

Abstract
There is an ongoing debate on whether acute human immunodeficiency virus infection is controlled by target cell limitation or by virus-specific cellular immunity. To resolve this question, we developed a novel mathematical modeling scheme which allows us to incorporate measurements of virus load, target cells, and virus-specific immunity and applied it to a comprehensive data set generated in an experiment involving rhesus macaques infected with simian immunodeficiency virus. Half of the macaques studied were treated during the primary infection period with reagents which block T-cell costimulation and as a result displayed severely impaired virus-specific immune responses. Our results show that early viral replication in normal infection is controlled to a large extent by virus-specific CD8+ T cells and not by target cell limitation.
The viremia in primary human immunodeficiency virus (HIV) infection is characterized by a steep initial increase rising to a peak after a few weeks, a postpeak decline, and an ultimate attainment of the so-called viral load set point (2, 4, 14, 15, 24, 25) (Fig. 1). Specific immune responses, especially virus-specific CD8+ T cells, have been proposed as important driving forces behind the postpeak decline (2, 17, 19, 21). In 1996, Phillips presented an alternative explanation for the postpeak decline. Using a mathematical model, he demonstrated that the observed pattern of early viral replication could be the result of the interaction between the virus and its target cells only (23). According to Phillips's explanation, the containment of viral replication at its peak and the postpeak decline of the virus load are due to an exhaustion of target cells.
FIG. 1.

Schematic of viremia (bold line), target cell (dashed line), and virus-specific CD8+-T-cell counts (dashed-dotted line) in primary HIV infection.
Previous studies investigated the contributions of the levels of cellular immunity on early viral replication experimentally by depleting CD8+ cells in simian immunodeficiency virus (SIV)-infected macaques (19, 26). These studies, however, are confounded by a potential effect of the CD8-depleting antibodies on the levels of target cells and innate immunity, as CD8-depleting antibodies may lead to increased homeostatic and antigen-driven proliferation of CD4+ T cells that constitute the main target cells of HIV and may deplete immune cells other than T cells, such as natural killer cells, which may also contribute to the control of viral replication (8a). As an alternative to CD8-depletion experiments, mathematical models can be employed to determine the roles of target cell limitation and virus-specific immune responses on viral replication. However, studies using mathematical models have remained equivocal so far (5, 27, 29).
Here, we sought to resolve the roles of target cell limitation and virus-specific immune responses in the control of early viral replication by analyzing primary SIVmac239 infection in rhesus macaques (Macacca mulatta), one of the most commonly used animal models for studies of AIDS pathogenesis (6). The advantages of an animal model for our purposes are the abilities to control for the timing of infection and to frequently monitor the virus load and the levels of target cells and virus-specific immune responses. In the experiment we analyze (8a), we interfered with the natural course of primary SIV infection in half of the animals by inhibiting the costimulatory signals T cells require to become activated by antigen-presenting cells and to initiate the differentiation of antigen-presenting cells and B cells. As a consequence, the treated animals have impaired SIV-specific CD8+-T-cell responses and delayed SIV-specific antibody responses. The resulting variance in the levels of SIV-specific immunity allowed us to investigate the determinants of primary SIV replication.
The modeling approach we pursued was to fit two simple models to the viremia measurements of the animals, one that describes the interaction between the virus and its target cells only and an extended model which additionally takes into account the inhibitory effect of virus-specific immune responses on viral replication. Measurements of the levels of target cells and virus-specific CD8+ T cells were incorporated into the model as components of the viral growth rate. Comparing the goodness of fit of the two models, we can determine whether the virus-specific CD8+-T-cell response plays a significant role in primary infection.
MATERIALS AND METHODS
Data.
The data set we analyzed was generated in an experiment in which eight rhesus macaques were infected with SIVmac239 (8a). From 1 day before infection until 27 days after infection, four animals were treated with CTLA4 immunoglobulin that blocks CD28/B7 interaction between T cells and dendritic cells, and thus inhibits T-cell activation and proliferation, and an anti-CD40L monoclonal antibody that inhibited the activation of dendritic cells and B cells by CD4+ T cells and the subsequent development of SIV-specific CD8+-T-cell responses and the production of anti-SIV antibodies. Plasma SIV RNA levels were quantified by real-time reverse transcriptase PCR (1, 12). The density of various lymphocyte subpopulations in the peripheral blood of the animals was quantified by multiparameter flow cytometry. The fraction of proliferating CD4+ and CD8+ T cells was assessed by staining for the nuclear antigen Ki67 which is expressed by cycling cells. SIV-specific CD8+ T cells were measured by using Mamu A*01 tetramers specific for immunodominant T-cell epitopes (SIV Gag181-189 and SIV Tat28-35). Further details on the experiment are published elsewhere (8a).
Figure 2 shows the measurements relevant for our study: the viremia, the density of proliferating CD4+ T cells (which constitute the most important target cells for virus replication), the density of proliferating CD8+ T cells (a potential surrogate marker for the SIV-specific CD8+-T-cell response during primary infection), and the sum of the densities of SIV Gag181-189- and SIV Tat23-35-specific CD8+ T cells in peripheral blood. Figure 2 gives important insights into the relationship between viral replication and the levels of target cells and virus-specific CD8+-T-cell responses. While in the treated animals Ki67+ CD4+ T cells decrease during the first 10 days of infection and remain low thereafter, they only temporarily attain low levels in the untreated animals. Furthermore, the virus-specific CD8+-T-cell response in the treated animals is weaker than in the untreated animals. As a consequence of these differences in the levels of target cells and virus-specific CD8+-T-cell responses, treated and untreated groups differ in their viremia. The peak viremia is significantly lower in the treated group than in the untreated group, while the postpeak viremia is significantly higher (8a). Intuitively, the higher peak viremia in the untreated animals could be attributed to higher target cell levels, and the lower postpeak viremia in the untreated group could be explained by the stronger virus-specific CD8+-T-cell responses. To quantitatively assess the competing effects of target cells and virus-specific CD8+ T cells on viral replication, we analyzed these data by using population dynamic models.
