]
- 188.Abadjieva A, Patel J, Webb M, Zinkevich V, Firman K. A deletion mutant of the type IC restriction endonuclease EcoR1241 expressing a novel DNA specificity. Nucleic Acids Res. 1993;21:4435–4443. doi: 10.1093/nar/21.19.4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Meister J, MacWilliams M, Hubner P, Jutte H, Skrzypek E, Piekarowicz A, Bickle TA. Macroevolution by transposition: drastic modification of DNA recognition by a type I restriction enzyme following Tn5 transposition. EMBO J. 1993;12:4585–4591. doi: 10.1002/j.1460-2075.1993.tb06147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.MacWilliams MP, Bickle TA. Generation of new DNA binding specificity by truncation of the type IC EcoDXXI hsdS gene. EMBO J. 1996;15:4775–4783. [PMC free article] [PubMed] [Google Scholar]
- 191.Janscak P, Bickle TA. The DNA recognition subunit of the type IB restriction-modification enzyme EcoAI tolerates circular permutions of its polypeptide chain. J. Mol. Biol. 1998;284:937–948. doi: 10.1006/jmbi.1998.2250. [DOI] [PubMed] [Google Scholar]
- 192.Lluch-Senar M, Luong K, Llorens-Rico V, Delgado J, Fang G, Spittle K, Clark TA, Schadt E, Turner SW, Korlach J, et al. Comprehensive methylome characterization of Mycoplasma genitalium and Mycoplasma pneumoniae at single-base resolution. PLoS. Genet. 2013;9:e1003191. doi: 10.1371/journal.pgen.1003191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Davis BM, Chao MC, Waldor MK. Entering the era of bacterial epigenomics with single molecule real time DNA sequencing. Curr. Opin. Microbiol. 2013;16:192–198. doi: 10.1016/j.mib.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Shen BW, Xu D, Chan SH, Zheng Y, Zhu Z, Xu SY, Stoddard BL. Characterization and crystal structure of the type IIG restriction endonuclease RM.BpuSI. Nucleic Acids Res. 2011;39:8223–8236. doi: 10.1093/nar/gkr543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Callahan SJ, Morgan RD, Jain R, Townson SA, Wilson GG, Roberts RJ, Aggarwal AK. Crystallization and preliminary crystallographic analysis of the type IIL restriction enzyme MmeI in complex with DNA. Acta Crystallogr. Sect. F. Struct. Biol. Cryst. Commun. 2011;67:1262–1265. doi: 10.1107/S1744309111028041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Morgan RD, Luyten YA. Rational engineering of type II restriction endonuclease DNA binding and cleavage specificity. Nucleic Acids Res. 2009;37:5222–5233. doi: 10.1093/nar/gkp535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Gough JA, Murray NE. Sequence diversity among related genes for recognition of specific targets in DNA molecules. J. Mol. Biol. 1983;166:1–19. doi: 10.1016/s0022-2836(83)80047-3. [DOI] [PubMed] [Google Scholar]
- 198.Nagaraja V, Shepherd JC, Bickle TA. A hybrid recognition sequence in a recombinant restriction enzyme and the evolution of DNA sequence specificity. Nature. 1985;316:371–372. doi: 10.1038/316371a0. [DOI] [PubMed] [Google Scholar]
- 199.Glover SW, Firman K, Watson G, Price C, Donaldson S. The alternate expression of two restriction and modification systems. Mol. Gen. Genet. 1983;190:65–69. doi: 10.1007/BF00330325. [DOI] [PubMed] [Google Scholar]
- 200.Firman K, Price C, Glover SW. The EcoR124 and EcoR124/3 restriction and modification systems: cloning the genes. Plasmid. 1985;14:224–234. doi: 10.1016/0147-619x(85)90006-x. [DOI] [PubMed] [Google Scholar]
- 201.Price C, Pripfl T, Bickle TA. EcoR124 and EcoR124/3: the first members of a new family of type I restriction and modification systems. Eur. J. Biochem. 1987;167:111–115. doi: 10.1111/j.1432-1033.1987.tb13310.x. [DOI] [PubMed] [Google Scholar]
- 202.Furuta Y, Kawai M, Uchiyama I, Kobayashi I. Domain movement within a gene: a novel evolutionary mechanism for protein diversification. PLoS. One. 2011;6:e18819. doi: 10.1371/journal.pone.0018819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Wilson GG. Amino acid sequence arrangements of DNA-methyltransferases. Methods Enzymol. 1992;216:259–279. doi: 10.1016/0076-6879(92)16026-g. [DOI] [PubMed] [Google Scholar]
- 204.Malone T, Blumenthal RM, Cheng X. Structure-guided analysis reveals nine sequence motifs conserved among DNA amino-methyltransferases, and suggests a catalytic mechanism for these enzymes. J. Mol. Biol. 1995;253:618–632. doi: 10.1006/jmbi.1995.0577. [DOI] [PubMed] [Google Scholar]
