

Abstract
Insulin resistance with its associated hyperglycemias represents one significant contributor to mortality in burned patients. A variety of cellular stress-signaling pathways are activated as a consequence of burn. A key player in the cellular stress response is the endoplasmic reticulum (ER). Here, we investigated a possible role for ER-stress pathways in the progression of insulin function dysregulation post-burn. Rats received a 60% TBSA thermal injury and a laparotomy was performed at 24, 72 and 192 h post-burn. Liver was harvested before and 1 min after insulin injection (1 IU/kg) into the portal vein and expression patterns of various proteins known to be involved in insulin and ER-stress signaling were determined by Western blotting. mRNA expression of Glucose-6-Phosphatase (G-6-P) and Glucokinase (GK) were determined by RT-PCR and fasting serum glucose and insulin levels by standard enzymatic and ELISA techniques, respectively. Insulin resistance indicated by increased glucose and insulin levels occurred starting 24 h post-burn. Burn injury resulted in activation of ER stress pathways, reflected by significantly increased accumulation of phospho-PERK and phosphor-IRE-1 leading to an elevation of phospho-JNK and serine phosphorylation of IRS-1 post-burn. Insulin administration caused a significant increase in tyrosine phosphorylation of IRS-1 leading to activation of the PI3K/Akt pathway in normal liver. Post-burn tyrosine phosphorylation of IRS-1 was significantly impaired associated with an inactivation of signaling molecules acting downstream of IRS-1 leading to significantly elevated transcription of G-6-P and significantly decreased mRNA expression of GK. Activation of ER-stress signaling cascades may explain metabolic abnormalities involving insulin action following burn.
Keywords: insulin resistance, PI3K/AKT, ER stress, burn injury
INTRODUCTION
Severe burns represent a devastating injury affecting nearly every organ system and leading to significant morbidity and mortality (1–3). One of the main contributors to adverse outcome of this patient population represents its profound metabolic changes associated with insulin resistance and hyperglycemia (3, 4). Maintaining euglycemia via insulin administration represents current standards in burn and trauma ICUs and has significantly improved the clinical outcome of these patients over the past years (5). However, associated hypoglycemic events and difficult blood glucose titrations reinforces the need for better understanding of the molecular mechanisms underlying insulin resistance post-burn potentially leading to novel therapeutic approaches further improving the prognosis of these patients.
Effects of insulin, in order to maintain normoglycemia, occur through the insulin signaling cascade (6). Upon binding to the α-subunit on the extracellular portion of its receptor, insulin induces auto-phosphorylation of the β-unit leading to conformational changes and phosphorylation of insulin receptor substrate (IRS)-1 at a critical tyrosine residue, which in turn leads to activation of the phosphatidylinositol-3 kinase (PI3K)/Akt pathway (7, 8). Several studies indicate the major role of PI3K in the regulation of metabolic actions of insulin signaling, including stimulation of glucose transport via phosphorylation of Akt and the resultant plasma membrane localization of the GLUT4 glucose transporter within skeletal muscle and adipose tissue (9, 10). Activation of AKT has also been shown to be of major importance for the regulation of hepatic glucose homeostasis via insulin signaling cascades, including inhibition of gluconeogenesis and glycogenolysis pathways and activation of glycolysis pathways via activation or inactivation of several key enzymes (11).
Recent work now suggests that stress-induced insulin resistance may be in part due to phosphorylation-based negative-feedback, which may uncouple the insulin receptor or insulin receptor-associated proteins from its downstream signaling pathways, altering insulin action (8). However, little is known about the complexity of molecular mechanisms underlying insulin resistance post-burn.
Over the past years, it has become increasingly clear that a variety of cellular stress signaling and inflammatory pathways are activated as a consequence of burn (12–14). A key player in the cellular stress response is the endoplasmic reticulum (ER), a membranous organelle that functions in synthesis and processing of secretory and membrane proteins (15, 16). ER stress is known to activate signaling pathways that result in major dysregulation of cellular homeostasis and may ultimately lead to apoptosis (5). Conditions that trigger ER stress include presence of various pro-inflammatory cytokines, viral infections, lipids, increased synthesis of secretory proteins, and expression of mutant or misfolded proteins (15), many of which are present following burn injury (5, 14, 17, 18).
Recent data now showed that ER stress may be associated with insulin resistance under conditions leading to hyperglycemia, such as obesity and type 2 diabetes (15). We thus decided to examine a possible role of ER-stress pathways in the pathogenesis of insulin dysregulation in the acute and post-acute phases in the liver using a rodent model of burn.
MATERIALS AND METHODS
All animal manipulations were approved by the Institutional Animal Care and Use Committee of the University of Texas Medical Branch at Galveston. The National Institutes of Health Guidelines for the Care and Use of Experimental Animals were met.