FIG. 2.

Measurements of viremia (a), Ki67+ CD4+ T cells (b), Ki67+ CD8+ T cells (c), and the sum of Gag181-189- and Tat28-35-specific CD8+ T cells (d) in treated and untreated animals. The period during which the costimulatory blocker was applied is shaded.
Models.
To determine the relative contributions of target cell limitation and cellular immunity to viral replication, it is necessary to keep track of the complex interaction between the virus, its target cells, and virus-specific immune cells. Population dynamic models are adequate tools for accomplishing this goal.
A common approach to investigate the roles of target cell limitation and virus-specific CD8+-T-cell responses in primary infection would be to construct a dynamic model describing the three populations in question—virus, target cells, virus-specific CD8+ T cells—and their interaction (Fig. 3a). The disadvantage of this approach is that one has to deal with a fairly large number of growth, death, and interaction terms, each associated with its own parameters. For the simplest model describing the interaction between the virus, its target cells, and the virus-specific CD8+ T cells, one has to deal with at least nine parameters (Fig. 3a offers a sample model). Furthermore, many interaction mechanisms,such as the depletion of CD4+ T cells or the stimulation of CD8+ T cells by the virus, are not well understood and their mathematical formulation will therefore be inherently speculative.
FIG. 3.

Diagrammatic comparison between the common approach (a) and our alternative approach (b). The common approach takes into account many aspects of the interaction between the virus, its target cells (mainly CD4+ T cells), and virus-specific CD8+-T-cell responses and therefore involves many (unknown) interaction mechanisms and parameters. Our alternative approach focuses on the effect of the target cells and virus-specific CD8+ T cells on virus replication and ignores all other aspects of the interaction, thus requiring many fewer parameters. The equations of population dynamic models that can be used for either approach are shown in order to illustrate the differences between the common approach and our alternative approach. The model of the common approach has four variables that denote the densities of susceptible and productively infected CD4+ T cells (T and T*, respectively) free virus (V), and virus-specific CD8+ T cells (E). The dot above the variables denotes the derivative with respect to time. This model assumes that susceptible CD4+ T cells are produced at a constant rate λ, die at a rate dT, and are infected by free virus at a rate βTV. Productively infected CD4+ T cells are assumed to die at the rate of δT and are cleared by virus-specific CD8+ T cells at a rate of kT*E. Free virus is produced by productively infected CD4+ T cells at a rate of πT* and is cleared at the rate of cV. Finally, virus-specific CD8+ T cells are assumed to arise at a rate σE and to die at a rate αE. The model used in the alternative approach has only one variable for free virus V. The growth rate g of the virus is a function of the density of susceptible CD4+ T cells (T) and the density of virus-specific CD8+ T cells (E). For the specific functions we assume for the viral growth rate g, see Appendix.
These disadvantages can be overcome by adopting an alternative approach. We decided not to describe all the aspects of the interaction between the virus and the immune cells involved in the infection but, rather, to focus on the effects that the target cells and the virus-specific CD8+ T cells exert on viral replication. We accomplish this by constructing models that describe the dynamics of the virus population only. Target cell and virus-specific CD8+-T-cell counts enter the model as components of the growth rate of the virus (Fig. 3b). This alternative approach reduces the number of parameters to the minimum required to study the role of target cells and specific CD8+ T cells on viral replication, while retaining the information contained in the target and immune cell measurements.
We constructed two models (see Appendix), one which describes the interaction between the virus and its target cells only (which we refer to as the target cell-based model) and an extended version of the target cell-based model, which additionally takes into consideration the potential effect of CD8+-T-cell responses (which we refer to as the immune control model). We fit the models to the virus load measurements between day 0 and day 28 after infection—a time interval which coincides with the period of treatment and is, therefore, convenient to define the period of primary infection. However, our results do not depend on this particular time interval. Comparing the fits of these two models using an F test (see Appendix) allows us to determine whether virus-specific CD8+ T cells play a significant role in controlling virus replication in primary infection.
In the immune control model, we need to incorporate measures of the virus-specific CD8+-T-cell response. Our data set contains two alternative measurements: (i) Gag181-189- and Tat23-35-specific CD8+ T cells and (ii) proliferating CD8+ T cells (as measured by the expression of the nuclear antigen Ki67). We prefer proliferating CD8+ T cells over Gag181-189- and Tat23-35-specific CD8+ T cells for several reasons. First, we do not know to what extent the Gag181-189- and Tat23-35-specific CD8+ T cells reflect the total extent of the CD8+-T-cell response in each animal. Although matched for Mamu A*01, these outbred animals differ at known major histocompatibility complex class I alleles as well as at potentially undefined major histocompatibility complex class I alleles (8a). Therefore, the extent to which the Gag181-189- and Tat23-35-specific CD8+ T cells reflect the total extent of the CD8+-T-cell response may differ from animal to animal. Moreover, we have evidence for immunologic escape from virus-specific CD8+-T-cell responses directed against the Tat23-35 epitope in all animals by day 20, and thus the Tat23-35-specific CD8+-T-cell response may no longer affect the prevailing virus population after this time point (8a). Here, we therefore present results based on proliferating CD8+ T cells. We emphasize, however, that a parallel analysis based on Gag181-189- and Tat23-35-specific CD8+ T cells yields similar results (see Appendix).
