

Abstract
The intestinal mucosal immune system is challenged with bacteria, viruses, and parasites, in addition to food and environmental antigens, that require dynamic immune responsiveness for homeostasis. One central signaling pathway is JAK-STAT, which regulates the adaptive and innate immune arms of mucosal immunity as well as epithelial repair and regeneration. Adaptive immunity includes lymphocyte mediated secretion of specific antibodies, while innate immune respones include secretion of non-antigen specific compounds. This review examines effects of specialized nutrition support on JAK-STAT in innate immune function and in lymphocyte modulation and epithelial antibody transport in gut-associated lymphoid tissue.
Keywords: JAK-STAT, STAT6, STAT4, Peyer patches, sIgA, pIgR, Paneth cells, goblet cells, mucosal immunity
Introduction
The human intestine contains large numbers of bacteria that exist in symbiosis with the host in times of health. Although the bacteria are in close proximity to the mucosa, robust mucosal immune functions mediate this symbiosis to prevent invasion of the intestinal tissue and the systemic circulation by bacteria. Barrier integrity is multifactorial and includes contributions from both the adaptive and the innate mucosal immune system.1 Innate defenses include the mucus covering of the epithelium produced by goblet cells,2,3 antimicrobial peptides and proteins produced and released by Paneth cells,4-7 tight junction proteins that bind the enterocytes to prevent passage of luminal components, and the commensal bacterial population themselves, which form an ecological barrier to pathogens. The adaptive immune system produces, transports and releases secretory IgA (sIgA) which work in concert with innate immunity as a complimentary but more specific arm of mucosal immunity.8-10
Our laboratory focuses on the effect of route and type of nutrition on mucosal immune function. Clinical studies demonstrate patients fed intravenously are at an increased risk of respiratory and intraabdominal infections compared with enteral fed patients. To investigate why this occurs, we established clinically relevant animals feeding models where nutrition is provided via the central vein or the gastrointestinal tract directly. More recently, these animal studies have included investigations of the role of Janus kinase-signal transducers and activators of transcription (JAK-STAT) pathways on intestinal immune function. This review discusses the recognized roles of JAK-STAT signaling in intestinal mucosal immune system in the regulation of both adaptive and innate mucosal immune function.
JAK-STAT Mechanisms of Action
The JAK-STAT signaling pathway was originally discovered in the early 1990s by Darnell and Stark. These investigators studied IFN activation of genes involved in immunity and identified STAT1 and STAT2. We now recognize JAK-STAT as one of the most important pleiotropic cascades employed by cells to transduce signals for hormones, growth factors, and cytokines.11-17 This pathway contains the Janus kinase (JAK) proteins JAK1, JAK2, JAK3, and TYK2 and the signal transducer and activator of transcription (STAT) proteins STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6. JAK-STAT provides the principle intracellular signaling mechanism required for a wide array of cytokines, including IL-2, IL-3, IL-4, IL-6, IL-7, IL-9, IL-11, IL-13, IL-15 IFN-γ, and others.16-18 In addition to cytokines, JAK-STAT signals the effects of chemokines, hormones and growth factors. In general, ligand binding induces JAK phosphorylation that become competent to subsequently phosphorylate residues on assorted STAT proteins, which otherwise remain latent in the cytoplasm. Among other functions, JAK-STAT signaling is implicated in cell development, cell growth and survival, and therefore has important implications in immune function. For example, STAT proteins play vital roles in the differentiation of T helper lymphocyte cells.
Adaptive Mucosal Immunity and the Mucosal Associated Lymphoid Tissue (MALT)
The mucosal immune system comprises ~60% of the body’s total immunity and produces ~80% of total immunoglobulin (Ig) per day.19 This system is tasked with defending the large mucosal surfaces of the body, including the respiratory tract, gastrointestinal tract, uro-genital tract, nasal passages, and mammary glands.20,21 To defend these surfaces, the hallmark molecule of acquired immunity is specific and non-specific secretory IgA (sIgA) at mucosal surfaces.22 sIgA neutralizes bacteria through opsonization providing immune exclusion. Bacteria are also bound and neutralized in the mucus layer, limiting their ability to directly attach and invade tissues. The adaptive mucosal immune system is organized into inductive sites, for antigen sensitization, and effector sites, where IgA is produced and released. Circulating naïve B and T cells enter the Peyer patches (PP) through interactions between L-selectin and α4β7 expressed on the circulating lymphocytes and mucosal addresin adhesion molecule-1 (MAdCAM-1) expressed on the high endothelial venules (HEV) of the PP.23-25 After stimulation and sensitization in PP, T and B lymphocytes migrate via lymphatics to the thoracic duct for distribution into the vascular system and homing to effector sites, such as the lamina propria of the intestine.26 Within these effector sites Th2 cytokines (IL-4, IL-5, IL-6, and IL-10) produced by the T lymphocytes stimulate maturation of B cells into competent IgA producing cells, i.e., plasma cells. These cells produce dimeric IgA, comprised of 2 IgA monomers linked by a small peptide called J-chain.27 The dimeric IgA produced in the lamina propria then undergoes transepithelial transport via polymeric immunoglobulin receptor (pIgR) expressed on the basal surface of mucosal enterocytes (Fig. 1). Within the lumen, sIgA provides protection at mucosal surfaces by attaching to bacteria and preventing bacterial attachment to the mucosa28 and by reducing expression of virulence factors by enteric pathogens.29 JAK-STAT signaling regulates several aspects of adaptive immunity, including Th differentiation, B cell maturation, and production of the transport protein pIgR.

Figure 1. Adaptive mucosal immune function in the small intestine. Naïve T and B lymphocytes enter the Peyer patches (PP) via interaction with their integrins L-selectin or α4β7 and MAdCAM-1 which is present on the high endothelial venules (HEV) of the PP. The cells are sampled and sensitized to antigen in Microfold (M) cells inside the PP. Sensitized cells return to the peripheral circulation via the thoracic duct and are distributed to other mucosal effector sites. Within these effector sites, Th2 cytokines (IL-4, IL-5, IL-6, and IL-10) produced by T lymphocytes stimulate maturation of B cells into competent IgA producing cells, i.e., plasma cells. These cells produce the principal molecule of adaptive immunity, dimeric IgA, in the lamina propria where it undergoes transepithelial transport via polymeric immunoglobulin receptor (pIgR) which is expressed on the basal surface of mucosal enterocytes.
