

Abstract
Phylogenetic relationships of the primarily wingless insects are still considered unresolved. Even the most comprehensive phylogenomic studies that addressed this question did not yield congruent results. To get a grip on these problems, we here analyzed the sources of incongruence in these phylogenomic studies by using an extended transcriptome data set. Our analyses showed that unevenly distributed missing data can be severely misleading by inflating node support despite the absence of phylogenetic signal. In consequence, only decisive data sets should be used which exclusively comprise data blocks containing all taxa whose relationships are addressed. Additionally, we used Four-cluster Likelihood Mapping (FcLM) to measure the degree of congruence among genes of a data set, as a measure of support alternative to bootstrap. FcLM showed incongruent signal among genes, which in our case is correlated neither with functional class assignment of these genes nor with model misspecification due to unpartitioned analyses. The herein analyzed data set is the currently largest data set covering primarily wingless insects, but failed to elucidate their interordinal phylogenetic relationships. Although this is unsatisfying from a phylogenetic perspective, we try to show that the analyses of structure and signal within phylogenomic data can protect us from biased phylogenetic inferences due to analytical artifacts.
Keywords: phylogenomics, ESTs, likelihood quartet mapping, conflicting hypotheses, Entognatha, Nonoculata, Ellipura, Protura, Diplura, Collembola, missing data
Introduction
Despite enormous efforts to resolve the tree of life, several deep nodes are still considered unresolved. A good example for such problems are the unresolved phylogenetic relationships of primarily wingless insects.
Most phylogenetic studies including multigene and phylogenomic analyses have recovered the monophyly of Hexapoda, the insect clade in a broad taxonomic sense (Regier et al. 2008, 2010; von Reumont et al. 2009, 2012; Meusemann et al. 2010; Trautwein et al. 2012). Furthermore, the monophyly of Ectognatha, which comprises insects in a strict taxonomic sense, namely jumping bristletails, silverfishes and firebrats, and winged insects, is well supported (reviewed in Grimaldi 2010; Trautwein et al. 2012). By contrast, phylogenetic relationships among the entognathous primarily wingless insects, the Protura (coneheads), Collembola (springtails), and Diplura (two-pronged bristletails), are unclear. Many authors consider these entognathous insects as being monophyletic, considering entognathy in which mouth parts are concealed in gnathal pouches (first discussed in detail by Hennig 1953) to have evolved in the last common ancestor of the three groups. Within Entognatha, either a clade uniting Protura and Collembola, referred to as Ellipura (Börner 1910), or a clade uniting Protura and Diplura, referred to as Nonoculata (Luan et al. 2005), has been proposed (Ellipura [Hennig 1953; Kristensen 1981, 1997; Shao et al. 1999; Bitsch and Bitsch 2000, 2004; Carapelli et al. 2000; Zhang et al. 2001]; Nonoculata [Giribet and Wheeler 2001; Giribet et al. 2004; Luan et al. 2005; Kjer et al. 2006; Mallatt and Giribet 2006; Misof et al. 2007; Dell’Ampio et al. 2009; von Reumont et al. 2009; Mallatt et al. 2010]). Other authors consider a paraphyly of Entognatha to be more likely, with Diplura as closest relatives to Ectognatha. Possible arguments for this hypothesis include the evolutionary origin of paired pretarsal claws and paired cerci (Kukalová-Peck 1987; Koch 1997; Beutel and Gorb 2006), the ultrastructure of the sperm (Dallai et al. 2011), and the differentiation process of the embryonic amnion (Machida 2006) in the last common ancestor of Diplura and Ectognatha.
Meusemann et al. (2010) and von Reumont et al. (2012) published the most relevant data sets and analyses covering the phylogenetic relationships among primarily wingless insects by including expressed sequence tag (EST) data of representatives of Protura, Collembola, and Diplura. Although both studies recovered the monophyly of Entognatha, Meusemann et al. found strong evidence for Protura and Diplura as closest relatives (i.e., Nonoculata) and von Reumont et al. for Protura and Collembola as closest relatives (i.e., Ellipura). These incongruent results are puzzling because taxon sampling of the primarily wingless insects is comparable in both studies, as well as the strategies used for orthology assignment, alignment masking, matrix optimization, and tree inference.
These special circumstances put us into the exceptionally favorable position to analyze possible sources of incongruence among these two large phylogenomic data sets. Most phylogenomic studies are based on concatenated supermatrices with low gene data coverage. Focusing on relationships among specific groups, many data blocks within such supermatrices therefore may not contain data for all taxa under consideration. Consequently, our starting hypothesis was that extensive missing data may mislead proper tree reconstruction. To tackle this problem, we complement the publicly available EST data of primarily wingless insects with additional EST data from representatives of Japygidae (Diplura) and Zygentoma (silverfishes and firebrats). We took particular care to concatenate a data set that contains only gene data blocks for which entognathous hexapods and outgroups had gene data coverage. We call such a data set in the following a decisive data set. Note that the term decisiveness has been used before in the context of phylogenomic data sets (Steel and Sanderson 2010; Sanderson et al. 2010), albeit based on a distinct criterion. The concatenated data set is the largest known data set covering primarily wingless insects. It was this data set that allowed us to analyze the effect of the observed uneven distribution of missing data on the extent of bootstrap support (BS). Complementary to the application of BS measures, we applied a Four-cluster Likelihood Mapping (FcLM) approach (Strimmer and von Haeseler 1997), which has been shown to be effective in disentangling signal among four groups of species. The application of bootstrapping and FcLM helped to assess the effect of the uneven distribution of missing data in indecisive data sets. Complementary to the previously mentioned analyses, we addressed the problem of incongruent signal among genes in a multigene data set by comparing tree reconstructions based on the entire decisive data set with tree reconstructions based on subsets of genes that support incongruent hypotheses. Altogether, our approach provides potential explanations for contradictory results among phylogenomic studies by pointing out underestimated sources of error and incongruence.
