

Abstract
The sequencing of multiple genomes of the same plant species has revealed polymorphic gene and exon loss. Genes associated with disease resistance are overrepresented among those showing structural variations, suggesting an adaptive role for gene and exon presence–absence variation (PAV). To shed light on the possible functional relevance of polymorphic coding region loss and the mechanisms driving this process, we characterized genes that have lost entire exons or their whole coding regions in 17 fully sequenced Arabidopsis thaliana accessions. We found that although a significant enrichment in genes associated with certain functional categories is observed, PAV events are largely restricted to genes with signatures of reduced essentiality: PAV genes tend to be newer additions to the genome, tissue specific, and lowly expressed. In addition, PAV genes are located in regions of lower gene density and higher transposable element density. Partial coding region PAV events were associated with only a marginal reduction in gene expression level in the affected accession and occurred in genes with higher levels of alternative splicing in the Col-0 accession. Together, these results suggest that although adaptive scenarios cannot be ruled out, PAV events can be explained without invoking them.
Keywords: exon deletion, presence–absence variation, whole genome evolution, transposable elements, adaptive evolution, Arabidopsis
Introduction
Intraspecies variation in gene content represents an important source of heterogeneity in the genome of a species and potentially contributes to an organism’s adaptability in response to external pressures (Feuk et al. 2006). Cataloguing significant gains and losses in coding regions within or between species will allow a deeper understanding of the mechanisms underlying the molecular evolution of genomes and can assist in identifying functional variation in agronomically elite varieties of staple crops (Wang, You, et al. 2013). To this end, several studies have examined polymorphic full or partial gene loss in several plant species. For instance, after resequencing 50 rice genomes, up to 1,327 possible gene loss events (2.4% of the total gene set) were detected relative to the Nipponbare reference accession (Xu et al. 2012). Significant intraspecies variation in gene content has also been reported in maize (Swanson-Wagner et al. 2010), sorghum (Zheng et al. 2011), and soybean (McHale et al. 2012). Previous studies in the model plant Arabidopsis thaliana, using resequencing microarrays and Illumina sequencing-by-synthesis reads, have also shown significant variations in total nuclear genome sequence among naturally occurring strains (Clark et al. 2007; Ossowski et al. 2008). A more recent study using 18 fully sequenced A. thaliana genomes found that, relative to the reference accession Col-0, 93.4% of proteins had intraspecies variation in their genes, inclusive of large deletions (Gan et al. 2011) with around 775 genes per accession found to have deletions spanning 50% or more of their coding region sequence (Gan et al. 2011). A comparison of 80 Arabidopsis genomes found that 9% of the total genes in A. thaliana showed presence–absence variation (PAV) averaging 444 absent genes per accession (Tan et al. 2012).
Characterization of coding region PAV has shown certain gene categories to be significantly enriched. For instance, 52 of the 154 nucleotide-binding site leucine-rich repeat (NBS-LRR) R (resistance) genes were found to be deleted in at least 1 of 50 rice cultivars (Xu et al. 2012). Similar overrepresentation of the R genes in A. thaliana has also been observed (Bakker et al. 2006; Shen et al. 2006), while in the soybean, genes enriched in structural variation are more likely to be involved in nucleotide binding and biotic defense (McHale et al. 2012). Enrichment of particular functional gene categories among genes affected by structural polymorphism suggests these structural polymorphisms may have a functional role, allowing accessions to be better adapted to the environmental conditions they face.
However, this hypothesis has not been explicitly tested. If significant polymorphic deletions are adaptive, we would expect that affected genes should show multiple signatures of being under selection. On the other hand, if structural polymorphisms mostly affect genes evolving under relaxed constraints, then their adaptive significance should be questioned.
Here we characterize genes affected by PAV spanning whole exons in A. thaliana, to investigate which genomic features, if any, are associated with these polymorphisms. Our results provide insights into the likely functional impact of structural variation in protein-coding genes.
Results
In order to characterize PAV in A. thaliana, we examined previously identified polymorphic deletions in 17 fully sequenced Arabidopsis accessions for which transcriptome data were available (Gan et al. 2011) (see Materials and Methods). We compiled a set of deletions that spanned entire exons in any of 17 accessions relative to the Col-0 reference genome. A subset of the annotated deletions was experimentally validated (Gan et al. 2011). To further rule out the possibility of wrongly identifying deletions due to differences between assemblies, exons were confirmed as missing by searching for homology between the Col-0 exon on all other accessions (see Materials and Methods).
A total of 794 exons were classified as missing in at least one of 17 accessions, corresponding to 411 genes (∼1.5% of the total gene set) including 81 genes where the full coding region was completely absent in at least one accession (supplementary table S1, Supplementary Material online). Exon losses are not uniformly distributed throughout the gene: missing exon sequences are more often found near the ends of each gene (supplementary fig. S1, Supplementary Material online).
Overall, ∼0.3% of the genes in each accession have at least one missing exon, representing between 10 and 50 kb of missing sequence per accession (supplementary table S2, Supplementary Material online). A total of 200 genes had exon loss affecting more than one accession, consistent with a previous study reporting a “common history” to deletion events in A. thaliana (Santuari et al. 2010).
Because partial deletions spanning whole exons might have distinct functional implications compared with full coding region deletions, the 330 genes with partial coding region loss spanning at least one full exon in at least one accession (exon PAV [E-PAV]) and the 81 genes with full coding region polymorphic deletions affecting at least one accession (full coding DNA sequence PAV [CDS-PAV]) were examined separately.
Genes Involved in Signal Transduction and Both Nucleotide and Protein Binding Are Overrepresented among PAV Genes
In order to characterize PAV genes, we first assessed whether these genes were overrepresented in particular gene classes or gene ontology (GO) categories. To do so, we used four classification schemes: “GO,” a condensed set of GO terms (GOslim), the Pfam protein domain database and the family classification scheme of (Gan et al. 2011) (see Materials and Methods). Of the 330 E-PAV genes, we found most to be poorly characterized with 50% of them having no associated GOslim term. The proportion of poorly characterized genes is greater among CDS-PAV genes, with more than 60% having no associated GOslim term for biological process. When examining genes with associated GOslim terms we found both E-PAV and CDS-PAV genes to be significantly enriched in genes associated with signal transduction and nucleotide binding (fig. 1 and supplementary fig. S2, Supplementary Material online). Furthermore, E-PAV genes also appear significantly enriched in genes associated with the GOslim term “other binding,” which includes proteins that bind to lipids, metal ions, and ATP, among other cofactors (fig. 1). Significant overrepresentation of functional categories among PAV genes is consistent with a previous assessment of large coding region indels in the soybean genome (McHale et al. 2012) and of whole gene deletions in A. thaliana (Tan et al. 2012). This is also observed when classifying genes using a broader set of GO rather than “GOslim” terms (supplementary fig. S3, Supplementary Material online).
Fig. 1.