Th1 and Th2 Differentiation Requires IL-4, IL-12, STAT4, and STAT6
Naïve CD4+ lymphocytes are classically understood to differentiate into one of two subsets: Th1 or Th2 cells.30,31 Each subset generates unique cytokine profiles with distinct functions.32 IL-12 and IL-4 are the key cytokines that are involved in the differentiation of Th1 and Th2 responses, respectively.33,34 Differentiated Th1 cells produce and release IL-2, TNF-α, and IFN-γ that promote cell-mediated immune function. Differentiated Th2 lymphocytes release IL-4, IL-5, IL-10, and IL-13, which promote B cell function and humoral mediated immunity. A balance between the Th2-type secreted cytokines (IL-4, IL-5, IL-6, and IL-10), which stimulate IgA production, and the Th1-type secreted cytokines (IFN and TNF), which inhibit IgA production, is required for appropriate humoral responsiveness (Fig. 2 and Table 1).
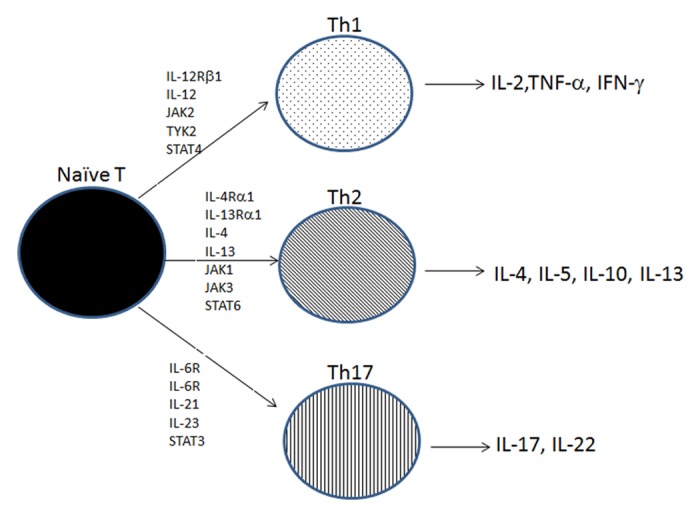
Figure 2. JAK-STAT and T helper cell differentiation of naïve T cells. Naïve T cells differentiate into several T helper cell subsets, Th1, Th2, of Th17. IL-12 is required for the differentiation into Th1 cells that secrete IFN-γ, TNF-α, and IL-2 to generate a response to intracellular pathogens. IL-4 is required for the differentiation into Th2 cells that secrete IL-4, IL-5, and IL-10 to generate defenses against Helminth infections. IL6, IL-21, and IL-23 are required for the differentiation into Th17 cells that secrete IL-17 and IL-22 to promote defense against extracellular pathogens.
Table 1. JAK-STAT interactions regulating mucosal immunity in the intestine.
| Receptor | Stimulus | JAK | STAT | Produces | Process |
|---|---|---|---|---|---|
|
IL-12Rβ1 |
IL-12 |
JAK2, TYK2 |
STAT3, STAT4 |
IL-2, TNF-α, IFN-γ |
Th1 differentiation, defense against intracellular pathogens |
|
IL-4Rα1 IL-13Rα1 |
IL-4, IL-13 |
JAK1, JAK3 |
STAT6 |
IL-4, IL-5, IL-10, IL-13 |
Th2 differentiation, mucus production, pIgR |
|
IL-6R |
IL-6, IL-21 IL-23 |
|
STAT3 |
IL-17, IL-17F, IL-22 |
Th17 differentiation, defense from extracellular pathogens, tissue inflammation |
|
IL-7Rα |
IL-7 |
JAK1, JAK3 |
STAT5a, STAT5b |
|
B cell maturation, T cell development |
|
IFNαR1 IFNαR2 |
IFNα |
JAK1, TYK2 |
STAT1, STAT2 |
|
Anti-viral defenses |
|
IFNγR1 IFNγR1 |
IFNγ | JAK1, JAK2 | STAT1 | Inflammatory and anti-bacterial defenses |
Complete development of the Th1 response requires IL-12 signaling of STAT4 while Th2 response requires IL-4 signaling of STAT6. Mouse studies using knockout mice demonstrate the importance of these two responses and the requirement for JAK-STAT signaling. Mice with insufficient expression of IL-12 or IL-12Rβ1 chain fail to generate Th1 cells, while mice with insufficient expression of IL-4 or IL-4Rα chain fail to generate a Th2 response.35-40 Furthermore, elevated activation of one response appears to suppress the other. For example, Th2 responses generate SOCS3 activation that inhibits Th1 signaling while Th1 responses prevent full activation of the transcription factor GATA3 that is necessary for Th2 differentiation.
STAT4, originally thought to be expressed only in lymphoid cells, is also expressed in monocytes, macrophages, and dendritic cells.41 Under the direction of microbial signals from the gut, dendritic cells and macrophages are activated and secrete IL-12. When antigen is sensed by these cells, IL-12 induces STAT4 phosphorylation that promotes maturation of naïve CD4+ cells into Th1 cells that produce and secrete IFN-γ, a pro-inflammatory cytokine42,43 (Fig. 2 and Table 1).
The cytokines IL-4 and IL-13 induce STAT6 phosphorylation and activation through several receptors depending on cell type. Type-II IL-4 receptors, expressed on non-hematopoietically derived cells, consist of IL-4Rα1 and IL-13Rα1 chains and are activated by both IL-4 or IL-13. Type-I receptors, expressed on hematopoietically derived cells such as lymphocytes, consist of IL-2Rγ and an IL-4Rα1 chain and are activated only by the cytokine IL-4.13 Following binding of IL-4 and IL-13 binding to the IL-4 receptor (IL-4R), activated JAK proteins then phosphorylate a tyrosine residue on STAT-6, which is otherwise latent in the cytoplasm.44 Uniquely, IL-4 and IL-13 induce tyrosine or serine phosphorylation and activation of STAT-6. Studies demonstrate STAT6 dependence for the maturation of CD4+ naïve cells into Th2 effector cells.45 Additionally, in the Peyer’s patches, IL-4 signaling via STAT 6 is essential for B cell development and class switching to IgA+ cells.
Th17 Differentiation Requires IL-6, TGFβ, and STAT3
Recent studies demonstrate additional T helper cell lineages, including the Th17 cells that selectively induce expression of the pro-inflammatory cytokines IL-17, IL-21, and IL-17F. Emerging data demonstrate the direct role of Th17 responses in protecting the host mucosal surfaces from extracellular pathogens, including bacteria and fungi.46-50 Complete development and differentiation of the Th17 response requires the cytokines IL-6, IL-21, and IL-23 and signaling through STAT3. Specifically, IL-6 binds the IL-6R inducing IL-21, which drives subsequent expression of IL-23R. Once expressed, IL-23R binds IL-23 produced and released by activated antigen presenting cells. IL-6 synergizes with TGF-β to promote expression of IL-17 and other Th17 lineage cytokines, including IL-22 and IL-17F. In the absence of IL-6, TGF-β promotes Foxp3 expression and expansion of the Treg lymphocytes, a T cell subset that provides intestinal immune homeostasis. The role of STAT3 in Th17 differentiation is demonstrated in mice where altered Th17 production is either impaired in mice that were abalted for T cell STAT3 expression or augmented in mice with overexpressed STAT3. CD4+ T cells are imperative in promoting the adaptive immune response by producing and secreting specific cytokine profiles that activate an appropriate immune response. Th1 cells provide protection from intracellular organisms, such as viruses, while Th2 responses are necessary to defend the mucosa from extracellular pathogens. Finally, Th17 cells provide defense against extracellular bacteria and fungi (Fig. 2 and Table 1).