Burn Injury
A well-established method was used to induce a full-thickness scald burn in our model (19). Sprague Dawley rats (n = 3 to 6 each for experimental and control groups), 325 to 350 grams, were allowed to acclimate for 1 week before conducting experiments. Rats were housed in an institutional animal care facility and received a high-protein diet (Ensure; Abbott Laboratories, IL, USA) and water ad libitum throughout the study. Ensure was administered 7 days before the study to adjust the animals to the liquid diet. Animals were anesthetized with general anesthesia (Ketamine 40 mg/kg body weight and Xylazine 5 mg/kg body weight, both injected intraperitoneally) and received analgesia (Buprenorphine 0.05 mg/kg body weight, injected subcutaneously). The dorsum of the trunk and the abdomen were shaved, and a 60% total body surface area (TBSA) burn was administered by placing the animals in a mold exposing defined areas of the skin of the back and abdomen under general anesthesia and analgesia. The mold was placed in 96–98°C water, scalding the back for 10 s and the abdomen for 2 s. This method delivers a full-thickness cutaneous burn as confirmed by histologic section. Lactated Ringers solution (40 ml/kg body weight) was administered intraperitoneally immediately after the burn for resuscitation. After burn and resuscitation, animals were observed, received oxygen and were then placed into cages. Sham animals were treated in an identical manner except that they were not subjected to the burn.
Experimental Design
Two different studies were performed. After deprivation of food overnight, animals included in the first study were anesthetized at 24, 72 and 192 h following the burn/sham procedure. Blood was withdrawn from the right femoral artery for glucose and insulin determinations and a laparotomy was performed and liver tissue was harvested and placed into liquid nitrogen. Blood was collected immediately after harvest and stored on ice until serum preparation. Serum was separated by centrifugation at 3000 rpm for 20 min at 4°C and stored at −80°C until analyzed. Tissue samples were processed for subsequent immunoblotting and real time quantitative RT-PCR as described below (Fig. 1A).
Figure 1. Study design.

Two different studies were performed. After deprivation of food overnight, animals included in the first study (A) were anesthetized at 24, 72 and 192 h following the burn/sham procedure and a laparotomy was performed and liver tissue was harvested for Western blot and RT-PCR analysis (n=6, each group and time point). Prior to tissue harvesting, blood was withdrawn from the right femoral artery for glucose and insulin determinations. Animals included in the second study (B) were subjected to an identical burn/sham procedure. In order to determine the effects of burn injury on hepatic insulin signaling pathways, a laparotomy was performed at 24, 72 and 192 h following the burn/sham procedure. At the respective time points, the right liver lobe was ligated, harvested and placed into liquid nitrogen for subsequent tissue preparations. Then, insulin (1 IU/kg) was injected directly into the portal vein and the remaining of the liver was harvested 1 min thereafter for subsequent tissue processing.
Animals included in the second study were subjected to an identical burn/sham procedure. In order to determine the effects of burn injury on hepatic insulin signaling pathways, a laparotomy was performed at 24, 72 and 192 h following the burn/sham procedure. At the respective time points, the right liver lobe was ligated, harvested and placed into liquid nitrogen for subsequent tissue preparations. Insulin (1 IU/kg, Novolin R, Novo Nordisk Pharma GmbH, Mainz, Germany) was subsequently injected directly into the portal vein and the remaining of the liver was harvested 1 min thereafter for subsequent tissue processing (Fig. 1B).
Fasting Serum Glucose and Insulin
Fasting insulin levels were measured using double-sandwich, enzyme-linked, immunosorbent assays (ELISA-technique, R&D-Systems Inc., Minneapolis, USA) according to the protocol of the manufacturer. Fasting glucose values were determined using hexokinase assays (R&D-Systems Inc., Minneapolis, USA).
SDS-Polyacrylamide Gel Electrophoresis and Immunoblotting
In order to determine the effect of burn injury on insulin signaling and ER-related signaling pathways in the liver, Western blot analysis was conducted. Liver tissue was homogenized and solubilized in 150 mM NaCl, 50 mM Tris-HCl, pH 7.8, 1% (w/v) Triton X-100, 1 mM EDTA, 0.5 mM phenylmethanesulfonyl fluoride and 13 Complete protease inhibitor mixture (Roche Molecular Biochemicals, Basel, Switzerland) for 5 min on ice and spun at 12,000 × g, and 20 mg of the resultant supernatant was run out on a 4–20% SDS-polyacrylamide gel and subsequently electrotransferred to nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA, USA). Nitrocellulose sheets were then probed with primary antibody against PKR-like ER-Kinase (PERK) (Santa Cruz Biotechnology, CA, USA), Thr980, 16F8-phosphorylated PERK (Cell Signaling Technology, Danvers, MA, USA), inositol requiring enzyme (IRE)-1, phosphorylated IRE-1 (Abcam, Cambridge, MA, USA), Seattle, WA, USA), c-Jun N-terminal Kinase (JNK), Thr183/185- phosphorylated-JNK, IRS-1, Ser612- phosphorylated IRS-1, Tyr612- phosphorylated IRS-1, PI3Kp85, phosphorylated PI3Kp85, protein kinase B (Akt), Thr308-phosphorylated AKT and Glycerinaldehyd-3-phosphat-Dehydrogenase (GAPDH) (all from Cell Signaling Technology, Danvers, MA, USA). Signals were amplified using horseradish peroxidase-conjugated secondary antibody and developed with chemiluminescent substrates (Pierce Biotechnology, Rockford, IL, USA). Results were quantified by using a densitometer to scan the blots (GENE GENIUS, Bio Imaging System; Syngene, Frederick, MD, USA).