Distribution of E-PAV genes (n = 330)—those with at least one, but not all, exons missing in at least one accession—by GOslim categories for molecular function (a), biological process (b) and cellular component (c), and by family (d). Both expected and observed number of E-PAV genes per category represented on each bar. Where there is a significant enrichment (P ≤ 0.05) between the amount of observed and expected E-PAV genes for a particular category, an asterisk is shown over the bars. Only categories with at least one E-PAV gene are shown.
When classifying genes by family, we observe an overrepresentation of members of the NBS-LRR family—involved in pathogen detection (DeYoung and Innes 2006)—among E-PAV genes (families “NBS-LRR active TNL,” adjusted P value = 8.57 × 10−35, and “NBS-LRR active CNL,” adjusted P value = 4.63 × 10−5; fig. 1), consistent with previous findings (Shen et al. 2006). Furthermore, when examining the 3,753 Pfam ID gene associations (supplementary figs. S4 and S5, Supplementary Material online), we observe an overrepresentation of members of the NB-ARC (APAF-1, R proteins, and CED-4) and LRR domain containing families (note that “NBS-LRR” refers to a composite of the NBS and LRR domains and that the NBS domain is also known as “NB-ARC” [McHale et al. 2006]). No enrichment of any particular gene family was observed among CDS-PAV genes (data not shown).
These significant enrichments in gene functional and domain annotations are in line with previous findings in Arabidopsis (Tan et al. 2012) and other plant species (Swanson-Wagner et al. 2010; Zheng et al. 2011; McHale et al. 2012) and have been proposed to reflect the adaptive role of large polymorphic deletions.
Genes Affected by PAV Show Signatures Consistent with Relaxed Selective Constraints
To determine whether PAV genes are generally associated with fast evolving proteins potentially under positive selection, we examined the rates of nonsynonymous to synonymous changes per gene (dN/dS). Using a randomization test, E-PAV genes were found to have a significantly higher dN/dS ratio compared with genes with all exons present, but only eight genes have a dN/dS ratio above 1 (fig. 2 and supplementary tables S1 and S3, Supplementary Material online). CDS-PAV genes had a nonsignificant increase in dN/dS compared with intact genes (those not affected by deletions spanning at least one exon in any accession; fig. 2 and supplementary table S3, Supplementary Material online).
Fig. 2.

Genetic features associated with intact (having no exons under P/A variation), E-PAV (having at least one, but not all, exons missing in at least one accession), and CDS-PAV (having the entire CDS missing in at least one accession) genes. From left to right, top to bottom: dN/dS, age, expression level, Tajima’s D, number of paralogs, and tau. See supplementary table S3, Supplementary Material online, for values of means and statistical analysis.
To further examine the selective pressures associated with PAV genes, we examined nucleotide diversity. We considered nucleotide diversity at both replacement sites and silent sites (defined as noncoding sites and the synonymous sites of protein-coding regions) for each gene, according to Gan et al. (2011). PAV genes were found to be associated with higher nucleotide diversity in both silent and replacement sites (supplementary table S3, Supplementary Material online).
Although higher dN/dS and nucleotide diversity are suggestive of relaxed selective constraints, this pattern is also consistent with a scenario of positive and/or balancing selection. To differentiate between these possible scenarios, Tajima’s D was calculated for each gene (see Materials and Methods). A threshold of ±2 was considered as the point at which D significantly departs from the null expectation of neutral evolution for any given gene. Of the 330 E-PAV genes with E-PAV, 24 have D < −2 and only 2 have D > 2 (AT1G12180, D = 2.17, and AT5G35460, D = 2.05, both of which are functionally uncharacterized). Among CDS-PAV genes, only seven have D < −2 and none have D > 2. Compared with the set of intact genes, there are no significant differences in the proportion of PAV genes either with D < 2 (randomization test P = 1 for both E- and CDS-PAV genes) or D > 2 (randomization test P = 0.93 and P = 1 for E- and CDS-PAV genes, respectively). As demographic characteristics of the Arabidopsis population may result in a shift in the average Tajima’s D among the general pool of genes, it is possible that these hard thresholds may not be informative. Indeed, we find that intact genes in Arabidopsis have the average Tajima’s D estimate shifted toward negative values. Thus, PAV genes could fall short of the hard threshold of +2 and still have a higher D estimate than the general pool of genes, suggestive of balancing selection. However, E-PAV genes do not show significant differences in Tajima’s D estimates compared with intact genes and CDS-PAV genes have; in fact, a significantly lower estimate of D (fig. 2 and supplementary tables S1 and S3, Supplementary Material online). It is possible that PAV genes may have a higher range of D values compared with intact genes, hiding a higher proportion of genes under positive and balancing selection that would not be reflected in overall changes in the mean. To test this, we compared the distributions of Tajima’s D estimates in the three sets of genes (intact, E-PAV, and CDS-PAV). However, we did not observe any evidence for increased dispersion in D among PAV genes (supplementary fig. S6, Supplementary Material online). To further examine this possibility, we examined the proportion of PAV genes below the fifth and above the 95th percentile of the “intact” distribution (D = −2.05 and 1.39, respectively). If a significantly higher proportion of E-PAV or CDS-PAV genes are found compared with the intact set at the positive end of the distribution, we can infer the existence of a detectable subset of PAV genes that may be undergoing balancing selection. However, such a pattern is clearly not observed—only 2.33% of E-PAV—and no CDS-PAV genes exceed the threshold value. At the opposite end of the distribution, we observe no overrepresentation in the proportion of E-PAV genes whose estimates of D are lower than the threshold (3.32%) although we do observe this for CDS-PAV genes (8.82%). This finding would suggest that a significant proportion of CDS-PAV genes might be undergoing stronger purifying or positive selection relative to intact genes. Together, these results suggest that although we cannot rule out the effect of balancing selection acting on a few individual PAV genes a general trend of balancing selection for PAV genes does not readily apply. The excess of negative D values among PAV genes coupled with the higher levels of nucleotide diversity and the significant increases in dN/dS ratios are consistent with a scenario of weaker purifying selection but could also be explained by positive selection.
We examined a number of parameters that have been previously associated by some studies with gene essentiality to further explore the functional importance of PAV genes, including a gene’s age (Chen, Trachana, et al. 2012) and the number of paralogs it has (Hanada et al. 2009; Makino et al. 2009), along with weaker associations such as expression level (Cherry 2010) and tissue specificity (Wolf et al. 2006).
