

Abstract
Cytokines play several roles in developing and/or reinforcing premature cellular senescence of young cells. One such cytokine, interleukin-6 (IL-6), regulates senescence in some systems in addition to its known functions of immune regulation and promotion of tumorigenesis. In this review, we describe recent advances in studies on the roles of IL-6 and its downstream signal transducer and activator of transcription 3 (STAT3) in regulating premature cellular senescence. IL-6/sIL-6Rα stimulation forms a senescence-inducing circuit involving the STAT3-insulin-like growth factor-binding protein 5 (IGFBP5) as a key axis triggering and reinforcing component in human fibroblasts. We describe how cytokines regulate the process of senescence by activating STAT3 in one system and anti-senescence or tumorigenesis in other systems. The roles of other STAT members in premature senescence also will be discussed to show the multiple mechanisms leading to cytokine-induced senescence.
Keywords: cellular senescence, cytokine, interleukin-6, STAT3, IGFBP5, tumorigenesis
Introduction
Both normal and tumor cells undergo senescence in response to various insults.1-4 Cellular senescence is a state characterized by the inability of cells to proliferate despite the presence of sufficient nutrients and mitogens while maintaining cell viability and metabolic activity.1-4 Cellular senescence is becoming recognized as an important program to escape from unregulated proliferation causing tumorigenesis, like the cell death program, apoptosis.
Recently, cytokines and chemokines, secreted by cells attacking target cells, by cells associated with chronic inflammation and by cells undergoing senescence, have been shown to trigger and/or reinforce the senescence process through various mechanisms.5,6
IL-6 is a multifunctional cytokine that regulates cell proliferation, survival, and differentiation, and enhances cellular function in multiple lineages of cells.7-10 IL-6 has been implicated in the pathogenesis of a variety of diseases often associated with inflammation, chronic immune diseases, tissue-infiltrating tumors, neurological diseases, and tissue aging.9,10 Because IL-6 and a soluble form of the IL-6Rα chain are secreted abundantly in tissues with inflammation, aging, and tumor infiltration, we became interested in the roles of IL-6 and its major signaling pathway involving signal transducer and activator of transcription 3 (STAT3) in the process of senescence and the balance between senescence and tumorigenesis.
In this review, we will describe how some cytokines, including IL-6, affect the senescence process and especially show the recent model on the IL-6/STAT3-induced senescence of human fibroblasts.11 The roles of other members of the STAT family also will be discussed.
Overall Pictures of Cellular Senescence
Normal human cells undergo senescence in response to various insults causing persistent DNA damage. Senescent cells generally have a flattened and enlarged morphology and show an increase in senescence-associated-β-galactosidase (SA-β-Gal) activity.1-4 An overview of premature cellular senescence is shown in Figure 1.

Figure 1. A general overview of premature cellular senescence. Three major types of senescence, replicative senescence, oncogene-induced senescence, and stress-induced senescence, are known. Any types of senescence-induction commonly involve persistent and excessive DNA damage response (DDR) and activate both p53- and RB-dependent pathways, resulting in senescence. DDR-independent mechanisms are also known. Senescent cells are active and secrete a number of cytokines, chemokines, and other molecules, which is termed senescence-associated secretory phenotype (SASP). Some factors in SASP affect the senescence-induction of those senescent cells and the surrounding cells.
Three different types of senescence have been characterized: replicative senescence, oncogene-induced senescence (OIS), and stress-induced senescence, including exposure to cytokines. Replicative senescence was first described by Hayflick and Moorhead in 1961,12 who demonstrated that after a finite number of cell divisions, normal human fibroblasts cultured in conditions that drive their continuous replication eventually reach an arrested state in which they do not respond to mitogenic stimuli despite their metabolic activity. This type of senescence is mainly caused by the DNA damage response (DDR) due to telomere shortening below a certain threshold.13 DDR is associated with the appearance of foci of γ-H2AX (a phosphorylated form of H2AX) and the DDR proteins including 53BP1.14 As a result of DDR, the DNA damage kinases, ATM and ATR are activated. After amplification of the DDR signals, ATM and ATR activate CHK1 and CHK2 kinases, which phosphorylate and activate CDC25 and p53, leading to transient growth arrest allowing cells to repair the DNA damage. When DNA damage exceeds a threshold, cells undergo either apoptosis or senescence. In addition to p53, the RB tumor suppressor and its signaling partners, including p16INK4a (a cyclin-dependent kinase inhibitor acting upstream of RB), are involved in replicative senescence. Activation of both p53 and p16INK4a-RB pathways is essential for induction of senescence in a variety of human cell lineages. The relative contribution of these pathways to senescence depends on the cell types.
Induction of senescence is also a frequent outcome of oncogenic mutations in normal cells. A number of actively mutated oncogenes, including Ras, Raf, MEK, and c-Myc, or inactivated tumor suppressor genes, including PTEN, have been shown to induce premature senescence.15,16 This type of senescence is called oncogene-induced senescence (OIS). OIS can occur independently of telomere shortening and induces senescence in young cells. Oncogene-induced senescence has been observed in tumor cells even at early stages.17,18
The third type of senescence is stress-induced premature senescence. The stress-induced senescence of normal cells is a response to stressors such as oxidative stress, ionizing/non-ionizing radiation, and DNA damaging reagents, including chemotherapeutic genotoxic drugs. Chronic stimulation of cells with cytokines causes enough stress to induce growth arrest or senescence.
These three types of senescence have very similar consequences, leading to persistent DNA damage and activation of the two pathways, p53-p21CIP1 and p16INK4a-pRB, which exert processes leading to growth arrest and senescence. The increase of reactive oxygen species (ROS) often causes DNA damage in different types of senescence.