After quantification, the intensities of the phosphorylated protein bands were divided by the total form of the respective protein or by the intensity of the respective GAPDH band. In those cases where a single blot was probed sequentially with more than one antibody, the nitrocellulose was stripped at 60°C for 30 min in stripping buffer (2% SDS, 100 mM β-mercaptoethanol, 62.5 mM Tris-HCl) before probing with the next antibody.
Real Time Quantitative RT-PCR
RNA samples for Real Time Analysis were quantified using a Nanodrop Spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and qualified by analysis on an RNA Nano chip using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Synthesis of cDNA was performed with 1 ug of total RNA in a 20 ul reaction using the reagents in the Taqman Reverse Transcription Reagents Kit from (Applied Biosystems, Foster City, CA, USA). Q-PCR amplifications (performed in triplicate) are done using 2 μl of cDNA in a total volume of 25 μl using SYBR green using the SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) as specified by the manufacturer. The final concentration of the probe is 250 nM and of the primers is 900 nM. Relative RT-QPCR assays are performed with 18S RNA as normalizer. Absolute analysis is performed using known amounts of a synthetic transcript of the gene of interest. All PCR assays are run in the ABI Prism 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, USA) and the conditions are as follows: (50°C, 2 min; 95°C, 10 min; then 40 cycles: 95°C, 15 sec; 59°C, 1 min). The SYBR Primers are: G6pc-539F (Sequence: AGGTGGTGGCTGGAGTCTTG) and G6pc-642R (Sequence: GAAGGTGATGAGACAGTACCTCTGG) for Glucose-6-Phosphatase; Gck-175F (Sequence: CCCACCTACGTGCGTTCC) and Gck-272R (Sequence: ACTTTGACCAGCATCACTCTGAAG) for Glucokinase (GK).
Data Analysis
All graphing and statistical calculations were performed using Origin Pro 8.0 (STATCON, Witzenhausen, Germany) and SigmaPlot 9.0 (Systat Software Inc, San Jose, CA, USA). Values are expressed as means ± S.E.M of at least three in vivo independent measurements per experimental group. Statistical difference between groups was determined using two or one-tailed Student’s t-test. Differences were considered significant at p< 0.05.
RESULTS
Hyperglycemia and hyperinsulinemia indicate insulin resistance following burn injury
Burn injury led to significantly elevated fasting serum levels of glucose and insulin starting at 24 h post-burn when compared to non-burned (sham) animals. Specifically, both, fasting glucose and insulin initially increased to values of 144.0 ± 17. 5 mg/dl and 32.4 ± 10.7 ng/ml, respectively, and subsequently decreased over time but remained significantly elevated for the time period studied, when compared to sham animals (106.9 ± 4.5 mg/dl and 5.4 ± 0.3 ng/ml, respectively) as shown in Figure 2A, B.
Figure 2. Hyperglycemia and hyperinsulinemia indicate insulin resistance following burn injury.

Fasting glucose (A) and fasting insulin levels (B) were determined at various time points post-burn. Throughout the figure, histograms depict serum concentrations of glucose or insulin at fasted levels. Results shown represent six different animals per group, as indicated in the main text. Bars represent means; error bars correspond to S.E.M. Asterisks denote statistical significance: p < 0.05 for every comparison between groups.
Thermal burn injury is associated with the hepatic ER stress response
Severe burn injury was associated with increased activation of ER stress pathways in liver tissue of burned rats, as reflected by the markedly increased accumulation of phospho-PERK and phospho-IRE-1 in livers isolated at 24 and 72 h post-burn (Fig. 3A, C). We also observed a decreased accumulation of both total PERK and total IRE-1 in liver tissue of burned rats at 192 h post-burn compared to non-burned animals (Fig. 3A, C). Utilization of GAPDH levels in these of subsequent experiments ensured us that the observed changes were not due unequal loading across lanes. Quantification of the above mentioned proteins revealed a significantly increased phosphorylation of PERK (Fig. 3B) at 24 and 72 h and showed a significant increase of phospho-IRE-1 (Fig. 3D) at 72 h after the initial burn compared to liver tissue from sham burned animals. At 192 h post-burn, however, no difference was found for phospho-PERK and phospho-IRE-1 between burn and sham burned animals. Interestingly, at 192 h post-sham procedure, a significantly increased accumulation of phospho-IRE-1 was found in hepatic tissue of sham animals when compared to the 24 and 72 h time point.
Figure 3. Severe burn injury is associated with activation of ER stress signaling pathways and phosphorylation of IRS-1 at its serine binding site.

