

Abstract
Nanopores were fabricated with an integrated microscale Pd electrode coated with either a hydrogen-bonding, or hydrophobic monolayer. Bare pores, or those coated with octane thiol, translocated single-stranded DNA with times of a few microseconds per base. Pores functionalized with 4(5)-(2-mercaptoethyl)-1H-imidazole-2-carboxamide slowed average translocation times, calculated as the duration of the event divided by the number of bases translocated, to about 100 microseconds per base at biases in the range of 50 to 80 mV.
Keywords: Nanopore, DNA translocation, DNA sequencing, slowing translocation, recognition tunneling
Purely electronic single-molecule DNA sequencing now appears to be possible using measured current blockades characteristic of blocks of bases passing through a nanopore.1 A key feature of this work was the use of a polymerase molecule as a “molecular ratchet” to slow the passage of the DNA to several ms per base.2 This stands in contrast to the very fast transit (a fraction of a microsecond per base) in nanopore experiments without such molecular ratchets.3 These speeds can be reduced a little by modifying the salt, solvent and temperature4 or by modifying the surface charge on the nanopore walls,5 but remain far too fast for current variations to be read at the pA levels of current discrimination required to decode sequence. Here, we investigate an alternative approach to slowing DNA translocation, the use of electrodes functionalized with molecules specifically designed to capture DNA bases.
We have been developing an alternative to ion-current readout which we call “Recognition Tunneling” (RT).6, 7 In RT (Figure 1) DNA bases are captured by recognition molecules that are strongly bonded to a pair of electrodes, and form complexes with DNA bases through weaker, non-covalent bonds. The electrodes are spaced closely enough (~2.5 nm8) that tunnel current flows when a base forms a bound complex with the recognition molecules in the tunnel junction. Characteristic fluctuations in the tunnel current signal the identity of the base9 and individual bases within a polymer chain are readily identified9, 10 so that the technique has the potential for better spatial resolution than ion-current readout. To be used in this application, the tunneling electrodes must be combined with a nanopore so that each base passes into the tunnel gap sequentially as the DNA is translocated through the nanopore. This paper describes another step forward in the development of this technology, which is the integration of a functionalized electrode with a nanopore and measurement of its effect on the translocation of single-stranded DNA molecules.
Figure 1.

Recognition tunneling: Two closely spaced electrodes are functionalized with recognition molecules (ICA) that form specific hydrogen bonds with DNA bases (shown here for thymine). Bonding is different for each base, resulting in characteristic current signals (I) when a bias (V) is applied across the gap.
We have investigated the unbinding kinetics of the recognition complex using dynamic force spectroscopy10 finding the off-rate of a DNA base, Koff ~ 0.3 s−1, and a key goal of the current work is to find out if these AFM-based measurements of binding kinetics are relevant to trapping of DNA bases in a functionalized nanopore. The slow off rate, as measured by AFM, implies that, without an applied external force, the DNA base-recognition molecule complex could remain bound in the tunnel junction for seconds. The long lifetime measured by AFM is probably a consequence of the effects of confinement.11 It follows that transport of DNA through a RT junction may be slowed down significantly simply as a result of the capture of each base while a single-stranded DNA molecules passes through the gap. The hope is that sequential capture of each of the bases will occur in turn because the on-rate for capture is quite large, as a result of the high effective concentration of the molecules taking part in the binding in the nanogap.9 We sought to test this slowing mechanism directly by using measurements of the duration of ion current blockades as DNA molecules of various length pass through a solid state nanopore incorporating an electrode funtionalized with recognition molecules (Figure 2b).
Figure 2.

Functionalizing a pore with a recognition molecule slows translocation: Translocation times for a 63 nt ssDNA were measured for a 4 nm diameter pore (inset in e) drilled into a 10 nm Pd film on top of an 18 nm SiN support (a), then when functionalized with 4(5)-(2-mercaptoethyl)-1H-imidazole-2-carboxamide (ICA – Figure 1) (b), and then again with the metal and functionalization removed (c). Examples of current blockades (at 50 mV bias) for the three cases are shown in d, e and f. The rise in background current in g is largely accounted for by the reduction in pore thickness. A scatter plot of the amplitude of the blockade vs. the log of the blockade times (g) shows that amplitudes are all quite similar, but the blockade times include many much longer events for the case of the functionalized pore (discrete values at small times reflect sampling). The blockade times (shown here for 70 mV bias, 1M KCl plus 1 mM phosphate buffer, pH 7.0) are approximately exponentially distributed (h) with 1/e times only a little longer than the instrumental limit for the metalized (green dots, N=321) and stripped pore (blue dots, N=464) but which approach 0.2 ms/nucleotide when the pore is functionalized (red dots, N=171).