We tested our model selection scheme by first applying it to the treated group. Due to the fact that the treated group had severely impaired specific immune responses to the virus, the contribution of CD8+-T-cell responses to viral replication should be negligible, i.e., the immune control model is not expected to fit significantly better than the target cell-based model.
RESULTS
Target cell limitation explains viral control in the treated animals.
Fitting the target cell-based model to the virus load measurements of the treated animals gives relatively good fits, given that our model is determined by two parameters only (Fig. 4, dashed lines). Including CD8+-T-cell responses into the analysis does not significantly improve the fits in three out of four animals (Fig. 4, solid lines; see Table 2). Thus, the impact of virus-specific CD8+-T-cell responses is negligible in these three animals. The animal REo6, for which the inclusion of the CD8+-T-cell response improves fit, is also the animal with the highest levels of proliferating CD8+ T cells, levels comparable to those of the untreated animals. As expected, we conclude that the interaction between the virus and its target cells alone can account for the pattern of viral replication observed in primary infection and that the CD8+-T-cell responses play a negligible role in controlling viral replication in the setting of a costimulatory blockade.
FIG. 4.

Fitting the target cell-based model (dashed lines) and the immune control model (solid lines) to the virus load of the treated animals. Only for animal REo6 does the immune control model fit significantly better.
TABLE 2.
Parameter estimates for the fits with the immune control modela
| Macaque | r (68% CI) | a′ (68% CI) | k (68% CI) | Residual sum of squares | Pb |
|---|---|---|---|---|---|
| Treated group | |||||
| RCr5 | 0.022 (0.017-0.023) | 0.63 (0.08-0.78) | 0.33 (0.18-0.56) | 0.14 | 0.015 |
| REo6 | 0.020 (0.019-0.028) | 0.07 (0.00-0.38) | 0.21 (0.11-0.24) | 0.56 | |
| REp6 | 0.023 (0.022-0.041) | 0.09 (0.00-0.60) | 0.22 (0.11-0.28) | 0.22 | |
| RIc6 | 0.039 (0.032-0.087) | 0.29 (0.00-0.86) | 0.43 (0.00-2.11) | 0.96 | |
| Mean ± SEc | 0.026 ± 0.005 | 0.27 ± 0.15 | 0.30 ± 0.06 | ||
| Untreated group | |||||
| RBm6 | 0.028 (0.001-0.042) | 0.00 (0.00-0.66) | 3.12 (0.23-4.19) | 1.38 | <0.001 |
| ROz5 | 0.018 (0.018-0.026) | 0.00 (0.00-0.00) | 0.45 (0.42-0.58) | 0.20 | <0.001 |
| RVy5 | 0.037 (0.010-0.047) | 0.00 (0.00-0.00) | 1.45 (0.36-2.15) | 9.44 | 0.014 |
| RYt5 | 0.035 (0.007-0.050) | 0.00 (0.00-0.00) | 0.84 (0.08-1.35) | 7.39 | 0.009 |
| Mean ± SE | 0.030 ± 0.005 | 0.00 ± 0.00 | 1.47 ± 0.68 |
r, production rater per target cell per day; a′, intrinsic death rate of infected cells per day; k, CTL killing rate per 100 Ki67+ CD8+ T cells per day.
P values are given only in instances of better fit of the immune control model.
SE, standard error of the means.
Target cell limitation does not explain viral control in the untreated animals.
The results for the untreated group are quite different. Fitting the target cell-based model yields very bad fits which do not account for the pattern in primary infection at all (Fig. 5, dashed lines). Thus, target cell limitation alone cannot account for primary virus replication. Intuitively, the reason behind the shortcomings of the target cell-based model is that the target cell levels do not decrease but are on average as high as before infection (Fig. 2b). The reason for the almost unchanged levels of proliferating CD4+ T cells may be that the depletion of virus-infected cells is balanced by antigen-driven proliferation of CD4+ T cells. Thus, the target cell-based model predicts a constantly growing virus population which is incompatible with the observed virus load in the untreated animals.
FIG. 5.

Fitting the target cell-based model (dashed lines) and the immune control model (solid lines) to the virus load of the untreated animals. The immune control model fits significantly better for all animals.
Including the effect of proliferating CD8+ T cells into our analysis improves the fits in all four animals significantly (Fig. 5, solid lines; see Table 2), which provides strong evidence for the crucial role of the SIV-specific CD8+-T-cell responses in the containment of early viral replication. The importance of SIV-specific CD8+ T cells is further supported by the finding that the postpeak decline in the viremia is positively correlated with the levels of proliferating CD8+ T cells (8a). An analysis based on Gag181-189- and Tat23-35-specific CD8+ T cells yields similar results (see Appendix and Fig. A1b).
FIG. A1.

