

Abstract
Sugar methyltransferases (MTs) are an important class of tailoring enzymes which catalyze the transfer of a methyl group from S-adenosyl-L-methionine to sugar-based N-, C- and O- nucleophiles. While sugar N- and C-MTs involved in natural product biosynthesis have been found to act on sugar nucleotide substrates prior to a subsequent glycosyltransferase reaction, corresponding sugar O-methylation reactions studied thus far occur after the glycosyltransfer reaction. Herein we report the first in vitro characterization using 1H-13C-gHSQC with isotopically-labeled substrates and the X-ray structure determination at 1.55 Å resolution of the TDP-3′-O-rhamnose-methyltransferase CalS11 from Micromonospora echinospora. This study highlights a unique NMR-based methyltransferase assay, implicates CalS11 to be a metal and general acid/base-dependent O-methyltransferase and, as a first crystal structure for a TDP-hexose-O-methyltransferase, presents a new template for mechanistic studies and/or engineering.
Keywords: sugar nucleotide, carbohydrate, natural product, S-adenosyl-L-methionine, X-ray crystallography
INTRODUCTION
The alkylation of sugars attached to natural products is a common late stage diversification tactic employed in nature (1). While such sugar C- or N-methylation typically occurs at the sugar nucleotide stage prior to the culminating glycosyltransferase-catalyzed attachment of the sugar to a given natural product aglycon, comparator O-methyltransferases (O-MTs) characterized in vitro thus far operate post-glycosylaton (i.e., after the unique functionalized sugar has been transferred to a natural product core aglycon). As examples of O-methylated sugar-containing natural products, methylated variants of L-rhamnose are found among a range of secondary metabolites including calicheamicin (Figure 1, 1, containing 3-O-methoxy-L-Rha) (2), steffimycin (Figure 1, 2, containing 2-O-methoxy-L-Rha) (3, 4), spinosad and elloramycin (Figure 1, 3 and 4 respectively, both containing 2,3,4-tri-O-methoxy-L-Rha) (5, 6). Our recent work on the reversibility of the glycosyltransferases involved in calicheamicin biosynthesis (7, 8) revealed the rhamnosyltransferase CalG1 to accept TDP-3-O-methoxy-rhamnose as a substrate, suggesting the putative rhamnosyl-O-methyltransferase encoded by the calicheamicin locus (CalS11) (2) to function at the sugar nucleotide level. This contrasts recent work on the three spinosad-associated rhamnose O-MTs (SpnH, SpnI and SpnK) which revealed these catalysts to operate in the more typical post-glycosylation style (9).
Figure 1.

Rhamnosylated natural products with the rhamnose moiety highlighted in blue. Structures 1, 2, 3 and 4 refer to calicheamicin, steffimycin, spinosad A and elloramycin, respectively. The CalS11-catalyzed methylation is highlighted in yellow.
Figure 3.
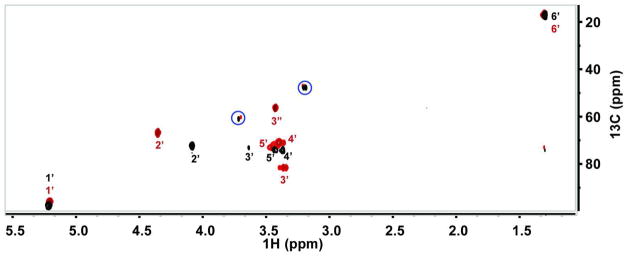
Overlay of 13C-1H HSQC spectrum before and after CalS11 reaction. Colors black and burgundy represent TDP-L-[U-13C]rhamnose and the corresponding 13C-methylated product, respectively. Blue circles denote peaks from ion pairing agent tetrabutylammonium bisulphate.
Figure 4.
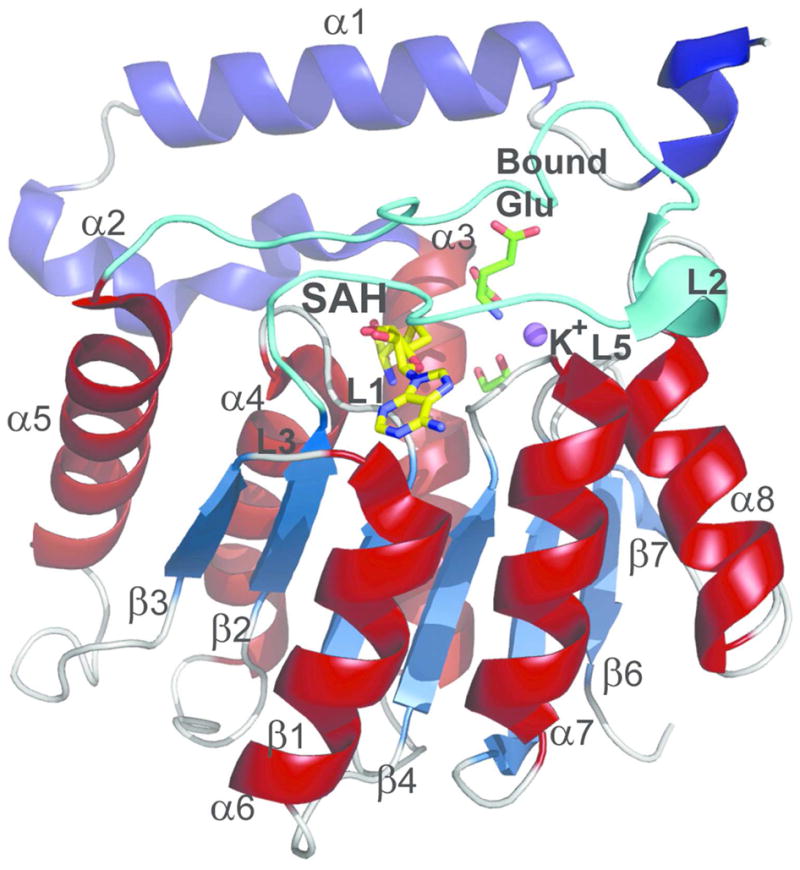
CalS11 monomer with secondary structural elements labeled. The N-terminal helices are colored blue and the helices from the core MT Rossmann fold are colored brick-red. The dynamic loop L2 is colored in cyan. SAH, bound glutamate and ethylene glycol are represented by stick models and are colored yellow and green, respectively. K+ ion is represented as a purple sphere.