Chemical modification of solid state nanopores has been used extensively for slowing translocation, either by modifying surface charge12 or by placing specific chemical traps, such as complimentary oligomers, on the pore surface.13 With extensive modification, it is even possible to stop the transport of DNA entirely.3 For RT, capture molecules, (4(5)-(2-mercaptoethyl)-1H-imidazole-2-carboxamide – ICA – Figure 1 shows a complex with dTMP) are designed to form multiple hydrogen bonds with all four DNA bases in complexes that bind a trapped analyte between a pair of functionalized electrodes (see Liang et al.14 for the other bonding arrangements). We are specifically interested in the case where the recognition molecules are attached to metal electrodes, in particular palladium, because it gives bigger tunneling signals than gold and is also more stable than gold because of its higher melting point.15 Evaporation of metal onto SiN pores has been used to reduce the pore size16 but it results in excessive noise owing to the accumulation of a large, dissipative double layer, the double-layer resistance falling with area.17 The solution to this problem is to reduce the size of the electrode material exposed to electrolyte, and this has been achieved in the past by coating most of the surface area of the electrode with PDMS.16, 18 Here, rather than coat the electrode, we have used microfabrication and e-beam lithography to fabricate small electrodes, achieving a reduction in noise similar to that obtained by coating the electrodes, but with a more easily fabricated structure. If the electrodes are small enough, there is essentially no difference in background noise between the metalized and non-metalized devices (cf. Figures 2d and f) We drilled pores through the electrodes (and the underlying SiN) using an electron beam, and measured translocation times for several different DNA molecules with different functionalizations of the metal part of the pore.
Results and discussion
We begin by describing an experiment in which we used the same Pd-SiN pore, measuring DNA translocation with the metal layer bare (Figure 2a), then again as functionalized with ICA molecules (Figure 2b) and then finally after the metal electrode was stripped away altogether (Figure 2c). 3 mm chips containing 30 nm thick silicon windows were purchased from Norcada (Alberta). Nanopores made with these membranes gave background currents in good agreement with theoretical estimates (Table 1) but the current blockade signals were too noisy for the small molecules used in this work. In order to improve the signals,19 we thinned a small window (area 10 x 10 microns) to 18 nm, into which a 10 nm thick Pd electrode was deposited (see methods). Pores drilled in these membranes gave much better signal-to-noise, but at the cost of a larger background leakage (data not shown) presumably because of damage caused by the thinning process. Pores were drilled using the electron beam in a F 2010 TEM, with the size of the pore monitored directly using the TEM CCD. Pore diameters were between 3 and 5 nm. The pores were not perfectly round (see the inset in Figure 2d) and quoted diameters are an average over the pore area.
Table 1.
Pore geometries and measured and calculated conductances in 1MKCl. The calculated conductance is based on electrolyte resistivity (G Calc. 1), and on solution of Poisson-Nernst-Planck-Stokes equations (G Calc. 2). These are in good agreement with measurements. There is a significant increase in conductance when even small amounts of DNA are added to the input reservoir.
| Pore Depth | Pore Diameter (nm) | G 1M KCl (nS) | G Calc. 1 (nS) | G Calc. 2 (nS) | G KCl+DNA (nS) |
|---|---|---|---|---|---|
| 30 nm SiN + 10 nm Pd | 3.0 | 2.9 | 2.57 | 2.56 | 4.0 |
| 10 nm SiN + 10 nm Pd | 4.2 | 9.7 | 9.58 | 9.53 | 48.5 |
After cleaning, the pores were mounted in a custom cell that sandwiched the chip between two PDMS gaskets, the cell filled with buffered 1M KCl solution on both sides, Ag/AgCl reference electrodes placed into the two chambers and current recorded using an Axon Axopatch 200B patch clamp amplifier. Table 1 lists the measured conductances of two pores of different geometry, together with conductance calculated using20
| (1) |
where d is the pore diameter, and L the pore length. σ is the specific conductance calculated from 6.02 x 1026 [μK + μCl] eC where e is the electronic charge, μK and μCl the electrophoretic mobilities of K and Cl ions (7.62 and 7.91x10−8 m2/Vs) and the numerical factor converts the concentration C (in moles/liter) to ion-pairs per cubic meter. The calculated (G calc1 – Table 1) and measured conductances (Table 1) are in good agreement. They are also in good agreement with a full solution of the Poisson-Nernst-Planck-Stokes equations (G calc2, Table 1) showing that electrophoresis alone accounts for most of the observed current (see the Supporting Information for more details).
When DNA (typically 5 to 100 nM concentration) was added to the negatively biased (cis) chamber, characteristic current blockades were observed (Figure 2d). Interestingly, we always observe an increase in the background ionic current after DNA is added to the input chamber (last column of Table 1), even before the characteristic current blockade spikes occur. This increase took longer to occur at low DNA concentrations, but, after an adequate wait, it was similar at all concentrations, implying that DNA was accumulated at the entrance of the pore.
