

Abstract
Members of the catechol diether class are highly potent non-nucleoside inhibitors of HIV-1 reverse transcriptase (NNRTIs). The most active compounds yield EC50 values below 0.5 nM in assays using human T-cells infected by wild-type HIV-1. However, these compounds like rilpivirine, the most recently FDA-approved NNRTI, bear a cyanovinylphenyl (CVP) group. This is an uncommon substructure in drugs that gives reactivity concerns. In the present work, computer simulations were used to design bicyclic replacements for the CVP group. The predicted viability of a 2-cyanoindolizinyl alternative was confirmed experimentally and provided compounds with 0.4-nM activity against the wild-type virus. The compounds also performed well with EC50 values of 10 nM against the challenging HIV-1 variant that contains the Lys103Asn/Tyr181Cys double mutation in the RT enzyme. Indolyl and benzofuranyl analogs were also investigated; the most potent compounds in these cases have EC50 values towards wild-type HIV-1 near 10 nM and high-nM activities towards the double-variant. The structural expectations from the modeling were much enhanced by obtaining an X-ray crystal structure at 2.88-Å resolution for the complex of the parent 2-cyanoindolizine 10b and HIV-1 RT. The aqueous solubilities of the most potent indolizine analogs were also measured to be ca. 40 µg/ml, which is similar to that for the approved drug efavirenz and ca. 1000-fold greater than for rilpivirine.
INTRODUCTION
Inhibitors of HIV-1 reverse transcriptase are central to anti-HIV therapy.1 Though there are five FDA-approved drugs in the non-nucleoside class,2 efavirenz (1) and rilpivirine (2) are particularly important as they are components of the one-a-day combination therapies Atripla and Complera.3 The other two components of the pills are the same, emtricitabine and tenofovir, which are in the nucleoside class of HIV-RT inhibitors. The goal of our research has been to discover new non-nucleoside inhibitors (NNRTIs) that may incorporate advantages for administration, formulation, diminished side effects, and activity towards variant strains of the virus. For example, issues with efavirenz include its daily dosage of 600 mg, poor activity towards HIV-1 variants containing the commonly occurring Lys103Asn (K103N) mutation in RT, and neurological side effects. The situation with rilpivirine is curious. Although it has much superior performance in cell-based assays than efavirenz, more virological failure is observed for patients using Complera than Atripla. 3,4 Another unusual feature of rilpivirine is its extremely low aqueous solubility (0.02 µg/ml)5 in comparison to the typical range of 4 – 4000 µg/ml for oral drugs.6
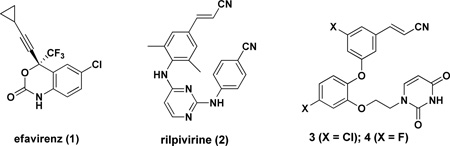
The challenges in developing new NNRTIs that represent an advance over existing compounds are great. One seeks simultaneously compounds that embody high potency towards the wild-type (WT) virus and numerous clinically observed variants, good pharmacological properties including solubility, an absence of structural features that may lead to rapid metabolism, and avoidance of toxicities stemming from reactive functional groups or metabolic products.7,8 A particularly promising class of NNRTIs that we have developed is catechol diethers including 3 and 4.9 3 appears to be the most potent anti-HIV agent ever reported with an EC50 of 0.055 nM in the standard MT-2 cell assay using wild-type HIV-1. The difluoro analog 4 is also extremely potent at 0.32 nM, has good potency towards variant strains containing the Y181C (16 nM) and K103N/Y181C (85 nM) mutations, and shows low cytotoxicity towards the T-cells (CC50 = 45 µM). It was also possible to obtain X-ray crystal structures of 3 and 4 in complex with WT HIV-RT.10 Thus, further structure-based design activities in the catechol diether series have a firm foundation.
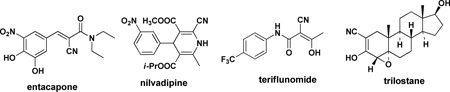
A structural feature in 3 and 4 as well as in rilpivirine that is addressed here is the cyanovinyl (CV) group. For most medicinal chemists viewing these structures, concern arises that the CV group may be sufficiently electrophilic to act as a “Michael acceptor” leading to potential covalent modification of proteins, nucleic acids, or other biological entities. Though in reality unsaturated nitriles are poor Michael acceptors that require reactive organometallic nucleophiles to undergo conjugate additions,11 the fact is almost no approved drugs contain a cyanovinyl group, and lack of precedent is often taken as a warning sign in drug discovery. When a search is done for a C=C-C≡N substructure in a comprehensive file containing the structures of ca. 1900 approved oral drugs,12 there are just five hits: rilpivirine, entacapone, nilvadipine, teriflunomide, and trilostane. For the latter two examples, the substructure only arises as a tautomer of an α-cyanoketone. Rilpivirine is unique in incorporating a simple CV group. By contrast, non-vinyl cyano groups are generally considered to be acceptable in drugs;13 there are 36 examples in the database.