Accumulation of phospho-PERK/PERK (A), phospho-IRE-1/IRE-1 (C), phospho-JNK/JNK (E) and phospho-IRS-1(Ser307)/IRS-1 (G) was determined in liver tissue isolated at 24, 72 and 192 h after burn or sham procedure. GAPDH served as control loading protein. Histograms depict intensities of the phosphorylated protein bands divided by the total form of the respective protein (B, D, F, H). Results shown represent three to five different animals per group, as indicated in the main text. Bars represent means; error bars correspond to S.E.M. Asterisks denote statistical significance: p < 0.05 for every comparison between groups.
Activation of JNK is associated with phosphorylation of IRS-1 at its serine binding site
We found phospho-JNK to be markedly increased only in liver tissue of animals subjected to burn injury at 24 and 72 h post-burn (Fig. 3E). Interestingly, at 192 h post-sham procedure, a markedly increased accumulation of phospho-JNK was found in hepatic tissue of sham animals when compared to the 24 and 72 h time point. Total JNK expression was noticeably decreased at 24 h post-burn when compared to controls (Fig. 3E). At 192 h however, an apparent increase in total JNK accumulation was observed in hepatic tissue of burned animals when compared to that of sham rats.
Quantification revealed a significantly elevated phosphorylation of JNK (Fig. 3F) at 24 and 72 h following burn, before decreasing to levels below these of sham-burned animals at 192 h post-burn. Activation of JNK was associated with hepatic phosphorylation of IRS-1 at its serine binding site, indicated by significant elevation of serine phosphorylated IRS-1 at 24, 72 and 192 h after the initial injury (Fig. 3G, H) when compared to sham animals. Sham animals, in contrast, revealed a slight decrease in hepatic serine phosphorylation of IRS-1 over time.
Since serine phosphorylation of IRS-1 has been found to prevent its activation by the classical phosphorylation of a tyrosine residue by the insulin receptor tyrosine kinase (20), we performed another study in order to determine the effects of burn injury on hepatic insulin signaling pathways.
Burn injury is associated with impaired activation of IRS-1 at its tyrosine binding site and inhibits PI3K/Akt signaling after insulin administration
Insulin administration directly into the portal vein of non-burn animals resulted in significantly increased tyrosine phosphorylation of IRS at all three time points studied. Liver tissue of burned animals, subjected to insulin administration, in contrast, did not show any significant activation of IRS-1 at its tyrosine binding site at 24 and 72 h post-burn (Fig. 4A, B). At 192 h, however, diminished tyrosine phosphorylation of IRS-1 in the burn group had subdued and burned animals showed similar activation of IRS-1 at its tyrosine binding site upon insulin administration as did sham animals (Fig. 4A, B). Total IRS-1 expression was found to be noticeably decreased at all time points studied, in both insulin treated and insulin untreated hepatic tissue of burned animals when compared to controls (Fig. 3G, 4A).
Figure 4. Burn injury is associated with impaired activation of IRS-1 at its tyrosine binding site and inhibits PI3K/Akt signaling after insulin administration.

Accumulation of hepatic phospho-IRS-1(Tyr612)/IRS-1(A) phospho PI3K/PI3K (C) and phospho-Akt/Akt (E) was determined in both, sham burned and burned animals before (-) and 1 min after insulin injection (+) at 24, 72 and 192 h post-burn or post-sham procedure. GAPDH served as control loading protein. Histograms depict intensities of the phosphorylated protein bands divided by the intensity of the total form of the respective protein band (B, D, F). Results shown represent three different animals per group, as indicated in the main text. Bars represent means; error bars correspond to S.E.M. Asterisks denote statistical significance: p < 0.05 for every comparison between groups.
Burn injury further led to diminished activation of signaling molecules acting downstream of IRS-1 that mediate the metabolic response to insulin, such as PI3K and AKT, while insulin administration directly into the portal vein of non-burned animals caused robust activation of the PI3K/AKT signaling pathway. Specifically, sham rats displayed significant increases in PI3K and AKT (Fig. 4D, F) phosphorylation upon injection of 1U insulin into the portal vein at 72 and 192 h and 24, 72 and 192 h post sham procedure, respectively. In burn animals, however, phosphorylation of PI3K and AKT (Fig. 4D, F) upon insulin administration was not significantly altered at 24, 72 h post-burn. Burn injury also led to increased accumulation of total AKT at 192 h post-burn compared to sham burned animals.
Burn injury leads to alterations in transcription of Glucokinase (GK) and Glucose-6-Phosphatase (G-6-P)
Hepatic GK and G-6-P are controlled primarily at the level of transcription (21). Insulin inhibits hepatic glucose production by stimulating GK transcription whereas G-6-P mRNA levels are increased as a result of fasting and lack of insulin action (22–24). Thus, we wished to assess the levels of the two key enzymes. Real-time RT-PCR analysis revealed a significantly decreased transcription of GK, a key enzyme for glycolysis in liver tissue at 72 and 192 h post-burn compared to sham animals. No difference was detected at 24 h between groups (Fig. 5A). mRNA expression of G-6-P was significantly increased in liver tissue of burned rats at the 24-h time point compared to sham animals. At 72 and 192 h post-burn no significant change on the transcription level of G-6-P between burned and non-burned animals was detected (Fig. 5B).