B Cell Maturation Requires STAT5 Activation by IL-7R
IL-7 is a cytokine that is integral to normal T and B cell development during lymphopoiesis and normal T and B cell function are essential to a proper antibody response by the mucosal immune system. IL-7 mediates its actions by binding to the IL-7R, consisting of the IL-7Rα chain and the γ chain (γc). Early stages of lymphocyte development require IL-7Rα, which is commonly expressed by lymphoid progenitor cells.51,52 After IL-7 binding to the IL-7Rα, JAK1 and JAK3 are activated53 and phosphorylate STAT5. After phosphorylation, STAT5 dimerizes and translocates to the cell nucleus. Ablation of the cytokine IL-7,51 the receptors IL-7Rα52 and γc,54,55 or the proteins JAK1 or JAK356 impairs normal B cell development in mice. STAT5 consists of two isoforms: STAT5A and STAT5B.57 Interestingly, B cell development is normal in mice lacking either isoform, but not both.58,59 However, complete STAT5 ablation results in mice with perinatal lethality associated with reduced T and B cell numbers and impaired maturation of B lymphocytes, αβ T lymphocytes, and γδ T lymphocytes.58,60 Studies using IL-7R−/− mice are characterized by reduced T-cell lymphopoiesis in the thymus and reduced B-cell lymphopoiesis in bone marrow. These studies demonstrate a direct role for IL-7R signaling through STAT5 in the process of T cell survival and B cell development that are essential to mounting a focused, adaptive response to antigen. Although the role of these pathways in sIgA synthesis have not been specifically investigated, JAK-STAT signaling is required for B cell maturation and plasma cell development.
sIgA Transport Protein pIgR Requires JAK1 and STAT6
The secretion of sIgA across the intestinal epithelium, via the polymeric immunoglobulin receptor (pIgR), is regulated by JAK-STAT. sIgA is the principle molecule of adaptive immunity secreted across the mucosa into the small intestinal lumen. sIgA provides antigen-specific and non-specific protection at the mucosal surfaces against ingested pathogens, microbes, and environmental antigen through several mechanisms, including immune exclusion and downregulation of bacterial virulence.
In vitro work demonstrates that the activated STAT6 forms dimmers, translocates to the nucleus where it binds specific DNA elements and activates transcription of several products, including pIgR.27,61-65 In addition to STAT6 mediation of pIgR by Th2 cytokines, proinflammatory cytokines also directly modulate the pIgR expression in mucosal epithelial cells.66,67 IL-4, IL-1, TNF-α, and IFN-γ all upregulate the expression of pIgR, demonstrating the necessity of this conserved immune protein during contrasting inflammatory profiles. Signaling with the cytokine IFN-γ receptor causes activation of STAT1 dimers, while signaling from IL-4 causes activation of STAT6
Since dietary and environmental antigens constantly stimulate the intestinal mucosa, our novel animal models study intestinal JAK-STAT signaling in the absence of enteral feeding, with the use of intravenously administered nutrition to prevent malnutrition. Initially this model was established to investigate changes in mucosal immunity that occur in clinical populations fed by intravenous nutrition. In these patients, the incidence of nosocomial pneumonia and intra-abdominal abscess is significantly greater with intravenously nutrition compared with enteral feeding, especially in the most critically ill. Animal studies demonstrate that the lack of enteral contents during intravenous feeding downregulates many aspects of intestinal immune function, including altered cytokines with reduced Th2 profiles, decreased T and B lymphocyte counts in both inductive and effector sites, reduced expression of the transport protein pIgR, and decreased levels of sIgA secretion at effector mucosal sites. Functionally, these changes result in reduced anti-viral and anti-bacterial defenses when mice are challenged with H1N1 virus and Pseudomonas aeruginosa, respectively.
Specific to JAK-STAT, these studies showed that parenteral nutrition—infusion of simple amino acids, sugars, minerals, and vitamins delivered directly into the central vein without enteral feeding—lowers tissue levels of IL-4, IL-13, pIgR, and luminal sIgA. Direct decreases in tissue levels of both phosphorylated JAK1 and STAT6 accompanied these changes. These data suggested the role of JAK-STAT signaling in adaptive immunity and the importance of phosphorylated JAK1 and STAT6 as mediators of pIgR transcription. A causative relationship was confirmed by administering exogenous IL-25 to the mice receiving intravenous nutrition. IL-25 stimulates production of the cytokines IL-4 and IL-13 by Th2 lymphocytes. Mice given intravenous nutrition with exogenous IL-25 had elevated tissue IL-4, IL-13, pIgR, and luminal sIgA that were associated with increases in phosphorylated JAK1 and STAT6.68
In another model examining the route and type of nutrition on mucosal defenses, animals were given the parenteral nutrition solution—comprised of simple amino acids, sugars, minerals, and vitamins—but delivered directly to the stomach using a gastrostomy tube. This route of feeding also lowers many parameters of intestinal immune function—since the diet contains only simple amino acids, sugars, minerals, and vitamins—but not to the degree associated with delivering the same solution into the vein without any enteral stimulation. Complex polyphenolic compounds, proanthocyanidins isolated from cranberry,69 were added to the elemental diet to stimulate the intestinal mucosa. That study observed the administration of elemental enteral diet alone decreased levels of IL-4, IL-13, pIgR, and sIgA with associated decreases in JAK1 and STAT6. The addition of proanthocyanidins to the elemental enteral diet increased IL-4, IL-13, pIgR, and sIgA, associated with increased JAK1 and STAT6 phosphorylation, suggesting the relationship between these cytokines, JAK-STAT proteins, and pIgR expression in the animal models.