In addition to DDR-induced p53 activation, DNA damage activates nuclear factor kappa B (NFκB) transcription factor,19 and p38 kinase is activated independently of DDR through NFκB and ROS.20 Both NFκB and active p38 cooperate in activation of a number of genes, mostly coding for cytokines, chemokines, receptors for cytokines and chemokines, and proteases.20 Some genes can be activated by either NFκB or p38 alone. The robust changes in secretion properties associated with the process of senescence are termed senescence-associated secretory phenotype (SASP)5,6 or senescence-messaging secretome (SMS).21 A recent study showed that persistent DNA damage is required to cause SASP.21 Factors in SASP include IL-1α, IL-1β, IL-6, a number of chemokines including CXCL8 (IL-8), CXCL-1, CCL-2, VEGF, CXCR2, and matrix metalloproteinases (MMPs).5,21-23 The spectrum of secreted factors varies, depending on cell types and senescence-inducing insults.5,24 SASP factors affect the behavior of neighboring cells. Some factors can promote tumorigenesis of premalignant tumor cells. Other factors, including IL-6 and IL-8, and other chemokines acting on the CXCR2 receptor, have been shown to reinforce the senescence process of cells producing these factors, depending on the cell types.22,23 Kuilman et al. showed that both IL-6 and IL-6Rα chain are induced by activated BRAF oncogene (BRAFV600E) in TIG3 human fibroblasts expressing hTERT in a C/EBPβ-dependent manner and required for maintaining or reinforcing oncogene-induced senescence.22 In a different strain of cells, Acosta et al. showed that CXCR2 and its ligands produced by senescent cells were required to maintain the senescence process.23 SASP also contributes to recruitment of immune cells for clearance of senescent cells, modulates tissue repair, and possibly affects aging of surrounding tissues.2
In addition to the molecules mentioned above, there are several molecules induced by cytokine signals and involved in the regulation of senescence. Promyelocytic leukemia protein (PML) is one of the targets of cytokine signals. Although the exact biochemical basis of PML tumor suppressor properties is unclear, PML and PML nuclear bodies (PML NBs) are involved in several tumor-suppressive pathways regulating apoptosis and senescence, including the p53-p21CIP1 and p16INK4a-pRB pathways. PML facilitates acetylation, protein stabilization, and phosphorylation-mediated activation of p53.25 Though it was known that the PML gene is a direct transcription target of p53, the cytokines interferon β (IFNβ) and IL-6 were shown to activate PML mRNA expression through STAT1 and STAT3, respectively.26-29 SOCS1 and SOCS3, both of which are target genes of some members of STAT family proteins, were shown to interact with p53 and enhance the transcriptional activity of p53, thereby contributing to cellular senescence.30-32
Role of IL-6 in Premature Senescence
Considering that both IL-6 and soluble IL-6Rα chains are produced at sufficient concentrations in various tissues in infection, inflammation, and aging and that a signal transducing receptor, gp130, of the IL-6R complex is expressed ubiquitously in most cells, it is possible that IL-6 and soluble IL-6Rα produced by surrounding cells affect the progress of senescence in certain cells that do not have IL-6Rα chains.
To understand the role of IL-6 in the course of physiological senescence of normal primary cells and premature senescence, we used human diploid fibroblast TIG3 cells. We first observed that both IL-6 and IL-6-Rα chain were expressed by old TIG3 cells, concomitantly with activated STAT3. This observation prompted us to examine the role of IL-6 in premature senescence of primary TIG3 fibroblasts. Exogenously added IL-6/soluble IL-6Rα actually triggered premature senescence of young TIG3 cells.
In the following sections, we first outline IL-6, the IL-6 receptor system, and its signaling pathways and then focus on the mechanisms by which IL-6/sIL-6Rα causes premature cellular senescence in one type of human fibroblast.
IL-6 Receptor System and Its Signaling Pathways
The IL-6 receptor system, its signal transduction pathways, and their major target genes are outlined in Figure 2A. IL-6 acts on cells as a dimer by binding to a specific IL-6 receptor (IL-6R) complex composed of two IL-6Rα chains (also known as IL-6Rα or CD126) and two signal-generating receptor β chain subunits, named gp130 (also known as IL-6Rβ or CD130). The gp130 receptor molecule is a common component of multiple receptors for the IL-6 family of cytokines, such as IL-6, oncostatin M, leukemia inhibitory factor, ciliary neurotrophic factor, and IL-11.9,10 IL-6Rα, which interacts with IL-6, is mainly expressed in hepatocytes and hematopoietic cells, such as some types of T cells, monocytes/macrophages, activated B cells, and neutrophils.9,10 In contrast, a wide range of cells express gp130. Interestingly, soluble isoforms of the IL-6Rα chains are generated by alternative splicing or by ectodomain shedding of the IL-6Rα chain.33-35 The latter process, including induced and constitutive types of ectodomain shedding, is mediated by a-disintegrin-and-metalloproteinase (ADAM) gene family members ADAM17 and ADAM10, respectively.33-35 Soluble IL-6Rα (sIL-6Rα) can work as a component of the IL-6R complex by binding to IL-6 and creating a complex with gp130 for IL-6 signaling. Because inflamed, infected, tumor-infiltrated, and aging tissues contain IL-6 and sIL-6Rα, both of which are produced or shed by myeloid lineage cells, most of the cells in such tissues with gp130 can be activated by IL-6 and sIL-6Rα.35,36 On interaction of IL-6 with IL-6Rα complex, gp130 molecules dimerize and the gp130-associated JAK family tyrosine kinases (JAK1, JAK2, and Tyk2) go through a conformational change, bringing the two JAKs close enough to phosphorylate each other and become activated. Then activated JAK kinases phosphorylate the cytoplasmic domain of gp130 at the specific tyrosine residues, which are required for activation of downstream signal transduction pathways.

Figure 2. (A) The IL-6 receptor system and its signaling pathways. The three major pathways are the STAT3-, the ERK-, and the PI3K/AKT-mediated pathways. These three pathways determine the response, depending on the cell context. STAT3 is modified by phosphorylation at Ser727 in addition to the phosphorylation at Tyr705. Several serine/threonine kinases for Ser727 are activated by IL-6. STAT3 is acetylated at multiple lysines by p300/CBP. These modifications affect the STAT3 activity with multiple mechanisms (not shown here). (B) Multiple layers of negative inhibitory loops determine the strength and duration of STAT3 activity. Newly synthesized SOCS3 restricts further activation of STAT3 by binding to tyrosine-phosphorylated gp130 and inhibiting JAK activity. SHP-2 also inhibits STAT3 activity by dephosphorylating phospho-Y705. Several other PTPases are known to dephosphorylate pY705 of STAT3 in the cytoplasm and nucleus. TC45 is a major nuclear PTPase for STAT3 and was recently shown to dephosphorylate phospho-Y705 of STAT3 in a phospho-Ser727-dependent manner.