After measurements with a bare Pd-SiN pore, the membrane was removed from the translocation cell, cleaned, and the Pd layer functionalized with ICA. The same pore was then remounted in the translocation cell and blockades recorded again. Much longer events were now observed (Fig. 2e). Since this was the same pore, these longer events must be a consequence of having functionalized the electrodes. Finally, a Piranha etch (caution, the use of Piranha can result in violent explosions) was used to strip away the metal electrode entirely and translocation measured again. Only rapid translocations were observed (Figure 2f) once the functionalized electrodes had been removed. Thus the effects of the chemical modification of the nanopore were completely reversed on stripping off the electrode. We can eliminate the possibility of major changes in nanopore geometry when the pore was subject to the various processing steps described above. As can be seen from Figs. 2d and 2e, the background ion current changes very little when the Pd electrode was functionalized, so the slowing of the translocation was not a result of occluding the pore. When the metal was stripped off, (Fig 2f) the current increased by about 68%. Equation 1 predicts a 52% increase, based on reducing the total pore length from 28 to 18 nm (metal plus SiN). Thus most of the observed increase in ion current background can be accounted for by the reduction of pore length, implying again that there were no large changes in pore geometry.
A summary of many blockade measurements from this one pore is given in Figs. 2g and h. A scatter plot of the blockade amplitude vs. translocation times is shown for all three experiments in Figure 2g (bare Pd – green points, ICA functionalized Pd – red points, stripped electrode – blue points). The distribution of blockade amplitudes remains essentially the same, supporting the conclusion that the pore diameter did not change significantly over the various processing steps. However, the distribution of translocation times is affected dramatically by functionalization. This is illustrated further by the histograms in Figure 2h where the distributions have been fitted by exponentials. The decay time of the distribution is increased by at least an order of magnitude when the ICA monolayer is present. The decay time of the fitted exponential for the ICA functionalized electrodes (red curve, Fig. 2h) is 13.2 ms, corresponding to 0.2 ms per base for the 63 nt DNA. This is much longer than the sub-microseconds per base reported for the translocation of double stranded DNA in SiN pores.3
Comparison with results for dsDNA is flawed by the fact that the bases in ssDNA are more accessible than those in dsDNA. This clearly plays a role as can be seen from the translocation times in the SiN (0.5 ms – blue curve, Fig. 2h) and bare metal (1 ms – green curve, Fig. h) pores for our ssDNA. These times are significantly longer than the instrumental response (data are rolled off at 5 kHz, i.e., 0.2 ms) so are significantly contributed to by the duration of a translocation event. 0.5 to 1 ms corresponds to 8 to 15 us per base, also longer than reported for dsDNA (though much faster ahan for the ICA functionalized pore). This suggests that increased interactions between the exposed bases and the pore surface already slow the translocation of ssDNA more than is the case for dsDNA, even without chemical modification, though the chemical modification causes a substantial additional slowing.
We next sought to test the specific nature of the molecular trapping. This was done by comparing several pores functionalized with either ICA (Figure 3a) or non-hydrogen bonding octanethiol (Figure 3b). The preparation and characterization of these monolayers is described in the Methods section. For these measurements, we used SiN membranes of 10 nm thickness (also from Norcada) onto which 10 by 10 micron Pd pads were deposited, again using TEM to drill 3 to 5 nm diameter pores. These thinner pores produced better signal to noise in the recorded blockade currents, and we were able to resolve two components in the distribution of blockade times when the Pd electrodes were functionalized with ICA. The shorter times were fitted by an exponential distribution and the longer ones by a Gaussian peak in the distribution (this resolution into two components was not possible with the data obtained from the thicker pores shown in Fig. 2). Examples of data from the thin pores are given in Figure 3c. The red curve is a fit consisting of an exponential plus a Gaussian peak to the data obtained with the ICA functionalized electrode. The blue curve is a single exponential fit to the data obtained with a OT functionalized pore. Clearly, a significant fraction of translocations are much longer in the case of ICA functionalization compared to OT functionalization. Figure 3d shows the peak time value, τp, of the fitted Gaussian peak for the ICA pores for both 120 nt and 63 nt ssDNA as a function of the bias applied across the pore (red (120 nt) and blue (63 nt) filled circles). In the case of the OT functionalized pores, this figure shows the decay time of the single exponential fits (open circles, 120 nt, crosses, 63 nt).
Figure 3.
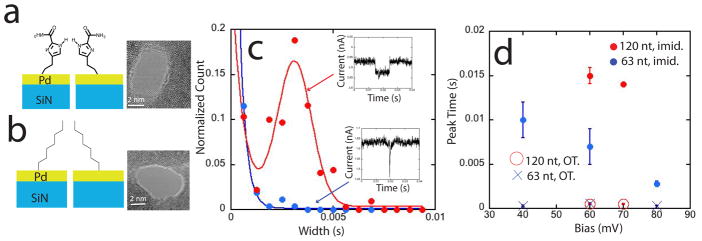
The slowing effect is specific to the hydrogen bonding molecule. Two pores of similar diameter functionalized with ICA (a) or octane thiol (OT) (b) produce very different distributions of translocation time as shown for a 63 nt ssDNA at a bias of 80 mV in (c). The OT functionalized pore (blue dots, N= 261) gives rise to an exponential distribution of decay times with a 1/e time of 0.3 ms. The ICA functionalized pore has about half of the events exponentially distributed (decay time = 0.6 ms) but, in these thinner nanopores (10 nm Pd, 10 nm SiN), a Gaussian peak of slower events is also observed (red curve and data points, N= 219) at 2.8 ms. Data for several pores and biases were fitted with an exponential (OT functionalization) or an exponential plus a Gaussian (ICA functionalization) and the peak values of the Gaussians or 1/e time of the exponentials are shown in (d). Functionalization with ICA increases translocation times by about an order of magnitude.