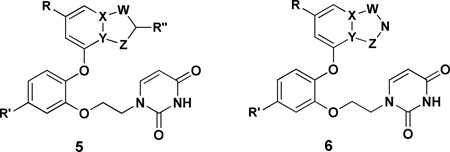
Thus, we set out to find a replacement for the cyanovinyl group in the catechol diethers. The prior results demonstrated the importance of the CV group to activity.9 The analog of 3 with the CV group truncated to just cyano has an EC50 of 14 nM towards the WT virus, and other compounds with the cyano of the CV group replaced by chlorine, OCH3, or CONH2 showed 100 to 1000-fold losses in activity. These findings are consistent with the modeling and crystallography,9,10 which document (a) close contact between the CV group and Trp229 in the NNRTI binding site, and (b) projection of the cyano group into the “tunnel” heading towards the polymerase active site with a water molecule hydrogen bonded to the cyano nitrogen atom. In order to replace these features and keeping in mind space constraints, it was decided to consider 6:5 heterobicyclic possibilities as indicated by 5 and 6. In 5, R” would likely be cyano, while in 6 the azole nitrogen would retain a hydrogen bond accepting site for a water molecule. In both cases, the choice of heteroatoms in the five-membered ring for optimal interaction with Trp229 was not obvious, nor was the effect of loss of the torsional degree of freedom associated with the CV group. In view of the wide range of options and the anticipated synthetic challenges, guidance was sought from computational analyses, specifically free energy perturbation calculations to predict relative free energies of binding for many alternatives.
COMPUTATIONAL DESIGN
The computational procedures that were utilized are exactly the same as in earlier studies.9,14 Starting from a crystal structure of the protein, the BOMB program7 was used to build initial structures for the desired protein-ligand complexes. Conjugate-gradient energy minimizations were performed to relax the structures and Monte Carlo free-energy perturbation (MC/FEP) were carried out to compute relative free energies of binding. The calculations were performed with the MCPRO program15 using the OPLS-AA force field for the protein,16 OPLS/CM1A for the ligands,17 and the TIP4P water model.18 The FEP calculations used 11 windows of simple overlap sampling, and many were also checked using 14 windows of double-wide sampling.19 The typical difference between the results was about 0.5 kcal/mol, which is a good measure of the statistical uncertainties. When both procedures were executed, the average of the results is reported. Each window covered at least 10 million configurations of equilibration and 10 million configurations of averaging. The unbound ligands and complexes were solvated in water caps with a 25-Å radius, amounting to ca. 2000 and 1250 water molecules, respectively. The 175 amino acid residues nearest to the ligand were included in the model for the complexes. A residue-based cutoff for non-bonded interactions was invoked at 10 Å. After short conjugate-gradient optimizations, the backbone atoms of the protein were not moved. The ligand and side chains with any atom within ca. 10 Å of the ligand were fully sampled. All water molecules were sampled using translations and rigid rotations.
The alternative heterocycles that were considered are designated 7a – 7k in Figure 1. They are typical 6:5 fused-ring possibilities, namely, indoles, indolizines, benzofurans, benzisoxazoles, and imidazopyridines. 7a, 7d, 7e, and 7b represent a ‘nitrogen walk’ around the 5-membered ring; the remaining isoindole isomer was excluded as such structures are labile.20 Furthermore, benzothiophenes were not considered as they appeared less promising in initial structure building than the benzofurans, and they are both prone to have metabolic liabilities.8 It was also anticipated that a substituent R” would be utilized as indicated in 5, so the methylated analogs 7l – 7r were modeled.
Figure 1.

Modeled compounds where Ar is the uracilylethoxyphenyl substructure of 5 or 6 with R’ = Cl. The number in parentheses is the computed relative free energy of binding (kcal/mol) with HIV-RT from the FEP calculations; the statistical uncertainty in the results is ±0.5 kcal/mol.
The structural situation is exemplified for the complex of 7l in Figure 2. The expectation from the modeling for 7a – 7r and the crystallography for 3 and 4,10 assuming 7l would bind at all, is that the indole fragment would make π contacts face-to-face with Tyr188 and edge-to-face with Tyr181 and Trp229. In addition, the methyl group or another substituent at C2 would project below the Phe227-Leu228-Trp229 loop into the tunnel heading towards the polymerase active site. The tunnel contains water molecules; however, the pocket below C3 of the indole is dry. Consequently, heterocycles with an unsaturated nitrogen in the C3 position, e.g., the benzimidazole corresponding to 7a or benzoxazole corresponding to 7f, were not considered. There is also no water in the small pocket between Pro95 and Tyr188. Thus, introductions of nitrogen atoms into the three possible sites for the six-membered ring were not evaluated.
Figure 2.

Illustration of a configuration from an MC simulation of the indole 7l bound to wild type HIV-1 reverse transcriptase. Carbon atoms of 7l are rendered in yellow. All water molecules and many residues in front of the ligand have been removed for clarity.
The results of the MC/FEP calculations are summarized in Figure 1. Many of the perturbations were made from 7a, which is taken as the reference compound for the listed relative free energies of binding, ΔΔGb. For methylated compounds, the perturbations were made to the desmethyl analog, e.g., 7m to 7b. One can attempt to rationalize the results after the fact, but even correct qualitative predictions in the absence of the computations are elusive owing to the numerous interactions involved. For example, it might seem to be unsurprising that 7b is computed to be less well bound than 7a since there is no water in the pocket for a hydrogen bond with the indole NH of 7b. However, the indole NH of 7a is also not able to participate in a hydrogen bond and the favorability of the electrostatic interactions with Trp229 and Tyr188 are unclear. The methylated 7c does bind better than 7b since the methyl group is placed in the pocket, though whether or not there is enough room is unclear without the result. The strong preference for the indolizine 7d over its isomer 7e is striking and must reflect electrostatic differences, which have arisen before for indolizine derivatives.21 Similarly, 7g is preferred over 7f. Furans are very poor hydrogen-bond acceptors, so the pattern is that it is beneficial to have a more acidic (positive) hydrogen near the tryptophan ring (7a, 7d, 7g) than a more electron rich atom. Continuing, introduction of a nitrogen atom at C2 of the 5-membered ring turned out to be detrimental in each case, 7h – 7k, by ca. 3 kcal/mol. In the MC simulations it is found that water in the tunnel does not penetrate past the Phe227-Trp229 loop, so a hydrogen bond with water is not made to the 2-position nitrogen atom. Thus, it appears that a cyano group at this position is needed to reach a water molecule in the tunnel.