Results
Orthology Assignment, Alignment, and Alignment Masking
Using the reference set of 1,886 1:1 orthologous genes (OGs), we identified between 52 and 682 putative 1:1 orthologous transcripts in the transcriptome assemblies of primarily wingless hexapods (table 1) and up to 1,886 for all taxa (supplementary table S1, Supplementary Material online). We excluded 20 OGs that were present in the five reference species but absent from all other species from subsequent analyses. After alignment masking (i.e., the exclusion of multiple sequence alignment sections in which sequence similarity cannot be distinguished from random similarity of sequences), the concatenated superalignment was composed of 73 taxa with a total alignment length of 881,235 amino acid sites, partitioned into 1,866 genes (supplementary fig. S1; for gene annotations, see supplementary table S2, Supplementary Material online).
Table 1.
Primarily Wingless Hexapod Species Included in This Study, and Their Number of OGs in the Original Supermatrix and in Three Data Subsets.
| Order | Family | Species | Source | No. of Contigs | Total no. of OGs | No. of OGs in M_Ento | No. of OGs in SOS | No. of OGs in SOSω |
|---|---|---|---|---|---|---|---|---|
| Protura | Acerentomidae | Acerentomon sp.a | NCBIa | 1,999 | 191 | 117 | 91 | 12 |
| Diplura | Campodeidae | Campodea fragilis | NCBI | 6,407 | 370 | 77 | 116 | 64 |
| Diplura | Japygidae | Megajapyx sp. | this study | 57,602 | 547 | 105 | 164 | 89 |
| Collembola | Neanuridae | Anurida maritima | NCBI | 3,504 | 328 | 55 | 105 | 60 |
| Collembola | Onychiuridae | Onychiurus arcticus | NCBI | 9,981 | 795 | 103 | 183 | 114 |
| Collembola | Isotomidae | Cryptopygus antarcticus | NCBI | 1,897 | 199 | 49 | 78 | 35 |
| Collembola | Isotomidae | Folsomia candida | NCBI | 5,967 | 442 | 60 | 122 | 78 |
| Collembola | Entomobryidae | Orchesella cincta | NCBI | 754 | 52 | 10 | — | — |
| Archaeognatha | Machilidae | Lepismachilis y-signata | NCBI | 2,288 | 270 | 60 | 107 | 54 |
| Zygentoma | Lepismatidae | Tricholepisma aurea | NCBI | 344 | 54 | 22 | — | — |
| Zygentoma | Lepismatidae | Thermobia domestica | this study | 45,358 | 682 | 96 | 194 | 124 |
Note.—M_Ento is the decisive data set in which all OGs are covered by Protura, Diplura, and Collembola; SOS and SOSω are indecisive to address the relationships of entognathous hexapod orders.
aAcerentomon sp.: erroneously assigned as A. franzi in Meusemann et al. (2010) and NCBI.
Relationships among Entognathous Hexapod Lineages
The data set M_Ento, which is decisive for addressing relationships among the three entognathous groups, Protura, Collembola, and Diplura (73 taxa, 117 genes, 32,883 aligned aa sites), moderately supported a clade Protura + Diplura (Nonoculata) (fig. 1). This is compatible with the results of the FcLM approach (topology T1 favored, fig. 2). Tree reconstruction supported Collembola as closest relatives to a clade comprising Nonoculata and Ectognatha. The clade Nonoculata + Ectognatha received moderate support (fig. 1; supplementary fig. S2, Supplementary Material online).
Fig. 1.

Simplified phylogenetic tree of the decisive data set M_Ento. Best ML tree (RAxML v.7.2.8, PROTCAT, LG + GAMMA), based on 117 OGs that are covered by Protura, Diplura, and Collembola. BS is derived from 1,000 bootstrap replicates. Rogue taxa (supplementary material [section 4], Supplementary Material online) were pruned prior to tree inference. The tree was rooted with Capitella sp. For the full tree, see supplementary figure S2, Supplementary Material online.
Fig. 2.

Results of the FcLM for all OGs in data set M_Ento. The chart shows the proportion of quartets (summed up for 117 OGs) that show predominant support for T1 ([Protura + Diplura] – [Collembola + remaining taxa], Nonoculata hypothesis, blue), T2 ([Protura + Collembola] – [Diplura + remaining taxa], Ellipura hypothesis, red), and T3 ([Diplura + Collembola] – [Protura + remaining taxa], yellow), see fig. 5. Quartets mapping in remaining Voronoi cells (gray) and T* (fig. 5) were not considered.