The Innate Intestinal Barrer
In addition to the transcytosis of sIgA across the enterocytes, the more basic aspect of intestinal mucosal defense is the epithelial barrier itself, which is collectively comprised of absorptive columnar enterocytes, mucin secreting goblet cells, antimicrobial secreting Paneth cells, hormone secreting enteroendocrine cells, and microfold (M) cells covering Peyer patches.70 These cells differentiate from pluripotent stem cells located at the base of intestinal crypts of Leiberkuhn and extend to form a continuous layer of columnar epithelial cells held together by tight-junction proteins. The total mucosal surface can reach 300 m2 in humans and is faced with maintaining a barrier between the host and over 100 trillion individual bacterial cells, parasites, environmental antigens, and toxins71 (Fig. 2).
The epithelium responds with specific cytokine profiles when exposed to potentially infectious organisms or harmful substances. For instance, viruses, pathogenic bacteria, and bacterial components stimulate various Toll-like receptors (TLRs) at the basolateral or apical surface of epithelial cells that leads to the release of classical pro-inflammatory Th1 cytokines, such as IFNs, inducing STAT1 activation.72 Since persistent inflammatory responses can be harmful and sometimes fatal, maintaining the necessary interactions for proper immune and inflammatory signaling is imperative (i.e., proper release of cytokine profiles in response to bacteria, virus, fungi, etc.). Many studies have investigated these interactions, and TLR signaling emerges as one factor that directly mediates this response.73,74 Classically, past studies focused on the role of the host in eliminating pathogenic bacteria, that required TLR signaling. Interestingly, recent studies demonstrate commensal bacteria also signal through TLR receptors. For example, the work from Medzhitov and colleagues demonstrate the necessity of TLR stimulation from commensal bacteria for the control of intestinal homeostasis and protection from injury.75 Specifically, they examined signaling from antibiotic-treated mice and demonstrated a necessary role for commensal bacteria in facilitating a proper response to stimuli since antibiotic treatment ablated certain cytokine signaling in the colon.
Viruses stimulate IFNα while bacterial components stimulate IFNγ.72,76 The IFNα receptor is comprised of IFNαR1 and IFNαR2 chains containing JAK1 and TYK2, where activation leads to STAT1:STAT2 dimerization and transcription of antiviral immune molecules. The IFNγ receptor is comprised of IFNγR1 and IFNγR2 chains containing JAK1 and JAK2, where activation induces STAT1:STAT1 dimerization and inflammatory and antibacterial immune responses. Other microbiome compositions, such as murine segmented filamentous bacteria, stimulate Th17 responses through TLR5 that are mediated through IL-17, IL-22, and IL-23, inducing STAT3 activation.77 Activation of STAT3 in the epithelium stimulates antimicrobial production, such as β-defensins and RegIIIγ, and inhibits NFκB activation that can induce proinflammatory tissue injury.78 Enteric parasites such as nematodes stimulate Th2 cytokine profiles, including IL-4, IL-9, IL-13, and IL-25, and are classically anti-inflammatory.79 Type-II IL-4 receptors, expressed on epithelial cells, consist of IL-4Rα1 and IL-13Rα1 chains and are activated by both IL-4 or IL-13.13 Epithelial responses to STAT6 activation during nematode infection includes downregulation of enterocyte ion absorption, increased mucous secretion by goblet cells, and release of antimicrobial compounds by Paneth cells, which aid in clearing parasites from the intestinal tract.
Goblet cells migrate to the intestinal villi tip every 3–5 d following differentiation before sloughing into the intestinal lumen. Goblet cell differentiation is stimulated by the Th2 cytokines, IL-4 and IL-13, which bind the Type-II IL-4 receptor and activate STAT6. Goblet cells produce and secrete mucin glycoproteins that are vital in maintaining barrier integrity and localize antimicrobial compounds at the epithelial surface. The primary secreted mucin in the small intestine is MUC2. Additionally, goblet cells also produce Trefoil factor 3 (TFF3) and Relmβ, compounds that are thought to stabilize the mucin layer and repair damaged epithelial cells. Cell culture lines demonstrate IL-4 and IL-13 upregulate TFF3 and MUC2 expression through STAT6 dependent mechanisms.80 In the colon, STAT3 activation stimulates MUC1, MUC3, and MUC13 via IL-22/JAK1.81
As noted, efficient clearance of nematode infection requires STAT6, where deletion of the IL-4Rα delays goblet cell expansion and slows parasite expulsion. Initial epithelial injury by parasites stimulates the release of the alarm cytokines IL-25 and IL-33, promoting Th2 lymphocyte expansion and elevated levels of IL-4, IL-5, IL-9, and IL-13. Specifically, IL-13 is required for these responses since exogenous IL-25 administration, which mimics mucosal responses to nematode, does not occur in IL-13 knockout mice.79 Furthermore macrophages and dendritic cells, stimulated by glycans and glycolipids on the parasites surface, amplify the Th2 cytokines, as well as eosinophils and basophil expansion.
Paneth cells, in contrast to goblet cells, remain at the base of the intestinal crypts near the stem cells and secrete numerous antimicrobial compounds, including lysozyme, secretory phospholipase A2(sPLA2), RegIIIγ, Angiogenin4, and defensins (cryptidins in mice).82 Interestingly, Paneth cells also contain elevated levels of TNF-α, IL-17, and IL-23 and are implicated in acute mucosal inflammation. During systemic TNF-α challenge, Paneth cells release large amounts of IL-17 into the mucosal immune system, making this cell type an interesting target for understanding systemic vs mucosal immune compartmentalization. The release of Paneth cell antimicrobial compounds into the intestinal lumen have broad spectrum activity against gram-positive and gram-negative bacteria and are the major strategy for regulation microbiome composition. Murine studies demonstrate IL-13 is also required for increased Paneth cell antimicrobial expression, where absence of IL-13−/− or IL-4R−/− inhibits responsiveness.83
Recently, we demonstrated intravenous nutrition—with a lack of enteral feeding—results in decreased tissue and luminal levels of IL-4 and IL-13, associated with reduced STAT6 phosphorylation, and decreased levels of both the Paneth cell product sPLA2 and the goblet cell product MUC2. These effects were reversible with the addition of intravenous IL-25 during intravenous feeding,84 similar to the mucosal response to nematode infection. Interestingly, our ex vivo studies demonstrate that the loss of Paneth and goblet cell products during intravenous feeding are associated with increased susceptibility to bacterial enteroinvasivness, however, these effects are reversible when intravenously fed animals are administered IL-25.84
JAK-STAT Mutations in Health
Inflammatory bowel disease
JAK-STAT signaling plays crucial roles in epithelial proliferation, differentiation, and apoptosis. States of intestinal hyper-inflammation, such as the inflammatory bowel diseases (IBD) ulcerative colitis (UC), Crohn disease (CD), and ankylosing spondylitis (AS), are associated with elevated STAT signaling.85 Since the epithelial barrier is comprised of non-hematopoietic derived cells which are in close proximity to the hematopoietically derived lymphocytes in the lamina propria, knockout studies were needed to elucidate epithelial specific STAT signaling.
