

Abstract
MicroRNAs (miRNA) are a novel class of small, noncoding RNA molecules that have gained the attention of many researchers in recent years due to their ability to posttranscriptionally regulate the expression of families of genes simultaneously. Their role in normal physiology and pathobiology is intriguing and their regulation in normal and disease states is fascinating. That the cells can return to a state of homeostasis when these small molecules are perturbed is truly remarkable given the multiple cellular targets of each miRNA and that many mRNAs are targeted by multiple miRNAs. Several reviews have covered aspects of miRNA function in biology and disease. Here, we review the role of miRNA in regulating the renin-angiotensin system, AGE/RAGE signalling, and under conditions of oxidative stress in the context of diabetic nephropathy.
1. Introduction
The World Health Organization states that ~347 million people, roughly 9.5% of the adult population, were suffering from diabetes in 2008 [1]. The incidence of diabetes is rapidly increasing with estimates suggesting that this number will almost double by 2030. Diabetes mellitus is a major cause of chronic kidney disease (CKD) worldwide and is associated with enhanced morbidity and mortality, in particular accelerated cardiovascular disease [2, 3].
Diabetic nephropathy (DN) is now the most common cause of end-stage renal failure in the Western world [4]. Clinical associations that frequently precede overt DN are hypertension and poor glycaemic control [5], although a subset of patients develop nephropathy despite the proper glycemic control [6] and normal blood pressure. Once nephropathy is established, blood pressure often rises further, but glycaemic control can paradoxically improve as a result of reduced renal insulin clearance [7]. It is postulated that the interplay between metabolic and hemodynamic pathways plays an important role in the development and progression of DN [8] (Figure 1). Increased systemic and intraglomerular pressure is associated with increased albuminuria and glomerular injury. Activation of the renin-angiotensin-aldosterone system (RAAS) has been recognized as a key component of DN progression. Additionally, chronic hyperglycemia promotes the generation of advanced glycation end-products (AGEs). It is widely accepted that AGEs mediate their effects both directly and indirectly through receptor-dependent mechanisms. The receptor for AGE (RAGE) acts as a signal transduction receptor, and the RAGE-AGE interaction activates multiple intracellular signalling pathways which increase the production of growth factors, inflammatory cytokines, and oxidative stress (Figure 1).
Figure 1.
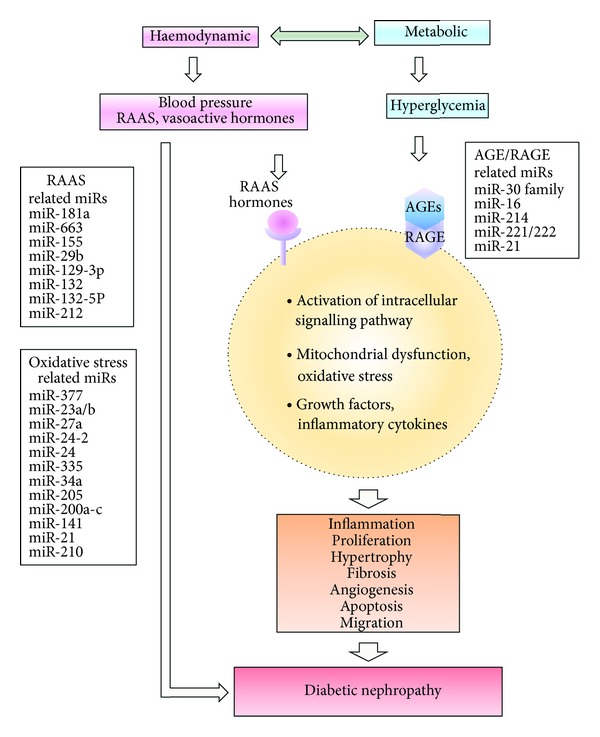
Schematic overview of mechanisms and microRNAs that are related to RAAS, AGE/RAGE, or oxidative stress contributing to diabetic nephropathy. AGE: advanced glycation end-product; miR: microRNA; RAAS: renin-angiotensin-aldosterone system; RAGE: receptor of AGE.
In recent times, a novel class of non-coding RNA, microRNA (miRNA; miR), has been found to be expressed in all tissues and plays important roles in tissue homeostasis and disease progression [9, 10]. Whilst the role of miRNA in the pathogenesis of DN has been extensively reviewed by others in relation to growth factors and fibrosis in DN, the focus of this review is on the role of miRNA in the renin-angiotensin system and the AGE/RAGE signalling pathway, and their downstream the mediators such as oxidative stress and the immune response in the context of diabetic nephropathy.
2. Biogenesis and Function of miRNA
miRNAs are a group of small (~22 nucleotide) single-stranded non-coding RNAs ubiquitously expressed in plants and animals where they act posttranscriptionally to modulate the expression of target genes [11, 12]. They were first discovered in 1993 when lin-14 protein expression was found to be regulated by the mature product of the lin-4 gene in Caenorhabditis Elegans (C. Elegans) [13]. The mechanism was found to rely upon sequence specificity in the 3′-untranslated region (UTR) of lin-14 to which lin-4 bound. Whilst this occurred with only partial complementarity, the binding occurred with sufficient affinity to result in inhibition of protein translation. At the time this was considered a peculiarity in the worm. It was not until 2000 when another such regulator, let-7, was discovered in C. Elegans [14], and soon after came the realisation that this form of regulation was conserved across many species, representing a general mechanism for regulating the expression of several genes.
miRNAs are found in intergenic sequences or on the antisense strand of genes and may possess their own promoter and regulatory sequences [15, 16]. Other miRNAs (almost 50% in the case of human miRNA) are found within gene sequences and are together regulated with their host gene [17–19]. Approximately half are found in polycistronic units from which the mature miRNAs are processed [16]. The primary transcript (pri-miRNA) is processed by a number of proteins including ribonuclease III, Drosha, and the RNA binding protein, DiGeorge syndrome critical region gene 8 (DGCR-8) protein, into a short hairpin RNA molecule termed the precursor miRNA (pre-miRNA) which is subsequently exported from the nucleus by exportin-5 [20–22]. Once in the cytosol, further processing is mediated by another ribonuclease III, Dicer, and this is followed by the incorporation of the mature strand of the duplex miRNA into the RNA-induced silencing complex (RISC) which includes the Argonaute family of proteins [23]. The miRNA-RISC complex stabilises the miRNA against nuclease attack and miRNA direct the complex to target RNA transcripts via sequence complimentarity between the miRNA seed sequence (2–8 nucleotides) and the miRNA recognition element (MRE) in the target transcript [20]. miRNAs modulate protein synthesis by binding to the 3′ UTR of mRNAs via incomplete base pairing [24] to the complementary seed sequence in the UTR, leading to translational repression. MREs are predominately located in the 3′ UTRs of mammalian mRNA, however; there is evidence indicating that miRNA can also mediate translational repression via binding 5′ UTRs or coding sequences [25, 26].
In plants and lower vertebrates where complete complimentarity exists between an miRNA seed sequence and the target mRNA, degradation of the target mRNA is induced. In contrast, miRNAs exercise finer control on protein expression in mammalian cells where they repress protein translation [27, 28], allowing for more comprehensive regulation of protein expression as any single miRNA can target many mRNAs, and any mRNA can be the target of several miRNAs [27]. This potential of miRNAs to regulate many genes adds significant complexity to the interpretation of many studies where the focus is on single genes. It is therefore critical to computationally detect the combination of miRNAs that target specific mRNA molecules. Such analyses often demand complex algorithms that need to be defined by high stringency levels to generate computational data that could then be assessed and validated in the wet lab. Added to this is the recent discovery of circulating miRNAs in microparticles [29] and the potential of these miRNAs to be delivered to sites distal to their generation.