Our tree reconstructions based on a selected optimal subset (SOS) extracted from a complete data matrix by optimizing information content and data saturation in iterative steps of gene and/or taxon exclusion (see MARE manual; Meusemann et al. 2010; Meyer and Misof 2010) (62 taxa, 253 genes, alignment length 55,429 aa positions) yielded monophyletic Entognatha with moderate support and Nonoculata with low support (fig. 3a and table 2; supplementary fig. S3a, Supplementary Material online). It should be kept in mind that this SOS is indecisive for addressing the relationships of Entognatha, with only one-third of all genes (79) of this data set being covered by all three entognathous groups (supplementary table S3, Supplementary Material online). The tree based on the data set SOSω, in which these 79 genes were removed to artificially create a maximally indecisive data set, showed Entognatha with strong support (table 2) and additionally, diplurans were paraphyletic with respect to Protura (fig. 3b; supplementary fig. S3b, Supplementary Material online). Both SOS data sets (11 taxa from the supermatrix, which included the collembolan Orchesella cincta were removed in the optimization process) did not contain any rogue taxa, that is, taxa that assume incongruent phylogenetic positions in a set of bootstrap trees (Aberer and Stamatakis 2011) (supplementary material [section 3], Supplementary Material online). Tree reconstructions of all data sets strongly supported monophyletic Ectognatha and monophyletic Hexapoda (table 2).
Fig. 3.
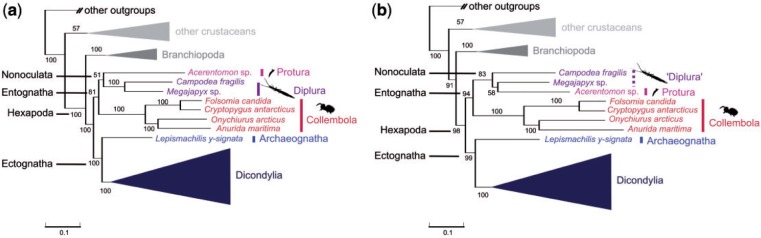
Simplified phylogenetic trees of data sets SOS (a) and SOSω (b). Best ML tree (RAxML v.7.2.8, PROTCAT, LG + GAMMA) (a) based on 253 OGs, 79 of which are covered by Protura, Diplura, and Collembola (SOS) and (b) based on 174 OGs, none of which are covered by Protura, Diplura, and Collembola (SOSω). BS is derived from 1,000 bootstrap replicates. Trees were rooted with Capitella sp. For the full trees, see supplementary figure S3a and S3b, Supplementary Material online.
Table 2.
BS (%) for Selected Clades in Tree Reconstructions with Various Data Sets.
| Clade | Data Set |
Data Subset of M_Ento |
||||||
|---|---|---|---|---|---|---|---|---|
| M_Ento | SOS | SOSω | Meusemann et al. (2010) | von Reumont et al. (2012) | M_Nono | M_Elli | M_DiCo | |
| Hexapoda | 100 (100) | 100 | 98 | 100 | 99 | 72 (100) | 100 (100) | 100 (100) |
| Diplura | 100 (100) | 100 | —a | N.A. | N.A. | 100 (100) | 100 (100) | 100 (100) |
| Collembola | 100 (100) | 100 | 100 | 100 | 100 | 100 (100) | 100 (100) | 100 (100) |
| (Protura, Diplura)b | 91 (96) | 51 | 83a | 100 | — | 100 (100) | — (−) | — (−) |
| (Protura, Collembola)c | — (−) | — | — | — | 98 | — (−) | 100 (100) | — (−) |
| (Diplura, Collembola) | — (−) | — | — | — | — | — (−) | — (–) | 99 (100) |
| Entognatha | — (−) | 81 | 94 | 86 | 98 | — (−) | — (−) | — (−) |
| ((Protura, Diplura), Ectognatha) | 80 (96) | — | — | — | — | 98 (100) | — (−) | — (−) |
| ((Collembola, Diplura), Ectognatha) | — (−) | — | — | — | — | — (−) | — (−) | 60 (83) |
| (Diplura, Ectognatha) | — (−) | — | — | — | — | — (−) | 66 (100) | — (−) |
| Ectognatha | 100 (100) | 100 | 99 | 100 | 100 | 100 (100) | 100 (100) | 95 (84) |
Note.—BS was assessed with RAxML from 1,000 bootstrap replicates (see Materials and Method). BS printed in brackets was assessed from partitioned ML analyses of data sets M_Ento, and its subsets using the Ufboot algorithm of IQ-TREE with 5,000 bootstrap replicates (supplementary material [section 5], Supplementary Material online). M_Ento is the decisive data set in which all OGs are covered by Protura, Diplura, and Collembola; SOS, SOSω, and the data sets from Meusemann et al. (2010; data set SOS, ML tree) and von Reumont et al. (2012; data set SOS, ML tree Set 1red) are indecisive to address the relationships of entognathous hexapod orders. M_Nono, M_Elli, and M_DiCo are subsets of M_Ento with predominant signal for different topologies and point out conflict of signal among genes.
aDiplurans are paraphyletic: Campodea + (Acerentomon,Megajapyx).
bNonoculata hypothesis.
cEllipura hypothesis.