Figure 5. Burn injury is associated with alterations in transcription of Glucokinase (GK) and Glucose-6-Phosphatase (G-6-P).
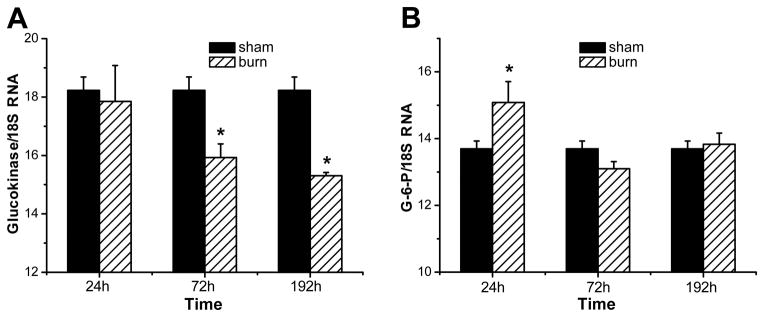
Total RNA was isolated from liver tissue of both burned and sham-burned and GK (A) and G-6-P (B) mRNA was assayed by real-time RT-PCR, with 18S ribosomal RNA (18S) used as endogenous control. Mean value of the target was divided by the mean value of the endogenous control to obtain a normalized mean quantity per sample. Results shown represent four different animals per group. Bars represent means; error bars correspond to S.E.M. Asterisks denote statistical significance: p < 0.05 for every comparison between groups.
DISCUSSION
Profound metabolic alterations post-burn associated with persistent changes in glucose metabolism and impaired insulin sensitivity significantly contribute to adverse outcome of burn patients (4, 25, 26). By enhancing adipose tissue lipolysis (27) and skeletal muscle proteolysis (28), stress mediators increase gluconeogenic substrates, including glycerol, alanine and lactate, thus augmenting hepatic glucose production in response to burn injury (29–31). Hyperglycemia fails to suppress hepatic glucose release during this time (32) and the suppressive effect of insulin on hepatic glucose release is attenuated, significantly contributing to post-burn hyperglycemia (33). Catecholamine-mediated enhancement of hepatic glycogenolysis, as well as direct sympathetic stimulation of glycogen breakdown, can further aggravate the hyperglycemia in response to stress (29). Some of these stress mediators, such as catecholamines and cytokines, have recently been shown to impair glucose disposal via alterations of the insulin signaling pathway and GLUT-4 translocation in muscle and adipose tissue, resulting in peripheral insulin resistance (16, 30, 34). Specifically, phosphorylation of IRS-1 at serine residues by various kinases may preclude its tyrosine phosphorylation by the insulin receptor tyrosine kinase, thus inhibiting insulin receptor signaling (35, 36). However, little is known about the complexity of altered signaling cascades underlying the impaired glucose metabolism in the liver post-burn.
Among these IRS-modifying enzymes, mounting evidence indicates that activation of c-JNK may be central in mediating insulin resistance in response to various stresses that occur in obesity and other conditions of insulin resistance (37). JNK was found to be activated upon specific stimuli, including presence of various cytokines, such as IL-6, IL-8, MCP-1 and TNF-α and internal cues, including ER stress (14, 15, 36, 38).
We recently showed that thermal injury leads to massive elevations of the above mentioned cytokines in rats for up to eight days post-burn indicating a profound inflammatory response (14). Here, we used a similar rodent model of burn injury to elucidate the molecular mechanisms underlying insulin resistance in the acute and post-acute phases following severe burn and investigated a possible role of ER stress pathways in the pathogenesis of insulin dysregulation in the liver post-burn.
As summarized in Figure 6, our results showed that fasting serum glucose and insulin levels are significantly elevated in rodents up to eight days after severe burn injury when compared to non-burned animals indicating insulin resistance. To examine whether ER stress is increased post-burn, we investigated, in a first study, the expression patterns of several molecular indicators of ER stress at various time points following burn injury. Severe burn injury was associated with increased activation of ER stress pathways in liver tissue of burned rats, as reflected by the markedly increased accumulation of phospho-PERK and phospho-IRE-1 in livers isolated at 24 and 72 h post-burn. ER stress is detected by transmembrane proteins which monitor the load of unfolded proteins in the ER lumen, and transmit this signal to the cytosol (16). Two of these proteins, inositol requiring enzyme-1 and PKR-like ER kinase, undergo oligomerization and phosphorylation in response to increased ER stress (16). Activity of JNK has already been shown to be increased through induction of ER stress pathways (39). Consistent with this phenomenon, we found total JNK activity, indicated by c-Jun phosphorylation, to be markedly elevated up to 72 h post burn injury. Active JNK is capable of phosphorylating IRS-1 at a specific serine residue, which has been found to prevent its activation by the classical phosphorylation of a tyrosine residue by the insulin receptor tyrosine kinase (20).
Figure 6. Molecular mechanisms underlying insulin resistance following thermal injury.