Comparison of the fits of the immune control model, based on different measures of SIV-specific CD8+-T-cell responses, in the treated animals (a) and in the untreated animals (b). Fits of the immune control model based on the sum of the Gag181-189- and Tat23-35-specific CD8+ T cells are shown as solid red lines, and fits of the immune control model based on Ki67+ CD8+ T cells are shown as solid black lines. Fits of the target cell-based model are shown as dashed lines.
Virus-infected cells are mainly killed by virus-specific CD8+ T cells.
Table 1 contains the parameter estimates for the target cell-based model. Since the target cell-based model is inadequate to account for the pattern of viral replication in the untreated group, we refrained from reporting parameter estimates for this group. The replication rate of the virus per target cell r was estimated to be 0.030 ± 0.005, which is consistent with the estimates from other studies of the replication rate of SIV and HIV (18, 20). According to this estimate, an increase of 33 in the number of target cells in 1 μl of blood leads to an increase of the viral growth rate of 1 log per day. The parameter a (that can be interpreted as the death rate of infected cells [11, 22, 28]) was estimated as 0.77 ± 0.16. This estimate of a corresponds to a half-life of infected cells of 0.9 ± 0.24 days, which is in agreement with the latest estimates (10).
TABLE 1.
Parameter estimates for the fits with the target cell-based modela
| Macaque | r (68% CI) | a (68% CI) | τ1/2 (days) | Residual sum of squares |
|---|---|---|---|---|
| Treated group | ||||
| RCr5 | 0.024 (0.022-0.033) | 1.09 (0.99-1.54) | 0.64 | 0.31 |
| REo6 | 0.028 (0.020-0.038) | 0.92 (0.38-1.21) | 0.75 | 2.07 |
| REp6 | 0.025 (0.022-0.030) | 0.54 (0.43-0.63) | 1.28 | 0.51 |
| RIc6 | 0.041 (0.031-0.093) | 0.52 (0.15-1.09) | 1.33 | 1.27 |
| Mean ± SEb | 0.030 ± 0.005 | 0.77 ± 0.16 | 1.00 ± 0.21 | |
| Untreated group | ||||
| RBm6 | NAc | NA | NA | 25.07 |
| ROz5 | NA | NA | NA | 15.36 |
| RVy5 | NA | NA | NA | 35.60 |
| RYt5 | NA | NA | NA | 33.47 |
r, production rate per target cell per day; a, death rate of infected cells per day; τ1/2, half-life of infected cells calculated from their death as τ1/2 = (ln 2)/a.
SE, standard error of the means.
NA, not available. Due to poor fits, the model parameters for animals in the untreated group could not be estimated.
Table 2 shows the estimates of the parameters of the immune control model and compares the goodness of the fits to those of the target cell-based model. In all four untreated animals, the immune control model fits significantly better. The estimate of the replication rate of the virus per target cell r does not differ significantly between the treated and untreated groups, indicating that the susceptibility of target cells and the rate at which target cells produce virus do not differ between the treated and untreated groups. The main difference between the two groups concerns the components of the death rate of infected cells. The intrinsic death rate of infected cells, a′, which comprises the rate of apoptosis and virus-induced killing, is significantly higher in the treated group than in the untreated group, for which a′ is consistently estimated as zero. This finding remains unchanged if we base our analysis on Gag181-189- and Tat23-35-specific CD8+ T cells instead of on proliferating CD8+ T cells. This result suggests that, during primary HIV infection, infected cells are not predominantly killed directly by the virus but mainly by cytotoxic T lymphocytes (CTL), as has been suggested earlier (16).
No impact of resting CD4+ T cells on viral replication.
To test for the relevance of viral replication in resting CD4+ T cells (as defined by CD4+ T cells that do not express Ki67) in primary infection, we constructed extensions of the target cell-based model and the immune control model that take into account the potential for viral replication in resting CD4+ T cells (see Appendix). We found that the inclusion of replication in resting CD4+ T cells did not explain early viral replication better than the target cell-based model or the immune control model (data not shown). Thus, even though the infection of resting cells is reported (9), according to our analysis, the contribution of the compartment of resting CD4+ T cells to viral replication is negligible. Furthermore, an extension of our analysis shows that a significant impact of antibody responses on viral containment in primary infection can also be ruled out (data not shown). This finding is consistent with low levels of antibodies in the treated animals and undetectable levels in the untreated animals during primary infection (8a).
DISCUSSION
In summary, our analysis shows that target cell limitation cannot explain the containment of viral replication observed during primary SIV infection. We also show that by including CD8+-T-cell responses into our analysis, we can account for the pattern of early viral replication significantly better. These results are robust with regard to changes of the time interval (day 0 to day 28) on which we based our analysis (data not shown). Moreover, we obtain similar results by using measures of Gag181-189- and Tat23-35-specific instead of proliferating CD8+ T cells (see Appendix), which suggests that proliferating CD8+ T cells may be an equivalent surrogate measure for the SIV-specific CD8+-T-cell response during primary or acute SIV infection.
Our study goes beyond CD8-depletion experiments and earlier quantitative studies in that we have carefully taken into account the impact of target cells on viral replication. The CD8-depletion study during primary SIV infection by Schmitz et al. (26), for example, did not measure the levels of proliferating CD4+ T cells. This omission makes solid conclusions about the role of target cell limitation and virus-specific CD8+ T-cell responses difficult. Similarly, the study by Stafford et al. (27), in which mathematical models were fitted to data on primary viremia in HIV-infected patients, did not take into consideration any measurements of target cells.