Incongruent Signal among Genes
Based on the M_Ento data set, the FcLM approach helped to identify a predominant signal for topology T1 (Protura + Diplura) – (Collembola + remaining taxa) in 51 genes (12,548 aligned aa positions) (data set M_Nono, derived from Nonoculata), a predominant signal for topology T2 (Protura + Collembola) – (Diplura + remaining taxa) in 35 genes (11,789 aligned aa positions) (data set M_Elli, derived from Ellipura), and a predominant signal for topology T3 (Diplura + Collembola) – (Protura + remaining taxa) in 31 genes (8,546 aligned aa positions) (data set M_DiCo) (fig. 4a and b). Tree inferences from data sets M_Nono, M_Elli, and M_DiCo (rogue taxa pruned, see Materials and Methods section) yielded maximal BS support for Nonoculata, Ellipura, and Diplura + Collembola, respectively (table 2; supplementary fig. S4, Supplementary Material online). However, although tree reconstruction of our data subsets M_Nono, M_Elli, and M_DiCo showed maximal BS support for incongruent topologies among the entognathous insect orders, the results from the FcLM approach indicated that signal for alternative topologies was present in all data sets (fig. 4a and b; supplementary table S4, Supplementary Material online), which is not reflected by the trees. To identify possible reasons for incongruent signal among genes, we assessed the correlation between functional classes of genes and the different phylogenetic hypotheses that are supported by the data subsets. We found no correlation (supplementary material [section 4], table S5 and fig. S5, Supplementary Material online). Additionally, we tested whether model misspecification can explain the observed incongruence among genes and analyzed the data set M_Ento and data subsets M_Nono, M_Elli, and M_Dico using partitioned phylogenetic analyses (Minh et al. 2013) with the best model selected for each gene (partition) separately (supplementary material [section 5], table S6, and figs. S6–S9, Supplementary Material online). With respect to the phylogenetic relationships addressed in our study, resulting topologies did not differ from unpartitioned analyses, and BS only differed to a minor degree (table 2).
Fig. 4.

Detailed results of the FcLM Mapping for all OGs included in data set M_Ento and data subsets M_Nono, M_Elli, M_DiCo. (a) Histogram of FcLM results. Each bar refers to an OG (for OG-IDs, see supplementary table S2, Supplementary Material online). Y axis: amount of quartets (in %), that predominantly support T1 ([Protura + Diplura] – [Collembola + remaining taxa], blue), T2 ([Protura + Collembola] – [Diplura + remaining taxa], red), and T3 ([Diplura + Collembola] – [Protura + remaining taxa], yellow), quartets that show ambiguous support are not considered (fig. 5). OGs with predominant support for T1 are classified into data set M_Nono (51 genes, 12,548 aligned aa positions); OGs with predominant support for T2 are classified into data set M_Elli; (35 genes, 11,789 aligned aa positions); OGs with predominant support for T3 are classified into data set M_DiCo (31 genes, 8,546 aligned aa positions). (b) FcLM results for data set M_Nono (left), M_Elli (middle), and M_DiCo (right). Each chart shows the proportion of quartets (summed up for the OGs included in the data sets) that show predominant support for T1, T2, and T3 (see above and fig. 5). Quartets that show ambiguous support (fig. 1) are not considered.
Discussion
The Importance of Data Set Decisiveness
Incongruences in proposed relationships among Protura, Collembola, and Diplura in the studies of Meusemann et al. (2010) and von Reumont et al. (2012), which both supported monophyly of Entognatha, motivated us to look for new approaches to uncover and analyze possible sources of incongruent signal in phylogenomic data sets.
Both SOS data sets in Meusemann et al. (2010) and von Reumont et al. (2012) were compiled with MARE (Meyer and Misof 2010) and were intended to address pancrustacean and arthropod relationships. Both data sets showed only low decisiveness for addressing the relationships of the three entognathous lineages: only 28 out of 128 genes in Meusemann et al. (2010) and 22 out of 316 genes in von Reumont et al. (2012) contained representatives of Protura, Diplura, and Collembola.
Despite low gene data coverage in both studies, the monophyly of Entognatha received high BS. By contrast, our decisive data set for addressing the relationships among these three insect orders lacks clear support for Entognatha (fig. 1). This puzzling result might be explained by the presence of an uneven distribution of missing data. We gained indirect evidence for this hypothesis with the analyses of the worst case data set SOSω. This data set is maximally indecisive for testing the monophyly of Entognatha, that is, none of the included genes were common to all three entognathous insect groups. Any inferred support for this clade in the SOSω analysis can be considered an artifact. Remarkably, bootstrapping delivered high, clearly artificial support for monophyletic Entognatha in the SOSω tree (fig. 3b).
We conclude from this indirect evidence that the support for Entognatha in Meusemann et al. (2010), von Reumont et al. (2012) and in our data set indecisive concerning this question (fig. 3a) probably results from an artificial signal due to uneven distribution of missing data (Philippe et al. 2011) among Protura, Diplura, and Collembola.
Based on the analyses of the decisive and indecisive data sets, we reject the hypothesis that missing data are unproblematic as long as many characters have been sampled overall (Wiens 2006). Missing data can be misleading as shown by the worst case SOSω data set analysis, in which relationships received high BS although the data set was maximally indecisive. Therefore, we strongly advocate the exclusive use of decisive data sets in phylogenomic studies.
Incongruent Signal between Genes in a Multigene Data Set
Even decisive data sets can contain incongruent signal (Degnan and Rosenberg 2009; Knowles 2009; Philippe et al. 2011). Using FcLM, we identified groups of genes that support different relationships of Protura, Collembola, and Diplura in the decisive data set M_Ento (fig. 4a and b). Additionally, we assessed conflict within the data with split analyses relying on NeighborNetworks (supplementary material [section 6] and figs. S10–S13, Supplementary Material online). This analysis corroborates the results of FcLM that all analyzed data sets did contain incongruent signal. Additional to the problem of indecisiveness discussed earlier, this incongruent signal among genes may partly be responsible for the contradictory results of Meusemann et al. (2010) and von Reumont et al. (2012). However, incongruent signal among genes is difficult to address and rectify. We analyzed two potential sources of conflict and can conclude that both can be excluded. First, we tested for homoplasy due to analogous selection regimes in functional complexes but found no correlation between predicted gene function and phylogenetic signal (supplementary material [section 4], fig. S5, and table S5, Supplementary Material online). Second, we were able to indirectly exclude model misspecifications as sources of incongruent signal because unpartitioned and partitioned maximum likelihood (ML) analyses yielded topologically congruent results and almost identical BS (table 2; supplementary material [section 5], table S6, and figs. S6–S9, Supplementary Material online). With respect to the FcLM, it may well be that this likelihood mapping approach selects sets of genes with congruent substitution processes. A possible solution, but certainly not a fully satisfying one, would be to increase the number of genes to minimize noise and confounding signal.