Murine enterocyte specific STAT5 deletion, which is normally activated by IL-22/JAK1, increases severity of DSS-colitis and delays wound healing following intestinal injury, perhaps through the loss of NFκB inhibition.86 In this model, epithelial STAT5 was required for tight junction barrier integrity. Similarly, epithelial deletion of STAT3, normally activated by IL-6/JAK2, results in greater DSS-colitis severity and impaired cellular stress responses related to epithelial restitution.87 Global knockout of STAT6 results in increased inflammation and crypt damage following DSS challenge, characterized by elevated nitrite/nitrate levels. While it remains unclear if hyperregulation of respective STAT pathways in these processes contribute toward or protect against epithelial pathogenesis in IBD, characterization of JAK-STAT signaling during the development and treatment of these intestinal diseases will further our understanding of the molecular signaling involved.
Hyper-IgE syndromes
Another disease associated with altered STAT signaling is hyperproduction of IgE. Production of IgE by plasma cells normally remains under tight regulation through stimulation by IL-4, IL-13, and IL-21 and suppression by IFN-γ, IL-10, and TGF-β. IL-4 binding of the IL-4R stimulates STAT6, leading to moderate production and release of IgE. Binding of IL-10 or IFN-γ to the IL-10R or IFN- γR stimulate STAT3, which inhibits STAT6 through STAT3 dependet mechanisms. The more recently identified role of IL-21 in IgE production in human B cells also signals through STAT3 and leads to more prominent IgE release than IL-4 or IL-13 under normal conditions.88 However, since STAT3 also has suppressive effects upon STAT6 mediated IgE production, STAT3 mutations lead to augmented IgE production by STAT6 and is characterized by hyper-IgE pathologies in humans. Patients carrying STAT3 mutations suffer from skin allergies, lung infections, elevated serum IgE, and abnormalities in connective tissues, skeletal muscle, and the vascular system. IFN-γ therapy in these patients produces mixed results.89
Therapeutic Interventions
Studies continue to define the role of the JAK-STAT signaling mutations and subsequent effects upon human health and disease, including immune diseases, cancer, and inflammatory disorders. These studies have identified mechanisms and pathways that may be amendable through pharmacologic intervention. Current clinical trials investigate the efficacy of inhibiting JAK protein signaling to ameliorate disease. However, one pediment remains the vast cross talk between diverse cytokines and JAK-STAT signaling machinery. Many therapies are not unique to a particular JAK resulting in unintended clinical consequences. Nevertheless, JAK inhibitor drugs that proved effective in laboratory studies are currently in phase I and II clinical trials. An example is the use of baricitinib, which targets JAK1/JAK2, in rheumatoid arthritis patients, and pacritinib, which targets advanced myeloid malignancies and chronic idiopathic myelofibrosis.
Conclusions
JAK-STAT signaling is one of the principal signaling pathways utilized by cytokine receptors. Since the discovery of this pathway, many studies elucidated the role of this pathway in many processes including cell development, growth and survival. We’ve described several roles of JAK-STAT signaling the intestinal mucosal immune system and its response to challenge by bacteria, viruses, and antigens found in intestinal lumen. Our studies on the route and type of nutrition have demonstrated reduced JAK-STAT signaling associated with loss of adaptive and innate mucosal immune function, which is reversible with exogenous stimulants. The JAK-STAT pathway is vital to T cell differentiation, B cell maturation and development, secretion of sIgA, and mucus and antibody production, which are required to maintain anti-viral and anti-bacterial defenses at the mucosal surface. Further elucidation of JAK-STAT signaling under normal and pathological conditions will better complete our understanding of intestinal immunity and homeostasis.
Acknowledgments
The project described was supported by Award Number I01BX001672 from the Biomedical Laboratory Research and Development Service of the VA Office of Research and Development. The contents of this article do not represent the views of the Veterans Affairs or the United States Government.
Glossary
Abbreviations:
- sIgA
secretory IgA
- Ig
immunoglobulin
- PP
Peyer patches
- MAdCAM-1
mucosal addressin adhesion molecule-1
- HEV
High endothelial venule
- pIgR
polymeric immunoglobulin receptor
- M
microfold
- TLR
toll like receptor
- AS
ankylosing spondylitis
- CD
Crohn disease
- IBD
inflammatory bowel disease
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/jak-stat/article/25530
References
- 1.McGuckin MA, Lindén SK, Sutton P, Florin TH. Mucin dynamics and enteric pathogens. Nat Rev Microbiol. 2011;9:265–78. doi: 10.1038/nrmicro2538. [DOI] [PubMed] [Google Scholar]
- 2.Johansson ME, Ambort D, Pelaseyed T, Schütte A, Gustafsson JK, Ermund A, et al. Composition and functional role of the mucus layers in the intestine. Cell Mol Life Sci. 2011;68:3635–41. doi: 10.1007/s00018-011-0822-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bergstrom KS, Kissoon-Singh V, Gibson DL, Ma C, Montero M, Sham HP, et al. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog. 2010;6:e1000902. doi: 10.1371/journal.ppat.1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ouellette AJ, Selsted ME. Paneth cell defensins: endogenous peptide components of intestinal host defense. FASEB J. 1996;10:1280–9. doi: 10.1096/fasebj.10.11.8836041. [DOI] [PubMed] [Google Scholar]