Our study, which used an approach very different from CD8-depletion models, largely agrees with the conclusions of Schmitz et al. (26) that virus-specific CD8+ T cells play a crucial role in primary SIV infection. However, our conclusions with regard to how early viral replication is controlled at its peak differ from those of Schmitz et al. (26). Schmitz et al. (26) find that the depletion of CD8+ cells affects neither the timing of the peak nor the peak level of viremia and, thus, rule out a role of the virus-specific CD8+-T-cell responses in the containment of viral replication at its peak. From our analysis, on the other hand, we conclude that viral containment at the peak is not due to target cell limitation.
Our finding that neither viral containment at the peak nor postpeak control of viral replication can be explained by target cell limitation in the untreated animals also disagrees with the conclusions of Stafford et al. (27) that target cell limitation could account for the pattern of viral replication during the first 100 days of infection. This discrepancy is due to the fact that the model employed by Stafford et al. (27) predicts that the levels of target cells decrease during primary infection, while we observed no such decrease in the untreated animals.
Our analysis is unable to identify the factors that control viral replication at its peak in the untreated group. We ruled out the possibility that containment of viral replication at its peak is due to target cell limitation in this group. Moreover, virus-specific CD8+-T-cell responses seem unlikely to be responsible for controlling SIV replication at the peak because they come up after the peak of the viremia. Thus, alternative factors may have to be invoked. It is possible, for example, that innate immune responses play a significant role in the control viral replication at its peak, which has been found to be the case in lymphocytic choriomeningitis virus infection in mice (3).
Even though we can show that virus-specific CD8+ T cells play a significant role in the control of primary infection, the prediction of the immune control model still deviates from the virus load observed. Several factors could account for the deviation. First, the measurements of target cells and virus-specific cellular immune responses in the blood of the animals may not reflect the levels of these cells in the entire animal. In particular, the redistribution of cells may confound our ability to predict viral replication. Second, our assumption that all proliferating CD4+ T cells are equally susceptible to infection may be overly simplistic. Should a subset of the proliferating CD4+ T cells, as for example SIV-specific T helper cells, be significantly more susceptible and productive than proliferating CD4+ T cells on average, our target cell-based model would have to be revised. Although there is evidence for increased susceptibility of HIV-specific CD4+ T cells during HIV infection (7), our target cell-based model would be inadequate only if specific CD4+ T cells were also shown to be substantially more productive than non-SIV-specific, proliferating CD4+ T cells. Third, the measured proliferating CD8+ T cells and Gag181-189- and Tat23-35-specific CD8+ T cells may not reflect the total extent of the overall cellular immune response. Last, immune responses other than cytotoxic T cells or antibodies, such as cytokines and chemokines released by T helper cells, may be at work. Since we did not measure cytokine or chemokine production in our experiments, we could not test for its impact on viral replication. In the future, experiments measuring levels of cytokines, chemokines, and virus-specific CD4+-T-cell responses combined with a quantitative analysis as presented here could address the detailed mechanisms of viral containment by the various branches of the immune system.
In the present analysis, we focused on primary infection and did not analyze the entire course of infection. A detailed analysis of all available data suggests that, especially during the later stages of infection, viral replication is determined by many factors, including antibodies and potentially unmeasured CD4+-T-cell-mediated responses (8a). Due to fully developed antibody responses and evident CTL escape in all the untreated animals after primary infection, our analysis is unlikely to yield the same straightforward results if applied to later stages of infection.
Given all of the caveats associated with the measurements on which we based our analysis and the limitation in our understanding of the dynamics of primary SIV infection in general, it is remarkable how well our extremely simple model accounts for the observed pattern of primary SIV replication. The insights into primary SIV infection gained from our study illustrate the power of mathematical models as tools for understanding the dynamics of viral infections.
Acknowledgments
We thank Alun Lloyd and Mark Tanaka for stimulating discussions.
We gratefully acknowledge the support of the Deutsche Forschungsgemeinschaft grant Re 1618/2-1 (R.R.R.) and the National Institutes of Health grants AI 49334 (R.A. and R.R.R.) and RO1 AI49155 (M.B.F. and S.I.S.). Mark Feinberg is an Elizabeth Glaser Scientist of the Pediatric AIDS Foundation.
APPENDIX
Target-cell-based model.
The target cell-based model is given by the following equation for the plasma viremia, v:
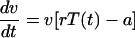 |
(1) |
Here, the dynamics of the virus is given by two terms: rT(t) which describes how the growth rate of the virus depends on the abundance of target cells, T(t), at the time t after infection, and a constant decay rate of viral RNA in plasma, a, which is approximately the death rate of infected cells (11, 22, 28). The parameter r is the replication rate of the virus per target cell per day.
We assume that proliferating CD4+ T cells constitute the main target cells of the virus. The function T(t) is constructed by interpolating the measurements of the density of Ki67+ CD4+ T cells in blood by a smooth function (a cubic spline).
The above differential equation can be solved analytically:
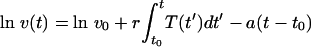 |
(2) |
Note that the above expression for the virus load is linear in the parameters r and a.
We fit the above expression to the logarithm of the observed virus load of each individual animal by separately minimizing the sum of squares of the residuals. This model (as all subsequent models) were fitted using the “optim()” routine of the R language of statistical computing (13) which allows us to confine the parameters to positive values. Bootstrap analysis was used to calculate 68% confidence intervals (CI) (8).
Immune control model.