Relationships of Protura, Collembola, and Diplura
Monophyly of Entognatha
The monophyly of Entognatha has never been maximally supported and this has not changed in our analyses (table 2). Studies encompassing representatives of Protura, Collembola, and Diplura are limited to only a few analyses (Colgan et al. 1998; Carapelli et al. 2000; Edgecombe et al. 2000; Giribet et al. 2001, 2005). Monophyletic Entognatha were recovered in all recent studies based on nuclear rRNA genes (Gao et al. 2008; Dell’Ampio et al. 2009; von Reumont et al. 2009; Mallatt et al. 2010). However, BS was low, which was either explained by character choice (Dell’Ampio et al. 2009) or the influence of nonstationary processes across taxa (von Reumont et al. 2009). From the morphological point of view, most apomorphies suggesting the monophyly of Entognatha represent reductions (malpighian papillae vs. tubules; reduction to loss of compound eyes). The only exception is the evolution of mouthparts that are concealed in gnathal pouches (Beutel and Gorb 2006). Diplura as closest relatives to Ectognatha is the only relation that contradicts monophyletic Entognatha, and for which morphological evidence has been published (Kukalová-Peck 1991; Koch 1997; Beutel and Gorb 2006; Dallai et al. 2011). In general, morphological support for any clade encompassing more than one of the entognathous lineages Protura, Diplura, and Collembola is weak, largely because character polarization is problematic. This is due to the lack of applicability of characters and/or missing comparative studies in the crustacean groups that are discussed to be most closely related to Hexapoda (Szucsich and Pass 2008).
Ellipura versus Nonoculata
Molecular analyses mostly support Nonoculata (Protura + Diplura) (Giribet et al. 2004; Luan et al. 2005; Kjer et al. 2006; Mallatt and Giribet 2006; Misof et al. 2007; Dell’Ampio et al. 2009; von Reumont et al. 2009; Mallatt et al. 2010; see Dell’Ampio et al. 2011 for a review) while most morphologists merge Protura and Collembola into Ellipura (Börner 1910; Hennig 1953; Kristensen 1981, 1997; Kukalová-Peck 1987; Bitsch and Bitsch 2000, 2004; Beutel and Gorb 2006). Molecular evidence for Ellipura is weak and limited to three mitochondrial single-gene analyses (Shao et al. 1999; Carapelli et al. 2000; Zhang et al. 2001), and morphological support for Nonoculata is nearly missing (Szucsich and Pass 2008). These controversies call for phylogenomic approaches.
The majority of the 117 genes that compose the decisive data set M_Ento contain predominant signal for Nonoculata (fig. 4a). Also, the FcLM analysis of M_Ento (fig. 2) and the phylogenetic tree of M_Ento (fig. 1) yielded monophyletic Nonoculata, albeit not being well supported. In summary, Nonoculata is slightly favored over Ellipura in our study, but the question of the phylogenetic relationships of the three entognathous hexapod orders remains unsettled.
Conclusions
Clades may be incorrect, even if receiving high BS support (e.g., monophyly of Entognatha in Meusemann et al. [2010], von Reumont et al. [2012], and in data sets SOS and SOSω of this study). This is a trivial conclusion and different reasons are mentioned in the literature (Lehtonen 2011, Simmons and Freudenstein 2011). We show that an uneven distribution of missing data (i.e., the use of indecisive data sets) can lead to strongly supported, yet incorrect, clades. To avoid misleading phylogenetic conclusions from seemingly robust trees based on phylogenomic data sets, we advise 1) using only data sets that are decisive for the phylogenetic question of interest, 2) including an alternative measure of support (Salichos and Rokas 2013); our method of choice was the FcLM approach, and 3) analyzing and documenting the inferred incongruence of signal between genes.
In our decisive data set, we found strong incongruence among genes that is neither correlated with functional classes of genes nor with model misspecifications in unpartitioned analyses. Based upon these notes of caution, we found no signal for the monophyly of Entognatha, and we found no strong signal for Ellipura or Nonoculata despite extending our data set with additional data from key taxa. In other words, the phylogeny and evolution of early hexapods remains enigmatic. Despite this, we show that there are valuable lessons to be learned from the analyses of phylogenomic data of primarily wingless insects, particularly in terms of incongruence among genes and data decisiveness.
Materials and Methods
Taxon Sampling and New Transcriptome Data
Our taxon sampling included 73 species: 46 hexapods, and, as outgroup species, 25 crustaceans, the chelicerate Ixodes scapularis, and the polychaete worm Capitella sp., both present in the reference set of taxa used for orthology assignment (discussed later). Transcriptome assemblies of 71 species were obtained from the Deep Metazoan Phylogeny database (http://www.deep-phylogeny.org/, last accessed November 4, 2013). We only used species for which more than 1,000 contigs were available (status: December 2011), with two exceptions: the springtail Orchesella cincta (Collembola, Entomobryidae, 754 contigs) and the silverfish Tricholepisma aurea (Zygentoma, Lepismatidae, 344 contigs), the only publicly available zygentoman transcriptome assembly (supplementary table S1, Supplementary Material online).