Compared with newer genes, older genes are more likely to be essential (Chen, Trachana, et al. 2012). After using the phylogenetic relationships of plant genomes to create a proxy for gene age, we observed that the 330 genes affected by E-PAV are more likely to be newer additions to the genome (fig. 2 and supplementary table S3, Supplementary Material online). It is also possible that E-PAV genes have a greater number of paralogous genes that might compensate for any loss of function. Consistent with this, we find that those genes with missing exons have higher number of paralogs compared with those genes with all exons present (fig. 2 and supplementary table S3, Supplementary Material online). However, the opposite result was observed when analyzing CDS-PAV genes—these have an average of 4.2 paralogs when compared with genes with no exon losses (fig. 2 and supplementary table S3, Supplementary Material online), suggesting their function is less essential. We then assessed the expression patterns of genes affected by exon presence–absence, because broadly and highly expressed genes are typically associated with higher levels of selection (Yang 2009). Using a randomization test, we found that genes with exon losses in one or more accessions, when compared with intact genes, had lower expression levels and higher tissue specificity (supplementary table S3, Supplementary Material online). In addition, we also observed that exons missing in at least one accession are, on average, shorter than exons present in all accessions (170 bp vs. 284 bp, randomization test P = 9.9e−5; supplementary table S3, Supplementary Material online). However, although exons affected by polymorphic deletions are shorter on average compared with nondeleted exons, E-PAV genes are longer than unaffected genes (2,360 bp compared with 2,142 bp, respectively; randomization test P = 0.008; supplementary table S3, Supplementary Material online). By contrast, CDS-PAV genes—where polymorphic deletions encompass the gene’s entire coding region—were found to be shorter than unaffected genes (640 bp compared with 2,142 bp, randomization test P = 9.9e−5; supplementary table S3, Supplementary Material online).
Overall, these findings show that although certain functional categories are overrepresented among genes with exon loss, more generally significant coding region loss is prevalent among novel, lowly expressed and poorly functionally characterized genes. These genes seem to have evolved more recently in the Arabidopsis genome and are likely to be under reduced selective constraint.
PAV Genes Are Located in Genomic Regions That Are Gene-Poor and Transposable Element-Rich
When characterizing the genomic context of genes affected by PAV, we found that genes with both exon and full coding region loss are separated by longer intergenic distances (fig. 3 and supplementary table S3, Supplementary Material online). Transposable element density around PAV genes was then assessed as gene-poor areas have been associated with a higher transposable element (TE) density (Wright et al. 2003). To do this, we used the reference accession (Col-0) and calculated TE density for each gene in all intergenic sequence in 1- to 100-kb windows centered on each gene’s midpoint by counting the number of bases found within TE annotations (see Materials and Methods). E-PAV genes were found to have an approximately 2-fold increase in the amount of bases annotated as a TE compared to genes that are intact in all accessions (e.g., TE sequence accounts for ∼30% of the nongenic sequence within a 10-kb window surrounding an E-PAV gene; fig. 3 and supplementary table S4, Supplementary Material online). Significant enrichment of specific TE superfamilies was also observed, notably, DNA transposons and LTR retrotransposons (supplementary table S4, Supplementary Material online).
Fig. 3.
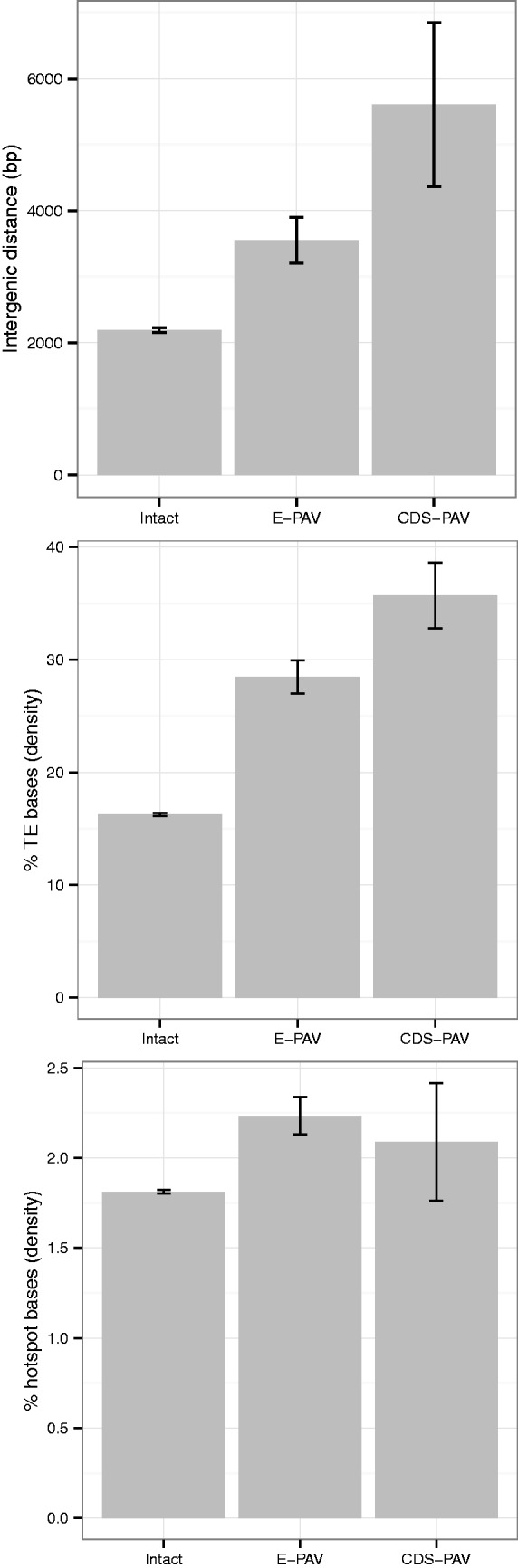
Genomic context for intact (having no exons under P/A variation), E-PAV (having at least one, but not all, exons missing in at least one accession), and CDS-PAV (having the entire CDS missing in at least one accession) genes. Averaged values for the genes in each set are given for, from top to bottom, the intergenic distance, the percentage of TE bases in the nongenic sequence of a 10-kb window centered on that gene’s midpoint, and the percentage of recombinogenic motifs in the genic sequence of a 1-kb window centered on that gene’s midpoint. See also supplementary tables 3 and 4 (Supplementary Material online) for the values of specific TE families and other window sizes.
In addition, we found that genes with missing exons have, on average, a shorter distance from the gene boundary to the nearest TE than those genes with all exons present (2.5 kb compared to 5.7 kb; randomization test, P = 9.9e−5; supplementary table S3, Supplementary Material online. When calculating the minimum distance to the nearest TE, classified by superfamily, E-PAV genes are significantly closer to every TE type: rolling circle TEs, DNA transposons, LTR retrotransposons, long interspersed elements (LINEs), and short interspersed elements (SINEs) (supplementary table S3, Supplementary Material online). Similar findings were obtained when analyzing TE content in the surrounding regions of CDS-PAV genes (supplementary table S3, Supplementary Material online).
Certain TE sequence motifs have been associated with recombination hotspots that could drive exon loss through promoting ectopic recombination events (Oliver and Greene 2009; Horton et al. 2012). To explore whether genes affected by PAV have a local enrichment for such hotspot motifs, we examined the density of these motifs both in and around genes (see Materials and Methods). However, we observed no significant differences in hotspot motif occupancy in the nongenic regions of windows surrounding E-PAV genes compared with intact genes (in window sizes of 1 to 100 kb centered on the gene’s midpoint; supplementary table S4, Supplementary Material online). Nevertheless, a significant enrichment in hotspot motif occupancy was observed in the genic sequence of all windows centered on E-PAV genes compared with those centered on intact genes (fig. 3 and supplementary table S4, Supplementary Material online). When comparing CDS-PAV genes to the intact set, we observed no consistent pattern of higher hotspot motif density within genic regions and only a marginally higher proportion of hotspot motifs in the nongenic regions that surround them, in windows up to 3 kb in size (P < 0.01; supplementary table S5, Supplementary Material online).