The Gaussian plus exponential fits, while clearly reasonable for the ICA data, contain relatively few points in any one measurement (cf. Figure 3c). We find that the median value of the blockade time duration distribution is quite close to the value of the Gaussian peak obtained from fits, so we have also analyzed all the distributions (ICA and OT functionalization) in terms of the median translocation time, τM. Table 2 shows the median translocation times (τM), the peak of the Gaussian distribution (τp) and the decay time of the exponential component (τ1/e) for a pair of pores of very similar diameter (3 and 3.3 nm). Clearly, the increase in translocation time when ICA molecules are used is a consequence of their specific chemistry and not just occlusion of the pore by the added molecular monolayer.
Table 2.
Example of fits to translocation time distributions for ICA and OT functionalized pores (63 nt ssDNA, bias = 80 mV). The additional Gaussian peak (τp) in the case of ICA includes about 40% of all translocation events, typical for all the ICA functionalized pores. The exponential component of the distribution is always somewhat longer that, but similar to the single exponential observed for OT pores. The median of the distribution is also shown (τM)
| Fuctionalization | N | τM (ms) | τ1/e (ms) | τp (ms) |
|---|---|---|---|---|
| ICA | 323 | 2.6 | 0.6±0.1 | 2.8±0.3 |
| OT | 261 | 0.3 | 0.3±0.1 | NA |
Is it possible that the increased times reflect transient sticking to the ICA layer without translocation of the entire molecule? Figure 4a shows τp for three different lengths of ssDNA at 60 and 70 mV bias (see Methods for the sequences). Within experimental errors, the translocation time increase linearly with molecular length, evidence that the entire molecule passes the pore. Evidently, the range of molecular lengths studied here is too small to manifest the non-linearities predicted by Muthukumar.21
Figure 4.
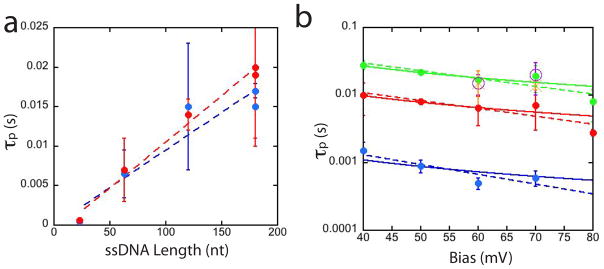
Translocation times in a functionalized pore depend weakly on bias but strongly on DNA length. (a) Translocation times increase linearly with length, evidence that complete molecules are translocating the functionalized pores. Blue dots are for 60 mV, red dots 70 mV. The dashed lines are linear fits to the data, the slope yielding a time of 0.1 ms/nt. Data are for 23, 63, 120 and 180 nt long ssDNA molecules. (b) Shows the value of the peak of the distribution of blockade times for the slowed molecules as a function of bias. Its dependence on bias can be fitted with either a 1/V dependence (friction-solid curves) or an exp (−V) dependence (activated unbinding-dashed curves). The three exponential fits are 0.007exp (− 0.04V) for 23 nt DNA, 0.03 exp (−0.03V) for 63 nt DNA and 0.08exp (−0.03V) for 180 nt DNA.
How do the translocation times compare to what is known about the kinetics of trapping in complexes like these? One might expect much slower dissociation kinetics in the nanopore where many interactions could occur between recognition molecules and the DNA. In fact, the translocation is much faster than the AFM data would predict. AFM force spectroscopy10 on molecular triplexes (recognition molecule – DNA base – recognition molecule) shows an activated dissociation process with an intrinsic off-rate at zero force of . In the Bell model,22 an applied force F, increases the dissociation rate according to where xTS parameterizes the potential landscape by the distance from the bound-state minimum energy to the transition state maximum energy. The AFM data were fitted with xTS = 0.8 nm. Using the result23 that the force applied to DNA in a nanopore is directly proportional to voltage according to F = 0.24 pN/mV, leads to FxTS = 0.19V where V is the bias applied across the nanopore in mV. With kT = 4.2 pN.nm, the predicted voltage dependence for the translocation time (inversely proportional to the off-rate) is ~ 3exp (−0.045V) where V is the applied bias in mV. τp is plotted vs V for three DNA lengths and exponential fits are shown as dashed lines in Figure 4b. While the exponents (0.03 to 0.04 mV−1) are similar to the value predicted by the AFM measurements, the prefactors are about two orders of magnitude smaller (0.007 to 0.08 vs 3 predicted). The bound complexes dissociate much more rapidly in the nanopore than they do in an AFM gap. In addition to the number of possible intermolecular interactions, another difference lies with how the dissociating force is applied. In the nanopore, the force is applied along the axis of the pore, while the bonds are likely to lie in directions different to that of the applied force. In the AFM experiments, the molecules are attached to PEG tethers, so the force should align more closely with the bond directions. On these grounds, one might expect the AFM dissociation rates to be faster, not slower. One possibility for explaining the shorter lifetime of the complexes in the nanopore is that the equilibrium-bonding configuration (i.e., full number of hydrogen bonds) is not achieved in the nanopore if the on-rates are too small for this this to occur. Experiments that scan a functionalized STM probe over DNA9 put an lower limit on Kon of about 10–100 s−1, but this may still be too slow to guarantee capture in the nanopore. The fact that about half the translocation events are not retarded lends support to this interpretation.