Finally, the results for introduction of a methyl group at C2 to yield 7l – 7r were interesting. In some cases, there is significant benefit, e.g., 7a going to 7l, while in others there is little change (7e to 7p, or 7g to 7r). In analyzing this, it appears that a key factor is the angle of projection of the methyl group into the tunnel. For 7l in the snapshot in Figure 2 the NH-C2-CH3 bond angle is 116° and the methyl group is directed upward above Cα of Phe227. The corresponding angle (C3-C2-CH3) in snapshots for 7m is near 130°, which brings the methyl group too close to the Cα atom. When 2-methylindole is optimized with OPLS/CM1A the corresponding angles are 120.1° and 130.2°. With ab initio calculations at the HF/6–311G(d) level the results are 120.6° and 130.2°, and with MP2/6-311G(d) optimizations they are 121.0° and 130.5°.22 For 2-methylbenzofuran, the O-C2-CH3 and C3-C2-CH3 angles are computed to be 118.3° and 129.7° with OPLS/CM1A. Thus, 7f benefits from addition of the methyl group in going to 7q, while the addition of the methyl group to 7g to give 7r is predicted to reduce the binding affinity. This reverses the preference for 7g over 7f.
SYNTHETIC CHEMISTRY
Based on the MC/FEP results in Figure 1, the synthetic efforts were directed at substituted indoles, indolizines, and benzofurans corresponding to 8, 10, and 12. A few of the isomers 9, 11, and 13 were also prepared. The full list of analogs that were synthesized is given in Table 1. While complete synthetic details and compound characterization are provided in the Supplementary Information, the synthetic routes for the key compounds with a cyano group in the 2-position of the bicyclic heterocycle are summarized here. The identity of all compounds was confirmed by 1H and 13C NMR and high-resolution mass spectrometry; purity was >95% as judged by high-performance liquid chromatography.
Table 1.
Inhibitory Activity (EC50) for HIV-1 and Cytotoxicity (CC50) in µM
| Compd | X | Y | Z | WT EC50a |
Y181C EC50a |
K103N/Y181C EC50a |
CC50b |
|---|---|---|---|---|---|---|---|
| 3 | 0.000055 | 0.049 | 0.220 | 10 | |||
| 4 | 0.00032 | 0.016 | 0.085 | 45 | |||
| 8a | H | H | H | 0.085 | ND | ND | 53 |
| 8b | Cl | H | H | 1.6 | NA | NA | 18 |
| 8c | H | CH3 | H | 0.39 | NA | NA | 15 |
| 8d | H | CN | H | 0.056 | 38 | 5.5 | 72 |
| 8e | H | CN | CH3 | 0.010 | NA | 0.80 | 1.2 |
| 8f | H | CN | Cl | 0.34 | 27 | 2.7 | 81 |
| 9a | H | H | CH3 | 6.0 | ND | ND | >100 |
| 9b | H | CH3 | H | NA | NA | NA | 17 |
| 10a | H | CH3 | H | 0.050 | ND | ND | 9.0 |
| 10b | H | CN | H | 0.00038 | 0.31 | 0.011 | >100 |
| 10c | H | CN | CH3 | 0.00090 | 0.80 | 0.032 | 3.0 |
| 10d | H | CN | F | 0.00070 | 0.17 | 0.068 | 40 |
| 10e | H | CN | Cl | 0.0037 | 0.22 | 0.022 | 39 |
| 10f | F | CN | H | 0.0027 | 0.60 | 0.22 | 100 |
| 10g | F | CN | CH3 | 0.0020 | 0.40 | 0.25 | 17 |
| 10h | F | CN | F | 0.00040 | 0.25 | 0.010 | 50 |
| 10i | F | CN | Cl | 0.0025 | 0.16 | 0.11 | 15 |
| 10j | 4-F | CN | H | 0.0020 | 9.0 | 0.22 | 35 |
| 10k | 4,5-diF | CN | H | 0.0032 | 1.2 | 0.50 | 19 |
| 10l | 4,5-diF | CN | F | 0.0068 | 1.2 | 0.23 | 54 |
| 10m | Cl | CN | H | 0.0051 | 1.8 | 0.90 | 55 |
| 10n | Cl | CN | Cl | 0.022 | 0.41 | 0.53 | 20 |
| 11a | F | CN | H | 0.017 | 6.9 | 1.3 | >100 |
| 12a | H | CN | H | 0.018 | NA | 0.42 | >100 |
| 12b | H | CN | CH3 | 0.21 | NA | 1.3 | >100 |
| 12c | H | CN | Cl | 0.019 | 1.9 | 0.26 | >100 |
| 12d | F | CN | H | 0.040 | 3.5 | 4.0 | >100 |
| 12e | F | CN | CH3 | 0.26 | NA | 5 | >100 |
| 12f | Cl | H | H | 0.800 | ND | ND | 19 |
| 12g | Cl | CH3 | H | 0.025 | NA | NA | 8.0 |
| 12h | Cl | CN | H | 0.0070 | NA | NA | 7.0 |
| 13a | Cl | H | H | 0.900 | ND | ND | 50 |
| nevirapine | 0.11 | NA | NA | >100 | |||
| efavirenz | 0.002 | 0.010 | 0.030 | 15 | |||
| etravirine | 0.001 | 0.008 | 0.005 | 11 | |||
| rilpivirine | 0.00067 | 0.00065 | 0.002 | 8 |
For 50% protection in MT-2 cells; antiviral curves used triplicate samples at each concentration. ND for not measured. NA for EC50 > CC50
For 50% inhibition of MT-2 cell growth; toxicity curves also used triplicate samples.