To further explore the potential biosynthetic divergence of CalS11 from the sugar O-MT postglycosylation paradigm, herein we report the in vitro characterization and X-ray structure determination of CalS11. This work supports the role of CalS11 as a rhamnose O-MT and specifically, as a TDP-3′-O-rhamnose-MT – the first confirmed sugar O-MT to function at the sugar nucleotide level. This work also reveals CalS11 to crystallize as a decamer and, consistent with this observation, the location and nature of the CalS11 decamer interface is unique in comparison to other MTs. The crystal structure of CalS11 in complex with S-adenosyl-L-homocysteine highlights a fairly typical MT monomeric structural fold wherein the putative CalS11 catalytic residues are highly conserved among sequence-related TDP-hexose and TylF family O-MTs. As the first biochemical and structural characterization for a TDP-sugar O-MT, this work may present a blueprint to enable the future engineering of sugar nucleotide-dependent MTs for combinatorial applications.
MATERIALS AND METHODS
Materials
E. coli B834(DE3)pLysS and BL21(DE3) competent cells were purchased from Invitrogen (Carlsbad, CA). The pET-28b E. coli expression vector and thrombin were purchased from Novagen (Madison, WI). Primers were purchased from Integrated DNA Technology (Coralville, IA). Pfu DNA polymerase was purchased from Stratagene (La Jolla, CA). Restriction enzymes and T4 DNA ligase were purchased from New England Biolabs (Ipswich, MA). Benzamidine sepharose was purchased from Pharmacia (Piscataway, NJ). PD-10 column [S-13C-methyl]-L-methionine was purchased from Cambridge Isotope Laboratories, (Andover, MA), and Ni-NTA superflow columns were purchased from GE Healthcare (Piscataway, NJ). All other chemicals were reagent grade or better and purchased from Sigma (St. Louis, MO). NMR experiments were performed in 5 mm NMR tubes and analyzed via a Varian 600 MHz equipped with HCX cryoprobe at UW NMRFAM. NMR data were processed using NMRPipe (10) and MestReNova. 13C-1H HSQC spectra of [S-13C-methyl]-L-methionine and [S-13C-methyl]adenosyl-L-methionine (13CH3-SAM) were acquired on a Varian 400 MHz equipped with a 5 mm OneNMR probe in the University of Kentucky College of Pharmacy NMR facility.
Protein Expression and Purification
The calS11 gene (2), amplified from the genomic DNA of Micromonospora echinospora, was separately cloned into vectors for both cell-based expression in E. coli, and expression in a wheat germ cell-free system. For E. coli expression, the calS11 gene was cloned into pET28a to provide a vector for the overproduction of N-terminal His6-CalS11 in E. coli BL21 (DE3). The culture was grown in the presence of 35 μg mL−1 of kanamycin at 37 °C to an OD600 of ~0.6, induced with 0.5 mM IPTG, and grown at 25 °C overnight. The cells were collected by centrifugation (30 min, 5000 rpm), resuspended in buffer A (20 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 7.8). The cells were lysed via incubation with 1 mg mL−1 lysozyme (~50,000 U mg−1; Sigma-Aldrich, St. Louis, MO, USA) for 30 min on ice followed by sonication (VirSonic 475; Virtis, Gardiner, NY; 100 W, 4 × 30 s pulses, ~1 min between pulses) on ice. N-His6-CalS11 was purified via affinity chromatography (5 mL HiTrap Ni-NTA chelating column, GE Healthcare, Piscataway, NJ) following standard protocol (50 mM sodium phosphate, 300 mM NaCl, pH 8.0 and a linear gradient of imidazole of 10–500 mM elution gradient) on an AKTA Purifier 10 (GE Healthcare) to provide ~60 mg of N-His6-CalS11. Buffer exchange of pooled fractions containing the purified protein was accomplished using a PD-10 column (GE Healthcare) eluted with 20 mM Tris-HCl, 100 mM NaCl, 5 mM CaCl2 pH 8. The N-His6-tag was subsequently removed by thrombin cleavage following standard protocol (Novagen, Madison, WI; 50 U of thrombin, 20 hrs, room temperature, 60 mgs of N-His6-CalS11, 3.5 mL total volume). This mixture was subsequently treated with 1 mL slurry of benzamidine sepharose (Pharmacia, NJ) to remove thrombin and the affinity tag cleaved CalS11 was purified via a second round of HiTrap Ni-NTA affinity chromatography. The flow through containing desired truncated CalS11 was concentrated and exchanged with storage buffer (25 mM Tris-HCl, pH 8) using PD-10 column. The purified CalS11 was concentrated to 11 mg mL−1, flash frozen in liquid nitrogen and stored at −80 °C. Protein concentrations were determined by Bradford assay (Bio-Rad, Hercules, CA, USA) using BSA as a standard.