Severe burn is associated activation of ER-stress related signaling pathways, indicated by activation of PERK and IRE-1. Phosphorylation of IRE-1 causes activation of c-JNK. Activation of JNK may then lead to phosphorylation of IRS-1 at serine residues which may preclude its tyrosine phosphorylation by the insulin receptor tyrosine kinase, thus resulting in impaired PI3K/Akt signaling and insulin resistance with its associated consequences.
Inhibition of JNK activity in obese mice with the synthetic inhibitor SP600125 has been recently found to reverse this ER stress–induced serine phosphorylation of IRS-1 (15, 40). Activation status of c-JNK thus represents a central and integrating mechanism linking ER-stress and intracellular glucose homeostasis, since serine phosphorylation of IRS-1 has been shown to impair insulin receptor signaling (20). In spite of the appreciable differences in total protein levels of the various signaling molecules examined, the fraction of activated protein relative to total clearly suggest trends that support our model presented in Figure 6.
In order to examine whether burn injury exerts its effects on post-burn hepatic insulin resistance via this mechanism, we performed a second study. At 24, 72 or 192 h following burn or sham procedure, the right liver lobe was ligated and the respective lobe was harvested and utilized as negative control for Western blot analysis. Then insulin was injected directly into the portal vein, the remaining of the liver was harvested 1 min thereafter, and the tyrosine phosphorylation status of IRS-1 was measured. Consistent with our hypothesis that burn injury may lead to impaired insulin receptor trafficking via ER stress-induced serine phosphorylation of IRS-1, we found that burned animals did not display any significant activation IRS-1 at its tyrosine binding after insulin administration. Also activation of signaling molecules acting downstream of IRS-1, such as PI3K and AKT did not reveal any activation at 24 and 72 h post-burn. Insulin administration directly into the portal vein of non-burn animals, however, led to robust activation of these proteins, indicated by tyrosine phosphorylation of IRS and phosphorylation of PI3K and AKT at all time points studied. Several studies indicate the major role of PI3 kinase in the regulation of metabolic actions of insulin signaling, including stimulation of glucose transport via phosphorylation of Akt and the resultant plasma membrane localization of the GLUT4 glucose transporter in skeletal muscle and adipose tissue (37, 41, 42). Thr308-phosphorylation of AKT is also known to be of major importance for the regulation of hepatic glucose homeostasis via insulin signaling cascades, including modulation of gluconeogenesis and glycolysis pathways via activation or inactivation of several key enzymes (11). Hepatic GK and G-6-P activities are reported to be controlled primarily at the level of transcription, being mainly regulated by insulin and glucagon (21). High insulin levels have been shown to inhibit hepatic glucose production by means of stimulation of GK gene transcription and inhibition of gluconeogenesis (21). Using Real-time RT-PCR analysis, we found an initial increase in G-6-P mRNA expression at 24 h post-burn and significantly decreased transcription of hepatic GK at 72 and 192 h after thermal injury. Hepatic GK has a major effect on glucose homeostasis and is a potential target for pharmacological treatment of type 2 diabetes, as evidenced by the fact that liver-specific GK-knockout mice exhibited mild hyperglycemia (43) and rats over expressing GK in the liver had reduced blood glucose (44). Low hepatic GK activity is also reported to favor the release of glucose synthesized by gluconeogenesis into the circulation (21).
G-6-P is one of the key enzymes that control gluconeogenesis and glucose output from the liver (21). Maitra and colleagues (45) demonstrated its critical role in the early hyperglycemic phase of hemorrhagic shock. Others found that G-6-P mRNA and activity are increased in liver during the course of fasting or in insulinopenic diabetes (22–24).
The demonstrated post-burn modulations of hepatic insulin signaling pathways with subsequent alterations in hepatic GK and G-6-P mRNA expression may thus significantly contribute to elevated systemic glucose levels observed in our study.
We found that activation of signaling cascades emanating from ER stress may explain, at least partially, the metabolic and molecular abnormalities regarding insulin action following severe burn. Thus, signaling molecules associated with ER stress may constitute adequate candidates for the development of therapeutic interventions that may limit insulin resistance following severe burn. The ER stress signaling cascades found in this study to be associated with hepatic metabolic dysregulation following severe burn and inflammatory response pathways converge at the IRS-1/PI3K/Akt hub. Therefore, targeting of these molecules may provide a viable and efficacious alternative for the management of patients post burn.
Acknowledgments
This study was supported by the American Surgical Association Foundation, grants from Shriners Hospitals for Children (8460 and 8660) and National Institutes of Health (RO1-GM56687, RO1-HD049471).