Src-homology 2 domain-containing protein tyrosine phosphatase 2 (SHP2) is recruited to the phosphorylated YSTV (pYSTV) motif in gp130 and phosphorylated and activated by JAK kinases, which in turn activate both the Ras-Raf-ERK1/2 pathway and the PI3K-AKT pathway though the GRB2-associated binding protein (Gab) family.9,10
Signal transducer and activator of transcription 3 (STAT3) and STAT1 are recruited to the phosphorylated YXXQ motifs in gp130 and phosphorylated by JAK kinases at the critical tyrosine residues, Y705 for STAT3 and Y701 for STAT1.9,10 The activated STAT3 and STAT1 dimerize with each other, making STAT3 or STAT1 homodimers and STAT3/STAT1 heterodimers. These activated STAT dimers enter the nucleus and bind to the specific DNA sequences in the regulatory regions of their target genes.37
In addition to phosphorylation of the critical tyrosine residues, both STAT3 and STAT1 are phosphorylated on Ser727 in the carboxyl-terminal transactivation domain38 by various serine/threonine kinases depending on the stimulus and cell type used.39-48 Although phosphorylation of Ser727 has been suggested to exert positive effects on STAT3-dependent gene activation,38,41,49 most likely through recruiting coactivator proteins,50-52 several reports have suggested that phosphorylation of Ser727 of STAT3 is required for distinct negative regulatory roles39,53 or for the role of STAT3 in the mitochondrial function of Ras-transformed cells.54 Yang et al. showed that STAT3 Ser727 phosphorylation in the nucleus is required for the recruitment of histone lysine methyltransferase SET9, which causes dimethylation of STAT3 at Lys140, leading to inhibition of STAT3 activity by suppressing the level of phospho-Tyr705.53
Another important role for phopho-Ser727 was recently shown by Wakahara et al.55 They showed that phospho-Ser727 has an intrinsic mechanism for shortening the duration of STAT3 activity independently of modification at Lys140 and that phopho-Ser727 leads to rapid dephosphorylation of phospho-Y705 largely through nuclear tyrosine phosphatase TC45, probably by modulating the TC45 activity.55 This finding revealed another layer of STAT3 regulation to give a maximum STAT3 activity for a proper duration, highlighting the role of regulated activity of tyrosine phosphatases. Previously, the two feedback inhibitory loops for STAT3 have been known.56 One inhibitory loop is mediated by SHP2, which is recruited to the pYSTV motif of gp130 and is phosphorylated on tyrosine residues by JAK kinases. In addition to activation of the downstream signaling cascade described above, the tyrosine phosphatase activity of SHP2 toward STAT3 also increases after tyrosine phosphorylation and then limits STAT3 activity.56 Another inhibitory loop is mediated by SOCS3.57 Socs3 mRNA is rapidly induced by both STAT3 and STAT1, and the SOCS3 protein interacts with the gp130 pYSTV motif through its SH2 domain and inhibits JAK activity through its kinase inhibitory region (KIR), thereby restricting further STAT3 activation.58,59 These three negative regulatory loops for STAT3 are depicted in Figure 2B.
STAT3 plays multiple roles depending on the context or condition of cells. In some conditions, STAT3 is involved in growth arrest and differentiation, and even in cell death.7,9,10 In other conditions, STAT3 is involved in proliferation and cell survival by activating pro-proliferative and pro-survival genes, including c-myc and cyclinD1, and anti-apoptotic bcl2, bclxl, or mcl1.9,10 In some conditions, persistently activated STAT3 can mediate tumorigenesis by protecting cells from apoptotic stimuli and by promoting cell-cycle progression in a variety of cancers and leukemias.60,61
As we learned, for IL-6 these three major pathways and multiple positive and negative regulators coordinately determine the function of IL-6 depending on the cellular context by activating a set of genes in a strictly regulated manner in terms of both strength and duration.
IL-6/STAT3 Regulates Multiple Processes Ranging from Premature Senescence11 to Tumorigenesis
As described earlier, Kuilman et al. first demonstrated the important roles of IL-6 produced by TIG3 fibroblasts that had been activated by oncogene BRAFV600E in further progression of senescence of such IL-6 producing cells.22 They showed that inductions of both C/EBPβ and IL-6 by BRAF were required for BRAF-induced senescence, suggesting that some factors of SASP or oncogene-induced secretory factors were essential for reinforcing cellular senescence.22
Normal human fibroblast TIG3 cells showed a senescent phenotype after about 55 population doublings (PD). At this stage, the older TIG3 cells showed constitutive expression of the mRNAs for IL-6 and IL-6Rα chain, while younger TIG3 cells at PD33 did not express either. Remarkably, STAT3 was also constitutively activated in older TIG3 cells without exogenous IL-6 stimulation. When IL-6 and soluble IL-6Rα were administered to cultures of young TIG3 cells at PD33, the cells showed the phenotypes of senescence, with growth arrest and SA-β gal activity, at around day 8. The current model of IL-6/STAT3-induced senescence in TIG3 fibroblasts11 is shown in Figure 3A. The levels of p53 protein first declined on day 2 and gradually increased thereafter. TIG3 fibroblasts with a p53 knockdown showed no sign of senescence and proliferated well in the presence of IL-6/sIL-6Rα, indicating that p53 was essential for the IL-6/sIL-6Rα-induced premature senescence.
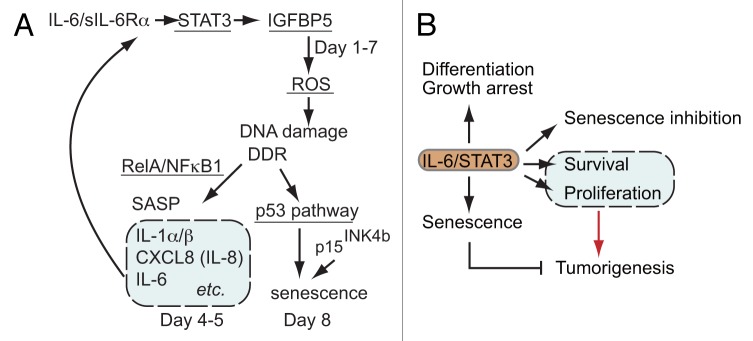
Figure 3. (A) A model of the senescence-inducing circuit involving the IL-6-STAT3-IGFBP5 axis. IGFBP5 produced in a STAT3-dependent manner causes the initial generation of ROS, subsequent DDR and SASP (expression of IL-1α, IL-1β, IL-6, and CXCL8). Prolonged expression of IGFBP5 caused by IL-6, together with other components of SASP, drives the circuit generating more ROS and severe DNA damage, leading to p53-dependent premature senescence. Inhibition of any constituent, STAT3, p53, ROS, IGFBP5, or RelA attenuates the IL-6/sIL-6Rα-induced premature senescence. The possible roles of the ERK1/2 and PI3K/AKT/mTOR-mediated pathways are discussed in the text. (B) The multiple roles of IL-6/STAT3 pathway. IL-6/STAT3 regulates multiple processes ranging from premature senescence to tumorigenesis.