Selenomethionyl CalS11 for initial crystallization trials and X-ray crystallographic structures was produced using standard cell-free production protocol of Center for Eukaryotic Structural Genomics (CESG) platform for cloning (11), protein expression (12), purification (13), and bioinformatics management (14). For this study, the calS11 gene was also amplified from genomic DNA and cloned into the pEU-His-FV vector using modifications of the protocol found in Blommel 2009 (11). Modifications included a two-step PCR protocol to introduce a N-terminal TEV protease cleavage site and the use of TaKaRa Ex Taq polymerase (Millipore) in the presence of 1% v/v DMSO in all PCR reactions to accommodate the GC-rich template. Cell-free expression was conducted on a 6×4 mL scale for 18 hours using WEPRO2240H extract and a Protemist100 protein synthesizer (CellFree Sciences, Yokohama, Japan). Selenomethionine was added to 0.6 mM, and the remaining 19 amino acids were added to 0.3 mM. His-tagged protein was purified by nickel affinity chromatography. The N-terminal His-tag was cleaved with TEV protease, and tag-free protein was isolated by subtractive nickel affinity chromatography. Size-exclusion chromatography provided additional purification and permitted exchange of CalS11 into the final protein buffer (5 mM HEPES, 50 mM NaCl, 0.3 mM TCEP, pH 7.0). CalS11 was concentrated to 16 mg mL−1, cryopreserved in 30 microliter beads by dropping into liquid nitrogen, and stored at −80 °C. Complete protocols are available from Target Track (http://sbkb.org/tt/) as target CESG-GO.111124. Plasmid pEU-His-FV is available from the PSI Materials Repository (http://dnasu.asu.edu/DNASU/GetCloneDetail.do?cloneid=84283).
Protein Crystallization
Initial crystallization screens were performed using an in house screen comprised of UW192, IndexHT, and Salt RX (Hampton Research, Aliso Viejo, CA) by the sitting drop method using a Mosquito® dispenser (TTP labTech Hertfordshire, United Kingdom). Crystal growth was monitored at 4°C and 20°C using Bruker Nonius Crystal Farm (Bruker AXS Inc., Madison, WI). Cell free synthesized SeMet-CalS11 crystals were grown by mixing 1 μl of protein sample solution (16 mg mL−1 CalS11), 3 mM tris(2-carboxyethyl)phosphine, 50 mM sodium chloride, 5 mM HEPES pH 8, and 1 μl of reservoir solution, 17.6% polyethylene glycol monomethyl ether 5,000, 160 mM potassium-glutamate, 100 mM 2-[bisamino]-2--1,3-propanediol pH 6.5, at 20 °C using the hanging drop method. Native CalS11 crystals were grown by mixing 1 μl of protein sample solution (11 mg mL−1 CalS11), 25 mM Tris, pH 8.0, 5 mg mL−1 calicheamicin T0 (8) and 1 μl of reservoir solution, 25% polyethylene glycol 3350, 0.2 M lithium sulfate, 100 mM Tris, pH 8.5, at 20 °C using the hanging drop method. Native CalS11 crystals were cryoprotected with 30% polyethylene glycol 3350, 200 mM lithium sulfate, and 100 mM Tris, pH 8.5 and SeMet-CalS11 crystals were cryoprotected with 20% ethylene glycol, 17.6% polyethylene glycol monomethyl ether, 160 mM potassium glutamate, 100 mM 2-[bisamino]-2-1,3-propanediol, pH 6.5 before flash freezing in liquid nitrogen for data collection.
Data Collection and Structure Refinement
X-ray diffraction data were collected at the Life Sciences Collaborative Access Team (LS-CAT) with an X-ray wavelength of 0.9794 Å, for the SeMet-CalS11 crystals, and 0.97872 Å, for the native CalS11 crystals, at the Advanced Photon Source at Argonne National Laboratory. Data sets were indexed and scaled using HKL2000 (15). For phasing experiments on SeMet-CalS11 (3TOS), phenix.Hyss (16) and ShelxD (17) were utilized for determining the selenium substructures, autoSHARP (18) for phasing, DM for density modification (19) and phenix.AutoBuild for automatic model building (16). For the native CalS11 structure, molecular replacement starting from SeMet-CalS11 (3TOS) was accomplished using phenix.AutoMR and phenix.AutoBuild. The structures were completed with alternating rounds of manual model building with COOT (20) and refinement with phenix.refine. Structure quality was assessed by Procheck (21) and Molprobity (22).
SAXS data
SAXS data were collected on the SIBYLS beamline using the high throughput data collection mode as described previously (23). Three concentrations of CalS11 were measured at 2.3 mg mL−1, 3.5 mg mL−1 and 7.0 mg mL−1. No concentration dependence was observed. Each concentration was exposed 4 times to X-rays with 0.5, 1, 2, and 5 second exposures. No radiation damage was observed through comparison of the 0.5 and 1 second exposures. For redundancy, two buffer blanks were collected - one prior to the concentration series and one after. Scattering profiles from both buffer blanks were subtracted from the SAXS data collected from samples and the resulting subtracted files were compared for agreement. The 2 and 5 second exposures were merged with the 1 second exposure to produce a final scattering curve for further analysis using the program PRIMUS (24). The scattering data was merged to obtain the highest signal to noise ratio and processed by GASBOR (25) to determine the three dimensional envelop. Based on the symmetry of crystal structure and the MW of the CalS11 decamer, P52 symmetry was imposed. Out of ten averaged GASBOR runs, eight models were selected to generate the final model based on their orientation with respect to P52 axis using program SUPCOMB (26).