Taken together, these results show that PAV genes are located in gene-poor and TE-rich regions of the genome, further supporting the hypothesis that PAV is associated with relaxed selective constraints. Enrichments of sequence motifs previously associated with recombination hotspots in or around PAV genes suggest that at least some exon deletion events may have resulted from recombination events involving these recombination hotspot motifs.
Exon Loss Is Associated with a Marginal Reduction in Expression Level
The aforementioned results suggest that E-PAV is associated with reduced selective constraints. To assess whether exon loss is likely to have resulted in reduced functionality for the genes affected, we compared expression levels for genes with and without missing exons across accessions. If exon loss causes or follows from diminished functionality by previous mutations, we would expect expression to be significantly reduced in those accessions affected by E-PAV. Using RNAseq transcription profiles for each Arabidopsis accession (Gan et al. 2011), we compared the expression patterns of individual genes in accessions affected by exon deletions with those accessions where the gene remained intact. To do this, we transformed expression data per accession to Z scores (Cheadle et al. 2003). We then looked only at those genes where exon loss had occurred in a single accession (210 genes). For each gene, we took 1) the expression level of that gene in the affected accession and 2) the mean expression level of that gene across the 17 unaffected accessions (the other 16 under study plus the reference genome, Col-0). We found that half of the genes examined had an expression level below this mean and 37% an expression level equal to it. However, on average, expression levels in the affected accession departed little from mean expression in unaffected accessions (0.15 standard deviations). In 27 genes (13% of cases), expression level in a gene affected by an exon deletion was higher than the mean expression across unaffected accessions with 14 cases showing a statistically significant difference (fig. 4 and supplementary table S6, Supplementary Material online). These 27 genes are generally poorly characterized with 12 having no functional category annotations. Most genes affected by exon deletions had low expression levels to begin with, although some exceptions are notable, such as rotamase CYP4 (AT3G62030; involved in a variety of cellular functions related to metabolism and response to several types of stress), which has an average expression level in the unaffected accessions of 400 rpkm, among the top 1% of genes with detectable expression in Col-0.
Fig. 4.
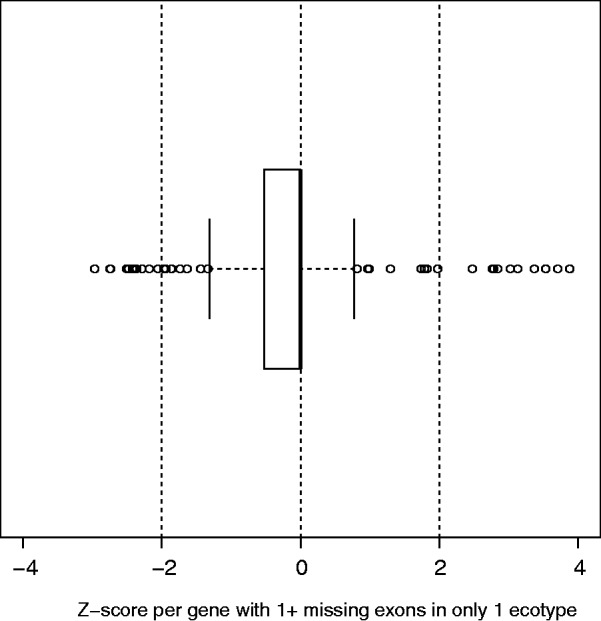
Distribution of Z scores for standardized transcript abundance data in the affected accession. Data show that 210 genes that have one or more missing exons in only one of 17 A. thaliana accessions (relative to Col-0).
It is possible that the moderate effect of exon loss on gene expression levels is explained by an overrepresentation of alternatively spliced exons among the set of missing exons. This would allow for the production of viable protein products in their absence. In order to test this, we quantified alternative splicing in 15,540 Arabidopsis genes, including 103 of the 330 E-PAV associated genes using a “comparable alternative splicing index” (see Materials and Methods), which corrects for the distorting effect of variation in transcript coverage among genes (reviewed in Chen, Tovar-Corona, et al. [2012]). E-PAV genes were found to have a significantly higher number of alternative splicing events compared with intact genes (3.35 and 1.13, respectively; randomization test P = 0.046).
Overall, these findings suggest that exon losses only have a marginal effect on the expression profile of genes in the accessions affected. The higher levels of alternative splicing among genes affected by exon loss raises the possibility that a significant proportion of lost exons are normally alternatively spliced, reducing selection pressure on these exons because a functional protein product would be produced in their absence anyway.
Discussion
Intraspecies structural variations in genes have been proposed to play an important role in the adaptation of particular populations to variation in environmental conditions (Feuk et al. 2006). Here we have characterized presence–absence coding sequence variation in 17 fully sequenced A. thaliana genomes, relative to the reference accession Col-0, affecting 411 genes including 81 instances of whole coding region deletions. We found a significant enrichment of genes associated with the GO terms for protein and nucleotide binding as well as signal transduction. Both gene family and Pfam annotation enrichment analysis revealed significant enrichments of gene members from the disease resistance associated NBS-LRR gene families. Significant deviations from random expectations have been observed in previous studies of PAV genes in plants, with similar overrepresentation of resistance-associated gene families among PAV genes. For instance, in sorghum (Zheng et al. 2011), PAV genes are enriched in nine Pfam categories, including the NB-ARC domain-containing family. In soybean (McHale et al. 2012), PAV-affected genes have also been found to be enriched for members of the NB-ARC family, and within the GO category of “defense response.” CDS-PAV genes have also been shown to deviate from random expectations in Arabidopsis (Tan et al. 2012), with the greatest significant enrichment in PAV genes also reported for those with NB-ARC domains.
These functional and/or gene family enrichments can be suggestive of an adaptive role for PAV events by aiding specific ecotypes in adapting to their local environment. Our results—showing that genes associated with, for example, resistance are more likely to be affected by PAV—are, at first glance, consistent with this hypothesis. In addition, we were able to confirm a previous report of CDS-PAV for three members of the R gene family—the single-exon gene AT5G05400 and the multiexon genes AT5G18350 and AT5G49140 (Shen et al. 2006)—a family known to have signatures of positive selection in A. thaliana (Mondragon-Palomino et al. 2002). However, comprehensive analysis for evidence of selection does not support this as a general interpretation.
dN/dS ratios are one of the most widely used estimates of selective pressure acting on protein coding genes with dN/dS >> 1 indicative but not a definitive signature of positive selection (Hurst 2002). Although there are, on average, a higher number of substitutions in E-PAV genes compared with intact genes, this is not a clear signature of adaptation and can suggest comparatively relaxed negative, rather than stronger positive, selection.