To take into account the potential inhibition of viral replication by specific CD8+ T cells, we extend the target cell-based model (equation 1) by the additional term kE(t):
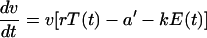 |
(3) |
E(t) is the interpolation function of the measurements of proliferating CD8+-T-cell densities in blood (as measured by the nuclear antigen Ki67), and the parameter k is the inhibition rate of viral replication per proliferating CD8+ T cell per day. The parameter a′ can be interpreted as the intrinsic death rate of infected cells, i.e., as the rate of apoptosis or virus-induced death. The total death rate of infected cells in the immune control model is given by a′ + kE(t) and depends on the level of the CD8+-T-cell response.
As with the target cell-based model (equation 1), the immune control model can be solved analytically:
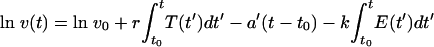 |
(4) |
and is fitted to the logarithm of the observed virus load by using a least square algorithm.
Model comparison.
We use the target cell-based model (equation 1) and the immune control model (equation 3) to determine whether the immune response plays a significant role in controlling the primary infection. We adopt a standard statistical strategy: first, we fit the two models by minimizing the residual sum of squares; then we determine whether the additional parameter in the immune control model (equation 3) (which measures the contribution of the CD8+-T-cell responses) explains a significant fraction of the variation in the viremia that is not explained by the target cell-based model (equation 1). Formally, we can test for the significance of “betterness” of the fit with the immune control model (equation 3) by performing an F test on the residual sum of squares normalized by the appropriate degrees of freedom:
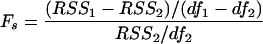 |
(5) |
Here, RSS1 and RSS2 denote the residual sums of squares resulting from a fit of the target cell-based model (equation 1) and the immune control model (equation 3), respectively, and df1 and df2 denote the corresponding degrees of freedom. Fitting eight data points with the target cell-based model (equation 1) that has two parameters, we have six degrees of freedom (df1 = 6), and since the immune control model (equation 3) has one more parameter than the target cell-based model (equation 1), we have df2 = 5. If Fs is larger than the critical value of the Fisher distribution F (df1, df2) = 6.61, where α = 0.05, then the virus-specific CD8+-T-cell response significantly contributes to the replication pattern of the virus in the primary infection.
Resting cell model.
To investigate the role of resting CD4+ T cells in viral replication, we extended the immune control model (equation 3) by the term r′R(t):
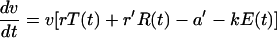 |
(6) |
Here, R(t) is the interpolation function of the measurements of nonproliferating CD4+ T cells (as measured by CD4+ T cells that do not display Ki67), and r′ is the production rate of the virus in resting CD4+ T cells (per cell per day). The production rate r′ of the virus in resting cells was estimated to be zero in all animals. Thus, viral replication in resting CD4+ T cells is negligible.
Analysis based on Gag181-189- and Tat28-35-specific CD8+ T cells.
The analysis presented in the main text is based on proliferating CD8+ T cells as a surrogate for the SIV-specific CD8+-T-cell response. Here, we present the results of an analysis based on the measurements of Gag181-189- and Tat23-35-specific CD8+ T cells. This alternative choice of a surrogate for the SIV-specific CD8+-T-cell responses affects only the fit of the immune control model and not the fit of the target cell-based model.
Figure A1a shows the fits of the immune control model based on the sum of Gag181-189- and Tat23-35-specific CD8+ T cells (solid red lines) to the treated animals. The fits based on the sum of Gag181-189- and Tat23-35-specific CD8+ T cells are very similar to the fits based on Ki67+ CD8+ T cells (solid black lines). Based on either of the two measurements of virus-specific CD8+-T-cell responses, the immune control model does not explain the pattern of viral replication significantly better than the target cell-based model (dashed lines) in three out of four animals (Tables 2 and A1).
Figure A1b shows the fits of the immune control model based on the sum of Gag181-189- and Tat23-35-specific CD8+ T cells (solid red lines) to the untreated animals and compares them with the fits based on Ki67+ CD8+ T cells (solid black lines). Except for animal RBm6, the fits of the immune control model based on the sum of the Gag181-189- and Tat23-35-specific CD8+ T cells are very similar to the fits of the same model based on Ki67+ CD8+ T cells.
Table A1 shows the parameter estimates of the immune control model based on the sum of Gag181-189- and Tat23-35-specific CD8+ T cells and compares the fit of the immune control model to the fit of the target cell-based model. In three out of four animals, the immune control model explains the pattern of viral replication significantly better than the target cell-based model. As in the analysis based on Ki67+ CD8+ T cells, the intrinsic death rate of infected cells, a′, is estimated as zero in all untreated animals, suggesting that, in primary infection, infected cells are killed mainly by CTL.
TABLE A1.