3. miRNA and the Renin-Angiotensin System in Kidney Disease
Blood pressure is regulated by the renin-angiotensin-aldosterone system. Renin production is the key regulatory step that initiates an enzymatic cascade that leads to angiotensin generation and the control of blood pressure in addition to fluid and electrolyte homeostasis [30]. Renin is synthesized and released by renal juxtaglomerular (JG) cells, which are located at the entrance to the glomerulus. In early embryonic development, renin producing cells are broadly distributed along intrarenal arteries in the glomerular mesangium and in a few developing tubules. With maturation the cells become progressively restricted to the afferent arterioles. Lineage studies demonstrate that this progressive restriction is achieved by differentiation of renin cells into vascular smooth muscle cells (VSMCs), mesangial cells, and a subset of tubular cells [31].
Sequeira-Lopez et al. were the first to address the role of miRNA in the maintenance of JG cells by generating mice with a conditional deletion of Dicer (Dicer cKO mice) specifically and exclusively in renin-expressing cells, resulting in selective ablation of miRNA generation only in renin cells and their descendants [32]. Deletion of Dicer resulted in severe reduction in the number of JG cells accompanied by decreased expression of the renin Ren1 and Ren2 genes, decreased plasma renin concentration (PRC), decreased BP, abnormal renal function, and striking nephrovascular abnormalities including striped corticomedullary fibrosis. This study clearly showed a critical role of miRNA in the orchestration of renal function and the importance of miRNA homeostasis for specific organ functions.
The kidney has long been invoked in the etiology of essential hypertension. This could involve alterations in expression of specific genes and miRNA. Marques and colleagues conducted for the first time transcriptome-wide study of differential expression of mRNAs and miRNA in kidneys of hypertensive subjects (15 untreated hypertensive and 7 normotensive white males) by microarray technology [33]. They confirmed differences of expression for nuclear receptor subfamily 4 group A member 1 (NR4A1), NR4A2, NR4A3, period circadian protein homolog 1 (PER1), and salt-inducible kinase 1 (SIK1) mRNAs and for the miRNAs hsa-miR-638 and hsa-let-7c by real-time quantitative PCR expression in the medulla. Functional experiments confirmed the predicted binding of hsa-let-7c to the 3′ untranslated region of NR4A2 mRNA. In the renal cortex they confirmed differences in expression of apoptosis-inducing factor, mitochondrion-associated, 1 (AIFM1), alpha-1-microglobulin/bikunin precursor (AMBP), apolipoprotein E (APOE), cluster of differentiation 36 (CD36), ephrin-B1 (EFNB1), NADH dehydrogenase (ubiquinone) complex I, assembly factor 1 (NDUFAF1), peroxiredoxin 5 (PRDX5), REN, renin binding protein (RENBP), solute carrier family 13 (sodium/sulfate symporters), member 1 (SLC13A1), syntaxin 4 (STX4), and troponin T type 2 (cardiac) (TNNT2) mRNAs, and the miRNAs hsa-miR-21, hsa-miR-126, hsa-miR-181a, hsa-miR-196a, hsa-miR-451, hsa-miR-638, and hsa-miR-663. Functional experiments demonstrated that hsa-miR-663 can bind to the REN and APOE 3′ untranslated regions regulating REN and APOE mRNA levels, whereas hsa-miR-181a regulated REN and AIFM1 mRNA. A major discovery was evidence for REN mRNA regulation via binding of miRNAs hsa-miR-181a and hsa-miR-663 to its 3′ UTR as the observed downregulation of these 2 miRNAs in hypertension could explain the elevation in intrarenal renin mRNA. Due to small sampling size, these findings need to be bolstered by the acquisition of suitable samples from larger cohorts of untreated hypertensive subjects in other settings.
Chronic kidney disease (CKD) is a known cardiovascular risk factor, and most patients with CKD die of cardiovascular disease (CVD) before reaching the need for dialysis [37]. Chen and colleagues measured vascular miRNAs in blood from 90 patients with CKD and found an association between decreased circulating levels and progressive loss of eGFR by multivariate analyses [34]. Expression of vascular miRNAs was decreased in the thoracic aorta of CKD rats compared to normal rats, with concordant changes in target genes of RUNX2, AT1R, and myocardin with no alteration in DROSHA or DICER, indicating that the low levels of expression are not due to altered intracellular processing. Furthermore, the expression of miR-155 was negatively correlated with calcification of the aorta, a process known to be preceded by vascular dedifferentiation in these animals. Overexpression of miR-155 in VSMC from CKD rats inhibited AT1R expression and decreased cellular proliferation, confirming a causative role of low miR155 in VSMC transformation to a more synthetic, proliferative phenotype. However, whether downregulation of these miRNAs is the cause or consequence of the widespread vascular phenotype abnormalities in patients with CKD remains to be determined.
Jeppesen and colleagues have shown that AngII regulates five miRNAs (miR-29b, -129-3p, -132, -132-5P, and -212) during in vitro stimulation of primary cardiac fibroblasts and of HEK293N cells overexpressing the AT1R [35]. Furthermore, Eskildsen and colleagues undertook a detailed analysis of potential miRNAs involved in AngII-mediated hypertension in rats and hypertensive patients, using miRNA microarray and qPCR analysis. miR-132 and miR-212 are highly increased in the heart, aortic wall, and kidneys of rats with hypertension (159 ± 12 mm Hg) and cardiac hypertrophy following chronic AngII infusion. In addition, activation of the endothelin receptor, another Gαq coupled receptor, also increased miR-132 and miR-212 [36]. A significant downregulation of miR-132 as well as a robust attenuation of miR-212 in human arteries from the ARB-treated patients was also observed, whereas treatment with β-blockers had no effect. In conclusion, miR-132 and miR-212 are upregulated in AngII-induced hypertension in organs associated with blood pressure control, possibly via the Gαq-dependent pathway.
Table 1 contains miRNAs that are known to be regulated by the renin-angiotensin system in the kidney and other tissues and associated with kidney disease, hypertension, and cardiovascular disease.
Table 1.
Relevant miRNA in RAAS.
| miRNAs | Tissue/organ/cell line | Source | Target | Functions | Reference |
|---|---|---|---|---|---|
| miR-181a | Kidney | Hypertensive patients | REN, AIFM1 | Hypertension | [33] |
| miR-663 | REN, APOE | ||||
|
| |||||
| miR-155 | Blood | CKD patients | AT1R | Hypertension, cardiovascular disease | [34] |
|
| |||||
| miR-29b, -129-3p, -132, -132-5P and -212 | HEK293N cells, cardiac fibroblasts | Human, Rat | Associated with cardiovascular disease | [35] | |
|
| |||||
| miR-132/212 | Heart, aortic wall, and kidneys | Rat | Associated with blood pressure control | [36] | |
| Artery | ARB-treated patients | ||||
AIFM1: apoptosis-inducing factor, mitochondrion-associated, 1; APOE: apolipoprotein E; AT1R: angiotensin II receptor, type 1; CKD: chronic kidney disease; HEK: human embryonic kidney cells; RAAS: renin-angiotensin-aldosterone system; REN: renin.