We further found that PAV genes have significantly higher nucleotide diversity both at silent and replacement sites. Both observations are suggestive of weaker purifying selection; however, they can also be expected if PAV genes were under higher balancing selection. Indeed, there is evidence to suggest that the diversity of resistance-associated genes is maintained by balancing selection (Van der Hoorn et al. 2002), which are overrepresented among PAV genes. Balancing selection has been proposed to stably maintain both the intact gene and the absent allele (Tan et al. 2012).
So, is balancing selection the most parsimonious explanation for why PAV genes are associated with higher nucleotide diversity? A classic scenario of transspecies polymorphism, associated with balancing selection, cannot be assessed given the limited sequence variation data available for A. lyrata, A. thaliana’s closest sequenced relative. It is possible that the “gene/exon present” and the “gene/exon absent” alleles are under selection to be maintained in different A. thaliana populations, allowing them to better adapt to their local environment. This would be consistent with the increase in nucleotide diversity, but this scenario cannot be distinguished from alternative neutral models. Conditional neutrality at PAV loci, where the functional gene has ceased to be adaptive in some but not all environments, cannot be ruled out (e.g., in the case of resistance genes where the corresponding pathogen is absent [Gos and Wright 2008]). In this case, the absent allele would have no selective advantage at any point but rather result from relaxed constraints associated with PAV genes in some Arabidopsis populations. Moreover, a model of generalized relaxed constraints affecting the PAV loci would also lead to increased nucleotide diversity and slight increases in dN/dS.
Tajima’s D, a comparison of two estimators of θ (the population mutation rate 4Neμ)—the number of segregating sites and the average number of pairwise differences between sequences (Tajima 1989)—offers a more reliable estimate of selective pressures acting on a gene as it incorporates information about the distribution of segregating alleles in a species. This allows more accurate estimations of the degree and direction of departure of sequence evolution from a neutral expectation (although nonselectionist interpretations of D are also possible, such as recent population expansion or bottlenecking for negative and positive D, respectively) (Tajima 1989). Tajima’s D values do not provide evidence for either E-PAV or CDS-PAV genes to be under balancing selection. Taking dN/dS, nucleotide diversity, and D estimates together, most PAV genes appear to be evolving under relaxed constraints.
A signature of relaxed selection associated with PAV genes is combined with a variety of features that have been associated with lower gene essentiality. We found that PAV genes have lower expression levels and higher tissue specificity; both of these features have been associated with higher rates of substitutions and reduced gene essentiality (Wolf et al. 2006; Cherry 2010). Older genes have been considered more essential (Chen, Trachana et al. 2012) and have been associated (in humans, flies and Aspergillus) with a higher expression level and stronger purifying selection (Wolf et al. 2009). We found that PAV genes are, on average, newer additions to the genome and that most exons affected by PAV do not have an orthologous exon in A. lyrata (663/794). We note that both E-PAV and CDS-PAV genes are enriched in reverse transcriptase domains (supplementary figs. S4 and S5, Supplementary Material online) and E-PAV genes for transposase domains (supplementary fig. S4, Supplementary Material online), suggesting exonization of TEs as the origin of some PAV-affected exons.
In addition, the fact that gene expression is only marginally reduced in accessions affected by exon deletion events suggests that the lost exons may only have had a limited impact on gene functionality. This is possibly explained in some cases by alternative splicing, which has already been associated with an increased frequency of exon loss in humans, mice, and rats—alternatively spliced forms are less likely to be conserved between species than constitutive exons (Modrek and Lee 2003). In A. thaliana, we found that genes with E-PAV are under weaker purifying selection and have a greater number of alternative splice events compared with intact genes. This observation suggests that alternatively spliced exons are likely to be under reduced selective constraints compared with constitutive exons, and thus whole exon deletions would have less of a detrimental effect than the loss of a constitutive exon. To the best of our knowledge, this is the first time that exon loss events have been associated with elevated alternative splicing levels within a species rather than between species.
The genomic context of genes has also been linked to both patterns of sequence evolution and features associated with gene essentiality. A recent study in A. thaliana has correlated the presence of TEs adjacent to genes with sequence variation within that gene (Wang, Weigel, et al. 2013), suggesting TEs tend to accumulate near genes under lower selective pressures located in regions with less efficient purging of TE sequence. Indeed, for our set of E-PAV genes, we find a higher density of TEs in the vicinity. In addition, we also find that genes undergoing PAV have an increased proportion of motifs associated with recombination hotspots within their sequence. Both findings are consistent with PAV events being associated with genes located in genomic regions evolving under reduced selective constraints. Moreover, higher TE content and hotspot motifs are consistent with the suggestion that unequal recombination between homologs may be a major mechanism for generating P/A polymorphisms (Tan et al. 2012). However, it should be noted that no recombinogenic motif is both necessary and sufficient for a recombination event to occur (Johnston and Cutler 2012), and as such, their connection, if any, to PAV remains speculative.
All of these features considered together suggest that although some individual deletions might have an adaptive value, overall coding region loss disproportionally affects genes under reduced selective pressures. So how are these results reconciled with the enrichment of certain gene families and GO functional terms? The enrichment of specific functional categories and gene families among PAV genes (fig. 1) leads to the implication of adaptive pressures favoring PAV on genes related to specific biological processes (Tan et al. 2012). However, as we have shown, PAV genes are associated with a variety of features suggestive of lower selective constraints. We argue that the enrichment of certain GO categories and/or gene families among genes associated with a particular genomic feature does not, by itself, allow us to draw conclusions about any adaptive processes these genes may be undergoing. Consistent with this, we find that intact genes associated with the gene categories in which PAV genes are enriched also show the same signatures of reduced selection (supplementary table S7, Supplementary Material online). This is notable for those sets of genes involved in, for example, signal transduction, nucleic acid binding, and the NBS-LRR family—categories enriched among PAV genes (fig. 1). For instance, if we compare the set of E-PAV genes to the set of genes with all exons present and the set of NBS-LRR genes to the set of genes belonging to other families, we find that both E-PAV and NBS-LRR genes are comparatively newer additions to the genome, have a higher dN/dS ratio, a higher number of alternative splicing events, a higher number of paralogs, a higher proportion of SNPs, and are found closer to TEs (supplementary table S7, Supplementary Material online). We note that the proportion of polymorphic sites is higher not only in PAV genes but in genes of that functional category. To demonstrate that PAV genes do not bias the comparison of, for example, the set of NBS-LRR genes to the set of genes belonging to other families, we repeat the analysis restricted to intact genes only and observe the same result (supplementary table S7, Supplementary Material online).