- 5.Ouellette AJ, Greco RM, James M, Frederick D, Naftilan J, Fallon JT. Developmental regulation of cryptdin, a corticostatin/defensin precursor mRNA in mouse small intestinal crypt epithelium. J Cell Biol. 1989;108:1687–95. doi: 10.1083/jcb.108.5.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harwig SS, Tan L, Qu XD, Cho Y, Eisenhauer PB, Lehrer RI. Bactericidal properties of murine intestinal phospholipase A2. J Clin Invest. 1995;95:603–10. doi: 10.1172/JCI117704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harwig SS, Eisenhauer PB, Chen NP, Lehrer RI. Cryptdins: endogenous antibiotic peptides of small intestinal Paneth cells. Adv Exp Med Biol. 1995;371A:251–5. doi: 10.1007/978-1-4615-1941-6_53. [DOI] [PubMed] [Google Scholar]
- 8.Li J, Kudsk KA, Gocinski B, Dent D, Glezer J, Langkamp-Henken B. Effects of parenteral and enteral nutrition on gut-associated lymphoid tissue. J Trauma. 1995;39:44–51, discussion 51-2. doi: 10.1097/00005373-199507000-00006. [DOI] [PubMed] [Google Scholar]
- 9.King BK, Li J, Kudsk KA. A temporal study of TPN-induced changes in gut-associated lymphoid tissue and mucosal immunity. Arch Surg. 1997;132:1303–9. doi: 10.1001/archsurg.1997.01430360049009. [DOI] [PubMed] [Google Scholar]
- 10.Zarzaur BL, Kudsk KA. The mucosa-associated lymphoid tissue structure, function, and derangements. Shock. 2001;15:411–20. doi: 10.1097/00024382-200115060-00001. [DOI] [PubMed] [Google Scholar]
- 11.Aaronson DS, Horvath CM. A road map for those who don’t know JAK-STAT. Science. 2002;296:1653–5. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- 12.Kisseleva T, Bhattacharya S, Braunstein J, Schindler CW. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene. 2002;285:1–24. doi: 10.1016/S0378-1119(02)00398-0. [DOI] [PubMed] [Google Scholar]
- 13.Mohr A, Chatain N, Domoszlai T, et al. Dynamics and non-canonical aspects of JAK/STAT signalling. Eur J Cell Biol. 2012;91:524–32. doi: 10.1016/j.ejcb.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 14.Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci. 2004;117:1281–3. doi: 10.1242/jcs.00963. [DOI] [PubMed] [Google Scholar]
- 15.Reich NC, Liu L. Tracking STAT nuclear traffic. Nat Rev Immunol. 2006;6:602–12. doi: 10.1038/nri1885. [DOI] [PubMed] [Google Scholar]
- 16.Schindler C, Darnell JE., Jr. Transcriptional responses to polypeptide ligands: the JAK-STAT pathway. Annu Rev Biochem. 1995;64:621–51. doi: 10.1146/annurev.bi.64.070195.003201. [DOI] [PubMed] [Google Scholar]
- 17.Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282:20059–63. doi: 10.1074/jbc.R700016200. [DOI] [PubMed] [Google Scholar]
- 18.Levy DE, Darnell JE., Jr. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–62. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 19.Brandtzaeg P, Halstensen TS, Kett K, Krajci P, Kvale D, Rognum TO, et al. Immunobiology and immunopathology of human gut mucosa: humoral immunity and intraepithelial lymphocytes. Gastroenterology. 1989;97:1562–84. doi: 10.1016/0016-5085(89)90406-x. [DOI] [PubMed] [Google Scholar]
- 20.Kudsk KA. Current aspects of mucosal immunology and its influence by nutrition. Am J Surg. 2002;183:390–8. doi: 10.1016/S0002-9610(02)00821-8. [DOI] [PubMed] [Google Scholar]
- 21.Kang W, Kudsk KA. Is there evidence that the gut contributes to mucosal immunity in humans? JPEN J Parenter Enteral Nutr. 2007;31:246–58. doi: 10.1177/0148607107031003246. [DOI] [PubMed] [Google Scholar]
- 22.Johnson CD, Kudsk KA. Nutrition and intestinal mucosal immunity. Clin Nutr. 1999;18:337–44. doi: 10.1016/S0261-5614(99)80012-0. [DOI] [PubMed] [Google Scholar]
- 23.Berg EL, McEvoy LM, Berlin C, Bargatze RF, Butcher EC. L-selectin-mediated lymphocyte rolling on MAdCAM-1. Nature. 1993;366:695–8. doi: 10.1038/366695a0. [DOI] [PubMed] [Google Scholar]
- 24.Berlin C, Berg EL, Briskin MJ, Andrew DP, Kilshaw PJ, Holzmann B, et al. Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell. 1993;74:185–95. doi: 10.1016/0092-8674(93)90305-A. [DOI] [PubMed] [Google Scholar]
- 25.Brandtzaeg P, Farstad IN, Haraldsen G. Regional specialization in the mucosal immune system: primed cells do not always home along the same track. Immunol Today. 1999;20:267–77. doi: 10.1016/S0167-5699(99)01468-1. [DOI] [PubMed] [Google Scholar]
- 26.Brandtzaeg P, Pabst R. Let’s go mucosal: communication on slippery ground. Trends Immunol. 2004;25:570–7. doi: 10.1016/j.it.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 27.Kaetzel CS, Blanch VJ, Hempen PM, Phillips KM, Piskurich JF, Youngman KR. The polymeric immunoglobulin receptor: structure and synthesis. Biochem Soc Trans. 1997;25:475–80. doi: 10.1042/bst0250475. [DOI] [PubMed] [Google Scholar]
- 28.Alverdy J. The effect of nutrition on gastrointestinal barrier function. Semin Respir Infect. 1994;9:248–55. [PubMed] [Google Scholar]
- 29.Alverdy JC, Laughlin RS, Wu L. Influence of the critically ill state on host-pathogen interactions within the intestine: gut-derived sepsis redefined. Crit Care Med. 2003;31:598–607. doi: 10.1097/01.CCM.0000045576.55937.67. [DOI] [PubMed] [Google Scholar]
- 30.Seder RA, Paul WE. Acquisition of lymphokine-producing phenotype by CD4+ T cells. Annu Rev Immunol. 1994;12:635–73. doi: 10.1146/annurev.iy.12.040194.003223. [DOI] [PubMed] [Google Scholar]
- 31.Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–73. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 32.Paul WE, Seder RA. Lymphocyte responses and cytokines. Cell. 1994;76:241–51. doi: 10.1016/0092-8674(94)90332-8. [DOI] [PubMed] [Google Scholar]
- 33.Rengarajan J, Szabo SJ, Glimcher LH. Transcriptional regulation of Th1/Th2 polarization. Immunol Today. 2000;21:479–83. doi: 10.1016/S0167-5699(00)01712-6. [DOI] [PubMed] [Google Scholar]
- 34.Murphy KM, Ouyang W, Farrar JD, Yang J, Ranganath S, Asnagli H, et al. Signaling and transcription in T helper development. Annu Rev Immunol. 2000;18:451–94. doi: 10.1146/annurev.immunol.18.1.451. [DOI] [PubMed] [Google Scholar]
- 35.Magram J, Sfarra J, Connaughton S, Faherty D, Warrier R, Carvajal D, et al. IL-12-deficient mice are defective but not devoid of type 1 cytokine responses. Ann N Y Acad Sci. 1996;795:60–70. doi: 10.1111/j.1749-6632.1996.tb52655.x. [DOI] [PubMed] [Google Scholar]
- 36.Magram J, Connaughton SE, Warrier RR, Carvajal DM, Wu CY, Ferrante J, et al. IL-12-deficient mice are defective in IFN gamma production and type 1 cytokine responses. Immunity. 1996;4:471–81. doi: 10.1016/S1074-7613(00)80413-6. [DOI] [PubMed] [Google Scholar]