For the 2-cyanoindoles 8 (Y = CN), the pathway is illustrated in Scheme 1. The syntheses began with an SNAr reaction between a 2-fluorobenzaldehyde and a 2-benzyloxyphenol to yield the phenoxybenzaldehyde scaffold.23 Cyclization to the indole was achieved using methyl azidoacetate,24 which was followed by reduction of the ester to the alcohol, and then oxidation to the aldehyde.25 After conversion of the aldehyde at C2 to the desired nitrile26 and Boc-protection of the indole nitrogen, the benzyl group was cleaved. To install the uracil containing side chain, the phenol was unmasked and alkylated with 3-benzoyl,1-bromoethyluracil. Finally, the Boc group was removed with tetra-n-butylammonium fluoride, and the uracil moiety was deprotected under basic conditions to afford the desired products. It may be noted that alternatives to the Boc-protection were considered via addition of methyl chloroformate or triisopropylsilyl chloride; however, in these cases, the deprotection step proved difficult.
Scheme 1. Synthesis of the 2-Cyanoindoles.

Reagents: (a) K2CO3, DMF, 100–120 °C, 12 h; (b) methyl azidoacetate, −40 °C, NaOMe, MeOH, 12 h; (c) xylene, reflux, 4 h; (d) LAH,THF, 0 °C, 0.5 h; (e) MnO2, DCM, rt, 24 h; (f) NH2 OSO3 H, MeOH, reflux, 0.5 h; (g) Cu(OAc)2, CH3 CN, reflux, 1 h; (h) 4-DMAP, Boc2O, THF, rt, 12 h; (i) H2 /Pd/C, MeOH, rt, 0.5 h; (j) 3-benzoyl,1-bromoethyluracil, K2CO3, DMF, 60 °C, 2 h; (k) TBAF, THF, reflux, 24 h; (l) NH3, MeOH, rt, 12 h.
For the indolizines 10 (Y = CN), comparatively easy installation of the desired cyano group at the 2-position was possible using acrylonitrile in the cyclization sequence,27 as summarized in Scheme 2. The resultant 8-bromo-2-cyanoindolizines underwent an SNAr reaction with a substituted 2-methoxyphenol to yield the diarylethers. Installation of the uracilylethoxy side chain then proceeded as in Scheme 1, following demethylation of the methoxy group with BBr3. The starting picolinaldehydes were obtained either by DIBALH reduction of the corresponding 2-methylester or SeO2 oxidation of the 2-methyl analog when Z was chlorine.
Scheme 2. Synthesis of the 2-Cyanoindolizines.

Reagents: (a) DABCO, neat, 2 h; (b) Ac2O, 100 °C, 2 h →140 °C, 16 h; (c) CuI, Cs2CO3, 2,2,6,6-tetramethyl-3,5-heptanedione, dioxane, 120 °C, 18 h; (d) BBr3, DCM, −78 °C → 0 °C, 3 h; (e) 3-benzoyl,1-bromoethyluracil, Cs2CO3 or K2CO3, DMF, 60 °C, 3 h; (f) NH4OH, DCM, 16 h.
For the 2-cyanobenzofurans 12 (Y = CN), the synthetic route in Scheme 3 begins with conversion of 2-hydroxybenzaldehyde derivatives to dibromovinyphenols using tetrabromomethane and triphenylphosphine (Scheme 3). A ligand-free, copper-catalyzed cross-coupling ensued to yield substituted 2-bromobenzofurans,28 which was followed by selective displacement of the 2-bromine with sodium cyanide to yield 2-cyanobenzofurans. The rest of the sequence then proceeded as for the 2-cyanoindolizines. Access to the starting aldehydes was more involved in this case requiring a five step procedure beginning with commercially available 2-bromo-6-methoxyanilines, as detailed in the Supplementary Information.
Scheme 3. Synthesis of the 2-Cyanobenzofurans.

Reagents: (a) PPh3, CBr4, DCM, 0°C → RT, 2 h; (b) CuI, K2CO3, 80 °C, 4 h; (c) NaCN, DMSO, 100 °C, 24 h; (d) CuI, Cs2CO3N,N-dimethylglycine hydrochloride, dioxane, 90 °C, 18 h; (e) BBr3, DCM, −78 °C → 0 °C, 3 h; (f) 3-benzoyl,1-bromoethyluracil, Cs2CO3 or K2CO3, DMF, 60 °C, 3 h; (g) NH4OH, DCM, 16 h.
ASSAY RESULTS
As in previous studies,6,9,14 activities against the IIIB strain of HIV-1 were measured using human MT-2 T-cells; EC50 values are obtained as the dose required to achieve 50% protection of the infected cells by the MTT colorimetric method.29,30 Activities against variant strains of the virus, which contain the clinically important Y181C and K103N/Y181C mutations in the RT enzyme, were also determined. CC50 values for inhibition of MT-2 cell growth by 50% are obtained simultaneously. The antiviral and toxicity curves used triplicate samples at each concentration. The results including those for several reference NNRTIs are listed in Table1.