Parameter estimates for the fits with the immune control model based on the sum of Gag181-189- and Tat28-35-specific CD8+ T cellsa
| Macaque | r (68% CI) | a′ (68% CI) | k (68% CI) | Residual sum of squares | Pb |
|---|---|---|---|---|---|
| Treated group | |||||
| RCr5 | 0.020 (0.018-0.021) | 0.46a (0.24-0.70) | 2.94 (1.82-3.52) | 0.04 | 0.003 |
| REo6 | 0.022 (0.018-0.027) | 0.26 (0.00-0.52) | 6.47 (2.48-9.72) | 1.28 | |
| REp6 | 0.024 (0.020-0.045) | 0.34 (0.00-0.66) | 2.16 (0.92-4.20) | 0.39 | |
| RIc6 | 0.038 (0.029-0.098) | 0.22 (0.00-0.63) | 2.54 (0.00-5.17) | 0.93 | |
| Mean ± SE | 0.026 ± 0.005 | 0.32 ± 0.06 | 3.53 ± 1.15 | ||
| Untreated group | |||||
| RBm6 | 0.007 (0.000-0.016) | 0.00 (0.00-0.00) | 2.43 (0.00-5.29) | 21.79 | |
| ROz5 | 0.016 (0.015-0.024) | 0.00 (0.00-0.33) | 1.33 (0.10-1.45) | 0.26 | <0.001 |
| RVy5 | 0.024 (0.010-0.032) | 0.00 (0.00-0.00) | 5.18 (2.51-7.92) | 9.08 | 0.012 |
| RYt5 | 0.028 (0.011-0.040) | 0.00 (0.00-0.00) | 7.08 (2.43-12.07) | 10.07 | 0.019 |
| Mean ± SE | 0.019 ± 0.005 | 0.000 ± 0.000 | 4.00 ± 1.51 |
r, production rate per target cell per day; a′, intrinsic death rate of infected cells per day; k, CTL killing rate per 100 tetramer-positive CD8+ T cells per day.
P values are given only in instances of better fit of the immune control model.
REFERENCES
- 1.Amara, R. R., F. Villinger, J. D. Altman, S. L. Lydy, S. P. O'Neil, S. I. Staprans, D. C. Montefiori, Y. Xu, J. G. Herndon, L. S. Wyatt, M. A. Candido, N. L. Kozyr, P. L. Earl, J. M. Smith, H. L. Ma, B. D. Grimm, M. L. Hulsey, J. Miller, H. M. McClure, J. M. McNicholl, B. Moss, and H. L. Robinson. 2001. Control of a mucosal challenge and prevention of AIDS by a multiprotein DNA/MVA vaccine. Science 292:69-74. [DOI] [PubMed] [Google Scholar]
- 2.Clark, S. J., M. S. Saag, W. D. Decker, S. Campbell-Hill, J. L. Roberson, P. J. Veldkamp, J. C. Kappes, B. H. Hahn, and G. M. Shaw. 1991. High titers of cytopathic virus in plasma of patients with symptomatic primary HIV-1 infection. N. Engl. J. Med. 324:954-960. [DOI] [PubMed] [Google Scholar]
- 3.Cousens, L. P., R. Peterson, S. Hsu, A. Dorner, J. D. Altman, R. Ahmed, and C. A. Biron. 1999. Two roads diverged: interferon alpha/beta- and interleukin 12-mediated pathways in promoting T cell interferon gamma responses during viral infection. J. Exp. Med. 189:1315-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daar, E. S., T. Moudgil, R. D. Meyer, and D. D. Ho. 1991. Transient high levels of viremia in patients with primary human immunodeficiency virus type 1 infection. N. Engl. J. Med. 324:961-964. [DOI] [PubMed] [Google Scholar]
- 5.De Boer, R. J., and A. S. Perelson. 1998. Target cell limited and immune control models of HIV infection: a comparison. J. Theor. Biol. 190:201-214. [DOI] [PubMed] [Google Scholar]
- 6.Desrosiers, R. C. 1990. The simian immunodeficiency viruses. Annu. Rev. Immunol. 8:557-578. [DOI] [PubMed] [Google Scholar]
- 7.Douek, D. C., J. M. Brenchley, M. R. Betts, D. R. Ambrozak, B. J. Hill, Y. Okamoto, J. P. Casazza, J. Kuruppu, K. Kuntsman, S. Wolinsky, Z. Grossman, M. Dybul, A. Oxenius, D. A. Price, M. Connors, and R. A. Koup. 2002. HIV preferentially infects HIV-specific CD4(+) T cells. Nature 417:95-98. [DOI] [PubMed] [Google Scholar]
- 8.Efron, B., and R. Tibshirani. 1986. Bootstrap methods for standard errors, confidence intervals, and other measures of statistical accuracy. Stat. Sci. 1:54-77. [Google Scholar]
- 8a.Garber, D. A., G. Silvestri, A. P. Barry, A. Fedanov, N. Kozyr, H. McClure, D. C. Montefiori, C. P. Larsen, J. D. Altman, S. I. Staprans, and M. B. Feinberg. 2004. Blockade of T cell costimulation reveals interrelated actions of CD4+ and CD8+ T cells in control of SIV replication. J. Clin. Investig. 113: 836-845. [DOI] [PMC free article] [PubMed]
- 9.Haase, A. T., K. Henry, M. Zupancic, G. Sedgewick, R. A. Faust, H. Melroe, W. Cavert, K. Gebhard, K. Staskus, Z. Q. Zhang, P. J. Dailey, H. H. Balfour, A. Erice, and A. S. Perelson. 1996. Quantitative image analysis of HIV-1 infection in lymphoid tissue. Science 274:985-989. [DOI] [PubMed] [Google Scholar]
- 10.Ho, D. D. 2002. HIV and lymphocyte dynamics. Vaccine 20:1933. [DOI] [PubMed] [Google Scholar]
- 11.Ho, D. D., A. U. Neumann, A. S. Perelson, W. Chen, J. M. Leonard, and M. Markowitz. 1995. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373:123-126. [DOI] [PubMed] [Google Scholar]