- 37.Wu C, Ferrante J, Gately MK, Magram J. Characterization of IL-12 receptor beta1 chain (IL-12Rbeta1)-deficient mice: IL-12Rbeta1 is an essential component of the functional mouse IL-12 receptor. J Immunol. 1997;159:1658–65. [PubMed] [Google Scholar]
- 38.Kühn R, Rajewsky K, Müller W. Generation and analysis of interleukin-4 deficient mice. Science. 1991;254:707–10. doi: 10.1126/science.1948049. [DOI] [PubMed] [Google Scholar]
- 39.Kopf M, Le Gros G, Bachmann M, Lamers MC, Bluethmann H, Köhler G. Disruption of the murine IL-4 gene blocks Th2 cytokine responses. Nature. 1993;362:245–8. doi: 10.1038/362245a0. [DOI] [PubMed] [Google Scholar]
- 40.Noben-Trauth N, Shultz LD, Brombacher F, Urban JF, Jr., Gu H, Paul WE. An interleukin 4 (IL-4)-independent pathway for CD4+ T cell IL-4 production is revealed in IL-4 receptor-deficient mice. Proc Natl Acad Sci U S A. 1997;94:10838–43. doi: 10.1073/pnas.94.20.10838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frucht DM, Aringer M, Galon J, Danning C, Brown M, Fan S, et al. Stat4 is expressed in activated peripheral blood monocytes, dendritic cells, and macrophages at sites of Th1-mediated inflammation. J Immunol. 2000;164:4659–64. doi: 10.4049/jimmunol.164.9.4659. [DOI] [PubMed] [Google Scholar]
- 42.Watford WT, Moriguchi M, Morinobu A, O’Shea JJ. The biology of IL-12: coordinating innate and adaptive immune responses. Cytokine Growth Factor Rev. 2003;14:361–8. doi: 10.1016/S1359-6101(03)00043-1. [DOI] [PubMed] [Google Scholar]
- 43.Thierfelder WE, van Deursen JM, Yamamoto K, Tripp RA, Sarawar SR, Carson RT, et al. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature. 1996;382:171–4. doi: 10.1038/382171a0. [DOI] [PubMed] [Google Scholar]
- 44.Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S, et al. Essential role of Stat6 in IL-4 signalling. Nature. 1996;380:627–30. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- 45.Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4:313–9. doi: 10.1016/S1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 46.Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2:933–44. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- 47.Harrington LE, Mangan PR, Weaver CT. Expanding the effector CD4 T-cell repertoire: the Th17 lineage. Curr Opin Immunol. 2006;18:349–56. doi: 10.1016/j.coi.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 48.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–4. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 49.Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24:677–88. doi: 10.1016/j.immuni.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 50.Chen Z, Laurence A, O’Shea JJ. Signal transduction pathways and transcriptional regulation in the control of Th17 differentiation. Semin Immunol. 2007;19:400–8. doi: 10.1016/j.smim.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.von Freeden-Jeffry U, Vieira P, Lucian LA, McNeil T, Burdach SE, Murray R. Lymphopenia in interleukin (IL)-7 gene-deleted mice identifies IL-7 as a nonredundant cytokine. J Exp Med. 1995;181:1519–26. doi: 10.1084/jem.181.4.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peschon JJ, Morrissey PJ, Grabstein KH, Ramsdell FJ, Maraskovsky E, Gliniak BC, et al. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J Exp Med. 1994;180:1955–60. doi: 10.1084/jem.180.5.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Foxwell BM, Beadling C, Guschin D, Kerr I, Cantrell D. Interleukin-7 can induce the activation of Jak 1, Jak 3 and STAT 5 proteins in murine T cells. Eur J Immunol. 1995;25:3041–6. doi: 10.1002/eji.1830251109. [DOI] [PubMed] [Google Scholar]
- 54.Cao X, Shores EW, Hu-Li J, Anver MR, Kelsall BL, Russell SM, et al. Defective lymphoid development in mice lacking expression of the common cytokine receptor gamma chain. Immunity. 1995;2:223–38. doi: 10.1016/1074-7613(95)90047-0. [DOI] [PubMed] [Google Scholar]
- 55.DiSanto JP, Müller W, Guy-Grand D, Fischer A, Rajewsky K. Lymphoid development in mice with a targeted deletion of the interleukin 2 receptor gamma chain. Proc Natl Acad Sci U S A. 1995;92:377–81. doi: 10.1073/pnas.92.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thomis DC, Gurniak CB, Tivol E, Sharpe AH, Berg LJ. Defects in B lymphocyte maturation and T lymphocyte activation in mice lacking Jak3. Science. 1995;270:794–7. doi: 10.1126/science.270.5237.794. [DOI] [PubMed] [Google Scholar]
- 57.Hennighausen L, Robinson GW. Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes Dev. 2008;22:711–21. doi: 10.1101/gad.1643908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hoelbl A, Kovacic B, Kerenyi MA, Simma O, Warsch W, Cui Y, et al. Clarifying the role of Stat5 in lymphoid development and Abelson-induced transformation. Blood. 2006;107:4898–906. doi: 10.1182/blood-2005-09-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moriggl R, Topham DJ, Teglund S, Sexl V, McKay C, Wang D, et al. Stat5 is required for IL-2-induced cell cycle progression of peripheral T cells. Immunity. 1999;10:249–59. doi: 10.1016/S1074-7613(00)80025-4. [DOI] [PubMed] [Google Scholar]
- 60.Yao Z, Cui Y, Watford WT, Bream JH, Yamaoka K, Hissong BD, et al. Stat5a/b are essential for normal lymphoid development and differentiation. Proc Natl Acad Sci U S A. 2006;103:1000–5. doi: 10.1073/pnas.0507350103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Igaz P, Tóth S, Falus A. Biological and clinical significance of the JAK-STAT pathway; lessons from knockout mice. Inflamm Res. 2001;50:435–41. doi: 10.1007/PL00000267. [DOI] [PubMed] [Google Scholar]
- 62.Johansen FE, Bosløven BA, Krajci P, Brandtzaeg P. A composite DNA element in the promoter of the polymeric immunoglobulin receptor regulates its constitutive expression. Eur J Immunol. 1998;28:1161–71. doi: 10.1002/(SICI)1521-4141(199804)28:04<1161::AID-IMMU1161>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 63.Schjerven H, Brandtzaeg P, Johansen FE. Mechanism of IL-4-mediated up-regulation of the polymeric Ig receptor: role of STAT6 in cell type-specific delayed transcriptional response. J Immunol. 2000;165:3898–906. doi: 10.4049/jimmunol.165.7.3898. [DOI] [PubMed] [Google Scholar]