ROS generation and the subsequent DNA damage response (DDR) were observed on day 2 and the levels of ROS increased thereafter, which were essential for IL-6/sIL-6Rα-induced senescence. Thus, IL-6/sIL-6Rα induced the premature senescence of fibroblasts in a ROS/DDR/p53-dependent manner. Both CDK inhibitor p16INK4a and p15INK4b were detected, suggesting that both the p53 and the RB-mediated pathways were involved in the IL-6/sIL-6Rα-induced premature senescence. As expected, STAT3 was essential for the ROS increase in the early phase and SA-β-galactosidase activity on day 8 in response to IL-6/sIL-6Rα. STAT3 activity was required for the mRNA expressions of IL-1α, IL-1β, IL-8, and IL-6 on days 4 and 5. RelA, a component of NFκB transcription factor, was required for both SASP and premature senescence in response to IL-6/sIL-6Rα, suggesting the role of SASP in the IL-6/sIL-6Rα-induced senescence of TIG3 fibroblasts.
We then investigated how IL-6/STAT3 trigger and eventually cause senescence of TIG3 cells after 8 days. There must be special mechanisms affecting cells for such a long period. The key molecule was sought and identified as IGFBP5, which was secreted by IL-6/sIL-6Rα-simulated TIG3 in a STAT3-dependent manner from the first days and continuously secreted until day 7. The kinetic pattern of IGFBP5 mRNA expression was different from that of SASPs such as IL-1α, IL-1β, IL-6, and CXCL8/IL-8, which peak on days 4 and 5. Exogenously added IGFBP5 increased ROS, DNA damage, SASP, and senescence. The knockdown of IGFBP5 in TIG3 cells inhibited the IL-6/sIL-6Rα-induced ROS increase and premature senescence. Thus IL-6/sIL-6Rα formed a senescence-inducing circuit involving the STAT3-IGFBP5 axis as a key triggering and reinforcing component in human diploid fibroblast TIG3 cells.
IGFBP5 has been shown to have multiple functions in various target cells.62 IGFBP5 was found to be involved in the replicative senescence of human endothelial cells (HUVECs), and IGFBP5 alone was sufficient to induce the premature senescence of HUVECs.63 In that case IL-6/STAT3 may trigger induction or reinforce the senescence process of HUVECs by activating the IGFBP5 gene. Recently, another human fibroblast, TIG1, derived from human female fetal lung, were shown to be induced to senescence by IL-6.64 It is possible that the senescence-inducing circuit involving IL-6-STAT3-IGFBP5 may not be limited to TIG3 cells.
However, in other premalignant or tumor cell systems, IL-6 and STAT3 often play opposite roles, inhibiting senescence, promoting tumorigenesis, and protecting cells from chemotherapy. Among the seven members of the STAT family, STAT3 is most important in determining whether immune responses in the tumor microenvironment promote or inhibit cancer (see the review by Yu et al.61). Recently Gilbert and Hemann showed that IL-6 has anti-senescence activity.65 By using mice bearing Eμ-myc-transformed lymphoma cells, they showed that paracrine factors, especially IL-6 and Timp-1, in the tumor microenvironment supported the survival of lymphoma cells following administration of the genotoxic chemotherapeutic drug doxorubicin.65 In the case of HCT116 colon cancer cells, Yun et al. showed that HCT116 cells responded to doxorubicin with enhanced proliferation and that secreted IL-6 and activated STAT3 were required for the proliferative response.66 When IL-6/STAT3 activity was inhibited by an anti-IL-6 antibody, an siRNA against gp130, or the use of dominant-negative STAT3, those cells developed premature senescence, indicating that IL-6-STAT3 signals activated by the drug treatment inhibited the drug-induced premature senescence of HCT116 cells in an autocrine manner. Tkach et al. studied the method of antitumor immunization using irradiated murine breast cancer cells expressing either dominant-negative STAT3 or parental breast cancer cells as immunogens and reported that compared with immunization with the parental cancer cells, immunization with STAT3-inhibited cells resulted in a more efficient immunotherapy against breast cancer cells through cytotoxic NK cells and CD4+ T cells.67 Because the breast cancer cells expressing dominant-negative STAT3 showed a senescent phenotype, it is likely that senescent tumor cells can elicit the anti-tumor immunity more efficiently than the parental cancer cells. Thus, the role of IL6/STAT3 really depends on the cell type or conditions including the environment and surrounding cells. This finding should be considered in developing a STAT3-targeting drug for anti-cancer activity. The various outcomes caused by IL-6/STAT3 are depicted in Figure 3B. We still do not understand well how IL-6/STAT3 determines the outcome from premature senescence to tumorigenesis.
Although we did not discuss the roles of IL-6-signaling pathways other than that mediated by STAT3, it is likely that both the ERK1/2-dependent pathway and PI3K/AKT/mTOR-dependent pathway play roles in both premature senescence and tumor development. Blagosklonny and his colleagues recently showed that activation of mTOR (mammalian target of rapamycin) by serum growth factors or oncogenes is required for the development of senescence using several different cell systems.68-71 It has been shown that both ERK1/2 and PI3K increase the activity of mTOR and mTOR drives senescence when cell cycle is blocked. Because growth factors in culture medium and exogenously added or secreted cytokines, including IL-6, can activate both ERK1/2 and PI3K/AKT/mTOR, it is quite likely that mTOR activity plays pro-senescent roles when cell cycle is blocked. As to the role of ERK in oncogene-induced senescence and replicative senescence, Deschênes-Simard et al. showed that the ERK/MAPK pathway is required for development of such senescence through promoting selective protein degradation.72 They termed this phenomenon senescence-associated protein degradation (SAPD).