The results for activity towards the wild-type virus are discussed first. The parent indole 8a turned out to be an 85-nM inhibitor, which is more potent than the drug nevirapine. Addition of a chlorine at C5 in the phenyl ring (8b) and of a 2-methyl group in the bicycle (8c) were not beneficial; however, the 2-cyano analog 8d did provide an improvement to 56 nM. Elaboration with a methyl group at the 6-position to yield 8e yielded a 10-nM NNRTI, while a chlorine at this position (8f) reduced the potency to 340 nM. Increased steric sensitivity is indicated in the pocket near Pro95 (Figure 2) compared to the case with the cyanovinylphenyl-containing catechol diethers,9 which would be consistent with the greater rigidity of the bicyclic alternatives. Only two of the isomeric indoles, 9a and 9b, were prepared. 9b, the isomer of 8c, was inactive, which is qualitatively consistent with the expectations from the MC/FEP results for 7l and 7m in Figure 1. For 9a, comparison can be made with 8a; the added methyl group reduces the activity ca. 70-fold, which is consistent with the predictions for 7a and 7m in Figure 1. In general, quantitative agreement is not expected between the MC/FEP results for relative free energies of binding and the EC50 results from the cell-based assays. Correct qualitative guidance is all that is needed for great facilitation of lead optimization.
The first two indolizines that were synthesized were 10a and 10b. The 2-cyano analog 10b was extremely potent at 0.38 nM (380 pM) and more than 100-fold more so than the methyl analog 10a. 10b also showed no cytotoxicity (CC50) up to the maximum tested concentration, 100 µM. This demonstrates that the design strategy of replacing the cyanovinylphenyl substituent could be very successful with proper choice of a cyano-substituted bicyclic heterocycle. A substituent in the 6-position of the indolizine was then explored with 10c – 10e. The methyl and fluoro derivatives, 10c and 10d, are high picomolar inhibitors, while the Z = Cl analog 10e was again less potent at 3.7 nM. Among the remaining indolizines, the difluoro compound 10h was the most potent with an EC50 of 0.4 nM; it also shows low cytotoxicity with a CC50 of 50 µM. Placement of a fluorine at C4 in the phenyl ring yielded 10j, which was found to have similar potency as the 5-fluoro analog, 10f. However, there was no added benefit of having fluorines at both C4 and C5 (10k, 10l). Only one isomeric indolizine was prepared, 11a. Consistent with the FEP results, it is less active than its isomer 10f, though the 6-fold difference is smaller than what might be expected from the results in Figure 1. This may stem from the computations being for the methyl (7o, 7p) rather than cyano compounds and/or limitations with the point-charge model in the force field and its accounting of the aryl-aryl interactions with Tyr181, Tyr188, and Trp229 (Figure 2).
The focus on the benzofurans was also for the 2-cyano substituted cases. The parent 12a emerged as an 18-nM inhibitor. Addition of the 6-methyl group to give 12b was now significantly detrimental (210 nM), while a chlorine at this position (12c) had no impact. For the fluoro analogs, 12d and 12e, addition of the 6-methyl group is again detrimental, and for the chloro compounds 12f – 12g the 100-fold benefit of progressing from hydrogen to methyl to cyano at the 2-position is apparent. Only one isomeric benzofuran 13a was prepared. Based onthe FEP results in Figure 1, a strong preference for the 12 series over 13 was not expected. However, the qualitative order is correct for the one case of 13a versus its isomer 12f. Overall, the FEP results were highly successful in narrowing the field of possible synthetic targets and in leading the discovery of the strikingly potent indolizines, especially 10b and 10h.
The performance of the compounds towards the mutant strains of HIV-1 is also recorded in Table 1. The variations in the results are difficult to rationalize with confidence in the absence of more experimental structural characterization of the complexes. However, the general pattern is that compounds that are more potent towards the wild type virus are also more potent towards the variants. Furthermore, the usual pattern with NNRTIs is that they are more potent towards the single Y181C variant than the double K103N/Y181C variant. Both variants are clinically important and the double mutant is viewed as being the most challenging variant. The K103N mutation is found in 57% of cases of NNRTI resistance and Y181C is found in 25% of cases.31 Thus, it was surprising to find that the new compounds are almost uniformly more active towards the K103N/Y181C than the Y181C variant. The EC50 values of 11 and 10 nM for 10b and 10h with the double variant are impressive and represent significant improvement over the results for 3 and 4. However, 3 and 4 perform ca. 10-fold better for the single mutant. Among the new compounds the greatest potency towards the Y181C variant is for 10d and 10i at 170 and 160 nM. It is easy to say that the present compounds are expected to lose potency towards Y181C variants owing to loss of aryl-aryl contacts indicated in Figure 2 and in the crystal structures for the complexes of 3 and 4.10 However, the reason for the greater loss for 10b and 10h is unclear, and the reason for the gain in activity for adding the K103N mutation is especially obscure.
CRYSTALLOGRAPHY
In order to begin to obtain a deeper understanding of the variations in activity for the indolizines, an X-ray crystal structure for 10b with WT HIV-1 RT was determined in the same manner as for 3 and 4.10 The best crystals obtained using the recombinant RT52A enzyme diffracted to amplitudes extending to 2.88 Å, and phases were determined via molecular replacement using the structure for 3 as the search model (PDB code: 4H4M).10 The overall conformation of the binding pocket is similar to the 4H4M structure in which the β6-β10-β9 sheet (p66: residues 105–110, 186–191, 178–183, respectively) and YMDD loop (p66: residues 183–186) shift in order to interact with the bound inhibitor. The crystal structure for the complex with 10b is illustrated in Figure 3; the positioning of the ligand and the contacts are predominantly as expected from Figure 2 with the cyano group projecting into the tunnel behind Phe227. An all-atom alignment comparing the inhibitor and non-nucleoside binding pocket of the 4H4M structure and the 10b:RT complex gives an rmsd of only 0.75 Å.