4. miRNAs in AGE/RAGE and Kidney Disease
Chronic hyperglycemia promotes the generation of advanced glycation end products (AGEs) as a result of sequential biochemical reactions involving nonenzymatic glycation of protein and lipids known as the Maillard reaction [8]. As a consequence of AGE formation, there is often concomitant liberation of reactive oxygen species (ROS) [38]. AGEs can induce expression of the MCP-1 in podocytes through activation of the AGE receptor (RAGE) and generation of intracellular reactive oxygen species (ROS) [39]. The induction of oxidative stress results in upregulation of nuclear factor (NF)-κB and various NF-κB-mediated proinflammatory genes, eventually leading to glomerular and tubulointerstitial injury. Therefore, AGE/RAGE and oxidative stress signalling are important in the progression of DN and targeting this axis by modulating the miRNAs that are involved is a potential new therapy.
There are very few studies examining the role of miRNA in AGE/RAGE signalling related to kidney disease. Most studies have focussed on the role of RAGE in the immune system or cancer because RAGE is a member of the immunoglobulin superfamily of cell surface molecules. When Dicer was selectively inactivated in mouse podocytes, multiple abnormalities were observed in glomeruli of mutant mice, including foot process effacement, irregular and split areas of the glomerular basement membrane, podocyte apoptosis and depletion, mesangial expansion, capillary dilation, and glomerulosclerosis [40]. Gene profiling by microarray analyses revealed upregulation of 190 genes in glomeruli isolated from mutant mice at the onset of proteinuria compared with control littermates. Target sequences for 16 miRNAs were significantly enriched in the 3′-untranslated regions of the 190 upregulated genes. Further, supporting the validity of the in silico analysis, 6 of the 8 top-candidate miRNAs were identified in miRNA libraries generated from podocyte cultures; these included miR-28, miR-34a, and four members of the miR-30 family, miR-30c-1, miR-30b, miR-30d, and miR-30c-2. Among the 15 upregulated target genes of the miR-30 miRNA family, RAGE, vimentin, heat-shock protein 20, and immediate early response 3 were known to be expressed in injured podocytes in experimental models and human kidney disease. RAGE and immediate early response 3 are known to mediate podocyte apoptosis, whereas vimentin and heat-shock protein 20 are involved in cytoskeletal structure [40]. The findings demonstrate the important roles of the mir-30 family in podocyte homeostasis and podocytopathies.
AGE/RAGE interaction induces inflammatory genes such as cyclooxygenase-2 (COX-2) [41]. S100b, a RAGE ligand, significantly increased COX-2 mRNA accumulation in THP-1 monocytes at 2 h via mRNA stability. S100b decreased occupancy of the DNA/RNA-binding protein, heterogeneous nuclear ribonuclear protein K (hnRNPK), at the COX-2 promoter but simultaneously increased its binding to the COX-2 3′-UTR. Additionally, S100b significantly downregulated the expression miR-16 which acts to destabilize COX-2 mRNA by binding to its 3′-UTR. hnRNPK knockdown increased miR-16 binding to COX-2 3′-UTR indicating a crosstalk between them. These results demonstrate that diabetic stimuli can efficiently stabilize inflammatory genes via opposing actions of key RNA-binding proteins and miRNA [41].
AGEs delay spontaneous apoptosis of monocytes and contribute to the development of inflammatory responses [42]. In genome-wide miRNA expression analysis significant upregulation of miR-214 was consistently observed in THP-1 and human monocytes treated with various AGEs. A striking increase in miR-214 was also detected in monocytes from patients with chronic renal failure. PTEN was identified as a target gene of miR-214. Overexpression of pre-miR-214 led to impaired PTEN expression and delayed apoptosis of THP-1 cells, whereas knockdown of miR-214 levels largely abolished AGE-induced cell survival. These findings define a role for miR-214-targeting of PTEN in AGE-induced monocyte survival [42].
High-mobility group box 1 protein (HMGB1) is a late inflammatory cytokine that signals danger to the immune system through the RAGE and Toll-like receptor [43]. miR-221 and miR-222 are involved in cell proliferation through the inhibition of the cell cycle regulator, p27kip1, in smooth muscle cells [44] and endothelial cells (ECs) [45]. HMGB1 increases the expression of miR-221 and miR-222 in primary cultures of excised papillary lesions and in an established papillary cancer cell line and overexpression of miR-222 and miR-221 caused by HMGB1 increases growth and motility in papillary thyroid cancer cells. Recently, it was reported that miR-21 and miR-221, which are tissue inhibitors of metalloproteinases (TIMP)3-targeting miRNA, were significantly upregulated in kidneys from diabetic mice compared to control littermates and in a mesangial cell line grown in high glucose conditions [46]. Mesangial expansion is one of the main characters in DN and is mainly due to accumulation of extracellular matrix (ECM). ECM turnover is regulated by TIMPs activities. In diabetic conditions, TIMP3 expression in kidney is strongly reduced, but the causes of this reduction are still unknown. miR-221/miR-222 cluster which has been examined in several cardiovascular disorders affects the angiogenic activity of stem cell factor (SCF) by targeting the receptor c-Kit [47]. High glucose levels elevate miR-221 and this correlates with decreased expression of c-kit and reduced EC migration [48]. miR-221/222 is also considered to be important in atherosclerosis. miR-221/222 targets NO synthase and transcription factor ETS-1 which are both major contributors to the atherosclerotic process. [47, 49, 50] Taken together, these results indicate that HMGB1/RAGE and miR-221/222 axis may be activated in the kidney and vasculature resulting in ECM accumulation and atherosclerosis in DN patients.
Table 2 lists the miRNAs that are known to be modulated by the AGE/RAGE in kidney, monocytes, VSMCs, and ECs and associated with kidney disease and vascular dysfunction.
Table 2.
Relevant miRNA in AGE/RAGE.
| miRNAs | Tissue/organ/cell line | Source | Target | Functions | Reference |
|---|---|---|---|---|---|
| miR-30 family | Podocyte | Mice | AGER, Vim, HSP20, Ier3 | Podocyte homeostasis and podocytopathies | [40] |
|
| |||||
| miR-16 | THP-1 monocytic cells | Human | COX-2 | Regulate inflammation | [41] |
|
| |||||
| miR-214 | THP-1 monocytic cells | Human | PTEN | Monocyte survival | [42] |
|
| |||||
| miR-221/222 | VSMCs | Rat | p27Kip1 and p57Kip2 | Cell proliferation | [44] |
|
| |||||
| miR-221/222 | Endothelial cells | Human | p27Kip1 and p57Kip2 | Cell proliferation | [45] |
|
| |||||
| miR-21 and miR-221 | MES 13 mesangial cells | Mice | Timp3 | DN progression | [46] |
| miR-21 | Kidney | Human | |||
|
| |||||
| miR-221/miR-222 | HUVECs | Human | c-Kit | Reduced EC migration | [48] |
AGE: advanced glycation end-product; AGER: advanced glycosylation end-product-specific receptor; c-kit: V-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog; COX: Cyclooxygenase; DN: diabetic nephropathy; EC: endothelial cells; HSP: Heat shock protein; HUVECs: human umbilical vein endothelial cells; Ier3: immediate early response 3; p27Kip1: Cyclin-Dependent Kinase Inhibitor 1B; p57Kip2: Cyclin-Dependent Kinase Inhibitor 1C; PTEN: phosphatase and tensin homolog; RAGE: Receptor for AGE; Timp: metallopeptidase inhibitor; Vim: Vimentin; VSMCs: vascular smooth muscle cells.