A more widely accepted view of the voltage dependence of the translocation time21, 24 is that τp varies as 1/V, a result that follows directly from Stoke’s law for viscous flow. This fits our data just as well as the activated binding model (solid lines in Figure 4b). Thus, while the activation energy could explain the slopes of the plots shown in Figure 4b, we do not have a satisfactory model of the association and dissociation kinetics of these complexes in the nanopore.
We have also measured capture rates in these functionalized pores. They are exponentially dependent on bias (data not shown) with absolute rates similar to those reported by Wanunu et al.25 for unfunctionalized pores. Wanunu et al. used salt gradients to enhance the electric fields that lead to threading of the DNA. The first step – electrostatic accumulation of DNA at the pore entrance – appears to be very efficient. Capture rates do not appear to depend strongly on DNA concentration given either an adequate waiting time and/or stirring of the solution in the input chamber. A clear signal of the accumulation of DNA is given by the increase in background current through the pore (Table 1), the conductance increasing more than 2x in most cases. We have shown in earlier work26 that accumulation of charged molecules at the entrance to a pore can lead to large enhancements of the ion current when electroosmotic flow is significant. However, simulations (Supporting Information) suggest than electroosmotic flow in negligible in the pores that are the subject of the current study. We have simulated various geometries for accumulated DNA at the pore entrance, but cannot account for the observed increase in current. However, the observation that this increase is eventually observed at almost any DNA concentration (with smaller concentrations taking much longer to manifest themselves) suggests that DNA is concentrated at the entrance to the pore, an important feature in that this concentration effect could lower the detection limit for nanopore devices.
Conclusions
In conclusion, we have shown that about half the population of molecules translocating a pore incorporating a thin electrode functionalized with specific recognition molecules are slowed dramatically. The slowing, although significant, is much less than suggested by AFM measurements of the lifetimes of the bound complexes, suggesting that the complexes in the nanopore are not in an equilibrium conformation compared to the complexes measured in force spectroscopy. While translocation times scale with DNA length, the translocation is surely stochastic at a base-to-base level. Nonetheless, the capture events depend on the specific chemistry of the recognition molecules, as slowing events are not observed when the pores are coated with a non hydrogen-bonding molecule.
Methods
Patterning Pd electrodes on membranes
The starting substrates used for fabrication of nanopores are standard TEM nitride membrane windows purchased from Norcada Inc (starting SiN thicknesses 30 nm and 10 nm). A small-area Pd electrode is deposited in the central free-standing region of the silicon nitride membranes using electron beam lithography (EBL). First an alphanumerical grid of location reference Au/Cr (35nm/5nm) markers are laid onto the chip by EBL. This grid is used to locate the position of the central membrane area accurately. AUTOCAD is used to design a pattern of 10μm x10μm squares and the pattern written on PMMA resist spin coated on the chips. 1nm Ti/9nm Pd is deposited using Lesker PVD75 Electron Beam Evaporator, followed by lift-off of PMMA resist to leave the metal pads on the membranes. The 30nm thick SiN membranes were thinned as follows: After exposure of the 10μm x10μm square pattern and development of exposed PMMA, the underlying SiN is etched using Oxford Instruments Plasmalab 80+RIE (F) to reduce the membrane thickness to 18nm. This is followed by metal deposition and lift off from the 10 micron square depression. To remove the Pd after measurements, chips were soaked in Piranha for 30 min, rinsed and used as described below.