The fact that we observed fewer PAV genes than a previous study examining 80 fully sequenced Arabidopsis genomes (n = 2,741; Tan et al. 2012) is likely due to differences in methodology. First, our analysis uses 17 genomes assembled using a combination of read-to-reference genome (Col-0) alignment and de novo approaches, and—importantly—for which transcriptome data were available (Gan et al. 2011), rather than the 80 accessions reported by Cao et al. (2011). Second, we use a more conservative methodology for defining significant deletions while Tan et al. (2012) define PAV genes using what is referred to as the “broad definition”: “one being found at a particular locus only in some genomes compared to the others.” This allows a gene to be called as a PAV gene even if a copy exists at a different locus. To minimize the inclusion of rearrangement events as deletions, Tan et al. (2012) examined their predicted PAV genes using BlastN against a reference accession, excluding from the “absent” category any gene with a counterpart that matches >50% of its length. Our definition of PAV is more restrictive as we only deemed an exon or gene to be deleted if genome alignments showed that the deletion spanned at least a whole exon or whole gene with not a single identifiable base remaining. Finally, the Tan et al. (2012) study used genomes assembled according to the TAIR8 annotated positions, whereas our data are assembled according to TAIR10. There is a small risk, therefore, of having incorporated now-obsolete gene models into their findings. Regardless of the methodological differences and the resulting variation in sample size, it is worth noting that our results are not in contradiction to those of previous studies examining PAV both in Arabidopsis and other species, as we find similar deviations from random expectations in the functional annotations of genes. Our analysis of sequence evolution and other genic features of PAV genes do not rule out the possibilities of conditional neutrality at PAV loci or that balancing selection may be acting on PAV genes, allowing adaptation to the environmental conditions of specific ecotypes. Instead, the findings presented show that PAV events can be explained by a nonadaptive interpretation where genes under reduced constraints are more susceptible to the spread of allele variants containing significant deletions.
In summary, our results suggest that although significant enrichment in functional categories among PAV genes was observed, most exon loss events are observed in newer, poorly functionally characterized genes associated with signatures linked to less essential genes evolving under lower purifying or balancing selection. This may reduce the potential functional relevance of structural variations within these genes. We conclude that although an adaptive model for PAV cannot be ruled out, the observed functional enrichments among PAV genes and increased nucleotide diversity can also be interpreted without invoking selection.
Materials and Methods
Genome Sequence and Annotations
Exon coordinates for A. thaliana strain Col-0 were obtained from The Arabidopsis Information Resource (TAIR) (ftp://ftp.arabidopsis.org/home/tair/Genes/TAIR10_genome_release/TAIR10_gff3/TAIR10_GFF3_genes.gff, dated 20 March 2012 [last accessed October 8, 2013]). The genomes of 17 A. thaliana accessions (Bur-0, Can-0, Ct-1, Edi-0, Hi-0, Kn-0, Ler-0, Mt-0, No-0, Oy-0, Rsch-4, Sf-2, Tsu-0, Wil-2, Ws-0, Wu-0, and Zu-0) were obtained from Gan et al. (2011). We did not use data from Po-0 because it has an unusually high frequency of heterozygosity and high similarity to Oy-0 (Gan et al. 2011). Each genome has been fully sequenced and assembled, using a combination of de novo assembly and read mapping to the reference accession, Col-0.
Detecting Missing Exons Relative to Col-0
For this analysis, we selected a set of deletions spanning at least one full exon in at least one accession relative to the Col-0 reference genome from a wider set of deletion events described by Gan et al. (2011). Exons absent in the Col-0 reference genome but present in other accessions are not included in any analysis. Confirmation of these deletions is described by the original authors who analyzed deletion breakpoints (Gan et al. 2011). In this data set, deletion breakpoints were estimated to within ∼30 bp, with left and right consensus sequences established by growing inward from these estimates using the read-mapping information. If there was a deletion, these two ends would overlap. Gan et al. (2011) confirmed this with alignments of the left and right consensus sequences, thus excluding errors of sequencing or misassembly. We further confirmed the presence or absence of each individual exon in each of 17 accessions relative to the Col-0 genome annotation using BlastN with default parameters (Altschul et al. 1990). Sequence alignments were obtained using the best hit homolog and the Smith–Waterman algorithm (fasta35 with parameters–a–A) (Pearson 2000). We confirmed an exon as missing if both 1) alignment could not be made and 2) if none of the nucleotide positions in the Col-0 exon mapped to any nucleotide in the accession.
Functional Category Enrichment Analysis
Four gene classification schemes were obtained. GOslim terms were obtained from TAIR (ftp://ftp.arabidopsis.org/home/tair/Ontologies/Gene_Ontology/ATH_GO_GOSLIM.txt, dated 9 July 2013 [last accessed October 8, 2013]), excluding terms unsupported by experimental or computational analysis, that is, evidence codes ND, NR, and NAS. GO term annotations were obtained from Ensembl BioMart (17 July 2013) (Smedley et al. 2009). “Pfam” terms were obtained from Pfam v27.0 (17 July 2013) (Punta et al. 2012). In addition, 7,119 genes were classified into 49 distinct families as in Gan et al. (2011). Statistical significance of the enrichment of both GOslim, GO terms, of Pfam class and family membership among both E-PAV and CDS-PAV-affected genes was assessed using Monte Carlo random sampling (1000 randomizations), with the P value of the enrichment of each category obtained using a Z test. The significance of individual categories was corrected for multiple testing by the Benjamini–Hochberg procedure.
Sequence Evolution Analysis
To approximate selective constraint on a gene, we calculated dN/dS. For each gene, we obtained a local alignment of the Col-0 CDS against its A. lyrata ortholog, using the Smith–Waterman algorithm (fasta35 with parameters–a–A) (Pearson 2000). dN/dS was calculated using the Yang and Nielson model, as implemented in the yn00 package of phylogenetic analysis by maximum likelihood (PAML) (Yang 2009). Using substitution estimates, as above, and SNP data from (Gan et al. 2011), we also estimated Tajima’s D (Tajima 1989) per gene. Nucleotide diversity is calculated according to Gan et al. (2011).
Paralog Number and Gene Age Annotations
Ortholog and paralog data were obtained from BioMart (Vilella et al. 2009). A proxy for gene age was established using taxonomic classifications, based on the phylostratigraphic method of (Domazet-Lošo et al. 2007). If a candidate ortholog was identified for each A. thaliana gene in any of 15 plant and algal species at a minimum identity of 30%, the gene was considered to be as old as the “broadest” taxonomic category held in common (see supplementary table S8, Supplementary Material online). This allowed us to make use of ortholog data despite divergence times relative to A. thaliana being known for only its closest relatives—at ∼5 million years for A. lyrata (Kuittinen et al. 2004) and 20 million years for Brassica rapa (Yang et al. 1999).
Gene Expression
Expression specificity was calculated as a tissue specificity index (tau) (Yanai et al. 2005), using the massively parallel signature sequencing (MPSS) database (Brenner et al. 2000; Meyers et al. 2004; Nakano et al. 2006). Expression levels were calculated using RNAseq transcript abundance data, as absolute read values corrected by sequence length in each accession (known as rpkm values: per gene, the number of reads per kilobase per million mapped reads) (Gan et al. 2011).