Other Mechanisms for Premature Senescence of Various Cells by Cytokines Especially Using STAT Family Proteins
A number of factors, both SASP and non-SASP factors, have been shown to be involved in replicative senescence or premature senescence in various types of cells with distinct mechanisms (Table 1). These factors include interferon β (IFN-β), IFN-γ, TNF-α, IL-1β, TGF-β, thrombopoietin, IGF1, IGF2, the IGFBP family members IGFBP3, IGFBP5, and IGFBP7, IL-6, and chemokines acting on CXCR2.11,22,23,63,73-81 As discussed earlier, only some of the secreted factors are involved in keeping or reinforcing the development of senescence, depending on the cell systems used. Hubackova et al. recently performed an assay of factors produced by three types of senescent BJ fibroblasts, replicative senescence, genotoxic stress-induced senescence, and activated H-RasV12E-induced senescence, for their senescence-inducing activity and found that IL-1β + TGFβ, which are commonly secreted by the three types of senescent cells, induced premature senescence of BJ cells by increasing Nox4 for ROS production.76
Table 1. Senescence-inducing cytokines and signaling molecules: cell systems and properties.
| Cytokine | Cell system | Mechanisms | References |
|---|---|---|---|
| IFNβ |
IMR90, IMR90+H-RasV12E |
DDR, ATM, p53-dependent senescence |
73 |
| Constitutively active STAT5 |
IMR90 BJ, IMR90 |
DDR, ATM, Chk2, p53 PML, reduced Myc, Rb |
83 and 84 |
| IGFBP5 |
HUVEC atherosclerotic lesion |
DDR, p53-dependent senescence IGFBP5 detected |
63 |
| IL-6/sIL-6Rα |
TIG3-BRAFV600E |
IL-6, induced by BRAFV600E-C/EBPβ, reinforce H-RasV12E-dependent senescence. IL-8 is also involved. |
22 |
| CXCR2 |
IMR90+MEK:ER |
MEK-induced CXCR2 reinforce MEK-dependent senescence |
23 |
| IFNβ |
Genotoxic drug-treated tumor cells BJ |
JAK/STAT-mediated PML1 induction contributes to drug-induced senescence |
26 and 27 |
| IL-6/sIL-6Rα |
TIG3 |
STAT3-IGFBP5 axis forms a senescence-inducing circuit involving ROS, DDR, p53, RelA and IL-6 |
11 |
| IL-1β+TGFβ |
BJ |
Secreted IL-1β+TGFβ induces senescence through increasing Nox4 |
76 |
| IL-22 |
Hepatic stellate cells |
STAT3-SOCS3 induces senescence and inhibits liver fibrosis. SOCS3 activates p53 by binding to p53. |
32 |
| IFNβ |
HPV-transformed keratinocytes |
JAK/STAT-induced PML together with p53 and p21CIP1 involved in IFNβ−induced senescence |
28 |
| TNFα+IFNγ | SV40-Tag-transformed β cell | Th1 cells specific to SV40 Tag causes senescence of target cells through STAT1, TNFR1, p16INK4a-dependent mechanism | 75 |
In a liver fibrosis model, Krizhanovsky et al. showed that during the course of CCl4-induced liver fibrosis, activated stellate cells showed senescence with a phenotype of enhanced secretion of cytokines, chemokines, and metalloproteinase 1 and 3 along with a reduction of collagen II, IV, and fibronectin.82 Interestingly, inhibition of the senescence phenotype using mice deficient for p53 increased the fibrotic area in the liver.82 They reported that recruitment and activation of NK cells enhanced the clearance of senescent cells. SASP seemed to be involved in both development of premature senescence of hepatic stellate cells and immune surveillance of those senescent cells. The role of STAT3 in the induction of premature senescence of hepatic stellate cells was recently shown. Kong et al. reported that exogenously administered IL-22 induced hepatic stellate cell senescence in a STAT3-dependent manner and limited liver fibrosis.32 They showed that STAT3-induced SOCS3 is required for p53 increase, and SOCS3 functions by interacting with p53, indicating the presence of another mechanism modulating p53 function by STAT3.
The roles of other STAT family members (and related molecules) have been shown to be involved in premature senescence. Mallette et al. reported that constitutively active STAT5 induced premature cellular senescence of human diploid fibroblasts through ATM and p53- and RB-dependent mechanisms.83,84 They further revealed the mechanism by which constitutively active STAT5 induced premature senescence of fibroblasts in a SOCS1-dependent manner and showed that STAT5-induced SOCS1 caused senescence by enhancing the transcriptional activity of p53 by directly interacting with p53 at the N-terminal region and with ATM at the C-terminal SOCS box, thereby enhancing p53 phosphorylation at Ser15.30
By using a mouse model in which the β-cell expressed simian virus 40 (SV40) large T antigen (Tag) creates β cell tumors, Braumüller et al. showed that TH1 cytokines IFNγ and TNFα cause premature senescence of β-cell cancers both in vitro and in vivo in a STAT1- and p16INK4a-RB-dependent fashion.75
These findings indicate that the multiple distinct mechanisms work to regulate cellular senescence.
Concluding Remarks and Perspective
Oncogenes, DNA damage, and chemotherapeutics induce premature senescence that is often reinforced by the senescence-associated secretome. Only some of the cytokines in the secretome as well as other cytokines not in the secretome, including IL-1, IL-6, TGF-β, IFN-β, and IGFBP5, can induce the premature senescence of young cells. We have described how IL-6/sIL-6Rα induces premature senescence in human primary fibroblasts and presented a model for the progression of premature senescence by IL-6/sIL-6Rα through a circuit involving STAT3-IGFBP5 as a critical axis. Considering that the levels of IL-6 and sIL-6Rα increase with aging and aging-associated diseases,85,86 it is possible that IL-6/sIL-6Rα in the environment contributes to the tissue aging process, and that the production of IL-6 and sIL-6Rα is not merely a result of aging, but may cause or enhance senescence in some conditions. Future research should address whether IL-6/STAT3 is really involved in the development of premature senescence in chronically inflamed or aging tissues in vivo in both human and murine systems. In other settings, IL-6 and STAT3 have been shown to be involved in the development of certain types of tumors. Effects of STAT3 on tumor initiation or early promotion are likely to be related to its ability to regulate the anti-apoptotic and proliferative genes. Some of STAT3-target genes are likely to inhibit different types of premature senescence to cause tumorigenesis. Recent study by Yoshimoto et al. showed that the enterohepatic circulation of deoxycholic acid, a gut bacterial metabolite known to cause DNA damage, provokes SASP phenotype in hepatic stellate cells, which in turn facilitate obesity-associated hepatocellular carcinoma development in mice after exposure to chemical carcinogen.87 Thus SASP in addition to inflammation shapes the cancer microenvironment. Future work should address whether inhibition of IL-6/STAT3 function can be used for anti-senescence or anti-aging of tissues as well as for inhibiting the formation of the cancer microenvironment. The matter of the surveillance of both normal and pre-malignant senescent cells is also important.