Drilling Pores
All pores are drilled using the electron beam in a JEOL 2010 TEM, with the size of the pore monitored directly using the TEM CCD. A test sample is first loaded and the objective lens is stigmated and focused onto the surface. The condenser aperture is introduced and the eucentric height is established by adjusting the height of the sample until the diffraction spot is eliminated. The surface is imaged at this point to check for stability. If the sample is found to be drifting, it is left to stabilize until there is minimal drift. At this point, the beam is focused to a tight spot under the highest magnification settings and a nanopore is drilled through the sample surface. The beam current and energy settings can be adjusted so as to drill pore gradually for a time of 30–45 s. The formation of the nanopore is indicated by the accompanying increase in electron flux crossing the sample. As soon as the pore is formed, the condenser is spread and an image of the fresh pore is captured via CCD camera. The beam is blanked immediately after this and the sample is taken out of the TEM. Pd coated pores form rather stable pores in which the opening is surrounded by single-crystal Pd (lattice fringes are clear in the images – inset in Figure 2d). For membranes thinner than 20 nm, successive imaging can introduce defects, which while too small to allow DNA translocation, can lead to an increased background ion current. This problem in minimized by drilling the pores at lower beam energy density (by using smaller apertures). Repeated imaging of the pore after drilling was avoided as this could cause the pores to change in size.
Cleaning and functionalizing devices
The chip is dipped briefly (less than a second) in piranha (a highly oxidizing mixture of Sulfuric Acid and 30% Hydrogen Peroxide in ratio of 3:1) and rinsed immediately with DI water. The chips are immersed in 0.1 mM ethanolic solutions of 4(5)-(2-mercaptoethyl)-1H-imidazole-2-carboxamide for 24 h. This long time was required in order to ensure that all parts of the exposed metal around the nanopore were functionalized. The solution was prepared with HPLC grade ethanol and degassed with argon.
The substrate was then rinsed gently with ethanol and dried under nitrogen prior to its use in experiments to prevent contamination of the translocation cell by excess reagent. The same protocol is used for preparing ethanolic solution of Octanethiol and for the corresponding functionalization. All chemical reagents (except for ICA) have been purchased from Sigma-Aldrich and used without further purification.
Characterizing monolayers
10 nm metal films (1 nm Ti, 9 nm Pd) were formed on 1 cm square silicon chips and functionalized by exactly the same procedure used for the small area Pd pads. This gave a large-area sample for characterization by standard methods. Ellipsometric measurements of SAM thicknesses were performed on a Rudolph Research AutoEL ellipsometer with a He-Ne laser light source, λ= 632.8 nm and an incidence angle of 70°. To calculate SAM thickness, a three phase model (ambient – organic film – Palladium) was used. The ellipsometric parameters of the bare Palladium substrate (obtained previously) were entered as set values and the refractive index of the organic film was assumed to be 1.46. Each sample surface was measured at five different spots and the average SAM thickness was calculated using a built in algorithm. Molecular modeling yields a length of 0.92 nm for the ICA molecule, while the measured thickness of the monolayer was 0.75 ± 0.12
XPS measurements were performed to examine the chemical state of thiol group sulfur using a VG ESCALAB 200i-XL photoelectron spectrometer. 15keV Al-Ka radiation (at 6× 10−10 mbar base pressure) was used as the source. High resolution spectra for the atomic core levels of Pd(3d), C(1s), N(1s) & S(2p) were obtained at a pass energy of 20eV followed by the recording of wide scan spectra at a pass energy of 150eV. CasaXPS software package was used for the curve fitting of S(2p) spectra and atomic concentration calculation. The binding energy of the S2p core level electrons is ~162eV, (indicated by a well fitted doublet in 2:1 ratio due to the two different spin states of S2p)referenced to C1s at ~284.80eV. This proves that the sulfur is bound to palladium. Measured elemental ratios are consistent with the composition of ICA (Table S1).
Contact angle goniometry was used to determine the hydrophilicity of the three surfaces. Measurements were performed at room temperature with a CAM200 Optical Contact Angle Meter from KSV Instruments Ltd. Freshly cleaned Pd films were hydrophilic, ICA functionalized film intermediate and alkanethiol films extremely hydrophobic (Table S2).
Infrared absorption spectra of ICA and OT monolayers were taken with a Thermo Nicolet 6700 FTIR (Thermo Fisher Scientific, MA) combined with Smart SAGA accessory(MCT/A detector), for 3,400–1,200 cm−1 region. The spectrum of powder sample was taken by the same instrument combined with Smart Orbit ATR accessory(DTGS KBr detector) over the same spectral range. Both of the spectrum were baseline corrected and smoothed using the OMNIC software package. The bulk FTIR spectra are well recapitulated in the monolayers (Figures S2 and S3). The following peak assignments were made after careful examination of the monolayer spectrum of ICA: C=C stretching: 1605cm−1; C=O stretching: 1677cm−1; CH2 asymmetric stretching & symmetric stretching, respectively: 2927cm−1, 2856cm−1; N-H stretching: 3024cm−1. Peak at 2332cm−1 is from atmospheric carbon-dioxide and that at 2058cm−1 may have appeared from some impurity. In the spectrum of OT monolayer, the important peaks to be assigned were CH2 asymmetric stretching & symmetric stretching, respectively: 2921cm−1, 2850cm−1.