TE and Hotspot Motif Density
TE coordinates for A. thaliana strain Col-0 were obtained from TAIR (file “TAIR10_Transposable_Elements,” dated 20 March 2012). For our analyses, we identified every instance of all 25 hotspot-associated motifs (of 5–9 bp) described by Horton et al. (2012) in the Col-0 reference genome. TE and hotspot motif density for each gene were calculated as the proportion of base pairs occupied by a TE or a hotspot motif within windows of size 1 to 100 kb centered on the nucleotide at the gene’s midpoint. Windows consist of both coding and noncoding sequence within a region of length (window size)/2 up- and downstream of the midpoint base. Both TE and hotspot motif density were calculated as the number of TE or motif bases, respectively, relative to the number of intergenic or genic bases contained within the window, rather than the total number of bases in the window.
Alternative Splicing Events
Alternative splicing events were identified using the methods described in Chen et al. (2011). In brief, the number of alternative splicing events per gene was identified by aligning expressed sequence tag (EST) data obtained from dbEST (Boguski et al. 1993) to the genome sequence (ftp://ftp.ncbi.nih.gov/repository/dbEST, last accessed May 1, 2011). Those ESTs aligning to regions with no annotated gene were excluded from the analysis. EST alignments were then used to create an exon template. Alternative splicing events per gene were identified by comparing alignment coordinates for each individual EST to exon annotations. As a low EST coverage can increase the number of falsely positive claims that an exon is constitutive, rather than spliced, we excluded genes with 10 or fewer ESTs. ESTs were assigned to genes using gene annotation coordinates. A comparable alternative splicing index that avoids transcript coverage biases was obtained using the transcript normalization method described in Kim et al. (2007). Briefly, for each gene 100 random samples of 10 ESTs were selected. Finally, the number of alternative splicing events were calculated for each random sample (as detailed earlier), with an overall average calculated per gene.
Randomization Test
A randomization test was used to obtain numerical P values to assess the statistical significance of any variation in the characteristics of PAV-affected genes compared with intact genes. In brief, we contrasted genomic feature parameters in E-PAV (n = 330) or CDS-PAV genes (n = 81) to the distribution of means of the same genomic feature in s = 10,000 randomly generated subsets of an equal number of genes drawn from the complete gene set. The numerical P value was calculated as follows: let q be the number of times the mean value of the PAV set exceeded the mean value of the randomly generated subset. Letting r = s − q, then the P value of this test is r + 1/s + 1.
Supplementary Material
Supplementary tables S1–S8 and figures S1–S5 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors acknowledge the valuable comments made by two anonymous reviewers. This work was supported by a University of Bath fee studentship to S.J.B., CONACyT scholarships to A.C.M. and J.M.T.C., a UK–China scholarship for excellence and University of Bath research studentship to L.C., a BBSRC grant (grant number BB/F022697/1) to P.X.K., and a Royal Society Dorothy Hodgkin Research Fellowship (DH071902), Royal Society research grant (grant number RG0870644), and a Royal Society research grant for fellows (grant number RG080272) to A.U.O.
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Bakker EG, Toomajian C, Kreitman M, Bergelson J. A genome-wide survey of R gene polymorphisms in Arabidopsis. Plant Cell. 2006;18:1803–1818. doi: 10.1105/tpc.106.042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boguski MS, Lowe TMJ, Tolstoshev CM. dbEST—database for expressed sequence tags. Nat Genet. 1993;4:332–333. doi: 10.1038/ng0893-332. [DOI] [PubMed] [Google Scholar]
- Brenner S, Johnson M, Bridgham J, et al. (24 co-authors) Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays. Nat Biotechnol. 2000;18:630–634. doi: 10.1038/76469. [DOI] [PubMed] [Google Scholar]
- Cao J, Schneeberger K, Ossowski S, et al. (17 co-authors) Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat Genet. 2011;43:956–963. doi: 10.1038/ng.911. [DOI] [PubMed] [Google Scholar]
- Cheadle C, Vawter MP, Freed WJ, Becker KG. Analysis of microarray data using Z score transformation. J Mol Diagn. 2003;5:73–81. doi: 10.1016/S1525-1578(10)60455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Tovar-Corona JM, Urrutia AO. Increased levels of noisy splicing in cancers, but not for oncogene-derived transcripts. Hum Mol Genet. 2011;20:4422–4429. doi: 10.1093/hmg/ddr370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Tovar-Corona JM, Urrutia AO. Alternative splicing: a potential source of functional innovation in the eukaryotic genome. Int J Evol Biol. 2012;2012:10. doi: 10.1155/2012/596274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W-H, Trachana K, Lercher MJ, Bork P. Younger genes are less likely to be essential than older genes, and duplicates are less likely to be essential than singletons of the same age. Mol Biol Evol. 2012;29:1703–1706. doi: 10.1093/molbev/mss014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JL. Expression level, evolutionary rate, and the cost of expression. Genome Biol Evol. 2010;2:757–769. doi: 10.1093/gbe/evq059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RM, Schweikert G, Toomajian C, et al. (18 co-authors) Common sequence polymorphisms shaping genetic diversity in Arabidopsis thaliana. Science. 2007;317:338–342. doi: 10.1126/science.1138632. [DOI] [PubMed] [Google Scholar]
- DeYoung BJ, Innes RW. Plant NBS-LRR proteins in pathogen sensing and host defense. Nat Immunol. 2006;7:1243–1249. doi: 10.1038/ni1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domazet-Lošo T, Brajković J, Tautz D. A phylostratigraphy approach to uncover the genomic history of major adaptations in metazoan lineages. Trends Genet. 2007;23:533–539. doi: 10.1016/j.tig.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- Gan X, Stegle O, Behr J, et al. (23 co-authors) Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature. 2011;477:419–423. doi: 10.1038/nature10414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gos G, Wright SI. Conditional neutrality at two adjacent NBS-LRR disease resistance loci in natural populations of Arabidopsis lyrata. Mol Ecol. 2008;17:4953–4962. doi: 10.1111/j.1365-294X.2008.03968.x. [DOI] [PubMed] [Google Scholar]
- Hanada K, Kuromori T, Myouga F, Toyoda T, Li WH, Shinozaki K. Evolutionary persistence of functional compensation by duplicate genes in Arabidopsis. Genome Biol Evol. 2009;1:409–414. doi: 10.1093/gbe/evp043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton MW, Hancock AM, Huang YS, et al. (13 co-authors) Genome-wide patterns of genetic variation in worldwide Arabidopsis thaliana accessions from the RegMap panel. Nat Genet. 2012;44:212–216. doi: 10.1038/ng.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst LD. The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet. 2002;18:486. doi: 10.1016/s0168-9525(02)02722-1. [DOI] [PubMed] [Google Scholar]