- 64.Kaetzel CS. The polymeric immunoglobulin receptor: bridging innate and adaptive immune responses at mucosal surfaces. Immunol Rev. 2005;206:83–99. doi: 10.1111/j.0105-2896.2005.00278.x. [DOI] [PubMed] [Google Scholar]
- 65.Martín MG, Wang J, Li TW, Lam JT, Gutierrez EM, Solorzano-Vargas RS, et al. Characterization of the 5′-flanking region of the murine polymeric IgA receptor gene. Am J Physiol. 1998;275:G778–88. doi: 10.1152/ajpgi.1998.275.4.G778. [DOI] [PubMed] [Google Scholar]
- 66.Johansen FE, Brandtzaeg P. Transcriptional regulation of the mucosal IgA system. Trends Immunol. 2004;25:150–7. doi: 10.1016/j.it.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 67.Kaetzel CS. The polymeric immunoglobulin receptor: bridging innate and adaptive immune responses at mucosal surfaces. Immunol Rev. 2005;206:83–99. doi: 10.1111/j.0105-2896.2005.00278.x. [DOI] [PubMed] [Google Scholar]
- 68.Heneghan AF, Pierre JF, Kudsk KA. IL-25 Improves IgA Levels During Parenteral Nutrition Through the JAK-STAT Pathway. Ann Surg. 2013 doi: 10.1097/SLA.0b013e318277ea9e. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pierre JF, Heneghan AF, Feliciano RP, Shanmuganayagam D, Krueger CG, Reed JD, et al. Cranberry Proanthocyanidins Improve Intestinal sIgA During Elemental Enteral Nutrition. JPEN J Parenter Enteral Nutr. 2013 doi: 10.1177/0148607112473654. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Langkamp-Henken B, Glezer JA, Kudsk KA. Immunologic structure and function of the gastrointestinal tract. Nutr Clin Pract. 1992;7:100–8. doi: 10.1177/0115426592007003100. [DOI] [PubMed] [Google Scholar]
- 71.DeWitt RC, Kudsk KA. The gut’s role in metabolism, mucosal barrier function, and gut immunology. Infect Dis Clin North Am. 1999;13:465–81, x. doi: 10.1016/S0891-5520(05)70086-6. [x.] [DOI] [PubMed] [Google Scholar]
- 72.Najjar I, Fagard R. STAT1 and pathogens, not a friendly relationship. Biochimie. 2010;92:425–44. doi: 10.1016/j.biochi.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–26. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 74.Medzhitov R. TLR-mediated innate immune recognition. Semin Immunol. 2007;19:1–2. doi: 10.1016/j.smim.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–41. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 76.Wang WL, Liu W, Gong HY, Hong JR, Lin CC, Wu JL. Activation of cytokine expression occurs through the TNFα/NF-κB-mediated pathway in birnavirus-infected cells. Fish Shellfish Immunol. 2011;31:10–21. doi: 10.1016/j.fsi.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 77.Jarnicki A, Putoczki T, Ernst M. Stat3: linking inflammation to epithelial cancer - more than a “gut” feeling? Cell Div. 2010;5:14. doi: 10.1186/1747-1028-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity. 2004;21:241–54. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 79.Zhao A, Urban JF, Jr., Sun R, Stiltz J, Morimoto M, Notari L, et al. Critical role of IL-25 in nematode infection-induced alterations in intestinal function. J Immunol. 2010;185:6921–9. doi: 10.4049/jimmunol.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Durual S, Blanchard C, Estienne M, Jacquier MF, Cuber JC, Perrot V, et al. Expression of human TFF3 in relation to growth of HT-29 cell subpopulations: involvement of PI3-K but not STAT6. Differentiation. 2005;73:36–44. doi: 10.1111/j.1432-0436.2005.07301006.x. [DOI] [PubMed] [Google Scholar]
- 81.Gersemann M, Becker S, Nuding S, Antoni L, Ott G, Fritz P, et al. Olfactomedin-4 is a glycoprotein secreted into mucus in active IBD. J Crohns Colitis. 2012;6:425–34. doi: 10.1016/j.crohns.2011.09.013. [DOI] [PubMed] [Google Scholar]
- 82.Porter EM, Bevins CL, Ghosh D, Ganz T. The multifaceted Paneth cell. Cell Mol Life Sci. 2002;59:156–70. doi: 10.1007/s00018-002-8412-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Steenwinckel V, Louahed J, Lemaire MM, Sommereyns C, Warnier G, McKenzie A, et al. IL-9 promotes IL-13-dependent paneth cell hyperplasia and up-regulation of innate immunity mediators in intestinal mucosa. J Immunol. 2009;182:4737–43. doi: 10.4049/jimmunol.0801941. [DOI] [PubMed] [Google Scholar]
- 84.Heneghan A, Pierre J, Gosain A, Kudsk KA. IL-25 Improves Luminal Innate Immunity and Barrier Function During Parenteral Nutrition. Ann Surg. 2013 doi: 10.1097/SLA.0b013e318284f510. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ohtani K, Ohtsuka Y, Ikuse T, Baba Y, Yamakawa Y, Aoyagi Y, et al. Increased mucosal expression of GATA-3 and STAT-4 in pediatric ulcerative colitis. Pediatr Int. 2010;52:584–9. doi: 10.1111/j.1442-200X.2009.03019.x. [DOI] [PubMed] [Google Scholar]
- 86.Gilbert S, Zhang R, Denson L, Moriggl R, Steinbrecher K, Shroyer N, et al. Enterocyte STAT5 promotes mucosal wound healing via suppression of myosin light chain kinase-mediated loss of barrier function and inflammation. EMBO Mol Med. 2012;4:109–24. doi: 10.1002/emmm.201100192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Willson TA, Jurickova I, Collins M, Denson LA. Deletion of intestinal epithelial cell STAT3 promotes T-lymphocyte STAT3 activation and chronic colitis following acute dextran sodium sulfate injury in mice. Inflamm Bowel Dis. 2013;19:512–25. doi: 10.1097/MIB.0b013e31828028ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Avery DT, Ma CS, Bryant VL, Santner-Nanan B, Nanan R, Wong M, et al. STAT3 is required for IL-21-induced secretion of IgE from human naive B cells. Blood. 2008;112:1784–93. doi: 10.1182/blood-2008-02-142745. [DOI] [PubMed] [Google Scholar]
- 89.King CL, Gallin JI, Malech HL, Abramson SL, Nutman TB. Regulation of immunoglobulin production in hyperimmunoglobulin E recurrent-infection syndrome by interferon gamma. Proc Natl Acad Sci U S A. 1989;86:10085–9. doi: 10.1073/pnas.86.24.10085. [DOI] [PMC free article] [PubMed] [Google Scholar]