- 205.Bujnicki JM, Radlinska M. Molecular evolution of DNA-(cytosine-N4) methyltransferases: evidence for their polyphyletic origin. Nucleic Acids Res. 1999;27:4501–4509. doi: 10.1093/nar/27.22.4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Radlinska M, Bujnicki JM. Site-directed mutagenesis defines the catalytic aspartate in the active site of the atypical DNA: m4C methyltransferase M.NgoMXV. Acta Microbiol. Pol. 2001;50:97–105. [PubMed] [Google Scholar]
- 207.Lunnen KD, Morgan RD, Timan CJ, Krzycki JA, Reeve JN, Wilson GG. Characterization and cloning of MwoI (GCN7GC), a new type-II restriction-modification system from Methanobacterium wolfei. Gene. 1989;77:11–19. doi: 10.1016/0378-1119(89)90354-5. [DOI] [PubMed] [Google Scholar]
- 208.Bujnicki JM. Sequence permutations in the molecular evolution of DNA methyltransferases. BMC Evol. Biol. 2002;2:3. doi: 10.1186/1471-2148-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Horton JR, Liebert K, Hattman S, Jeltsch A, Cheng X. Transition from nonspecific to specific DNA interactions along the substrate-recognition pathway of dam methyltransferase. Cell. 2005;121:349–361. doi: 10.1016/j.cell.2005.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Pues H, Bleimling N, Holz B, Wolcke J, Weinhold E. Functional roles of the conserved aromatic amino acid residues at position 108 (motif IV) and position 196 (motif VIII) in base flipping and catalysis by the N6-adenine DNA methyltransferase from Thermus aquaticus. Biochemistry. 1999;38:1426–1434. doi: 10.1021/bi9818016. [DOI] [PubMed] [Google Scholar]
- 211.Labahn J, Granzin J, Schluckebier G, Robinson DP, Jack WE, Schildkraut I, Saenger W. Three-dimensional structure of the adenine-specific DNA methyltransferase M.Taq I in complex with the cofactor S-adenosylmethionine. Proc. Natl Acad. Sci. USA. 1994;91:10957–10961. doi: 10.1073/pnas.91.23.10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Schluckebier G, Kozak M, Bleimling N, Weinhold E, Saenger W. Differential binding of S-adenosylmethionine S-adenosylhomocysteine and Sinefungin to the adenine-specific DNA methyltransferase M.TaqI. J. Mol. Biol. 1997;265:56–67. doi: 10.1006/jmbi.1996.0711. [DOI] [PubMed] [Google Scholar]
- 213.Bitinaite J, Maneliene Z, Menkevicius S, Klimasauskas S, Butkus V, Janulaitis A. Alw26I, Eco31I and Esp3I–type IIs methyltransferases modifying cytosine and adenine in complementary strands of the target DNA. Nucleic Acids Res. 1992;20:4981–4985. doi: 10.1093/nar/20.19.4981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Zhu Z, Samuelson JC, Zhou J, Dore A, Xu SY. Engineering strand-specific DNA nicking enzymes from the type IIS restriction endonucleases BsaI, BsmBI, and BsmAI. J. Mol. Biol. 2004;337:573–583. doi: 10.1016/j.jmb.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 215.Malygin EG, Evdokimov AA, Hattman S. Dimeric/oligomeric DNA methyltransferases: an unfinished story. Biol. Chem. 2009;390:835–844. doi: 10.1515/BC.2009.082. [DOI] [PubMed] [Google Scholar]
- 216.Marks P, McGeehan J, Wilson G, Errington N, Kneale G. Purification and characterisation of a novel DNA methyltransferase, M.AhdI. Nucleic Acids Res. 2003;31:2803–2810. doi: 10.1093/nar/gkg399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Callow P, Sukhodub A, Taylor JE, Kneale GG. Shape and subunit organisation of the DNA methyltransferase M.AhdI by small-angle neutron scattering. J. Mol. Biol. 2007;369:177–185. doi: 10.1016/j.jmb.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Janulaitis A, Petrusyte M, Maneliene Z, Klimasauskas S, Butkus V. Purification and properties of the Eco57I restriction endonuclease and methylase–prototypes of a new class (type IV) Nucleic Acids Res. 1992;20:6043–6049. doi: 10.1093/nar/20.22.6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Wah DA, Hirsch JA, Dorner LF, Schildkraut I, Aggarwal AK. Structure of the multimodular endonuclease FokI bound to DNA. Nature. 1997;388:97–100. doi: 10.1038/40446. [DOI] [PubMed] [Google Scholar]
- 220.Wah DA, Bitinaite J, Schildkraut I, Aggarwal AK. Structure of FokI has implications for DNA cleavage. Proc. Natl Acad. Sci. USA. 1998;95:10564–10569. doi: 10.1073/pnas.95.18.10564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Xu SY, M.R.B.J. (USA patent no. 6593122). Ref Type: Patent www.patentstorm.us/patents/6593122.