5. miRNAs in Oxidative Stress and Kidney Disease
Reactive oxygen species (ROS) is a collective term that includes a number of reactive and partially reduced oxygen (O2) metabolites. Some of them are free radicals, such as superoxide anion (O2 −) and hydroxyl radicals (•OH) that are extremely reactive molecular species with an unpaired electron in their outer orbital. A number of studies have demonstrated that both type 1 and type 2 diabetes are associated with overproduction of oxygen-derived free radicals. Increased ROS production and reduced levels of antioxidants culminate with an in increased level of oxidative stress leading to oxidative damage to cellular components [51]. Among the many enzymatic systems implicated in ROS generation in vascular tissues, enzymes of the mitochondrial respiratory chain (complexes I and III), xanthine oxidase, uncoupled nitric oxide synthase, and nicotinamide adenine dinucleotide phosphate reduced form (NADPH) oxidase (NOX) appear to be particularly important [52–54]. Increased NOX-mediated superoxide production has been reported in experimental models of diabetes and occurs in parallel with upregulation of NOX1 and NOX4 [55, 56]. NOX-mediated generation of superoxide is an important mediator of matrix accumulation, renal fibrosis, and podocyte injury in DN [57].
miR-377 is upregulated in spontaneous and STZ-induced mouse models of DN and in mesangial cells exposed to high glucose and TGF-β1 [58]. Stable overexpression of miR-377 in mesangial cells increased fibronectin synthesis. Furthermore, genes potentially relevant to the pathogenesis of DN were confirmed experimentally, including the cytoskeletal regulator p21-activated kinase 1 (PAK1) and superoxide dismutases (SOD1/SOD2) that catalyse ROS, which accumulates in response to hyperglycemia [58].
It has previously been shown that upregulation of the miR-23a~27a~24-2 cluster induces caspase-dependent and caspase-independent cell death in human embryonic kidney cells via c-jun N-terminal kinase (JNK) with increases in ROS and the release of proapoptotic factors such as cytochrome c (cyt c) in addition to apoptosis-inducing factor (AIF) from the intermembrane space of mitochondria to the cytosol [59]. In order to better understand the molecular mechanism responsible for miR-23a~27a~24-2 cluster-induced cell death, gene expression profiling was performed in control and miR-23a~27a~24-2 cluster overexpressing HEK293T cells. This revealed miR-23a~27a~24-2 cluster-induced apoptosis was associated with endoplasmic reticulum (ER) stress and the unfolded protein response (UPR) pathways in HEK293T cells. Overexpression of the miR-23a~27a~24-2 cluster resulted in ER stress and altered mitochondrial membrane permeability and this was further established by increased intracellular and mitochondrial calcium levels in HEK293T cells [60].
There have been reports that miR-23b levels were increased in the kidneys of TGF-β1 transgenic mice and rats following subtotal nephrectomy, which was found to be localized to podocytes and tubular epithelium by in situ hybridization. miR-23b was also upregulated by TGF-β1 in cultured renal epithelial cells. In vitro gain and loss-of-function studies pointed to several miR-23b targets, including TGF-β receptor type II, SMAD3, and TGF-β1 itself, suggesting a negative feedback loop-regulating TGF-β1 signaling. Modulation of miR-23b in cultured podocytes altered expression of WT1, nephrin, and podocin and also influenced motility of cultured tubular cells [61].
Studies have found that in the aged organs, including the kidney, expression of antioxidant enzymes such as SOD, catalase, Gpx, and peroxiredoxins is downregulated [62, 63] thus leading to reduced antioxidant capacity. From bioinformatic analysis of the miRNA expression profile of young and old rat kidneys, mitochondrial SOD2 and thioredoxin reductase 2 (Txnrd2) are potential targets of miR-335 and miR-34a, respectively, as aging mesangial cells exhibited significant upregulation of miR-335 and miR-34a and marked downregulation of SOD2 and Txnrd2. Further studies confirmed SOD2 and Txnrd2 as target genes of miR-335 and miR-34a which coincided with ROS generation [64].
Within the mitochondria, uncoupling protein 2 (UCP2) has recently been reported as a negative regulator of ROS generation [65]. Its ablation leads to marked increase of oxidative stress in several cell types [66]. In the stroke-prone spontaneously hypertensive rat (SHRsp) kidneys, severe renal damage along with increased rate of inflammation and oxidative stress was observed [67]. UCP2 gene and protein levels were downregulated paralleled by differential expression of kidney miR-24 and -34a, which were identified to target the UCP2 gene. The silencing of the UCP2 gene in renal mesangial cells led to increased rate of ROS generation, increased inflammation and apoptosis, reduced cell vitality, and increased necrosis, suggesting that UCP2 is critical in preventing oxidative stress damage in renal mesangial cells [67].
Specific miRNAs, including miR-205 and the miR-200 family (miR-200a–c, miR-141, and miR-429), were shown to mediate epithelial-to-mesenchymal transition (EMT) in response to TGF-β1 in Madin-Darby canine kidney cells [68]. EMT is thought to be an important event driving renal fibrosis. Downregulation of these miRNAs relieves their cooperative repression of the mesenchymal transcription factors ZEB1 and SIP1 that, in turn, are free to inhibit E-cadherin expression promoting epithelial dedifferentiation.
Muratsu-Ikeda et al. assessed changes in miRNA expression in the cultured renal tubular cell line HK-2 under hypoxia-reoxygenation-induced oxidative stress or ER stress using miRNA microarray assay and real-time RT-PCR. Among altered miRNA expression, miR-205 was markedly decreased in both stress conditions. Functional analysis revealed that decreased miR-205 led to an increase in cell susceptibility to oxidative and ER stresses and that this increase was associated with the induction of intracellular ROS and suppression of antioxidant enzymes. Furthermore, miR-205 bound to the 3′-UTR of the prolyl hydroxylase 1 (PHD1/EGLN2) gene and suppressed the transcription level of EGLN2, which modulates both intracellular ROS level and ER stress state [69].
miRNA profiling of HUVEC treated for 8 and 24 hrs with 200 μM hydrogen peroxide (H2O2) showed that miR-200c and the cotranscribed miR-141 increased more than eightfold. The other miR-200 family members were also induced, albeit at a lower level [70]. miR-200c overexpression in HUVEC recapitulates many aspects of the oxidative stress-induced phenotype, since it induces cell growth arrest, apoptosis, and cellular senescence. All these effects are mediated, at least in part, by the inhibition of the target ZEB1 by the miR-200 family [70]. miR-200 family induction following H2O2 exposure has been confirmed in different cell lines such as human and mouse immortalized fibroblasts, colon carcinoma (CT26), mammary gland epithelial cells (NMuMG) and human cell lines, melanoma cells (MDA-MB-435S), kidney cells (293T), breast adenocarcinoma (MDA-MB-436 and BT-549), and ovarian adenocarcinoma (SKOV3). Notably, in all these cell lines, all miR-200 family members were upregulated [71]. In a recent study, an analysis of miRNAs upregulated in diabetic mouse heart compared to control was performed revealing that miR-200c and miR-141 were among the most upregulated [72]. The authors show that miR-141 targets the inner mitochondrial membrane phosphate transporter, solute carrier family 25 member 3 (Slc25a3), which provides inorganic phosphate to the mitochondrial matrix and is essential for ATP production, suggesting an important role of miR-200 family in mitochondrial responses involved in cardiac diseases associated with diabetes and obesity.