Acknowledgments
This work was supported in part by the Ministry of Education, Culture, Sports, Science, and Technology of Japan, the Japan Health Foundation, and the Kampou Science Foundation. The authors gratefully acknowledge their appreciation to members of the Nakajima laboratory for helpful discussions and to Junko Kanbara for excellent technical and secretarial assistance.
Glossary
Abbreviations:
- sIL-6Rα
soluble IL-6 receptor α
- IGFBP5
insulin-like growth factor-binding protein 5
- SASP
senescence-associated secretory phenotype
- PD
population doubling
- ROS
reactive oxygen species
- DDR
DNA damage response
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/jak-stat/article/25763
References
- 1.Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463–79. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–56. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sikora E, Arendt T, Bennett M, Narita M. Impact of cellular senescence signature on ageing research. Ageing Res Rev. 2011;10:146–52. doi: 10.1016/j.arr.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Ohtani N, Hara E. Roles and mechanisms of cellular senescence in regulation of tissue homeostasis. Cancer Sci. 2013;104:525–30. doi: 10.1111/cas.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009;9:81–94. doi: 10.1038/nrc2560. [DOI] [PubMed] [Google Scholar]
- 7.Nakajima K, Yamanaka Y, Nakae K, Kojima H, Ichiba M, Kiuchi N, et al. A central role for Stat3 in IL-6-induced regulation of growth and differentiation in M1 leukemia cells. EMBO J. 1996;15:3651–8. [PMC free article] [PubMed] [Google Scholar]
- 8.Kortylewski M, Heinrich PC, Mackiewicz A, Schniertshauer U, Klingmüller U, Nakajima K, et al. Interleukin-6 and oncostatin M-induced growth inhibition of human A375 melanoma cells is STAT-dependent and involves upregulation of the cyclin-dependent kinase inhibitor p27/Kip1. Oncogene. 1999;18:3742–53. doi: 10.1038/sj.onc.1202708. [DOI] [PubMed] [Google Scholar]
- 9.Hirano T, Nakajima K, Hibi M. Signaling mechanisms through gp130: a model of the cytokine system. Cytokine Growth Factor Rev. 1997;8:241–52. doi: 10.1016/S1359-6101(98)80005-1. [DOI] [PubMed] [Google Scholar]
- 10.Hirano T, Ishihara K, Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. 2000;19:2548–56. doi: 10.1038/sj.onc.1203551. [DOI] [PubMed] [Google Scholar]
- 11.Kojima H, Kunimoto H, Inoue T, Nakajima K. The STAT3-IGFBP5 axis is critical for IL-6/gp130-induced premature senescence in human fibroblasts. Cell Cycle. 2012;11:730–9. doi: 10.4161/cc.11.4.19172. [DOI] [PubMed] [Google Scholar]
- 12.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 13.d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–8. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 14.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–45. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 15.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/S0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 16.Serrano M, Blasco MA. Cancer and ageing: convergent and divergent mechanisms. Nat Rev Mol Cell Biol. 2007;8:715–22. doi: 10.1038/nrm2242. [DOI] [PubMed] [Google Scholar]
- 17.Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–5. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- 18.Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- 19.Janssens S, Tschopp J. Signals from within: the DNA-damage-induced NF-kappaB response. Cell Death Differ. 2006;13:773–84. doi: 10.1038/sj.cdd.4401843. [DOI] [PubMed] [Google Scholar]
- 20.Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30:1536–48. doi: 10.1038/emboj.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodier F, Coppé JP, Patil CK, Hoeijmakers WA, Muñoz DP, Raza SR, et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009;11:973–9. doi: 10.1038/ncb1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–31. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 23.Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–18. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 24.Coppé JP, Patil CK, Rodier F, Krtolica A, Beauséjour CM, Parrinello S, et al. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS One. 2010;5:e9188. doi: 10.1371/journal.pone.0009188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 2000;406:207–10. doi: 10.1038/35021000. [DOI] [PubMed] [Google Scholar]
- 26.Novakova Z, Hubackova S, Kosar M, Janderova-Rossmeislova L, Dobrovolna J, Vasicova P, et al. Cytokine expression and signaling in drug-induced cellular senescence. Oncogene. 2010;29:273–84. doi: 10.1038/onc.2009.318. [DOI] [PubMed] [Google Scholar]
- 27.Hubackova S, Novakova Z, Krejcikova K, Kosar M, Dobrovolna J, Duskova P, et al. Regulation of the PML tumor suppressor in drug-induced senescence of human normal and cancer cells by JAK/STAT-mediated signaling. Cell Cycle. 2010;9:3085–99. doi: 10.4161/cc.9.15.12521. [DOI] [PubMed] [Google Scholar]
- 28.Chiantore MV, Vannucchi S, Accardi R, Tommasino M, Percario ZA, Vaccari G, et al. Interferon-β induces cellular senescence in cutaneous human papilloma virus-transformed human keratinocytes by affecting p53 transactivating activity. PLoS One. 2012;7:e36909. doi: 10.1371/journal.pone.0036909. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Hubackova S, Krejcikova K, Bartek J, Hodny Z. Interleukin 6 signaling regulates promyelocytic leukemia protein gene expression in human normal and cancer cells. J Biol Chem. 2012;287:26702–14. doi: 10.1074/jbc.M111.316869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calabrese V, Mallette FA, Deschênes-Simard X, Ramanathan S, Gagnon J, Moores A, et al. SOCS1 links cytokine signaling to p53 and senescence. Mol Cell. 2009;36:754–67. doi: 10.1016/j.molcel.2009.09.044. [DOI] [PubMed] [Google Scholar]
- 31.Mallette FA, Calabrese V, Ilangumaran S, Ferbeyre G. SOCS1, a novel interaction partner of p53 controlling oncogene-induced senescence. Aging (Albany NY) 2010;2:445–52. doi: 10.18632/aging.100163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kong X, Feng D, Wang H, Hong F, Bertola A, Wang FS, et al. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology. 2012;56:1150–9. doi: 10.1002/hep.25744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rose-John S, Waetzig GH, Scheller J, Grötzinger J, Seegert D. The IL-6/sIL-6R complex as a novel target for therapeutic approaches. Expert Opin Ther Targets. 2007;11:613–24. doi: 10.1517/14728222.11.5.613. [DOI] [PubMed] [Google Scholar]