Translocation measurements
The nanopore chip was sandwiched between two PDMS gaskets containing 500 ul liquid reservoirs in contact with the chip faces. Ag/AgCl electrodes, freshly made by oxidizing Ag wire with bleach, were dipped into each fluid reservoir. The reservoirs were cleaned by sonication in a 1:1 isopropanol/water mixture. The salt solution used for all translocation measurements was 1M KCl + 1mM Phosphate buffer, pH 7.4. All solvents were sonicated under an argon flow to remove oxygen. The piranha cleaning step described above resulted in good wetting of the pores. It was effective even in the case of the ICA and OT functionalized pores where functionalization followed the piranha cleaning step. After the flow cell is assembled under optical microscope and wetted, the entire set up is placed inside a Faraday cage. Current Voltage measurements are made using an Axon Axopatch 200B patch clamp amplifier. The amplifier applies a constant potential across the pore and measures the current necessary to maintain the potential drop. The current signal is captured in real time using LABVIEW software. At the outset of the experiment, the baseline current values are noted for various voltages so as to establish the open pore conductance. Once this is established, the analyte molecules are introduced in the cis side of device.
DNA Oligomers
23nt and 63nt samples were custom synthesized by Integrated DNA Technologies (IDT).
23nt 5′-TGG AGT GTG ACA ATG GTG TTT GT-3′
63nt 5′-CCT CGC ATG ACT CAA CTG CCT GGT GAT ACG AGG ATG GGC ATG CTC TTC CCG ACG GTA TTG GAC-3′
120nt and 180nt were from IBA-Lifesciences.
120nt 5′-CCT CGC ATG ACT CAA CTG CCT GGT GAT ACG AGG ATG GGC ATG CTC TTC CCG ACG GTA TTG GAC CCT CGC ATG ACT CAA CTG CCT GGT GAT ACG AGG ATG GGC ATG CTC TTC CCG ACG GTA-3′
180nt 5′-CCT CGC ATG ACT CAA CTG CCT GGT GAT ACG AGG ATG GGC ATG CTC TTC CCG ACG GTA TTG GAC CCT CGC ATG ACT CAA CTG CCT GGT GAT ACG AGG ATG GGC ATG CTC TTC CCG ACG GTA TTG GAC CCT CGC ATG ACT CAA CTG CCT GGT GAT ACG AGG ATG GGC ATG CTC TTC CCG ACG-3′
Oligonucleotides were HPLC purified by the vendors. Sequences were chosen for minimal secondary structure using mFold.
Calculations of the ionic current: were based on the solution of Poisson-Nernst Planck-Stokes equations,27 using COMSOL 4.3a, mimicking closely the conditions of the experiment. Distributions of the charges, electric fields and various components of current in the pore are discussed in the Supporting Information.
Supplementary Material
Acknowledgments
We benefitted from the advice of J. He. This work was supported in part by a DNA sequencing technology grant from the NHGRI, HG 006323.
Footnotes
Supporting Information Available: XPS, contact angle and FTIR data. This information is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Manrao EA, Derrington IM, Laszlo AH, Langford KW, Hopper MK, Gillgren N, Pavlenok M, Niederweis M, Gundlach JH. Reading DNA at Single-Nucleotide Resolution with a Mutant MspA Nanopore and phi29 DNA Polymerase. Nature Biotechnol. 2012;30:349–353. doi: 10.1038/nbt.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cherf GM, Lieberman KR, Rashid H, Lam CE, Karplus K, Akeson M. Automated Forward and Reverse Ratcheting of DNA in Nanopore at 5-A Precision. Nature Biotechnol. 2012;14:344–348. doi: 10.1038/nbt.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keyser U. Controlling Molecular Transport through Nanopores. J Roy Soc Interface. 2011;8:1369–1378. doi: 10.1098/rsif.2011.0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fologea D, Uplinger J, Thomas B, McNabb DS, Li J. Slowing DNA Translocation in a Solid-State Nanopore. Nano Lett. 2005;5:1734. doi: 10.1021/nl051063o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen P, Gu J, Brandin E, Kim Y, Wand Q, Branton D. Probing Single DNA Molecule Transport using Fabricated Nanopores. Nano Lett. 2004;4:2293. doi: 10.1021/nl048654j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Branton D, Deamer D, Marziali A, Bayley H, Benner SA, Butler T, Di Ventra M, Garaj S, Hibbs A, Huang X, et al. Nanopore Sequencing. Nature Biotechnology. 2008;26:1146–1153. doi: 10.1038/nbt.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lindsay S, He J, Sankey O, Hapala P, Jelinek P, Zhang P, Chang S, Huang S. Recognition Tunneling. Nanotechnology. 