- 12.Hofmann-Lehmann, R., R. K. Swenerton, V. Liska, C. M. Leutenegger, H. Lutz, H. M. McClure, and R. M. Ruprecht. 2000. Sensitive and robust one-tube real-time reverse transcriptase-polymerase chain reaction to quantify SIV RNA load: comparison of one- versus two-enzyme systems. AIDS Res. Hum. Retrovir. 16:1247-1257. [DOI] [PubMed] [Google Scholar]
- 13.Ihaka, R., and R. Gentleman. 1996. R: a language for data analysis and graphics. J. Comput. Graph. Stat. 5:299-314. [Google Scholar]
- 14.Kahn, J. O., and B. D. Walker. 1998. Acute human immunodeficiency virus type 1 infection. N. Engl. J. Med. 339:33-39. [DOI] [PubMed] [Google Scholar]
- 15.Kaufmann, G. R., P. Cunningham, A. D. Kelleher, J. Zaunders, A. Carr, J. Vizzard, M. Law, and D. A. Cooper. 1998. Patterns of viral dynamics during primary human immunodeficiency virus type 1 infection. J. Infect. Dis. 178:1812-1815. [DOI] [PubMed] [Google Scholar]
- 16.Klenerman, P., R. E. Phillips, C. R. Rinaldo, L. M. Wahl, G. Ogg, R. M. May, A. J. McMichael, and M. A. Nowak. 1996. Cytotoxic T lymphocytes and viral turnover in HIV type 1 infection. Proc. Natl. Acad. Sci. USA. 93:15323-15328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koup, R. A., and D. D. Ho. 1994. Shutting down HIV. Nature 370:416. [DOI] [PubMed] [Google Scholar]
- 18.Little, S. J., A. R. McLean, C. A. Spina, D. D. Richman, and D. V. Havlir. 1999. Viral dynamics of acute HIV-1 infection. J. Exp. Med. 190:841-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matano, T., R. Shibata, C. Siemon, M. Connors, H. C. Lane, and M. A. Martin. 1998. Administration of an anti-CD8 monoclonal antibody interferes with the clearance of chimeric simian/human immunodeficiency virus during primary infections of rhesus macaques. J. Virol. 72:164-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nowak, M. A., A. L. Lloyd, G. M. Vasquez, T. A. Wiltrout, L. M. Wahl, N. Bischofberger, J. Williams, A. Kinter, A. S. Fauci, V. M. Hirsch, and J. D. Lifson. 1997. Viral dynamics of primary viremia and antiretroviral therapy in simian immunodeficiency virus infection. J. Virol. 71:7518-7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ogg, G. S., X. Jin, S. Bonhoeffer, P. R. Dunbar, M. A. Nowak, S. Monard, J. P. Segal, Y. Cao, S. L. Rowland-Jones, V. Cerundolo, A. Hurley, M. Markowitz, D. D. Ho, D. F. Nixon, and A. J. McMichael. 1998. Quantitation of HIV-1 specific cytotoxic T lymphocytes and plasma load of viral RNA. Science 279:2103-2106. [DOI] [PubMed] [Google Scholar]
- 22.Perelson, A. S., A. U. Neumann, M. Markowitz, J. M. Leonard, and D. D. Ho. 1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 271:1582-1586. [DOI] [PubMed] [Google Scholar]
- 23.Phillips, A. N. 1996. Reduction of HIV concentration during acute infection: independence from a specific immune-response. Science 271:497-499. [DOI] [PubMed] [Google Scholar]
- 24.Piatak, M., Jr., M. S. Saag, L. C. Yang, S. J. Clark, J. C. Kappes, K. C. Luk, B. H. Hahn, G. M. Shaw, and J. D. Lifson. 1993. High levels of HIV-1 in plasma during all stages of infection determined by competitive PCR. Science 259:1749-1754. [DOI] [PubMed] [Google Scholar]
- 25.Schacker, T., A. C. Collier, J. Hughes, T. Shea, and L. Corey. 1996. Clinical and epidemiologic features of primary HIV infection. Ann. Intern. Med. 125:257-264. [DOI] [PubMed] [Google Scholar]
- 26.Schmitz, J. E., M. J. Kuroda, S. Santra, V. G. Sasseville, M. A. Simon, M. A. Lifton, P. Racz, K. Tenner-Racz, M. Dalesandro, B. J. Scallon, J. Ghrayeb, M. A. Forman, D. C. Montefiori, E. P. Rieber, N. L. Letvin, and K. A. Reimann. 1999. Control of viremia in simian immunodeficiency virus infection by CD8(+) lymphocytes. Science 283:857-860. [DOI] [PubMed] [Google Scholar]
- 27.Stafford, M. A., L. Corey, Y. Z. Cao, E. S. Daar, D. D. Ho, and A. S. Perelson. 2000. Modeling plasma virus concentration during primary HIV infection. J. Theor. Biol. 203:285-301. [DOI] [PubMed] [Google Scholar]
- 28.Wei, X., S. K. Gosh, M. E. Taylor, V. A. Johnson, E. A. Emini, P. Deutsch, J. D. Lifson, S. Bonhoeffer, M. A. Nowak, B. H. Hahn, M. S. Saag, and G. M. Shaw. 1995. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 373:117-122. [DOI] [PubMed] [Google Scholar]
- 29.Wick, D., S. G. Self, and L. Corey. 2002. Do scarce targets or T killers control primary HIV infection? J. Theor. Biol. 214:209-214. [DOI] [PubMed] [Google Scholar]