We generated new transcriptome data for Megajapyx sp. (Diplura, Japygidae) and the firebrat Thermobia domestica (Packard 1837) (Zygentoma, Lepismatidae) (table 1). Extraction of RNA, complementary deoxyribonucleic acid (cDNA) library construction, library normalization, and 454 pyrosequencing of ∼1,000,000 ESTs per species using the GS-FLX Titanium System, ROCHE were carried out at the Max Planck Institute for Molecular Genetics (MPIMG), Berlin, Germany. Vector clipping, trimming, and soft masking of raw reads and assembly into contigs was conducted at the Center for Integrative Bioinformatics (CIBIV), Vienna, Austria. Steps at the MPIMG and the CIBIV were done as described in von Reumont et al. (2012) and Simon et al. (2012), for details see supplementary material (section 1; Supplementary Material online). Raw sequence reads were deposited at the National Center for Biotechnology Information (NCBI), Sequence Read Archive (accession numbers Megajapyx sp.: SRR400673; T. domestica: SRR400672). Transcriptome assemblies of Megajapyx sp. (accession numbers JT047774–JT094274) and T. domestica (accession numbers T494145–JT533227) were deposited at the Transcriptome Shotgun Assembly (TSA) Database, NCBI Bioproject ID PRJNA81579 and PRJNA81581 (http://www.ncbi.nlm.nih.gov/bioproject, last accessed November 4, 2013). For submission, we excluded contigs shorter than 200 bp, according to the submission guidelines; the full transcriptome assemblies are available at http://zfmk.de/bioinformatics/Full_Transcriptome_Assemblies.zip (last accessed November 4, 2013).
Orthology Assignment
To identify 1:1 OGs in our transcriptome assemblies, we used the Hidden Markov Model based Search for Orthologs using Reciprocity (HaMStR) pipeline (Ebersberger et al. 2009; http://www.deep-phylogeny.org/hamstr/, last accessed November 4, 2013), version 4. As reference set for clusters of OGs, we used a set of 1,886 1:1 OGs (represented by amino acid sequences) based on five reference species (supplementary material [section 2] and table S2, Supplementary Material online). We defined orthology being present if bi-directional best hits were found between our transcript sequences and the reference species Daphnia pulex, Ixodes scapularis, Apis mellifera, and Capitella sp.
Alignment, Alignment Masking, and Concatenation
We aligned amino acid sequences using MAFFT L-INS-i (Katoh and Toh 2008) v.6.850 for each gene separately. Afterwards, randomly similar aligned sections were identified with a modified version of ALISCORE (Misof B and Misof K 2009; Kück et al. 2010; Meusemann et al. 2010; for modifications, see Meusemann et al. 2010) using the following options: default sliding window size; -r: maximum number of pairwise sequence comparisons; -e: special scoring for gappy amino acid data. Identified randomly similar aligned sections were masked with ALICUT v.2.0 (Kück 2009; www.utilities.zfmk.de, last accessed November 4, 2013). Masked alignments were concatenated into supermatrices with FASconCAT v.1.0 (Kück and Meusemann 2010).
Design of Decisive and Indecisive Data Sets
We extracted all genes from the supermatrix that contain at least one representative of each 1) Protura, 2) Diplura, 3) Collembola, and 4) remaining species to generate a decisive data set among entognathous lineages. The resulting data set is called M_Ento.
We generated two additional data subsets from the original supermatrix: 1) A so-called selected optimal subset (SOS), generated with MARE v.0.1.2-rc (Meyer and Misof 2010; http://mare.zfmk.de, last accessed November 4, 2013), applying taxon weighting -t 1.5. This approach is analogous to Meusemann et al. (2010) and von Reumont et al. (2012). 2) From this SOS data set, we compiled a data set called SOSω by removing all genes that were covered by all three entognathous lineages to receive a maximally indecisive “worst case” data set in which each gene contained maximally two entognathous lineages.
Four-Cluster Likelihood Mapping
Additional to tree reconstruction with BS, we applied the FcLM approach using the M_Ento data set (Strimmer and von Haeseler 1997). We binned sequenced species into four clusters: 1) Protura (1 species), 2) Diplura (2 species), 3) Collembola (5 species), and 4) remaining species (65 species) (supplementary table S1, Supplementary Material online). Next, we 1) estimated the tree-likeness of each gene, that is the amount of quartets that showed support for one out of the three possible topologies and 2) evaluated which of the three possible topologies was supported by the majority of those quartets (predominant support): T1 (Protura + Diplura) and (Collembola + remaining taxa), T2 (Protura + Collembola), and (Diplura + remaining taxa), or T3 (Diplura + Collembola) and (Protura + remaining taxa) (fig. 5). The competing hypotheses of Meusemann et al. (2010) and von Reumont et al. (2012) are represented by either T1 (Nonoculata hypothesis) or T2 (Ellipura hypothesis); the third topology T3 does not represent a currently debated hypothesis. FcLM was conducted using TREE-PUZZLE v.5.2 (Schmidt et al. 2002; http://www.tree-puzzle.de, last accessed November 4, 2013), applying the BLOSUM62 substitution matrix (Henikoff S and Henikoff JG 1992) as the BLOSUM62 substitution matrix is implemented in the software MARE (Meyer and Misof, 2010; http://mare.zfmk.de, last accessed November 4, 2013).
Fig. 5.