Preparation of [S-13C-methyl]adenosyl-L-methionine (13CH3-SAM)
Isotopically-labeled 13CH3-SAM was prepared using 1.1 mM [S-13C-methyl]-L-methionine, 1.2 equivalent of ATP and 50 μg methionine adenosyltransferase (MAT) from Sulfolobus solfataricus (27) in 20 mM NaH2PO4 buffer, 10 mM MgCl2, 100 mM KCl, pH 8.0 at 37 °C for 1 hr. Under these conditions, quantitative conversion to the desired product was observed based upon HPLC (Figure S3, top-panel) and HSQC (Figure 3). The crude reaction mixture was lyophilized and dissolved in 100 μl D2O and directly used in the subsequent CalS11 reaction.
CalS11 Reaction
In a 5 mm NMR tube, a mixture of 1 mM TDP-L-[U-13C]rhamnose, 1.1 mM 13CH3-SAM (Figure S4) and 50 μg of the purified CalS11 in a final volume of 500 μL of 20 mM NaH2PO4 buffer prepared with D2O, pH 8, was incubated at 30 °C for 1 hr. The progress of the reaction was monitored by sensitivity enhanced 1H-13C-gHSQC (Figure 3). NMR chemical shift assignments were made using 2D-planes of 3D- 1H-13C–HCCH-TOCSY and 1H-13C–HCCH-COSY from Varian BioPack. To assess metal dependency, three separate reactions were monitored by 1H-13C-gHSQC (after 1 hr and over-night incubation at 30 °C), each containing commercially available SAM (Sigma, St. Louis, MO) and 12 μg of purified CalS11 in a final volume of 250 μL 25 mM KH2PO4, pH 8 in a 3 mm NMR tube. The first of these contained no additional additives, the second included 10 mM EDTA, and the latter was conducted in the presence of 5 mM MgCl2.
RESULTS AND DISCUSSION
In vitro Characterization
TDP-3-methoxy-rhamnose biosynthesis en route to calicheamicin parallels TDP-rhamnose (TDP-Rha) biosynthesis in many other bacteria (3–6) and requires five enzymes - TDP-4,6-dehydratase, CalS3; TDP-3,4-epimerase, CalS1; TDP-3-ketoreductase, CalS2; and TDP-3-O-methyltransferase, CalS11 (Figure 2). While TDP-Rha biosynthesis in a variety of bacteria has been well-studied, studies of subsequent rhamnose methylation in secondary metabolism (e.g., in spinospin and elloramycin) revealed methylation to occur after the glycosyltransfer reaction (9, 28). The application of TDP-L-[U-13C]rhamnose and 13CH3-SAM (Figure S4) as substrates enabled a precise 1H-13C-HSQC experiment (Figure 3) to directly assess the progress of the CalS11 reaction and provide definitive characterization of the reaction products. This study revealed the first definitive evidence that CalS11 acts at the sugar nucleotide level. Of specific importance in this analysis is the emerging methoxy 1H and 13C signature at 3.43 ppm and 58.64 ppm, respectively (Table 1/Figure 3). Figure S2 highlights a subsequent comparison of three reactions (with commercially available SAM as the donor), also monitored via NMR, to address the metal dependency of CalS11. Specifically, a comparison of product formation of a standard reaction containing no additives (no detectable product at 1 hr and ~30% conversion after 12 hr), a comparator reaction containing 5 mM MgCl2 (~60% at 1 hr and nearly quantative after 12 hr), and a parallel reaction containing 10 mM EDTA (no product at 1 hr or 12 hr), clearly supports the participation of divalent metal in catalysis and is consistent with an observed metal-binding ligand within the determined CalS11 structure.
Figure 2.
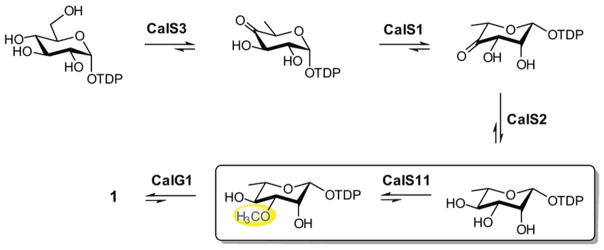
The biosynthetic pathway of TDP-methoxy-rhamnose in M. echinospora en route to calicheamicin production.
Table 1.
1H and 13C chemical shift values of TDP-L-[U-13C]rhamnose and the corresponding 13C-methylated product.
| Atom | dTDP-rhamnose 1H, 13C (ppm) |
dTDP-3-methoxy-rhamnose 1H, 13C (ppm) |
|---|---|---|
| H1′, C1′ | 5.20, 98.07 | 5.20, 98.27 |
| H2′, C2′ | 4.07, 73.62 | 4.35, 69.40 |
| H3′, C3′ | 3.62, 74.46 | 3.34, 83.91 |
| H3″, C3″ | 3.43, 58.64 | |
| H4′, C4′ | 3.35, 74.24 | 3.40, 73.33 |
| H5′, C5′ | 3.43, 75.39 | 3.44, 75.24 |
| H6′, C6′ | 1.30, 19.36 | 1.31, 19.47 |
Overall Structure
The crystal structure of CalS11 was determined at 1.55 Å and 2.2 Å resolution, respectively, in two different crystal forms. The 1.55 Å crystal structure (PDB 3TOS) has one decamer in the asymmetric unit while the 2.2 Å structure (PDB 4GF5) contained two. Both belonged to space group P1 and contained one molecule of S-adenosyl homocysteine (SAH) per monomer. In addition, 3TOS contained one molecule of glutamic acid and one K+ bound per subunit. Of the 257 possible residues in CalS11, only the first 7 residues were not modeled because of insufficient electron density. The final structures were refined to an Rcryst and Rfree of 14.6% and 17.3% (PDB 3TOS) and 15.9% and 21.3% (PDB 4GF5), respectively (Table 2). Unless otherwise specified, the discussion herein refers to 1.55 Å structure (PDB 3TOS).