On the contrary to the role for miR-200 family in oxidative stress, miR-200 family has been reported for antifibrotic roles in DN. The miR-200 family also plays an important role in EMT, which is considered to mediate production of renal fibroblasts, in part by targeting ZEB1/2, the transcriptional repressors of E-cad [68, 77, 78]. On the other hand, another group has demonstrated that TGF-β activated Akt in glomerular mesangial cells by inducing miR-200b and miR-200c, both of which target FOG2, an inhibitor of phosphatidylinositol 3-kinase activation [79], suggesting the role of miR-200 family on glomerular mesangial hypertrophy in the progression of DN.
The contrasting findings above highlight the complex nature of miRNA research. Some of the differences may relate to variances in experimental models and/or conditions; however, one often overlooked explanation is that some effects of miRNA and inhibitors are likely to be indirect in nature. Our understanding of miRNA function is continually evolving. Recent evidence demonstrates regulation of gene expression via deadenylation, by altering message stability, and by effects on transcription. Bioinformatics utilising pathway analysis will be needed to better understand the crosstalk between factors that drive many downstream processes and how those processes ultimately impact the expression of individual genes.
miRNA profiling of rat VSMCs treated with 200 μM H2O2 for 6 hours revealed an upregulation of miR-21 [73]. This study showed that miR-21 participates in H2O2-mediated gene regulation via its target programmed cell death 4 (PDCD4) and transcription factor AP-1 pathway activity. Elevated miR-21 is thought to contribute to atherosclerosis by directly targeting PPARα, leading to an increased inflammatory response in ECs. Inhibition of miR-21 causes activation of AP-1 as well as upregulation of proinflammatory factors such as VCAM-1 and MCP-1 [80]. miR-21 can also act as an inhibitor of angiogenesis by reducing EC proliferation, migration, and tube formation in culture via inhibition of RhoB. miR-21 can also increase nitric oxide (NO) in HUVECs exposed to shear stress via increased phosphorylation of nitric oxide synthase (NOS) and decreased apoptosis [74]. This activity is balanced by the ability of miR-21 to repress SOD-2 which is important in antioxidant defence. Furthermore, miR-21 is elevated in angiogenic precursor cells by asymmetrical dimethylarginine (ADMA) which is a powerful NOS inhibitor, ultimately resulting in elevated intracellular reactive oxygen (ROS) species [75]. Increased miR-21 levels were recently demonstrated in renal cortex in db/db mice [81]. Knockdown of miR-21 decreased mesangial expansion, collagen I/IV, and FN1 expression and reduced macrophage infiltration and TNFα and MCP-1 expression. The gene expression changes were replicated in vitro in both PTC and mesangial cells (MCs) with miR-21 overexpression enhancing fibrogenesis via a mechanism which in part involved the direct targeting of SMAD7. Interestingly, miR-21 targets PTEN and also leads to decreased mesangial expansion in db/db mice [82]. miR-21 has also been implicated in regulation of TGF-β signalling in a number of animal models of tubulointerstitial fibrosis and associated renal dysfunction. In one such model, SMAD7 overexpression in the rat unilateral ureteral obstruction model has restored miR-21 expression to normal levels with congruent improvements in renal pathology [83]. In line with a profibrotic role for miR-21, upregulation of this miRNA is inhibited by SMAD3 deletion in an obstructive nephropathy model [84]. Furthermore, regulation of PDCD4 by miR-21 enhances podocyte apoptosis and loss in conjunction with increased tubular epithelial cells survival against growth arrest signals [85, 86].
miR-210 appears to function as master regulator of the hypoxic response as it was found to be upregulated by hypoxia in virtually all the cell types tested to date [87, 88]. Recent data demonstrate that Hif1α can block both mitochondrial respiration via the electron transport chain (ETC) through transcriptional activation of miR-210 in some cell types [87, 88]. Upregulation of miR-210 along with VEGF and VEGFR2 expression was confirmed in renal ischemia/reperfusion (I/R) injury of male Balb/c mice. Furthermore, overexpression of miR-210 in HUVEC-12 cells enhanced VEGF and VEGFR2 expression and promoted angiogenesis in matrigel in vitro. These results suggest that miR-210 may be involved in targeting the VEGF signaling pathway to regulate angiogenesis after renal I/R injury [76].
Table 3 summarises the miRNAs that are regulated by oxidative stress in the kidney and other tissues and associated with kidney disease and vascular dysfunction resulting from excessive ROS production.
Table 3.
Relevant miRNA in oxidative stress.
| miRNAs | Tissue/organ/cell line | Source | Target | Functions | Reference |
|---|---|---|---|---|---|
| NHMC | Human | SOD1/2, PAK1 | Fibronectin synthesis | [58] | |
| miR-377 | MES 13 mesangial cells | Mouse | |||
| Kidney | Mouse | ||||
|
| |||||
| miR-23a~27a~24-2 | HEK293T | Human | ER stress and UPR pathways-associated apoptosis | [60] | |
|
| |||||
| miR-23b | Renal epithelial cells, podocyte | Mice, Rat | TGF-β receptor type II, SMAD3, TGF-β1 | Negative feedback loop-regulating TGF-β1 signaling | [61] |
|
| |||||
| miR-335 | Kidney, primary mesangial cells | Rat | SOD2 | Renal aging | [64] |
| miR-34a | Txnrd2 | ||||
|
| |||||
| miR-24 miR-34a |
Kidney, CRL 2573 mesangial cell | Rat | UCP2 | Oxidative stress damage | [67] |
|
| |||||
| miR-205 | HK-2 renal tubular cell | Human | PHD1/EGLN2 | Antioxidative and anti-ER stress | [69] |
|
| |||||
| miR-200c | HUVEC | Human | ZEB1 | Cell growth arrest, apoptosis, and cellular senescence | [70] |
|
| |||||
| miR-200a/-141 | Fibroblasts, | Mice | p38α | Enhanced oxidative stress, tumorigenesis, and chemosensitivity | [71] |
| CT26 colon carcinoma, | Mice | ||||
| NMuMG mammary gland epithelial cells, | Mice | ||||
| MDA-MB-435S melanoma cells, | Human | ||||
| 293T kidney cells, | Human | ||||
| MDA-MB-436 and BT-549 breast adenocarcinoma, | Human | ||||
| SKOV3 ovarian adenocarcinoma | Human | ||||
|
| |||||
| miR-200c/-141 | Heart | Mice | Slc25a3 | Decreased mitochondrial ATP production | [72] |
| HEK293 | Human | ||||
|
| |||||
| miR-21 | VSMCs | Rat | PDCD4 | Cellular injury | [73] |
|
| |||||
| miR-21 | HUVECs | Human | PTEN | Increased NO production, reduced apoptosis | [74] |
|
| |||||
| miR-21 | Angiogenic progenitor cell | Human | SOD2 | Increased intracellular ROS concentration and impaired NO bioavailability | [75] |
|
| |||||
| miR-210 | Kidney | Mice | Activation of VEGF Signaling pathway | [76] | |
| HUVECs | Human | ||||
ATP: adenosine-triphosphate; ER: endoplasmic reticulum; HEK: human embryonic kidney cells; HUVECs: human umbilical vein endothelial cells; NF-kB: nuclear factor k-light-chain-enhancer of activated B cells; NHMC: normal human mesangial cells; NO: nitric oxide; PAK1: P21 protein (Cdc42/Rac)-activated kinase 1; PDCD4: programmed cell death 4; PHD1/EGLN2: prolyl hydroxylase 1; PTEN: phosphatase and tensin homolog; ROS: reactive oxygen; Slc25a3: solute carrier family 25 member 3; SOD: superoxide dismutase; TGF-β: transforming growth factor-β; Txnrd2: thioredoxin reductase 2; UCP2: uncoupling protein 2; UPR: unfolded protein response; VEGF: vascular endothelial growth factor; VEGF: vascular endothelial growth factor; ZEB1: zinc finger E-box-binding homeobox 1.