2D simplex graph. Voronoi cells are areas, in which quartets show predominant or maximal support for either of the three topologies T1, T2, T3, or in which quartets show ambiguous support T12, T13, T23, and T*. For further explanations, refer to Strimmer and von Haeseler (1997, fig. 3). Voronoi cell corresponding to T1 (blue): quartets show support for (Protura + Diplura) – (Collembola + remaining taxa); Voronoi cell corresponding to T2 (red): quartets show support for (Protura + Collembola) – (Diplura + remaining taxa); Voronoi cell corresponding to T3 (yellow): quartets show support for (Diplura + Collembola) – (Protura + remaining taxa); Voronoi cells corresponding to T12, T13, T23 (gray) do not show clear support for T1, T2, and T3; in T* all topologies are equally likely.
For each gene in the data set M_Ento, we calculated the proportions of quartets that predominantly supported either topology T1, T2, or T3. According to the topology that was supported by the majority of quartets, we classified each gene into one of three groups, supporting Nonoculata, Ellipura, or Diplura + Collembola (fig. 5 and supplementary table S4, Supplementary Material online). Quartets for which the support remained ambiguous (T12, T23, T13, and T*; fig. 5) were not used for classification (see supplementary fig. S14 [Supplementary Material online] for the results with all quartets). All classified genes (supplementary table S4, Supplementary Material online) were subsequently concatenated into three submatrices called M_Nono (genes supporting Nonoculata), M_Elli (genes supporting Ellipura), and M_DiCo (genes supporting Diplura + Collembola).
Phylogenetic Tree Inference
ML tree reconstruction was done from all data sets: M_Ento, M_Nono, M_Elli, and M_DiCo, SOS, and SOSω (discussed earlier). We estimated evolutionary models for each data set with ModelGenerator v.0.85 (Keane et al. 2006). The best fitting model was selected based upon the Akaike Information Criterion (AIC; Akaike 1974). ML trees were inferred with RAxML (Stamatakis 2006), v.7.2.8-ALPHA, HYBRID (Ott et al. 2007; Pfeiffer and Stamatakis 2010) using the CAT model of rate heterogeneity (Stamatakis 2006) and the LG protein substitution matrix (Le and Gascuel 2008). Final tree searches were conducted under the GAMMA model of rate heterogeneity (Yang 1996). Bootstrap analyses were performed with the rapid algorithm (Stamatakis 2006), which also included subsequent searches for the best scoring ML tree. We obtained BS for each node from 1,000 rapid bootstrap replicates, and checked a posteriori if sufficient bootstrap trees were computed using the bootstopping criteria (Pattengale et al. 2010, default settings). ML analyses were conducted on a Linux cluster at the Cologne High Efficient Operating Platform for Science (CHEOPS), Regionales Rechenzentrum Köln (RRZK), using eight nodes with 12 cores each.
After tree inference, we scrutinized our trees for rogue taxa (Aberer et al. 2013; Aberer and Stamatakis 2011, see supplementary material [section 3], figs. S2 and S4, table S7, Supplementary Material online, for details and results). We removed sequences corresponding to taxa that were identified as rogues from the concatenated alignments and repeated the tree inferences. All trees were edited with Treegraph v.2.0 (Stöver and Müller 2010), and rooted with Capitella sp. Data sets are deposited at Dryad: http://doi.org/10.5061/dryad.mk8p7 (last accessed November 4, 2013).
Supplementary Material
Supplementary material (sections 1–6), tables S1–S7, and figures S1–S14 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
K.M. and B.Mi. provided EST data for Thermobia domestica, and G.P., E.D.A. and N.U.S. for Megajapyx sp. J.B. and M.P. wrote Perl scripts for the analysis pipeline and B.Me. adopted the FcLM approach. Processing and sequence assembly of EST data were performed by I.E. and A.v.H. The study was conceived by B.Mi., K.M., and E.D.A. Orthology assignment and subsequent analyses were conducted by K.M. Rogue taxa analyses were performed by A.J.A. and A.S. Partitioned ML analyses were provided by B.Q.M. The manuscript was written by K.M., N.U.S., R.S.P., E.D.A., and B.Mi with useful comments and revisions from A.J.A., B.Q.M., M.G.W., A.v.H., I.E., A.S., and G.P. All authors read and approved the final manuscript. The authors thank Martin Streinzer for help in collecting Megajapyx sp. They acknowledge Michael Kube and Richard Reinhardt (MPIMG, Berlin, Germany) for extraction of RNA, generating cDNA libraries, and ESTs. They thank Sascha Strauss for help with processing and assembling the EST data and John Plant (University of Vienna, Austria) for examining the English. They acknowledge the Cologne High Efficient Operating Platform for Science (CHEOPS, HPC cluster at the RRZK, University of Cologne, Cologne, Germany; available from: http://rrzk.uni-koeln.de/cheops.html) for the opportunity to perform analyses. Finally, the authors thank two anonymous reviewers for helpful comments that considerably improved the manuscript. This work was supported by the Austrian Science Foundation (FWF) grant P 20497-B17 to E.D.A., N.U.S. and G.P., the German Science Foundation (DFG): priority program SPP 1174 “Deep Metazoan Phylogeny” (http://www.deep-phylogeny.org), and by institutional funding of the Heidelberg Institute for Theoretical Studies. A.v.H. and I.E. were funded by the German Science Foundation (DFG) grant HA1628/9. A.v.H. and B.Q.M. were supported by the Austrian Science Foundation (FWF) grant I760. K.M. and B.M. were funded by the German Science Foundation (DFG) grant MI 649/6.