- 222.Kong H, Morgan RD, Maunus RE, Schildkraut I. A unique restriction endonuclease, BcgI, from Bacillus coagulans. Nucleic Acids Res. 1993;21:987–991. doi: 10.1093/nar/21.4.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Kong H, Roemer SE, Waite-Rees PA, Benner JS, Wilson GG, Nwankwo DO. Characterization of BcgI, a new kind of restriction-modification system. J. Biol. Chem. 1994;269:683–690. [PubMed] [Google Scholar]
- 224.Kong H, Smith CL. Substrate DNA and cofactor regulate the activities of a multi-functional restriction-modification enzyme, BcgI. Nucleic Acids Res. 1997;25:3687–3692. doi: 10.1093/nar/25.18.3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Kong H. Thesis/Dissertation. Boston University; 1998. Characterization of a new restriction-modification system, the BcgI system of Bacillus coagulans; pp. 1–130. [Google Scholar]
- 226.Marshall JJ, Halford SE. The type IIB restriction endonucleases. Biochem. Soc. Trans. 2010;38:410–416. doi: 10.1042/BST0380410. [DOI] [PubMed] [Google Scholar]
- 227.Sears LE, Zhou B, Aliotta JM, Morgan RD, Kong H. BaeI, another unusual BcgI-like restriction endonuclease. Nucleic Acids Res. 1996;24:3590–3592. doi: 10.1093/nar/24.18.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Marshall JJ, Gowers DM, Halford SE. Restriction endonucleases that bridge and excise two recognition sites from DNA. J. Mol. Biol. 2007;367:419–431. doi: 10.1016/j.jmb.2006.12.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Marshall JJ, Smith RM, Ganguly S, Halford SE. Concerted action at eight phosphodiester bonds by the BcgI restriction endonuclease. Nucleic Acids Res. 2011;39:7630–7640. doi: 10.1093/nar/gkr453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Smith RM, Marshall JJ, Jacklin AJ, Retter SE, Halford SE, Sobott F. Organization of the BcgI restriction-modification protein for the cleavage of eight phosphodiester bonds in DNA. Nucleic Acids Res. 2013;41:391–404. doi: 10.1093/nar/gks1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Smith RM, Jacklin AJ, Marshall JJ, Sobott F, Halford SE. Organization of the BcgI restriction-modification protein for the transfer of one methyl group to DNA. Nucleic Acids Res. 2013;41:405–417. doi: 10.1093/nar/gks1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Piekarowicz A, Golaszewska M, Sunday AO, Siwinska M, Stein DC. The HaeIV restriction modification system of Haemophilus aegyptius is encoded by a single polypeptide. J. Mol. Biol. 1999;293:1055–1065. doi: 10.1006/jmbi.1999.3198. [DOI] [PubMed] [Google Scholar]
- 233.Vitor JM, Morgan RD. Two novel restriction endonucleases from Campylobacter jejuni. Gene. 1995;157:109–110. doi: 10.1016/0378-1119(94)00668-i. [DOI] [PubMed] [Google Scholar]
- 234.Cesnaviciene E, Petrusyte M, Kazlauskiene R, Maneliene Z, Timinskas A, Lubys A, Janulaitis A. Characterization of AloI, a restriction-modification system of a new type. J. Mol. Biol. 2001;314:205–216. doi: 10.1006/jmbi.2001.5049. [DOI] [PubMed] [Google Scholar]
- 235.Vitor JMB A.P.M.M.V.J. Helicobacter pylori type IIG restriction and modification loci. Helicobacter. 2011;16:98. [Google Scholar]