6. Future Perspectives
Considerable progress has been made in identifying a number of important roles for miRNA in various biological processes and in disease. There is much excitement at the prospect that some miRNAs appear to be important to the regulation of several related processes in diabetes and its complications, including the modulation of the RAAS and oxidative stress pathways. The number of miRNAs relevant to these conditions is constantly increasing (Tables 1–3). It is encouraging that in some cases restoring the expression of a dysregulated miRNA can attenuate or even reverse disease. Our initial understanding of gene regulation has continued to change from simple concepts in terms of protein factors sitting on DNA to complex epigenetics involving chromatin dynamics and multiple histone and DNA modifications. Even this complexity has been superseded by the ability of miRNA to modulate the expression of multiple targets posttranscriptionally. Whilst the biology around miRNA continues to generate new and interesting findings, the challenge of the future is to translate some of the exciting experimental findings to the potential therapeutic interventions.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Danaei G, Finucane MM, Lu Y, et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2·7 million participants. The Lancet. 2011;378(9785):31–40. doi: 10.1016/S0140-6736(11)60679-X. [DOI] [PubMed] [Google Scholar]
- 2.Stenvinkel P. Chronic kidney disease: a public health priority and harbinger of premature cardiovascular disease. Journal of Internal Medicine. 2010;268(5):456–467. doi: 10.1111/j.1365-2796.2010.02269.x. [DOI] [PubMed] [Google Scholar]
- 3.Sarnak MJ, Levey AS, Schoolwerth AC, et al. Kidney disease as a risk factor for development of cardiovascular disease: a statement from the American heart association councils on kidney in cardiovascular disease, high blood pressure research, clinical cardiology, and epidemiology and prevention. Circulation. 2003;108(17):2154–2169. doi: 10.1161/01.CIR.0000095676.90936.80. [DOI] [PubMed] [Google Scholar]
- 4.Gilbertson DT, Liu J, Xue JL, et al. Projecting the number of patients with end-stage renal disease in the United States to the year 2015. Journal of the American Society of Nephrology. 2005;16(12):3736–3741. doi: 10.1681/ASN.2005010112. [DOI] [PubMed] [Google Scholar]
- 5.Cull C, Manley S, Frighi V, Holman R, Turner R. UK Prospective Diabetes Study (UKPDS). X. Urinary albumin excretion over 3 years in diet-treated Type 2, (non-insulin-dependent) diabetic patients, and association with hypertension, hyperglycaemia and hypertriglyceridaemia. Diabetologia. 1993;36(10):1021–1029. [PubMed] [Google Scholar]
- 6.Shamoon H, Duffy H, Fleischer N, et al. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. New England Journal of Medicine. 1993;329(14):977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 7.Amico JA, Klein I. Diabetic management in patients with renal failure. Diabetes Care. 1981;4(3):430–434. doi: 10.2337/diacare.4.3.430. [DOI] [PubMed] [Google Scholar]
- 8.Cooper ME. Interaction of metabolic and haemodynamic factors in mediating experimental diabetic nephropathy. Diabetologia. 2001;44(11):1957–1972. doi: 10.1007/s001250100000. [DOI] [PubMed] [Google Scholar]
- 9.Soifer HS, Rossi JJ, Sætrom P. MicroRNAs in disease and potential therapeutic applications. Molecular Therapy. 2007;15(12):2070–2079. doi: 10.1038/sj.mt.6300311. [DOI] [PubMed] [Google Scholar]
- 10.Stefani G, Slack FJ. Small non-coding RNAs in animal development. Nature Reviews Molecular Cell Biology. 2008;9(3):219–230. doi: 10.1038/nrm2347. [DOI] [PubMed] [Google Scholar]
- 11.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friedman RC, Farh KK-H, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Research. 2009;19(1):92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- 14.Reinhart BJ, Slack FJ, Basson M, et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans . Nature. 2000;403(6772):901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- 15.Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294(5543):853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- 16.Lee Y, Kim M, Han J, et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO Journal. 2004;23(20):4051–4060. doi: 10.1038/sj.emboj.7600385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baskerville S, Bartel DP. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA. 2005;11(3):241–247. doi: 10.1261/rna.7240905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim Y-K, Kim VN. Processing of intronic microRNAs. EMBO Journal. 2007;26(3):775–783. doi: 10.1038/sj.emboj.7601512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Research. 2004;14(10 A):1902–1910. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 21.Gregory RI, Yan K-P, Amuthan G, et al. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432(7014):235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- 22.Lee Y, Ahn C, Han J, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425(6956):415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 23.Chendrimada TP, Gregory RI, Kumaraswamy E, et al. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436(7051):740–744. doi: 10.1038/nature03868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Small EM, Olson EN. Pervasive roles of microRNAs in cardiovascular biology. Nature. 2011;469(7330):336–342. doi: 10.1038/nature09783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hausser J, Syed AP, Bilen B, Zavolan M. Analysis of CDS-located miRNA target sites suggests that they can effectively inhibit translation. Genome Research. 2013;23(4):604–615. doi: 10.1101/gr.139758.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee I, Ajay SS, Jong IY, et al. New class of microRNA targets containing simultaneous 5′-UTR and 3′-UTR interaction sites. Genome Research. 2009;19(7):1175–1183. doi: 10.1101/gr.089367.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466(7308):835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shyu A-B, Wilkinson MF, Van Hoof A. Messenger RNA regulation: to translate or to degrade. EMBO Journal. 2008;27(3):471–481. doi: 10.1038/sj.emboj.7601977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diehl P, Fricke A, Sander L, et al. Microparticles: major transport vehicles for distinct microRNAs in circulation. Cardiovascular Research. 2012;93(4):633–644. doi: 10.1093/cvr/cvs007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lorenzen JM, Haller H, Thum T. MicroRNAs as mediators and therapeutic targets in chronic kidney disease. Nature Reviews Nephrology. 2011;7(5):286–294. doi: 10.1038/nrneph.2011.26. [DOI] [PubMed] [Google Scholar]
- 31.Sequeira López MLS, Pentz ES, Nomasa T, Smithies O, Gomez RA. Renin cells are precursors for multiple cell types that switch to the renin phenotype when homeostasis is threatened. Developmental Cell. 2004;6(5):719–728. doi: 10.1016/s1534-5807(04)00134-0. [DOI] [PubMed] [Google Scholar]