References
- 1.Thombs BD, Singh VA, Milner SM. Children under 4 years are at greater risk of mortality following acute burn injury: evidence from a national sample of 12,902 pediatric admissions. Shock. 2006;26:348–352. doi: 10.1097/01.shk.0000228170.94468.e1. [DOI] [PubMed] [Google Scholar]
- 2.Lorente JA, Vallejo A, Galeiras R, Tomicic V, Zamora J, Cerda E, de la Cal MA, Esteban A. Organ dysfunction as estimated by the sequential organ failure assessment score is related to outcome in critically ill burn patients. Shock. 2009;31:125–131. doi: 10.1097/SHK.0b013e31817fc3ef. [DOI] [PubMed] [Google Scholar]
- 3.Herndon DN, Tompkins RG. Support of the metabolic response to burn injury. Lancet. 2004;363:1895–1902. doi: 10.1016/S0140-6736(04)16360-5. [DOI] [PubMed] [Google Scholar]
- 4.McCowen KC, Malhotra A, Bistrian BR. Stress-induced hyperglycemia. Crit Care Clin. 2001;17:107–124. doi: 10.1016/s0749-0704(05)70154-8. [DOI] [PubMed] [Google Scholar]
- 5.Gauglitz GG, Herndon DN, Jeschke MG. Insulin resistance postburn: underlying mechanisms and current therapeutic strategies. J Burn Care Res. 2008;29:683–694. doi: 10.1097/BCR.0b013e31818481ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.White MF. The insulin signalling system and the IRS proteins. Diabetologia. 1997;40 (Suppl 2):S2–17. doi: 10.1007/s001250051387. [DOI] [PubMed] [Google Scholar]
- 7.Kahn CR, White MF, Shoelson SE, Backer JM, Araki E, Cheatham B, Csermely P, Folli F, Goldstein BJ, Huertas P. The insulin receptor and its substrate: molecular determinants of early events in insulin action. Recent Prog Horm Res. 1993;48:291–339. doi: 10.1016/b978-0-12-571148-7.50015-4. [DOI] [PubMed] [Google Scholar]
- 8.Le Roith D, Zick Y. Recent advances in our understanding of insulin action and insulin resistance. Diabetes Care. 2001;24:588–597. doi: 10.2337/diacare.24.3.588. [DOI] [PubMed] [Google Scholar]
- 9.Chang L, Chiang SH, Saltiel AR. Insulin signaling and the regulation of glucose transport. Mol Med. 2004;10:65–71. doi: 10.2119/2005-00029.Saltiel. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodrigues AL, De Souza EP, Da Silva SV, Rodrigues DS, Nascimento AB, Barja-Fidalgo C, De Freitas MS. Low expression of insulin signaling molecules impairs glucose uptake in adipocytes after early overnutrition. J Endocrinol. 2007;195:485–494. doi: 10.1677/JOE-07-0046. [DOI] [PubMed] [Google Scholar]
- 11.Yu XX, Pandey SK, Booten SL, Murray SF, Monia BP, Bhanot S. Reduced adiposity and improved insulin sensitivity in obese mice with antisense suppression of 4E-BP2 expression. Am J Physiol Endocrinol Metab. 2008;294:E530–539. doi: 10.1152/ajpendo.00350.2007. [DOI] [PubMed] [Google Scholar]
- 12.Ballard-Croft C, Maass DL, Sikes P, White J, Horton J. Activation of stress-responsive pathways by the sympathetic nervous system in burn trauma. Shock. 2002;18:38–45. doi: 10.1097/00024382-200207000-00008. [DOI] [PubMed] [Google Scholar]
- 13.Jeschke MG, Gauglitz GG, Song J, Kulp GA, Finnerty CC, Cox RA, Barral JM, Herndon DN, Boehning D. Calcium and ER stress mediate hepatic apoptosis after burn injury. J Cell Mol Med. 2009 doi: 10.1111/j.1582-4934.2008.00644.x. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gauglitz GG, Song J, Herndon DN, Finnerty CC, Boehning D, Barral JM, Jeschke MG. Characterization of the inflammatory response during acute and post-acute phases after severe burn. Shock. 2008;30:503–507. doi: 10.1097/SHK.0b013e31816e3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 16.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 17.Finnerty CC, Herndon DN, Przkora R, Pereira CT, Oliveira HM, Queiroz DM, Rocha AM, Jeschke MG. Cytokine expression profile over time in severely burned pediatric patients. Shock. 2006;26:13–19. doi: 10.1097/01.shk.0000223120.26394.7d. [DOI] [PubMed] [Google Scholar]
- 18.Gauglitz GG, Herndon DN, Kulp GA, Meyer WJ, 3rd, Jeschke MG. Abnormal insulin sensitivity persists up to three years in pediatric patients post-burn. J Clin Endocrinol Metab. 2009;94:1656–1664. doi: 10.1210/jc.2008-1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herndon DN, Wilmore DW, Mason AD., Jr Development and analysis of a small animal model simulating the human postburn hypermetabolic response. J Surg Res. 1978;25:394–403. doi: 10.1016/s0022-4804(78)80003-1. [DOI] [PubMed] [Google Scholar]
- 20.Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 21.Jung UJ, Lee MK, Park YB, Jeon SM, Choi MS. Antihyperglycemic and antioxidant properties of caffeic acid in db/db mice. J Pharmacol Exp Ther. 2006;318:476–483. doi: 10.1124/jpet.106.105163. [DOI] [PubMed] [Google Scholar]