Figure 3.

Stereo view of the crystal structure for 10b complexed with HIV-RT. Numerous contacts in the binding site are apparent and consistent with those in the crystal structures for 3 and 4 and in the modeled structure in Figure 2. The PDB code is 4MFB.
In the computer simulations and the crystal structure for 3 and 10b there is some variation in the conformation of the uracilylethoxy group where the dihedral angle for the connection to the central phenyl ring (C2-C1-O-C) can either be near 180° (anti as in Figure 3) or nearer syn as in the case for 3.10 The change is accompanied by some differences in hydrogen bonding between the uracil ring and Lys102, Lys103, and Pro236. In the present case, the C2 carbonyl oxygen of the uracil is 3.1 Å from the backbone N of Lys103, and N3 of the uracil is 3.4 Å from the backbone carbonyl oxygen of Pro236. However, the latter pair is not oriented well for hydrogen bonding, as illustrated in Figure 4. This region is solvent exposed and significant exchange of protein-ligand, protein-water, and ligand-water hydrogen bonds is observed in the MC simulations.
Figure 4.

A space-filling rendering made from the crystal structure of 10b bound to HV-RT. Carbon atoms of 10b are in yellow. Notable points are the hydrogen bond between a uracil oxygen atom and the backbone NH of Lys103, the aryl-aryl contacts with Tyr181 and Trp229, and the orientation of Lys103 including the contact of the Cβ atom with the inhibitor.
There are additional points of note. The orientation of Tyr181 is the same in Figure 2 and Figure 3 with the tyrosine ring and the central ring of the inhibitor perpendicular, while in the crystal structures for 3 and 4 they are parallel owing to 90° rotation of the tyrosine ring. In both cases the interaction is offset so it is not an optimal aryl-aryl interaction. However, the edge-to-face interaction between Tyr181 and the indolizine 6-membered ring of 10b does appear to be ideal and is such that a substituent could not be placed at C6 of the indolizine (Z in 10) without steric conflict with the tyrosine ring. The tight packing is apparent in Figure 4. It seems likely that placement of a substituent at C6 would be accompanied by rotation of the tyrosine ring to the parallel geometry observed for the complexes of 3 and 4. Thus, the adverse effects of the Tyr181Cys mutation on potency for the indolizines can be attributed primarily to the loss of the aryl-aryl interaction between the bicyclic heterocycle and Tyr181.
Turning to Lys103, the orientation of its side chain is the same in the three crystal structures and from the MC modeling of the complexes. The side chain extends into the entrance channel of the NNRTI binding site with the ammonium group solvent exposed (Figure 3 and Figure 4). The shortest contact between the side chain and the inhibitors is between Cβ and C5 in the central ring at 3.8 to 4.1 Å separation. A substituent on C5 (X in 8 – 13) would also be in van der Waals contact with Cβ of Lys103. It is unclear based on the present structures why conversion of Lys103 to Asn is beneficial for the heterobicyclic catechol diethers and not so for the cyanovinylphenyl analogs. Crystal structures for additional complexes such as for 3 and 10b with Y181C, K103N, and K103N/Y181C HIV-RT are much desired to provide clarification.
A final structural point concerns the positioning of the cyano groups of the inhibitors in the tunnel. Figure 5 provides two views comparing the crystal structures for 3 and 10b. They show that there are some differences such that the indolizine makes closer contact with Trp229 and that the cyano group of 3 extends further into the tunnel. As a result the cyano nitrogen atom of 3 is only 3.6 Å from the ammonium nitrogen of Lys223 (Figure 5b), while the corresponding distance in the complex for 10b is 5.8 Å. In the crystal structure for 4, the side chain of Lys223 has a conformational kink that increases the separation to 6.5 Å (PDB code: 4H4O). There is water in this region that promotes the variety of possible arrangements. In any event, ion-dipole interactions with Lys223 or direct hydrogen bonding to water or Lys223 are expected to be contributing to the observed strong benefits of the cyano groups to the antiviral activity.
Figure 5.

Two views comparing the crystal structures for 3 and 10b. In A, the difference in conformation of the uracilylethoxy side chain is apparent, while B illustrates that the cyano group of 3 extends farther into the tunnel region and closer to Lys223.
SOLUBILITY
Poor solubility, which has been a problem for many NNRTIs, often leads too low bioavailability, difficulties in formulation, and high dosages.6,32,33 The issue reflects the hydrophobic nature of the NNRTI binding site. In the design of the catechol diethers, attention was paid to having both good potency and aqueous solubility. In our laboratory, solubilities are measured using a shake-flask procedure.6,34 The compounds are dissolved in Britton-Robinson buffer and stirred in vials for 48 hours at 25 °C. The pH of the buffer solutions is 6.5 as measured by a Corning General Purpose pH Combination probe (4136L21). The solution containing excess solid is filtered using a Whatman Mini-UniPrep syringeless filter device with a 0.45 µm pore size, and the supernatant is analyzed by UV-vis spectrophotometry (Agilent 8453).
As shown in Table 2, the solubilities for the drugs etravirine and rilpivirine are below 1 µg/ml, which is well outside the normal range of 4 – 4000 µg/ml for oral drugs.6,33 The solubilities of the most potent indolizine analogs, 10b and 10h, are both ca. 40 µg/ml. 4, the cyanovinylphenyl analog of 10h, is 4-fold less soluble at 11 µg/ml. As also shown in Table 2, there is reasonable inverse correlation of the observed solubilities with computed octanol-water partition coefficients, which are readily obtained using ChemDraw.38 Thus, it was expected that the new compounds would have solubilities somewhat less than for nevirapine, but much greater than for etravirine or rilpivirine.