- 32.Sequeira-Lopez MLS, Weatherford ET, Borges GR, et al. The microRNA-processing enzyme dicer maintains juxtaglomerular cells. Journal of the American Society of Nephrology. 2010;21(3):460–467. doi: 10.1681/ASN.2009090964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marques FZ, Campain AE, Tomaszewski M, et al. Gene expression profiling reveals renin mRNA overexpression in human hypertensive kidneys and a role for microRNAs. Hypertension. 2011;58(6):1093–1098. doi: 10.1161/HYPERTENSIONAHA.111.180729. [DOI] [PubMed] [Google Scholar]
- 34.Chen NX, Kiattisunthorn K, O'Neill KD, et al. Decreased microRNA is involved in the vascular remodeling abnormalities in chronic kidney disease (CKD) PloS ONE. 2013;8 doi: 10.1371/journal.pone.0064558.e64558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jeppesen PL, Christensen GL, Schneider M, et al. Angiotensin II type 1 receptor signalling regulates microRNA differentially in cardiac fibroblasts and myocytes. British Journal of Pharmacology. 2011;164(2):394–404. doi: 10.1111/j.1476-5381.2011.01375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eskildsen TV, Jeppesen PL, Schneider M, et al. Angiotensin II regulates microRNA-132/-212 in hypertensive rats and humans. International Journal of Molecular Sciences. 2013;14:11190–11207. doi: 10.3390/ijms140611190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Go AS, Chertow GM, Fan D, McCulloch CE, Hsu C-Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. New England Journal of Medicine. 2004;351(13):1296–1370. doi: 10.1056/NEJMoa041031. [DOI] [PubMed] [Google Scholar]
- 38.Fu M-X, Wells-Knecht KJ, Blackledge JA, Lyons TJ, Thorpe SR, Baynes JW. Glycation, glycoxidation, and cross-linking of collagen by glucose: kinetics, mechanisms, and inhibition of late stages of the Maillard reaction. Diabetes. 1994;43(5):676–683. doi: 10.2337/diab.43.5.676. [DOI] [PubMed] [Google Scholar]
- 39.Gu L, Hagiwara S, Fan Q, et al. Role of receptor for advanced glycation end-products and signalling events in advanced glycation end-product-induced monocyte chemoattractant protein-1 expression in differentiated mouse podocytes. Nephrology Dialysis Transplantation. 2006;21(2):299–313. doi: 10.1093/ndt/gfi210. [DOI] [PubMed] [Google Scholar]
- 40.Shi S, Yu L, Chiu C, et al. Podocyte-selective deletion of dicer induces proteinuria and glomerulosclerosis. Journal of the American Society of Nephrology. 2008;19(11):2159–2169. doi: 10.1681/ASN.2008030312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shanmugam N, Reddy MA, Natarajan R. Distinct roles of heterogeneous nuclear ribonuclear protein K and microRNA-16 in cyclooxygenase-2 RNA stability induced by S100b, a ligand of the receptor for advanced glycation end products. Journal of Biological Chemistry. 2008;283(52):36221–36233. doi: 10.1074/jbc.M806322200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li L-M, Hou D-X, Guo Y-L, et al. Role of microRNA-214-targeting phosphatase and tensin homolog in advanced glycation end product-induced apoptosis delay in monocytes. Journal of Immunology. 2011;186(4):2552–2560. doi: 10.4049/jimmunol.1001633. [DOI] [PubMed] [Google Scholar]
- 43.Mardente S, Mari E, Consorti F, et al. HMGB1 induces the overexpression of miR-222 and miR-221 and increases growth and motility in papillary thyroid cancer cells. Oncology Reports. 2012;28:2285–2289. doi: 10.3892/or.2012.2058. [DOI] [PubMed] [Google Scholar]
- 44.Liu X, Cheng Y, Zhang S, Lin Y, Yang J, Zhang C. A necessary role of miR-221 and miR-222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circulation Research. 2009;104(4):476–486. doi: 10.1161/CIRCRESAHA.108.185363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Togliatto G, Trombetta A, Dentelli P, Rosso A, Brizzi MF. MIR221/MIR222-driven post-transcriptional regulation of P27KIP1 and P57KIP2 is crucial for high-glucose- and AGE-mediated vascular cell damage. Diabetologia. 2011;54(7):1930–1940. doi: 10.1007/s00125-011-2125-5. [DOI] [PubMed] [Google Scholar]
- 46.Fiorentino L, Cavalera M, Mavilio M. Regulation of TIMP3 in diabetic nephropathy: a role for microRNAs. Acta Diabetologica. 2013 doi: 10.1007/s00592-013-0492-8. [DOI] [PubMed] [Google Scholar]
- 47.Poliseno L, Tuccoli A, Mariani L, et al. MicroRNAs modulate the angiogenic properties of HUVECs. Blood. 2006;108(9):3068–3071. doi: 10.1182/blood-2006-01-012369. [DOI] [PubMed] [Google Scholar]
- 48.Li Y, Song Y-H, Li F, Yang T, Lu YW, Geng Y-J. microRNA-221 regulates high glucose-induced endothelial dysfunction. Biochemical and Biophysical Research Communications. 2009;381(1):81–83. doi: 10.1016/j.bbrc.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suárez Y, Fernández-Hernando C, Pober JS, Sessa WC. Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circulation Research. 2007;100(8):1164–1173. doi: 10.1161/01.RES.0000265065.26744.17. [DOI] [PubMed] [Google Scholar]
- 50.Zhu N, Zhang D, Chen S, et al. Endothelial enriched microRNAs regulate angiotensin II-induced endothelial inflammation and migration. Atherosclerosis. 2011;215(2):286–293. doi: 10.1016/j.atherosclerosis.2010.12.024. [DOI] [PubMed] [Google Scholar]
- 51.Bashan N, Kovsan J, Kachko I, Ovadia H, Rudich A. Positive and negative regulation of insulin signaling by reactive oxygen and nitrogen species. Physiological Reviews. 2009;89(1):27–71. doi: 10.1152/physrev.00014.2008. [DOI] [PubMed] [Google Scholar]
- 52.Kaneto H, Katakami N, Kawamori D, et al. Involvement of oxidative stress in the pathogenesis of diabetes. Antioxidants and Redox Signaling. 2007;9(3):355–366. doi: 10.1089/ars.2006.1465. [DOI] [PubMed] [Google Scholar]
- 53.Gill PS, Wilcox CS. NADPH oxidases in the kidney. Antioxidants and Redox Signaling. 2006;8(9-10):1597–1607. doi: 10.1089/ars.2006.8.1597. [DOI] [PubMed] [Google Scholar]
- 54.Palicz A, Foubert TR, Jesaitis AJ, Marodi L, McPhail LC. Phosphatidic acid and diacylglycerol directly activate NADPH oxidase by interacting with enzyme components. Journal of Biological Chemistry. 2001;276(5):3090–3097. doi: 10.1074/jbc.M007759200. [DOI] [PubMed] [Google Scholar]
- 55.Sedeek M, Callera G, Montezano A, et al. Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: implications in type 2 diabetic nephropathy. American Journal of Physiology. Renal Physiology. 2010;299(6):F1348–F1358. doi: 10.1152/ajprenal.00028.2010. [DOI] [PubMed] [Google Scholar]
- 56.Forbes JM, Coughlan MT, Cooper ME. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes. 2008;57(6):1446–1454. doi: 10.2337/db08-0057. [DOI] [PubMed] [Google Scholar]
- 57.Eid AA, Gorin Y, Fagg BM, et al. Mechanisms of podocyte injury in diabetes role of cytochrome P450 and NADPH oxidases. Diabetes. 2009;58(5):1201–1211. doi: 10.2337/db08-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Q, Wang Y, Minto AW, et al. MicroRNA-377 is up-regulated and can lead to increased fibronectin production in diabetic nephropathy. FASEB Journal. 2008;22(12):4126–4135. doi: 10.1096/fj.08-112326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chhabra R, Adlakha YK, Hariharan M, Scaría V, Saini N. Upregulation of miR-23a∼27a∼24-2 cluster induces caspase-dependent and -independent apoptosis in human embryonic kidney cells. PLoS ONE. 2009;4(6) doi: 10.1371/journal.pone.0005848.e5848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chhabra R, Dubey R, Saini N. Gene expression profiling indicate role of ER stress in miR-23a~27a~24-2 cluster induced apoptosis in HEK293T cells. RNA Biology. 2011;8(4):648–664. doi: 10.4161/rna.8.4.15583. [DOI] [PubMed] [Google Scholar]