- 34.Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. 2011;1813:878–88. doi: 10.1016/j.bbamcr.2011.01.034. [DOI] [PubMed] [Google Scholar]
- 35.Chalaris A, Garbers C, Rabe B, Rose-John S, Scheller J. The soluble Interleukin 6 receptor: generation and role in inflammation and cancer. Eur J Cell Biol. 2011;90:484–94. doi: 10.1016/j.ejcb.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 36.Taga T, Hibi M, Hirata Y, Yamasaki K, Yasukawa K, Matsuda T, et al. Interleukin-6 triggers the association of its receptor with a possible signal transducer, gp130. Cell. 1989;58:573–81. doi: 10.1016/0092-8674(89)90438-8. [DOI] [PubMed] [Google Scholar]
- 37.Darnell JE., Jr. STATs and gene regulation. Science. 1997;277:1630–5. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 38.Wen Z, Zhong Z, Darnell JE., Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–50. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- 39.Chung JK, Uchida E, Grammer TC, Blenis J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol Cell Biol. 1997;17:6508–16. doi: 10.1128/mcb.17.11.6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boulton TG, Zhong Z, Wen Z, Darnell JE, Jr., Stahl N, Yancopoulos GD. STAT3 activation by cytokines utilizing gp130 and related transducers involves a secondary modification requiring an H7-sensitive kinase. Proc Natl Acad Sci U S A. 1995;92:6915–9. doi: 10.1073/pnas.92.15.6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abe K, Hirai M, Mizuno K, Higashi N, Sekimoto T, Miki T, et al. The YXXQ motif in gp 130 is crucial for STAT3 phosphorylation at Ser727 through an H7-sensitive kinase pathway. Oncogene. 2001;20:3464–74. doi: 10.1038/sj.onc.1204461. [DOI] [PubMed] [Google Scholar]
- 42.Fu AK, Fu WY, Ng AK, Chien WW, Ng YP, Wang JH, et al. Cyclin-dependent kinase 5 phosphorylates signal transducer and activator of transcription 3 and regulates its transcriptional activity. Proc Natl Acad Sci U S A. 2004;101:6728–33. doi: 10.1073/pnas.0307606100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yokogami K, Wakisaka S, Avruch J, Reeves SA. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr Biol. 2000;10:47–50. doi: 10.1016/S0960-9822(99)00268-7. [DOI] [PubMed] [Google Scholar]
- 44.Kojima H, Sasaki T, Ishitani T, Iemura S, Zhao H, Kaneko S, et al. STAT3 regulates Nemo-like kinase by mediating its interaction with IL-6-stimulated TGFbeta-activated kinase 1 for STAT3 Ser-727 phosphorylation. Proc Natl Acad Sci U S A. 2005;102:4524–9. doi: 10.1073/pnas.0500679102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohkawara B, Shirakabe K, Hyodo-Miura J, Matsuo R, Ueno N, Matsumoto K, et al. Role of the TAK1-NLK-STAT3 pathway in TGF-beta-mediated mesoderm induction. Genes Dev. 2004;18:381–6. doi: 10.1101/gad.1166904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sato N, Kawai T, Sugiyama K, Muromoto R, Imoto S, Sekine Y, et al. Physical and functional interactions between STAT3 and ZIP kinase. Int Immunol. 2005;17:1543–52. doi: 10.1093/intimm/dxh331. [DOI] [PubMed] [Google Scholar]
- 47.Wierenga AT, Vogelzang I, Eggen BJ, Vellenga E. Erythropoietin-induced serine 727 phosphorylation of STAT3 in erythroid cells is mediated by a MEK-, ERK-, and MSK1-dependent pathway. Exp Hematol. 2003;31:398–405. doi: 10.1016/S0301-472X(03)00045-6. [DOI] [PubMed] [Google Scholar]
- 48.Jain N, Zhang T, Kee WH, Li W, Cao X. Protein kinase C delta associates with and phosphorylates Stat3 in an interleukin-6-dependent manner. J Biol Chem. 1999;274:24392–400. doi: 10.1074/jbc.274.34.24392. [DOI] [PubMed] [Google Scholar]
- 49.Shen YH, Schlessinger K, Zhu XJ, Meffre E, Quimby F, Levy DE, et al. Essential role of STAT3 in postnatal survival and growth revealed by mice lacking STAT3 serine 727 phosphorylation. Mol Cell Biol. 2004;24:407–19. doi: 10.1128/MCB.24.1.407-419.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schuringa JJ, Schepers H, Vellenga E, Kruijer W. Ser727-dependent transcriptional activation by association of p300 with STAT3 upon IL-6 stimulation. FEBS Lett. 2001;495:71–6. doi: 10.1016/S0014-5793(01)02354-7. [DOI] [PubMed] [Google Scholar]
- 51.Lufei C, Koh TH, Uchida T, Cao X. Pin1 is required for the Ser727 phosphorylation-dependent Stat3 activity. Oncogene. 2007;26:7656–64. doi: 10.1038/sj.onc.1210567. [DOI] [PubMed] [Google Scholar]
- 52.Lee H, Herrmann A, Deng J-H, Kujawski M, Niu G, Li Z, et al. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell. 2009;15:283–93. doi: 10.1016/j.ccr.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang J, Huang J, Dasgupta M, Sears N, Miyagi M, Wang B, et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc Natl Acad Sci U S A. 2010;107:21499–504. doi: 10.1073/pnas.1016147107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–6. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wakahara R, Kunimoto H, Tanino K, Kojima H, Inoue A, Shintaku H, et al. Phospho-Ser727 of STAT3 regulates STAT3 activity by enhancing dephosphorylation of phospho-Tyr705 largely through TC45. Genes Cells. 2012;17:132–45. doi: 10.1111/j.1365-2443.2011.01575.x. [DOI] [PubMed] [Google Scholar]
- 56.Schmitz J, Dahmen H, Grimm C, Gendo C, Müller-Newen G, Heinrich PC, et al. The cytoplasmic tyrosine motifs in full-length glycoprotein 130 have different roles in IL-6 signal transduction. J Immunol. 2000;164:848–54. doi: 10.4049/jimmunol.164.2.848. [DOI] [PubMed] [Google Scholar]