Table 2.
Summary of crystal parameters, data collection, and refinement statistics. Values in parentheses are for the highest resolution shell.
| CalS11 (4GF5) | CalS11 (3TOS) | |
|---|---|---|
| Crystal parameters | ||
| Space group | P1 | P1 |
| Unit-cell parameters (Å) | a = 75.11, b = 106.24, c = 184.60 | a = 78.32, b = 106.08, c = 106.33 |
|
| ||
| Data collection statistics | ||
| Wavelength (Å) | 0.97872 | 0.9794 |
| Resolution range (Å) | 50.00-2.20 (2.24-2.20) | 50.00-1.55 (1.58-1.55) |
| No. of reflections (measured/unique) | 254196/254196 | 417415/417415 |
| Completeness (%) | 94.8 (62.9) | 96.8 (94.6) |
| Rmerge* | 0.110 (0.484) | 0.079 (0.297) |
| Redundancy | 3.9 (3.7) | 3.9 (3.8) |
| Mean I/sigma (I) | 6.30 (2.36) | 10.00 (4.26) |
|
| ||
| Refinement and model statistics | ||
| Resolution range (Å) | 49.32-2.20 | 43.10-1.55 |
| No. of reflections (work/test) | 254162/1899 | 412070/1875 |
| Rcryst§ | 0.159 (0.196) | 0.146 (0.166) |
| Rfree¶ | 0.213 (0.254) | 0.173 (0.251) |
| RMSD bonds (Å) | 0.007 | 0.019 |
| RMSD angles (°) | 1.186 | 1.716 |
| B factor (protein/solvent) (Å2) | 15.1/16.98 | 13.2/33.1 |
| No. of protein atoms | 40128 | 21291 |
| No. of waters | 2734 | 5202 |
| No. of auxiliary molecules | 20 SAH, 28 Sulfide | 10 SAH, 10 Potassium ion, 10 Glutamic Acid, 14 1,2-Ethanediol |
|
| ||
| Ramachandran plot (%) | ||
| Favorable region | 97.5 | 97.5 |
| Additional allowed region | 2.5 | 2.5 |
| Disallowed region | 0.0 | 0.0 |
| PDB ID | 4GF5 | 3TOS |
Rmerge = Σh Σi | Ii (h) − <I(h)>| / ΣhΣi Ii(h), where Ii(h) is the intensity of an individual measurement of the reflection and <I(h)> is the mean intensity of the reflection.
Rcryst = Σh ||Fobs| − |Fcalc|| / Σh |Fobs|, where Fobs and Fcalc are the observed and calculated structure-factor amplitudes, respectively.
Rfree was calculated as Rcryst using 5.0 % of randomly selected unique reflections that were omitted from the structure refinement.
Each CalS11 monomer folds into a single globular domain responsible for binding SAM and the substrate. The domain exhibits a common tertiary structure consisting of a core α/β Rossmann fold characteristic of all other SAM-dependent MTs (29) and an α-helical N-terminal domain containing two α-helices, α1 and α2 that forms the top of the active site cavity (colored blue in Figure 4). In the overall subunit structure, a central parallel β-sheet (β1–β7) is located between helices (α3 and α8) (Figure 4). The β-sheet has strand topology (β3, β2, β1, β4, β5, β7, β6) containing a single antiparallel strand (β7) flanked on both sides by helices. This is consistent with a class I MT fold (30), which is described as a ‘doubly wound-open α/β sandwich’. A loop, L2 (colored cyan in Figure 4), between the strand β2 and helix α5, forms a lid on the SAH binding site of the globular domain which, analogous to MT homologs, commonly serves as a gate for the co-factor entry and product release.
While most natural product MTs exists as dimers, to our knowledge, this is the first decameric MT structure (Figure 5A). In this decameric form, also confirmed in solution by small angle X-ray scattering (SAXS) (Figure 5B), each monomer interacts with three other monomers via two distinct interfaces. The first interface, S1 (Figures 5C & 5D), is 2615 Å2 involving ~64 residues, accounting for 20% solvent accessible surface area of each subunit, and involves intra-subunit interaction associated with helices α1- α5 and intercalation of helices, α1 and α2 between the neighboring subunits (Figure 4). Some of the residues from the N-terminal α2 of the intercalating subunit form part of the lining of the active-site. The second interface, S2 (Figures 5C & 5D), is 1350 Å2 involving ~36 residues and accounting for 10.7% of the solvent-accessible area of the subunit, and includes loop interactions among the outer end of the β-sheet of Rossmann fold (29) of one subunit with β6 of other. The putative S1 and S2 interfaces observed here are distinct from O-MT structures described thus far and may serve as a potential distinguishing feature of the NDP-sugar O-MTs.
Figure 5.

A. Decamer structure of CalS11. B. CalS11 in solution by SAXS. C & D. Illustration of subunit-subunit interactions. Six subunits are illustrated with three represented as surface representations (two green and one blue) and three represented as ribbon cartoons (two pink and one red). For the red subunit, the major interaction surface is S1 and the minor is S2.