Table 2.
Experimental Aqueous Solubility at pH 6.5 (S) and Computed ClogP
CONCLUSION
Computer simulations guided the discovery of potent, novel inhibitors of HIV-1 reverse transcriptase. MC/FEP calculations were used to analyze possible replacements for a cyanovinylphenyl group that has been featured in prior NNRTIs, but that possesses reactivity concerns. The results indicated that indoles, indolizines, and benzofurans as represented by 8, 10, and 12 would be the most viable. The predictions were validated by synthesis and assaying that yielded inhibitors of the wild-type virus with EC50 values of 10 nM or lower in all three series. The most active compounds are the indolizines 10b and 10h which are strikingly potent with EC50 values of 0.4 nM towards the wild-type virus and 10 nM towards a difficult variant strain bearing K103N and Y181C mutations in the RT enzyme. The structural characterization was much enhanced by obtaining an X-ray crystal structure for 10b in complex with HIV-RT at 2.88-Å resolution. The structure confirmed the expected placement of the inhibitor in the binding site, the extensive aryl-aryl interactions with Tyr181, Tyr188, and Trp229, and the extension of the cyano group of 10b towards Lys223 in the tunnel region. The contact between Tyr181 and the C6-C7 region of the indolizine appears to be optimal and can explain the significant loss in activity of 10b and 10h towards the Y181C variant. However, the structural origin of the ca. 30fold gain in activity upon addition of the K103N mutation is unclear and makes further crystallographic investigation desirable. The aqueous solubilities of 10b and 10h were also measured and, at ca. 40 µg/ml, they are in the normal range for oral drugs. In comparison to the FDA-approved drug efavirenz, 10b and 10h show improved activity towards the wild-type virus and the K103N/Y181C variant, diminished activity towards the Y181C variant, lower cytotoxicity, and similar solubility. In comparison to etravirine and rilpivirine, the new NNRTIs show similar activity towards the wild-type virus and the K103N/Y181C variant, poorer activity towards the Y181C variant, reduced cytotoxicity, and at least 100-fold greater solubility. Overall, the valuable role that FEP calculations can play in lead optimization has been further illustrated,7 equivalents for a cyanovinylphenyl group have been reported, and novel, potent anti-HIV agents with low cytotoxicity and good solubility have been discovered and structurally characterized.
Supplementary Material
ACKNOWLEDGMENTS
Gratitude is expressed to the National Institutes of Health (AI44616, GM32136, GM49551) for research support and for a fellowship to KMF (AI104334). Dr. Xiaoling Wu is thanked for assistance with the NMR spectroscopy. Receipt of the following reagents through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH is also greatly appreciated: MT-2 cells, catalog #237, and nevirapine-resistant HIV-1 (N119), catalog #1392, from Dr. Douglas Richman; HTLV-IIIB/H9, #398, from Dr. Robert Gallo; and HIV-1IIIB (A17 Variant) from Dr. Emilio Emini.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
Synthetic details, NMR and HRMS spectral data for compounds in Tables 1, and crystallographic details. The crystal structure data for the complex of 10b and HIV-RT have been deposited in the RCSB Protein Data Bank with the code 4MFB. These materials are available free of charge via the Internet at http://pubs.acs.org .
REFERENCES
- 1.Asahchop EL, Wainberg MA, Sloan RD, Tremblay CL. Antimicrob. Agents Chemother. 2012;56:5000–5008. doi: 10.1128/AAC.00591-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhan P, Chen X, Li D, Fang Z, De Clercq E, Liu X. Med. Res. Rev. 2013;33(S1):E1–E72. doi: 10.1002/med.20241. [DOI] [PubMed] [Google Scholar]
- 3.Permpalung N, Putcharoen O, Avihingsanon A, Ruxrungtham K. Expert Opin. Pharmacother. 2012;13:2301–2317. doi: 10.1517/14656566.2012.729040. [DOI] [PubMed] [Google Scholar]
- 4.James C, Preininger L, Sweet M. Am. J. Health-Syst. Pharm. 2012;69:857–861. doi: 10.2146/ajhp110395. [DOI] [PubMed] [Google Scholar]
- 5.Janssen PAJ, Lewi PJ, Arnold E, Daeyaert F, de Jonge M, Heeres J, Koymans L, Vinkers M, Guillemont J, Pasquier E, Kukla M, Ludovici D, Andries K, de Bethune M-P, Pauwels R, Das K, Clark AD, Jr., Frenkel YV, Hughes SH, Medaer B, De Knaep F, Bohets H, De Clerck F, Lampo A, Williams P, Stoffels P. J. Med. Chem. 2005;48:1901–1919. [Google Scholar]
- 6.Bollini M, Cisneros JA, Spasov KA, Anderson KS, Jorgensen WL. Bioorg. Med. Chem. Lett. 2013;23:5213–5216. doi: 10.1016/j.bmcl.2013.06.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jorgensen WL. Acc. Chem. Res. 2009;42:724–733. doi: 10.1021/ar800236t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stepan AF, Walker DP, Bauman J, Price DA, Baillie TA, Kalgutkar AS, Aleo MD. Chem. Res. Toxicol. 2011;24:1345–1410. doi: 10.1021/tx200168d. [DOI] [PubMed] [Google Scholar]