2010;21:262001–262013. doi: 10.1088/0957-4484/21/26/262001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang S, He J, Zhang P, Gyarfas B, Lindsay S. Analysis of Interactions in a Molecular Tunnel Junction. J Am Chem Soc. 2011;133:14267–14269. doi: 10.1021/ja2067737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang S, Huang S, Liu H, Zhang P, Akahori R, Li S, Gyarfas B, Shumway J, Ashcroft B, He J, et al. Chemical Recognition and Binding Kinetics in a Functionalized Tunnel Junction. Nanotechnology. 2012;23:235101–235115. doi: 10.1088/0957-4484/23/23/235101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang S, He J, Chang S, Zhang P, Liang F, Li S, Tuchband M, Fuhrman A, Ros R, Lindsay SM. Identifying Single Bases in a DNA Oligomer with Electron Tunneling. Nature Nanotechnology. 2010;5:868–873. doi: 10.1038/nnano.2010.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friddle RW, Noy A, De Yoreoa James J. Interpreting the Widespread Nonlinear Force Spectra of Intermolecular Bonds. Proc Natl Acad Sci (USA) 2012;109:13573–13578. doi: 10.1073/pnas.1202946109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wanunu M, Meller A. Chemically Modified Solid State Nanopores. Nano Lett. 2007;7:1580–1585. doi: 10.1021/nl070462b. [DOI] [PubMed] [Google Scholar]
- 13.Iqbal SM, Akin D, Bashir R. Solid State Nanopore Channels with DNA Selectivity. Nature Nanotechnology. 2007;2:243–248. doi: 10.1038/nnano.2007.78. [DOI] [PubMed] [Google Scholar]
- 14.Liang F, Li S, Lindsay S, Zhang P. Synthesis, Physicochemical Properties, and Hydrogen Bonding of 4(5)-Substituted-1H-imidazole-2-carboxamide, A Potential Universal Reader for DNA Sequencing by Recognition Tunneling. Chem Eur J. 2012;18:5998–6007. doi: 10.1002/chem.201103306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang S, Sen S, Zhang P, Gyarfas B, Ashcroft B, Lefkowitz S, Peng H, Lindsay S. Palladium Electrodes for Molecular Tunnel Junctions. Nanotechnology. 2012;23:425202. doi: 10.1088/0957-4484/23/42/425202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei R, Pedone D, Zurner A, Doblinger M, Rant U. Fabrication of Metallized Nanopores in Silicon Nitride Membranes for Single Molecule Sensing. Small. 2010;6:1406–1414. doi: 10.1002/smll.201000253. [DOI] [PubMed] [Google Scholar]
- 17.Hassibi A, Navid R, Dutton RW, Lee TH. Comprehensive study of noise Processes in Electrode Electrolyte Interfaces. J App Phys. 2004;96:1074–1082. [Google Scholar]
- 18.Tabard-Cossa V, Trivedi D, Wiggin M, Jetha NN, Marziali A. Noise Analysis and Reduction in Solid State Nanopores. Nanotechnology. 2007;18:305505–305511. [Google Scholar]
- 19.Wanunu M, Dadosh T, Ray V, Jin J, McReynolds L, Drndic M. Rapid Electronic Detection of Probe-Specific MicroRNAs using Thin Nanopore Sensors. Nature Nanotechnology. 2010;5:807–814. doi: 10.1038/nnano.2010.202. [DOI] [PubMed] [Google Scholar]
- 20.Healy K, Scheidt B, Morrison AP. Solid State Nanopore Technologies for Nanopore-based DNA analysis. Nanomedicine. 2007:875–897. doi: 10.2217/17435889.2.6.875. [DOI] [PubMed] [Google Scholar]
- 21.Muthukumar M. Polymer Translocation through a Hole. J Chem Phys. 1999;111:10371–10374. [Google Scholar]
- 22.Bell GI. Models for the Specific Adhesion of Cells to Cells. Science. 1978;200:618–627. doi: 10.1126/science.347575. [DOI] [PubMed] [Google Scholar]
- 23.Keyser UF, Koelman BN, van Dorp S, Krapf D, Smeets RMM, Lemay SG, Dekker NH, Dekker C. Direct Force Measurements on DNA in a Solid-State Nanopore. Nature Physics. 2006;2:473–477. [Google Scholar]
- 24.Kasianowicz JJ, Brandin E, Branton D, Deamer DW. Characterization of Individual Polynucleotide Molecules using a Membrane Channel. Proc Nat Acad Sci. 1996;93:13770–13773. doi: 10.1073/pnas.93.24.13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wanunu M, Morrison W, Rabin Y, Grosberg AY, Meller A. Electrostatic Focusing of Unlabelled DNA into Nanoscale Pores using a Salt Gradient. Nature Nanotechnology. 2010;5:60–65. doi: 10.1038/nnano.2009.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park JH, He J, Gyarfas B, Lindsay S, Krstić PS. DNA Translocating Through a Carbon Nanotube Can Increase Ionic Current. Nanotechnology. 2012;23:455107–455113. doi: 10.1088/0957-4484/23/45/455107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pang P, He J, Park JH, Krstic PS, Lindsay S. Origin of Giant Ionic Currents in Carbon Nanotube Channels. ACS Nano. 2011;5:7277–7283. doi: 10.1021/nn202115s. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.