Active Site
The SAM/SAH binding site is located in the C-terminal end of the cleft formed by the β strands and this position is conserved in all MTs (31). SAH is partially exposed to solvent and bound to the enzyme through an extensive hydrogen bond network and van der Waals interactions (Figure 6A). The CalS11-SAH interaction is mainly provided by residues in the loops L1 (between β1 and α4), L2 (between β2 and α5) and L3 (between β3 and helix α6). Specifically, residues in L1 interact with the amino acid moiety and the ribose, L2 associates with the ribose and adenine ring, and L3 affiliates with the adenine ring of SAH (Figure 4). Loop L2 (residues 109–135) (colored cyan in Figures 4 & 6A) also facilitates the co-factor entry and SAH release. Structural comparison and primary sequence analysis of other SAM-methyltransferases reveal L1, which contains a glycine-rich consensus sequence (XGly77XXXGly81) and an acidic residue in loop L3 (Asp166 in CalS11) to be highly conserved.
Figure 6.
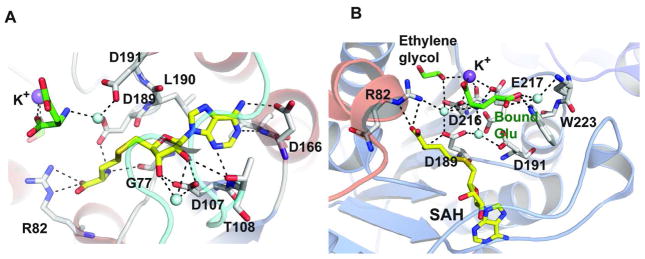
A. Residues involved in SAH binding. B. Residues involved in the interaction of bound glutamate (colored green) and K+. SAH, bound glutamate and ethylene glycol are represented as stick models and are colored yellow and green, respectively. Na+ ion is represented as a purple sphere and water molecules as light blue spheres.
While attempts to obtain a complex with bound TDP-Rha (in the presence or absence of SAM) by co-crystallization failed, two ligand surrogates - glutamic acid and ethylene glycol – were found to occupy the putative substrate-binding site of CalS11. The bound glutamic acid surrogate forms direct H-bonding interactions with Trp223, Asp216 and Glu217 as well as water mediated interactions with Asp189 and Asp191 (Figure 6B). In addition, a potassium ion was found coordinated by Asp216, Glu217, glutamic acid, ethylene glycol and water (Figure 6B). Based on a putative SN2 mechanism, the position of the glutamic acid in the substrate binding site, and the structural conservation of the active residues in the closest homolog NovP, Asp191 is most suited as the putative CalS11 catalytic base. Interestingly, the CalS11 potassium ion coordinating residues (Asp216 and Glu217) are highly conserved among other O-MTs (Figure S1). Assuming the observed potassium ion engages some of the residues involved in CalS11 magnesium ion binding, the metal-active distances (SAH sulfur-metal = 9.6 Å; Asp191 carboxylate-metal = 6.9 Å) are such that protein reorganization, perhaps driven via substrate binding, would be required for direct metal participation in catalysis.
Structurally-related proteins
While this work represents the first reported NDP-6-deoxy-hexose-O-MT structure, a CalS11-based structural similarity search returned more than 200 hits with P-values less than 0.0001 (using the jFATCAT-rigid algorithm). Upon closer analysis, the structurally-related proteins display little or no overall sequence identity to each other, but most share a common SAM-dependent MT Rossmann fold. Many members also contain additional domains outside the core MT structure that play a role in substrate recognition or alternative functions. The structural similarity to CalS11 is mostly restricted to the Rossmann fold and comparators within this set share only 6–19% sequence identities with CalS11. Table S1 summarizes a selection of structurally- and functionally-related CalS11 proteins involved in natural product biosynthesis. The scope of the following discussion is focused upon the closest comparator NovP, the novobiocin L-noviose-4′-O-MT.
Structural alignment of CalS11 with NovP (Figure 7A) highlights conservation of residues in the active site where the putative catalytic base, conserved in both enzymes, is located within loop L3 of CalS11 structure. This putative catalytic base is Asp198 in NovP and, based upon structural correlation, Asp191 in CalS11 (Figure 7B). In addition, loop L2, which forms a lid over the cosubstrate SAM in NovP, is also conserved in CalS11 and likely serves a similar gating function. In NovP, this loop is a flexible helix while in CalS11, this substructure is a dynamic loop (Figure 7A). In addition, while both CalS11 and NovP contain a conserved acid-rich motif near the active the site (Asp216 and Glu217 in CalS11, Figure 7B), only the CalS11 structure revealed a bound metal.
Figure 7.
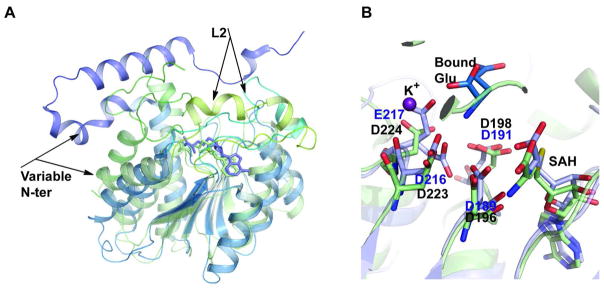
Structural overlay of CalS11 with related MTs. A. Overlay with NovP, where NovP is colored light green and CalS11 is colored light-blue. B. Overlay of active-site residues of NovP and CalS11 with CalS11-bound potassium ion represented by the violet sphere. In A, the variable N-terminal secondary structures and the lids in NovP and CalS11 are labeled. In B, CalS11 active-site residues are highlighted in blue.