- 236.Jurenaite-Urbanaviciene S, Serksnaite J, Kriukiene E, Giedriene J, Venclovas C, Lubys A. Generation of DNA cleavage specificities of type II restriction endonucleases by reassortment of target recognition domains. Proc. Natl Acad. Sci. USA. 2007;104:10358–10363. doi: 10.1073/pnas.0610365104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Boyd AC, Charles IG, Keyte JW, Brammar WJ. Isolation and computer-aided characterization of MmeI, a type II restriction endonuclease from Methylophilus methylotrophus. Nucleic Acids Res. 1986;14:5255–5274. doi: 10.1093/nar/14.13.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Morgan RD, Bhatia TK, Lovasco L, Davis TB. MmeI: a minimal Type II restriction-modification system that only modifies one DNA strand for host protection. Nucleic Acids Res. 2008;36:6558–6570. doi: 10.1093/nar/gkn711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Nakonieczna J, Kaczorowski T, Obarska-Kosinska A, Bujnicki JM. Functional analysis of MmeI from methanol utilizer Methylophilus methylotrophus, a subtype IIC restriction-modification enzyme related to type I enzymes. Appl. Environ. Microbiol. 2009;75:212–223. doi: 10.1128/AEM.01322-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Dryden DT, Edwardson JM, Henderson RM. DNA translocation by type III restriction enzymes: a comparison of current models of their operation derived from ensemble and single-molecule measurements. Nucleic Acids Res. 2011;39:4525–4531. doi: 10.1093/nar/gkq1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Schwarz FW, van AK, Toth J, Seidel R, Szczelkun MD. DNA cleavage site selection by Type III restriction enzymes provides evidence for head-on protein collisions following 1D bidirectional motion. Nucleic Acids Res. 2011;39:8042–8051. doi: 10.1093/nar/gkr502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Morgan RD, Dwinell EA, Bhatia TK, Lang EM, Luyten YA. The MmeI family: type II restriction-modification enzymes that employ single-strand modification for host protection. Nucleic Acids Res. 2009;37:5208–5221. doi: 10.1093/nar/gkp534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Halford SE. Hopping, jumping and looping by restriction enzymes. Biochem. Soc. Trans. 2001;29:363–374. doi: 10.1042/bst0290363. [DOI] [PubMed] [Google Scholar]
- 244.Halford SE, Bilcock DT, Stanford NP, Williams SA, Milsom SE, Gormley NA, Watson MA, Bath AJ, Embleton ML, Gowers DM, et al. Restriction endonuclease reactions requiring two recognition sites. Biochem. Soc. Trans. 1999;27:696–699. doi: 10.1042/bst0270696. [DOI] [PubMed] [Google Scholar]
- 245.Smith RM, Diffin FM, Savery NJ, Josephsen J, Szczelkun MD. DNA cleavage and methylation specificity of the single polypeptide restriction-modification enzyme LlaGI. Nucleic Acids Res. 2009;37:7206–7218. doi: 10.1093/nar/gkp790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Smith RM, Josephsen J, Szczelkun MD. An Mrr-family nuclease motif in the single polypeptide restriction-modification enzyme LlaGI. Nucleic Acids Res. 2009;37:7231–7238. doi: 10.1093/nar/gkp795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.vanAelst K, Sisakova E, Szczelkun MD. DNA cleavage by Type ISP Restriction-Modification enzymes is initially targeted to the 3′-5′ strand. Nucleic Acids Res. 2013;41:1081–1090. doi: 10.1093/nar/gks1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Sisakova E, van AK, Diffin FM, Szczelkun MD. The Type ISP Restriction-Modification enzymes LlaBIII and LlaGI use a translocation-collision mechanism to cleave non-specific DNA distant from their recognition sites. Nucleic Acids Res. 2013;41:1071–1080. doi: 10.1093/nar/gks1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Smith RM, Josephsen J, Szczelkun MD. The single polypeptide restriction-modification enzyme LlaGI is a self-contained molecular motor that translocates DNA loops. Nucleic Acids Res. 2009;37:7219–7230. doi: 10.1093/nar/gkp794. [DOI] [PMC free article] [PubMed] [Google Scholar]