- 57.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–65. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 58.Babon JJ, Kershaw NJ, Murphy JM, Varghese LN, Laktyushin A, Young SN, et al. Suppression of cytokine signaling by SOCS3: characterization of the mode of inhibition and the basis of its specificity. Immunity. 2012;36:239–50. doi: 10.1016/j.immuni.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kershaw NJ, Murphy JM, Liau NP, Varghese LN, Laktyushin A, Whitlock EL, et al. SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat Struct Mol Biol. 2013;20:469–76. doi: 10.1038/nsmb.2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19:2474–88. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- 61.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beattie J, Allan GJ, Lochrie JD, Flint DJ. Insulin-like growth factor-binding protein-5 (IGFBP-5): a critical member of the IGF axis. Biochem J. 2006;395:1–19. doi: 10.1042/BJ20060086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim KS, Seu YB, Baek SH, Kim MJ, Kim KJ, Kim JH, et al. Induction of cellular senescence by insulin-like growth factor binding protein-5 through a p53-dependent mechanism. Mol Biol Cell. 2007;18:4543–52. doi: 10.1091/mbc.E07-03-0280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Udono M, Kadooka K, Yamashita S, Katakura Y. Quantitative analysis of cellular senescence phenotypes using an imaging cytometer. Methods. 2012;56:383–8. doi: 10.1016/j.ymeth.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 65.Gilbert LA, Hemann MT. DNA damage-mediated induction of a chemoresistant niche. Cell. 2010;143:355–66. doi: 10.1016/j.cell.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yun UJ, Park SE, Jo YS, Kim J, Shin DY. DNA damage induces the IL-6/STAT3 signaling pathway, which has anti-senescence and growth-promoting functions in human tumors. Cancer Lett. 2012;323:155–60. doi: 10.1016/j.canlet.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 67.Tkach M, Coria L, Rosemblit C, Rivas MA, Proietti CJ, Díaz Flaqué MC, et al. Targeting Stat3 induces senescence in tumor cells and elicits prophylactic and therapeutic immune responses against breast cancer growth mediated by NK cells and CD4+ T cells. J Immunol. 2012;189:1162–72. doi: 10.4049/jimmunol.1102538. [DOI] [PubMed] [Google Scholar]
- 68.Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008;7:3355–61. doi: 10.4161/cc.7.21.6919. [DOI] [PubMed] [Google Scholar]
- 69.Demidenko ZN, Shtutman M, Blagosklonny MV. Pharmacologic inhibition of MEK and PI-3K converges on the mTOR/S6 pathway to decelerate cellular senescence. Cell Cycle. 2009;8:1896–900. doi: 10.4161/cc.8.12.8809. [DOI] [PubMed] [Google Scholar]
- 70.Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY) 2010;2:344–52. doi: 10.18632/aging.100160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Blagosklonny MV. Cell cycle arrest is not senescence. Aging (Albany NY) 2011;3:94–101. doi: 10.18632/aging.100281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Deschênes-Simard X, Gaumont-Leclerc MF, Bourdeau V, Lessard F, Moiseeva O, Forest V, et al. Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes Dev. 2013;27:900–15. doi: 10.1101/gad.203984.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Moiseeva O, Mallette FA, Mukhopadhyay UK, Moores A, Ferbeyre G. DNA damage signaling and p53-dependent senescence after prolonged beta-interferon stimulation. Mol Biol Cell. 2006;17:1583–92. doi: 10.1091/mbc.E05-09-0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim KS, Kang KW, Seu YB, Baek SH, Kim JR. Interferon-gamma induces cellular senescence through p53-dependent DNA damage signaling in human endothelial cells. Mech Ageing Dev. 2009;130:179–88. doi: 10.1016/j.mad.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 75.Braumüller H, Wieder T, Brenner E, Aßmann S, Hahn M, Alkhaled M, et al. T-helper-1-cell cytokines drive cancer into senescence. Nature. 2013;494:361–5. doi: 10.1038/nature11824. [DOI] [PubMed] [Google Scholar]
- 76.Hubackova S, Krejcikova K, Bartek J, Hodny Z. IL1- and TGFβ-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine 'Bystander senescence'. Aging (Albany, NY Online) 2012;4:932–51. doi: 10.18632/aging.100520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Besancenot R, Chaligné R, Tonetti C, Pasquier F, Marty C, Lécluse Y, et al. A senescence-like cell-cycle arrest occurs during megakaryocytic maturation: implications for physiological and pathological megakaryocytic proliferation. PLoS Biol. 2010;8:e1000476. doi: 10.1371/journal.pbio.1000476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chakravarthy MV, Abraha TW, Schwartz RJ, Fiorotto ML, Booth FW. Insulin-like growth factor-I extends in vitro replicative life span of skeletal muscle satellite cells by enhancing G1/S cell cycle progression via the activation of phosphatidylinositol 3′-kinase/Akt signaling pathway. J Biol Chem. 2000;275:35942–52. doi: 10.1074/jbc.M005832200. [DOI] [PubMed] [Google Scholar]
- 79.Hernandez L, Kozlov S, Piras G, Stewart CL. Paternal and maternal genomes confer opposite effects on proliferation, cell-cycle length, senescence, and tumor formation. Proc Natl Acad Sci U S A. 2003;100:13344–9. doi: 10.1073/pnas.2234026100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Debacq-Chainiaux F, Pascal T, Boilan E, Bastin C, Bauwens E, Toussaint O. Screening of senescence-associated genes with specific DNA array reveals the role of IGFBP-3 in premature senescence of human diploid fibroblasts. Free Radic Biol Med. 2008;44:1817–32. doi: 10.1016/j.freeradbiomed.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 81.Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132:363–74. doi: 10.1016/j.cell.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–67. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mallette FA, Gaumont-Leclerc MF, Huot G, Ferbeyre G. Myc down-regulation as a mechanism to activate the Rb pathway in STAT5A-induced senescence. J Biol Chem. 2007;282:34938–44. doi: 10.1074/jbc.M707074200. [DOI] [PubMed] [Google Scholar]
- 84.Mallette FA, Gaumont-Leclerc MF, Ferbeyre G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 2007;21:43–8. doi: 10.1101/gad.1487307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Daynes RA, Araneo BA, Ershler WB, Maloney C, Li GZ, Ryu SY. Altered regulation of IL-6 production with normal aging. Possible linkage to the age-associated decline in dehydroepiandrosterone and its sulfated derivative. J Immunol. 1993;150:5219–30. [PubMed] [Google Scholar]
- 86.Ershler WB, Keller ET. Age-associated increased interleukin-6 gene expression, late-life diseases, and frailty. Annu Rev Med. 2000;51:245–70. doi: 10.1146/annurev.med.51.1.245. [DOI] [PubMed] [Google Scholar]
- 87.Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]