In recent years, structures of several sugar MTs have been solved. These include MTs that act upon sugars appended to aglycons such as RebM, rebeccamycin D-glucose-4′-O-MT (32), MycE (33) and NovP (34), as well as MTs that act upon sugar nucleotides, including DesVI, TDP-D-desosamine 3′-N,N-di-MT (35), TylM1, a TDP-D-mycaminose 3′-N,N-di-MT (36) and TcaB9, a TDP-D-tetronitrose C-3′-MT (37). Yet, despite the plethora of available sequences and structures, there remains a lack of any clear structural signature which denotes nucleophile specificity (O-, N-, C-MTs), sugar regiospecificity (2-, 3-, or 4-X-MTs where X is O-, N-, or C-), the nature of the aglycon (i.e., MTs which act upon sugars appended to NDP or aglycon) or sugar configuration (e.g, D- and/or L- as well as 2-, 3-, 4- and/or 6-stereochemistry). Thus, while the current CalS11 study stands as the first biochemical and structural characterization of a nucleotide sugar O-MT, more structures are needed to delineate broader general structure-activity MT correlations and/or develop predictive models relevant to this important enzyme family (38).
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health grants U01GM098248 (GNP), CA84374 (JST) and the National Center for Advancing Translational Sciences (UL1TR000117). Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. NMR experiments were carried at the National Magnetic Resonance Facility at Madison (NMRFAM), which is supported by National Institutes of Health (P41RR02301, P41GM66326, P41GM66326, P41RR02301, RR02781, RR08438), National Science Foundation (DMB-8415048, BIR-9214394), U.S. Department of Agriculture and UW-Madison, WI grants. The Life Sciences Collaborative Access Team (LS-CAT) has been supported by Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor.
Footnotes
Conflict of Interest
J.S.T. is cofounder of Centrose, Madison, WI.
This includes a CalS11 sequence alignment and table of structurally-related proteins, the key CalS11 metal dependency NMR spectra, and MAT reaction HPLC, HRMS and NMR characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Singh S, Phillips GN, Jr, Thorson JS. The structural biology of enzymes involved in natural product glycosylation. Nat Prod Rep. 2012;29:1201–1237. doi: 10.1039/c2np20039b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahlert J, Shepard E, Lomovskaya N, Zazopoulos E, Staffa A, Bachmann BO, Huang K, Fonstein L, Czisny A, Whitwam RE, Farnet CM, Thorson JS. The calicheamicin gene cluster and its iterative type I enediyne PKS. Science. 2002;297:1173–1176. doi: 10.1126/science.1072105. [DOI] [PubMed] [Google Scholar]
- 3.Gullon S, Olano C, Abdelfattah MS, Brana AF, Rohr J, Mendez C, Salas JA. Isolation, characterization, and heterologous expression of the biosynthesis gene cluster for the antitumor anthracycline steffimycin. Appl Environ Microbiol. 2006;72:4172–4183. doi: 10.1128/AEM.00734-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luzhetskyy A, Mayer A, Hoffmann J, Pelzer S, Holzenkamper M, Schmitt B, Wohlert SE, Vente A, Bechthold A. Cloning and heterologous expression of the aranciamycin biosynthetic gene cluster revealed a new flexible glycosyltransferase. Chem Bio Chem. 2007;8:599–602. doi: 10.1002/cbic.200600529. [DOI] [PubMed] [Google Scholar]
- 5.Ramos A, Lombo F, Brana AF, Rohr J, Mendez C, Salas JA. Biosynthesis of elloramycin in Streptomyces olivaceus requires glycosylation by enzymes encoded outside the aglycon cluster. Microbiology. 2008;154:781–788. doi: 10.1099/mic.0.2007/014035-0. [DOI] [PubMed] [Google Scholar]
- 6.Waldron C, Matsushima P, Rosteck PR, Jr, Broughton MC, Turner J, Madduri K, Crawford KP, Merlo DJ, Baltz RH. Cloning and analysis of the spinosad biosynthetic gene cluster of Saccharopolyspora spinosa. Chem Biol. 2001;8:487–499. doi: 10.1016/s1074-5521(01)00029-1. [DOI] [PubMed] [Google Scholar]
- 7.Zhang CS, Griffith BR, Fu Q, Albermann C, Fu X, Lee IK, Li LJ, Thorson JS. Exploiting the reversibility of natural product glycosyltransferase-catalyzed reactions. Science. 2006;313:1291–1294. doi: 10.1126/science.1130028. [DOI] [PubMed] [Google Scholar]
- 8.Zhang CS, Bitto E, Goff RD, Singh S, Bingman CA, Griffith BR, Albermann C, Phillips GN, Thorson JS. Biochemical and structural insights of the early glycosylation steps in calicheamicin biosynthesis. Chem Biol. 2008;15:842–853. doi: 10.1016/j.chembiol.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim HJ, White-Phillip JA, Ogasawara Y, Shin N, Isiorho EA, Liu HW. Biosynthesis of spinosyn in Saccharopolyspora spinosa: synthesis of permethylated rhamnose and characterization of the functions of SpnH, SpnI, and SpnK. J Am Chem Soc. 2010;132:2901–2903. doi: 10.1021/ja910223x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 11.Blommel PG, Martin PA, Seder KD, Wrobel RL, Fox BG. High throughput protein expression and purification: Methods and protocols. Meth Mol Biol. 2009;498:55–73. doi: 10.1007/978-1-59745-196-3_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Makino S, Goren MA, Fox BG, Markley JL. Cell-free protein synthesis technology in NMR high-throughput structure determination. Methods Mol Biol. 2010;607:127–147. doi: 10.1007/978-1-60327-331-2_12. [DOI] [PubMed] [Google Scholar]