- 22.Gardner LB, Liu Z, Barrett EJ. The role of glucose-6-phosphatase in the action of insulin on hepatic glucose production in the rat. Diabetes. 1993;42:1614–1620. doi: 10.2337/diab.42.11.1614. [DOI] [PubMed] [Google Scholar]
- 23.Argaud D, Zhang Q, Pan W, Maitra S, Pilkis SJ, Lange AJ. Regulation of rat liver glucose-6-phosphatase gene expression in different nutritional and hormonal states: gene structure and 5′-flanking sequence. Diabetes. 1996;45:1563–1571. doi: 10.2337/diab.45.11.1563. [DOI] [PubMed] [Google Scholar]
- 24.Haber BA, Chin S, Chuang E, Buikhuisen W, Naji A, Taub R. High levels of glucose-6-phosphatase gene and protein expression reflect an adaptive response in proliferating liver and diabetes. J Clin Invest. 1995;95:832–841. doi: 10.1172/JCI117733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guvener M, Pasaoglu I, Demircin M, Oc M. Perioperative hyperglycemia is a strong correlate of postoperative infection in type II diabetic patients after coronary artery bypass grafting. Endocr J. 2002;49:531–537. doi: 10.1507/endocrj.49.531. [DOI] [PubMed] [Google Scholar]
- 26.Christiansen C, Toft P, Jorgensen HS, Andersen SK, Tonnesen E. Hyperglycaemia and mortality in critically ill patients. A prospective study. Intensive Care Med. 2004;30:1685–1688. doi: 10.1007/s00134-004-2325-2. [DOI] [PubMed] [Google Scholar]
- 27.Wolfe RR, Herndon DN, Jahoor F, Miyoshi H, Wolfe M. Effect of severe burn injury on substrate cycling by glucose and fatty acids. N Engl J Med. 1987;317:403–408. doi: 10.1056/NEJM198708133170702. [DOI] [PubMed] [Google Scholar]
- 28.Gore DC, Jahoor F, Wolfe RR, Herndon DN. Acute response of human muscle protein to catabolic hormones. Ann Surg. 1993;218:679–684. doi: 10.1097/00000658-199321850-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson LE, van Soeren MH. Insulin resistance and hyperglycemia in critical illness: role of insulin in glycemic control. AACN Clin Issues. 2004;15:45–62. doi: 10.1097/00044067-200401000-00004. [DOI] [PubMed] [Google Scholar]
- 30.Gearhart MM, Parbhoo SK. Hyperglycemia in the critically ill patient. AACN Clin Issues. 2006;17:50–55. doi: 10.1097/00044067-200601000-00007. [DOI] [PubMed] [Google Scholar]
- 31.Carlson GL. Insulin resistance and glucose-induced thermogenesis in critical illness. Proc Nutr Soc. 2001;60:381–388. doi: 10.1079/pns200193. [DOI] [PubMed] [Google Scholar]
- 32.Wolfe RR, Durkot MJ, Allsop JR, Burke JF. Glucose metabolism in severely burned patients. Metabolism. 1979;28:1031–1039. doi: 10.1016/0026-0495(79)90007-6. [DOI] [PubMed] [Google Scholar]
- 33.Cree MG, Zwetsloot JJ, Herndon DN, Qian T, Morio B, Fram R, Sanford AP, Aarsland A, Wolfe RR. Insulin sensitivity and mitochondrial function are improved in children with burn injury during a randomized controlled trial of fenofibrate. Ann Surg. 2007;245:214–221. doi: 10.1097/01.sla.0000250409.51289.ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hunt DG, Ivy JL. Epinephrine inhibits insulin-stimulated muscle glucose transport. J Appl Physiol. 2002;93:1638–1643. doi: 10.1152/japplphysiol.00445.2002. [DOI] [PubMed] [Google Scholar]
- 35.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 36.Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307) J Biol Chem. 2000;275:9047–9054. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- 37.Hotamisligil GS, Budavari A, Murray D, Spiegelman BM. Reduced tyrosine kinase activity of the insulin receptor in obesity-diabetes. Central role of tumor necrosis factor-alpha. J Clin Invest. 1994;94:1543–1549. doi: 10.1172/JCI117495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 40.Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ruderman NB, Kapeller R, White MF, Cantley LC. Activation of phosphatidylinositol 3-kinase by insulin. Proc Natl Acad Sci U S A. 1990;87:1411–1415. doi: 10.1073/pnas.87.4.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Folli F, Saad MJ, Backer JM, Kahn CR. Insulin stimulation of phosphatidylinositol 3-kinase activity and association with insulin receptor substrate 1 in liver and muscle of the intact rat. J Biol Chem. 1992;267:22171–22177. [PubMed] [Google Scholar]
- 43.Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 1999;274:305–315. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- 44.Ferre T, Pujol A, Riu E, Bosch F, Valera A. Correction of diabetic alterations by glucokinase. Proc Natl Acad Sci U S A. 1996;93:7225–7230. doi: 10.1073/pnas.93.14.7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maitra SR, Wang S, El-Maghrabi MR, Henry MC. Regulation of liver and kidney glucose-6-phosphatase gene expression in hemorrhage and resuscitation. Acad Emerg Med. 2000;7:731–738. doi: 10.1111/j.1553-2712.2000.tb02259.x. [DOI] [PubMed] [Google Scholar]