- 61.Shang H, Nitsche E, Jing X. Inhibition of TGF-b signaling by miR-23b. Journal of the American Society of Nephrology. 2008;(143A) [Google Scholar]
- 62.Rao G, Xia E, Richardson A. Effect of age on the expression of antioxidant enzymes in male Fischer F344 rats. Mechanisms of Ageing and Development. 1990;53(1):49–60. doi: 10.1016/0047-6374(90)90033-c. [DOI] [PubMed] [Google Scholar]
- 63.Meng Q, Wong YT, Chen J, Ruan R. Age-related changes in mitochondrial function and antioxidative enzyme activity in fischer 344 rats. Mechanisms of Ageing and Development. 2007;128(3):286–292. doi: 10.1016/j.mad.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 64.Bai X-Y, Ma Y, Ding R, Fu B, Shi S, Chen X-M. miR-335 and miR-34a promote renal senescence by suppressing mitochondrial antioxidative enzymes. Journal of the American Society of Nephrology. 2011;22(7):1252–1261. doi: 10.1681/ASN.2010040367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mattiasson G, Sullivan PG. The emerging functions of UCP2 in health, disease, and therapeutics. Antioxidants and Redox Signaling. 2006;8(1-2):1–38. doi: 10.1089/ars.2006.8.1. [DOI] [PubMed] [Google Scholar]
- 66.Pi J, Bai Y, Daniel KW, et al. Persistent oxidative stress due to absence of uncoupling protein 2 associated with impaired pancreatic β-cell function. Endocrinology. 2009;150(7):3040–3048. doi: 10.1210/en.2008-1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Di Castro S, Scarpino S, Marchitti S, et al. Differential modulation of uncoupling protein 2 in kidneys of stroke-prone spontaneously hypertensive rats under high-salt/low-potassium diet. Hypertension. 2013;61:534–541. doi: 10.1161/HYPERTENSIONAHA.111.00101. [DOI] [PubMed] [Google Scholar]
- 68.Gregory PA, Bert AG, Paterson EL, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nature Cell Biology. 2008;10(5):593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 69.Muratsu-Ikeda S, Nangaku M, Ikeda Y, et al. Downregulation of miR-205 modulates cell susceptibility to oxidative and endoplasmic reticulum stresses in renal tubular cells. PloS ONE. 2012;7 doi: 10.1371/journal.pone.0041462.e41462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Magenta A, Cencioni C, Fasanaro P, et al. MiR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death and Differentiation. 2011;18(10):1628–1639. doi: 10.1038/cdd.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mateescu B, Batista L, Cardon M, et al. MiR-141 and miR-200a act on ovarian tumorigenesis by controlling oxidative stress response. Nature Medicine. 2011;17(12):1627–1635. doi: 10.1038/nm.2512. [DOI] [PubMed] [Google Scholar]
- 72.Baseler WA, Thapa D, Jagannathan R, Dabkowski ER, Croston TL, Hollander JM. miR-141 as a regulator of the mitochondrial phosphate carrier (Slc25a3) in the type 1 diabetic heart. American Journal of Physiology. Cell Physiology. 2012;303:C1244–C1251. doi: 10.1152/ajpcell.00137.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lin Y, Liu X, Cheng Y, Yang J, Huo Y, Zhang C. Involvement of MicroRNAs in hydrogen peroxide-mediated gene regulation and cellular injury response in vascular smooth muscle cells. Journal of Biological Chemistry. 2009;284(12):7903–7913. doi: 10.1074/jbc.M806920200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Weber M, Baker MB, Moore JP, Searles CD. MiR-21 is induced in endothelial cells by shear stress and modulates apoptosis and eNOS activity. Biochemical and Biophysical Research Communications. 2010;393(4):643–648. doi: 10.1016/j.bbrc.2010.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fleissner F, Jazbutyte V, Fiedler J, et al. Short communication: asymmetric dimethylarginine impairs angiogenic progenitor cell function in patients with coronary artery disease through a MicroRNA-21-Dependent mechanism. Circulation Research. 2010;107(1):138–143. doi: 10.1161/CIRCRESAHA.110.216770. [DOI] [PubMed] [Google Scholar]
- 76.Liu F, Lou Y-L, Wu J, et al. Upregulation of microRNA-210 regulates renal angiogenesis mediated by activation of VEGF signaling pathway under ischemia/perfusion injury in vivo and in vitro. Kidney and Blood Pressure Research. 2012;35(3):182–191. doi: 10.1159/000331054. [DOI] [PubMed] [Google Scholar]
- 77.Park S-M, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes and Development. 2008;22(7):894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. Journal of Biological Chemistry. 2008;283(22):14910–14914. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Park JT, Kato M, Yuan H, et al. FOG2 protein down-regulation by transforming growth factor-beta1-induced microRNA-200b/c leads to Akt kinase activation and glomerular mesangial hypertrophy related to diabetic nephropathy. The Journal of Biological Chemistry. 2013;288:22469–22480. doi: 10.1074/jbc.M113.453043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhou J, Wang K-C, Wu W, et al. MicroRNA-21 targets peroxisome proliferators-activated receptor-α in an autoregulatory loop to modulate flow-induced endothelial inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(25):10355–10360. doi: 10.1073/pnas.1107052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhong X, Chung AC, Chen HY, et al. miR-21 is a key therapeutic target for renal injury in a mouse model of type 2 diabetes. Diabetologia. 2013;56:663–674. doi: 10.1007/s00125-012-2804-x. [DOI] [PubMed] [Google Scholar]
- 82.Zhang Z, Peng H, Chen J, et al. MicroRNA-21 protects from mesangial cell proliferation induced by diabetic nephropathy in db/db mice. FEBS Letters. 2009;583(12):2009–2014. doi: 10.1016/j.febslet.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 83.Chung ACK, Dong Y, Yang W, Zhong X, Li R, Lan HY. Smad7 suppresses renal fibrosis via altering expression of TGF-β/Smad3-regulated microRNAs. Molecular Therapy. 2013;21:388–398. doi: 10.1038/mt.2012.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhong X, Chung ACK, Chen H-Y, Meng X-M, Lan HY. Smad3-mediated upregulation of miR-21 promotes renal fibrosis. Journal of the American Society of Nephrology. 2011;22(9):1668–1681. doi: 10.1681/ASN.2010111168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Godwin JG, Ge X, Stephan K, Jurisch A, Tullius SG, Iacomini J. Identification of a microRNA signature of renal ischemia reperfusion injury. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(32):14339–14344. doi: 10.1073/pnas.0912701107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Saal S, Harvey SJ. MicroRNAs and the kidney: coming of age. Current Opinion in Nephrology and Hypertension. 2009;18(4):317–323. doi: 10.1097/MNH.0b013e32832c9da2. [DOI] [PubMed] [Google Scholar]
- 87.Chan SY, Loscalzo J. MicroRNA-210: a unique and pleiotropic hypoxamir. Cell Cycle. 2010;9(6):1072–1083. doi: 10.4161/cc.9.6.11006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Devlin C, Greco S, Martelli F, Ivan M. MiR-210: more than a silent player in hypoxia. IUBMB Life. 2011;63(2):94–100. doi: 10.1002/iub.427. [DOI] [PMC free article] [PubMed] [Google Scholar]