- 13.Vinarov DA, Newman CL, Tyler EM, Markley JL, Shahan MN. Wheat germ cell-free expression system for protein production. Curr Prot Prot Sci. 2006;(Unit 5.18) doi: 10.1002/0471140864.ps0518s44. [DOI] [PubMed] [Google Scholar]
- 14.Zolnai Z, Lee PT, Li J, Chapman MR, Newman CS, Phillips GN, Jr, Rayment I, Ulrich EL, Volkman BF, Markley JL. Project management system for structural and functional proteomics: Sesame. J Struct Funct Genomics. 2003;4:11–23. doi: 10.1023/a:1024684404761. [DOI] [PubMed] [Google Scholar]
- 15.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 16.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 18.delaFortelle E, Bricogne G. Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Method Enzymol. 1997;276:472–494. doi: 10.1016/S0076-6879(97)76073-7. [DOI] [PubMed] [Google Scholar]
- 19.Cowtan KD, Main P. Phase combination and cross validation in iterated density-modification calculations. Acta Crystallogr D. 1996;52:43–48. doi: 10.1107/S090744499500761X. [DOI] [PubMed] [Google Scholar]
- 20.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 21.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. Procheck - a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 22.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, II, Tsutakawa SE, Jenney FE, Jr, Classen S, Frankel KA, Hopkins RC, Yang S-j, Scott JW, Dillard BD, Adams MWW, Tainer JA. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Meth. 2009;6:606–612. doi: 10.1038/nmeth.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Konarev PV, Petoukhov MV, Volkov VV, Svergun DI. ATSAS 2.1, a program package for small-angle scattering data analysis. J Appl Crystallogr. 2006;39:277–286. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petoukhov MV, Svergun DI. New methods for domain structure determination of proteins from solution scattering data. J Appl Crystallogr. 2003;36:540–544. [Google Scholar]
- 26.Kozin MB, Svergun DI. Automated matching of high- and low-resolution structural models. J Appl Crystallogr. 2001;34:33–41. [Google Scholar]
- 27.Porcelli M, Cacciapuoti G, Carteni-Farina M, Gambacorta A. S-adenosylmethionine synthetase in the thermophilic archaebacterium Sulfolobus solfataricus: Purification and characterization of two isoforms. FEBS J. 1988;177:273–280. doi: 10.1111/j.1432-1033.1988.tb14373.x. [DOI] [PubMed] [Google Scholar]
- 28.Patallo EP, Blanco G, Fischer C, Brana AF, Rohr J, Mendez C, Salas JA. Deoxysugar methylation during biosynthesis of the antitumor polyketide elloramycin by Streptomyces olivaceus: Characterization of three methyltransferase genes. J Biol Chem. 2001;276:18765–18774. doi: 10.1074/jbc.M101225200. [DOI] [PubMed] [Google Scholar]
- 29.Rossmann MG, Moras D, Olsen KW. Chemical and biological evolution of nucleotide-binding protein. Nature. 1974;250:194–199. doi: 10.1038/250194a0. [DOI] [PubMed] [Google Scholar]
- 30.Schubert HL, Blumenthal RM, Cheng X. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem Sci. 2003;28:329–335. doi: 10.1016/S0968-0004(03)00090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin JL, McMillan FM. SAM (dependent) I AM: the S-adenosylmethionine-dependent methyltransferase fold. Curr Opin Struct Biol. 2002;12:783–793. doi: 10.1016/s0959-440x(02)00391-3. [DOI] [PubMed] [Google Scholar]
- 32.Singh S, Mccoy JG, Zhang C, Bingman CA, Phillips GN, Thorson JS. Structure and mechanism of the rebeccamycin sugar 4′-O-methyltransferase RebM. J Biol Chem. 2008;283:22628–22636. doi: 10.1074/jbc.M800503200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akey DL, Li S, Konwerski JR, Confer LA, Bernard SM, Anzai Y, Kato F, Sherman DH, Smith JL. A new structural form in the SAM/metal-dependent O methyltransferase family, MycE from the mycinamicin biosynthetic pathway. J Mol Biol. 2011;413:438–450. doi: 10.1016/j.jmb.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gómez García I, Stevenson CEM, Usón I, Freel Meyers CL, Walsh CT, Lawson DM. The crystal structure of the novobiocin biosynthetic enzyme NovP: the first representative structure for the TylF O-methyltransferase superfamily. J Mol Biol. 2010;395:390–407. doi: 10.1016/j.jmb.2009.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burgie ES, Holden HM. Three-dimensional structure of DesVI from Streptomyces venezuelae: a sugar N,N-dimethyltransferase required for dTDP-desosamine biosynthesis. Biochemistry. 2008;47:3982–3988. doi: 10.1021/bi800063j. [DOI] [PubMed] [Google Scholar]
- 36.Carney AE, Holden HM. Molecular architecture of TylM1 from Streptomyces fradiae: an N,N-dimethyltransferase involved in the production of dTDP-D-mycaminose. Biochemistry. 2010;50:780–787. doi: 10.1021/bi101733y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bruender NA, Thoden JB, Kaur M, Avey MK, Holden HM. Molecular architecture of a C-3′-methyltransferase involved in the biosynthesis of D-tetronitrose. Biochemistry. 2010;49:5891–5898. doi: 10.1021/bi100782b. [DOI] [PubMed] [Google Scholar]
- 38.Liscombe DK, Louie GV, Noel JP. Architectures, mechanisms and molecular evolution of natural product methyltransferases. Nat Prod Rep. 2010;29:1238–1250. doi: 10.1039/c2np20029e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.