- Johnston HR, Cutler DJ. Population demographic history can cause the appearance of recombination hotspots. Am J Hum Genet. 2012;90:774–783. doi: 10.1016/j.ajhg.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Magen A, Ast G. Different levels of alternative splicing among eukaryotes. Nucleic Acids Res. 2007;35:125–131. doi: 10.1093/nar/gkl924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuittinen H, de Haan AA, Vogl C, Oikarinen S, Leppala J, Koch M, Mitchell-Olds T, Langley CH, Savolainen O. Comparing the linkage maps of the close relatives Arabidopsis lyrata and A. thaliana. Genetics. 2004;168:1575–1584. doi: 10.1534/genetics.103.022343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino T, Hokamp K, McLysaght A. The complex relationship of gene duplication and essentiality. Trends Genet. 2009;25:152–155. doi: 10.1016/j.tig.2009.03.001. [DOI] [PubMed] [Google Scholar]
- McHale L, Tan X, Koehl P, Michelmore R. Plant NBS-LRR proteins: adaptable guards. Genome Biol. 2006;7:212. doi: 10.1186/gb-2006-7-4-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHale LK, Haun WJ, Xu WW, Bhaskar PB, Anderson JE, Hyten DL, Gerhardt DJ, Jeddeloh JA, Stupar RM. Structural variants in the soybean genome localize to clusters of biotic stress-response genes. Plant Physiol. 2012;159:1295–1308. doi: 10.1104/pp.112.194605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers BC, Tej SS, Vu TH, Haudenschild CD, Agrawal V, Edberg SB, Ghazal H, Decola S. The use of MPSS for whole-genome transcriptional analysis in Arabidopsis. Genome Res. 2004;14:1641–1653. doi: 10.1101/gr.2275604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modrek B, Lee CJ. Alternative splicing in the human, mouse and rat genomes is associated with an increased frequency of exon creation and/or loss. Nat Genet. 2003;34:177–180. doi: 10.1038/ng1159. [DOI] [PubMed] [Google Scholar]
- Mondragon-Palomino M, Meyers BC, Michelmore RW, Gaut BS. Patterns of positive selection in the complete NBS-LRR gene family of Arabidopsis thaliana. Genome Res. 2002;12:1305–1315. doi: 10.1101/gr.159402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano M, Nobuta K, Vemaraju K, Tej SS, Skogen JW, Meyers BC. Plant MPSS databases: signature-based transcriptional resources for analyses of mRNA and small RNA. Nucleic Acids Res. 2006;34:D731–D735. doi: 10.1093/nar/gkj077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver KR, Greene WK. Transposable elements: powerful facilitators of evolution. Bioessays. 2009;31:703–714. doi: 10.1002/bies.200800219. [DOI] [PubMed] [Google Scholar]
- Ossowski S, Schneeberger K, Clark RM, Lanz C, Warthmann N, Weigel D. Sequencing of natural strains of Arabidopsis thaliana with short reads. Genome Res. 2008;18:2024–2033. doi: 10.1101/gr.080200.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson WR. Flexible sequence similarity searching with the FASTA3 Program Package. Methods Mol Biol. 2000;132:185–219. doi: 10.1385/1-59259-192-2:185. [DOI] [PubMed] [Google Scholar]
- Punta M, Coggill PC, Eberhardt RY, et al. (16 co-authors) The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santuari L, Pradervand S, Amiguet-Vercher A-M, Thomas J, Dorcey E, Harshman K, Xenarios I, Juenger T, Hardtke C. Substantial deletion overlap among divergent Arabidopsis genomes revealed by intersection of short reads and tiling arrays. Genome Biol. 2010;11:R4. doi: 10.1186/gb-2010-11-1-r4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Araki H, Chen L, Chen JQ, Tian D. Unique evolutionary mechanism in R-genes under the presence/absence polymorphism in Arabidopsis thaliana. Genetics. 2006;172:1243–1250. doi: 10.1534/genetics.105.047290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley D, Haider S, Ballester B, Holland R, London D, Thorisson G, Kasprzyk A. BioMart—biological queries made easy. BMC Genomics. 2009;10:22. doi: 10.1186/1471-2164-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson-Wagner RA, Eichten SR, Kumari S, Tiffin P, Stein JC, Ware D, Springer NM. Pervasive gene content variation and copy number variation in maize and its undomesticated progenitor. Genome Res. 2010;20:1689–1699. doi: 10.1101/gr.109165.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S, Zhong Y, Hou H, Yang S, Tian D. Variation of presence/absence genes among Arabidopsis populations. BMC Evol Biol. 2012;12:86. doi: 10.1186/1471-2148-12-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Hoorn RAL, De Wit PJGM, Joosten MHAJ. Balancing selection favors guarding resistance proteins. Trends Plant Sci. 2002;7:67–71. doi: 10.1016/s1360-1385(01)02188-4. [DOI] [PubMed] [Google Scholar]
- Vilella AJ, Severin J, Ureta-Vidal A, Heng L, Durbin R, Birney E. EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009;19:327–335. doi: 10.1101/gr.073585.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Weigel D, Smith LM. Transposon variants and their effects on gene expression in Arabidopsis. PLoS Genet. 2013;9:e1003255. doi: 10.1371/journal.pgen.1003255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, You FM, Lazo GR, Luo M-C, Thilmony R, Gordon S, Kianian SF, Gu YQ. PIECE: a database for plant gene structure comparison and evolution. Nucleic Acids Res. 2013;41:D1159–D1166. doi: 10.1093/nar/gks1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Carmel L, Koonin EV. Unifying measures of gene function and evolution. Proc R Soc B. 2006;273:1507–1515. doi: 10.1098/rspb.2006.3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Novichkov PS, Karev GP, Koonin EV, Lipman DJ. The universal distribution of evolutionary rates of genes and distinct characteristics of eukaryotic genes of different apparent ages. Proc Natl Acad Sci U S A. 2009;106:7273–7280. doi: 10.1073/pnas.0901808106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SI, Agrawal N, Bureau TE. Effects of recombination rate and gene density on transposable element distributions in Arabidopsis thaliana. Genome Res. 2003;13:1897–1903. doi: 10.1101/gr.1281503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Liu X, Ge S, et al. (25 co-authors) Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat Biotechnol. 2012;30:105–111. doi: 10.1038/nbt.2050. [DOI] [PubMed] [Google Scholar]
- Yanai I, Benjamin H, Shmoish M, et al. (12 co-authors) Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics. 2005;21:650–659. doi: 10.1093/bioinformatics/bti042. [DOI] [PubMed] [Google Scholar]
- Yang H. In plants, expression breadth and expression level distinctly and non-linearly correlate with gene structure. Biol Direct. 2009;4:45. doi: 10.1186/1745-6150-4-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YW, Lai KN, Tai PY, Li WH. Rates of nucleotide substitution in angiosperm mitochondrial DNA sequences and dates of divergence between Brassica and other angiosperm lineages. J Mol Evol. 1999;48:597–604. doi: 10.1007/pl00006502. [DOI] [PubMed] [Google Scholar]
- Zheng L-Y, Guo X-S, He B, et al. (11 co-authors) Genome-wide patterns of genetic variation in sweet and grain sorghum (Sorghum bicolor) Genome Biol. 2011;12:R114. doi: 10.1186/gb-2011-12-11-r114. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.