- 9.Bollini M, Domaoal RA, Thakur VV, Gallardo-Macias R, Spasov KA, Anderson KA, Jorgensen WL. J. Med. Chem. 2011;54:8582–8591. doi: 10.1021/jm201134m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frey KM, Bollini M, Mislak AC, Cisneros JA, Gallardo-Macias R, Jorgensen WL, Anderson KA. J. Am. Chem. Soc. 2012;134:19501–19503. doi: 10.1021/ja3092642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fleming FF, Wang Q. Chem. Rev. 2003;103:2035–2077. doi: 10.1021/cr020045d. [DOI] [PubMed] [Google Scholar]
- 12.Dahlgren MK, Schyman P, Tirado-Rives J, Jorgensen WL. J. Chem. Inf. Model. 2013;53:1191–1199. doi: 10.1021/ci4001597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fleming FF, Yao L, Ravikumar PC, Funk L, Shook BC. J. Med. Chem. 2010;53:7902–7917. doi: 10.1021/jm100762r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jorgensen WL, Bollini M, Thakur VV, Domaoal RA, Spasov K, Anderson KS. J. Am. Chem. Soc. 2011;133:15686–15696. doi: 10.1021/ja2058583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jorgensen WL, Tirado-Rives J. J. Comput. Chem. 2005;26:1689–1700. doi: 10.1002/jcc.20297. [DOI] [PubMed] [Google Scholar]
- 16.Jorgensen WL, Maxwell DS, Tirado-Rives J. J. Am. Chem .Soc. 1996;118:11225–11236. [Google Scholar]
- 17.Jorgensen WL, Tirado-Rives J. Proc. Natl. Acad. Sci. U.S.A. 2005;102:6665–6670. doi: 10.1073/pnas.0408037102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 19.for a review, see. Jorgensen WL, Thomas LT. J. Chem. Theory Comput. 2008;4:869–876. doi: 10.1021/ct800011m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heugebaert TSA, Roman BI, Sevens CV. Chem Soc. Rev. 2012;41:5626–5640. doi: 10.1039/c2cs35093a. [DOI] [PubMed] [Google Scholar]
- 21.Kim JT, Hamilton AD, Bailey CM, Domaoal RA, Wang L, Anderson KS, Jorgensen WL. J. Am. Chem. Soc. 2006;128:15372–15373. doi: 10.1021/ja066472g. [DOI] [PubMed] [Google Scholar]
- 22.Frisch MJ. Gaussian 09, Revision A.02. Wallingford, CT: Gaussian, Inc; 2009. [Full reference is given in the Supporting Information.] [Google Scholar]
- 23.Whitlock GA, Blagg J, Fish PV. Bioorg. Med. Chem. Lett. 2008;18:596–599. doi: 10.1016/j.bmcl.2007.11.080. [DOI] [PubMed] [Google Scholar]
- 24.Bamberg JT, O’Yang C, Sui M, Zhao S-H. 2010 US7741326 B2. [Google Scholar]
- 25.Zheng C, Lu Y, Zhang J, Chen X, Chai Z, Ma W, Zhao G. Chem. Eur. J. 2010;16:5853–5857. doi: 10.1002/chem.201000130. [DOI] [PubMed] [Google Scholar]
- 26.(a) Ryabova SY, Alekseeva LM, Lisitsa EA, Granik VG. Russ. Chem. Bull., Int. Ed. 2006;55:1248–1254. [Google Scholar]; (b) Attanasi O, Palma P, Serra-Zanetti F. Synthesis. 1983;9:741–742. [Google Scholar]
- 27.Bode ML, Kaye PT. J. Chem. Soc. Perkin Trans. 1993;1:1809–1813. [Google Scholar]
- 28.(a) Newman SG, Aureggi V, Bryan CS, Lautens M. Chem. Commun. 2009:5236–5238. doi: 10.1039/b912093a. [DOI] [PubMed] [Google Scholar]; (b) Zhou W, Chen W, Wang L. Org. Biomol. Chem. 2012;10:4172–4178. doi: 10.1039/c2ob25356a. [DOI] [PubMed] [Google Scholar]
- 29.Lin TS, Luo MZ, Liu MC, Pai SB, Dutschman GE, Cheng YC. Biochem. Pharmacol. 1994;47:171–174. doi: 10.1016/0006-2952(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 30.Ray AS, Yang Z, Chu CK, Anderson KS. Antimicrob. Agents Chemother. 2002;46:887–891. doi: 10.1128/AAC.46.3.887-891.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Béthune M-P. Antiviral Res. 2010;85:75–90. doi: 10.1016/j.antiviral.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 32.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 33.Jorgensen WL, Duffy EM. Adv. Drug Deliv. Rev. 2002;54:355–366. doi: 10.1016/s0169-409x(02)00008-x. [DOI] [PubMed] [Google Scholar]
- 34.Baka E, Comer JEA, Takács-Novák K. J. Pharm. Biomed. Anal. 2008;46:335–341. doi: 10.1016/j.jpba.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 35.Morelock MM, Choi LL, Bell GL, Wright JL. J. Pharm. Sci. 1994;83:948–952. doi: 10.1002/jps.2600830706. [DOI] [PubMed] [Google Scholar]
- 36.Weuts I, Van Dycke F, Voorspoels J, de Cort S, Stokbroekx S, Leemans R, Brewster ME, Xu D, Segmuller B, Turner YTA, Roberts CJ, Davies MC, Qi S, Craig DQM, Reading M. J. Pharm. Sci. 2011;100:260–274. doi: 10.1002/jps.22242. [DOI] [PubMed] [Google Scholar]
- 37.Sun L-Q, Qin B, Huang L, Qian K, Chen C-H, Lee K-H, Xie L. Bioorg. Med. Chem. Lett. 2012;22:2376–2379. doi: 10.1016/j.bmcl.2012.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.ChemDraw. CambridgeSoft Inc.: Cambridge, MA; 2013. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.