References
- Aberer AJ, Komprass D, Stamatakis A. Pruning Rogue Taxa improves phylogenetic accuracy: an efficient algorithm and web service. Syst Biol. 2013;62(1):162–166. doi: 10.1093/sysbio/sys078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aberer AJ, Stamatakis A. A simple and accurate method for rogue taxon identification. IEEE BIBM. 2011;2011:118–122. [Google Scholar]
- Akaike H. A new look at the statistical model identification. IEEE Trans Aut Control. 1974;19:716–723. [Google Scholar]
- Bitsch C, Bitsch J. The phylogenetic interrelationships of the higher taxa of apterygote hexapods. Zool Scr. 2000;29:131–156. [Google Scholar]
- Bitsch C, Bitsch J. Phylogenetic relationships of basal hexapods among the mandibulate arthropods: a cladistic analysis based on comparative morphological characters. Zool Scr. 2004;33:511–550. [Google Scholar]
- Beutel RG, Gorb SN. A revised interpretation of attachment structures in Hexapoda with special emphasis on Mantophasmatodea. Arthropod Syst Phylogeny. 2006;64:3–25. [Google Scholar]
- Börner C. Die phylogenetische Bedeutung der Protura. Sdr Biolog Centralbl. 1910;30:633–641. [Google Scholar]
- Carapelli A, Frati F, Nardi F, Dallai R, Simon C. Molecular phylogeny of the apterygote insects based on nuclear and mitochondrial genes. Pedobiologia. 2000;44:361–373. [Google Scholar]
- Colgan DJ, McLauchlan A, Wilson GDF, Livingston S, Macaranas J, Edgecombe D, Cassis G, Gray MR. Histone H3 and U2 snRNA DNA sequences and arthropod molecular evolution. Aust J Zool. 1998;46:419–437. [Google Scholar]
- Dallai R, Mercati D, Carapelli A, Nardi F, Machida R, Sekiya K, Frati F. Sperm accessory microtubules suggest the placement of Diplura as the sister-group of Insecta s.s. Arthropod Struct Dev. 2011;40:77–92. doi: 10.1016/j.asd.2010.08.001. [DOI] [PubMed] [Google Scholar]
- Degnan JH, Rosenberg NA. Gene tree discordance, phylogenetic inference, and the multispecies coalescent. Trends Ecol Evol. 2009;24:332–340. doi: 10.1016/j.tree.2009.01.009. [DOI] [PubMed] [Google Scholar]
- Dell’Ampio E, Szucsich NU, Carapelli A, Frati F, Steiner G, Steinacher A, Pass G. Testing for misleading effects in the phylogenetic reconstruction of ancient lineages of hexapods: influence of character dependence and character choice in analyses of 28S rRNA sequences. Zool Scr. 2009;38:155–170. [Google Scholar]
- Dell’Ampio E, Szucsich NU, Pass G. Protura and molecular phylogenetics: status quo of a young love. Soil Organisms. 2011;83:347–358. [Google Scholar]
- Ebersberger I, Strauss S, Von Haeseler A. HaMStR: profile hidden Markov model based search for orthologs in ESTs. BMC Evol Biol. 2009;9:157. doi: 10.1186/1471-2148-9-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgecombe GD, Wilson GDF, Colgan DJ, Gray MR, Cassis G. Arthropod cladistics: combined analysis of Histone H3 and U2 snRNA sequences and morphology. Cladistics. 2000;16:155–203. doi: 10.1111/j.1096-0031.2000.tb00352.x. [DOI] [PubMed] [Google Scholar]
- Gao Y, Bu Y, Luan Y. Phylogenetic relationships of basal hexapods reconstructed from nearly complete 18S and 28S rRNA gene sequences. Zool Sci. 2008;25:1139–1145. doi: 10.2108/zsj.25.1139. [DOI] [PubMed] [Google Scholar]
- Giribet G, Edgecombe GD, Carpenter JM, D’Haese CA, Wheeler WC. Is Ellipura monophyletic? A combined analysis of basal hexapod relationships with emphasis on the origin of insects. Org Div Evol. 2004;4:319–340. [Google Scholar]
- Giribet G, Edgecombe GD, Wheeler WC. Arthropod phylogeny based on eight molecular loci and morphology. Nature. 2001;413:157–161. doi: 10.1038/35093097. [DOI] [PubMed] [Google Scholar]
- Giribet G, Richter S, Edgecombe GD, Wheeler WC. The position of crustaceans within Arthropoda—evidence from nine molecular loci and morphology. In: Koenemann S, Jenner RA, editors. Crustacea and arthropod relationships. Crustacean issues 16: Festschrift for F. R. Schram. Boca Raton (FL): Taylor & Francis; 2005. pp. 307–352. [Google Scholar]
- Giribet G, Wheeler WC. Some unusual small-subunit ribosomal DNA sequences of metazoans. Am Mus Novit. 2001;3337:1–14. [Google Scholar]
- Grimaldi DA. 400 million years on six legs: on the origin and early evolution of Hexapoda. Arthropod Struct Dev. 2010;39:191–203. doi: 10.1016/j.asd.2009.10.008. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig W. Kritische Bemerkungen zum phylogenetischen System der Insekten. Beitr Entomol Sonderheft. 1953;3:1–85. [Google Scholar]
- Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 2008;9:286–298. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- Keane TM, Creevey CJ, Pentony MM, Naughton TJ, McInerney JO. Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol Biol. 2006;6:29. doi: 10.1186/1471-2148-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjer KM, Carle FL, Litman J, Ware J. A molecular phylogeny of Hexapoda. Arthropod Syst Phylogeny. 2006;64:3–44. [Google Scholar]
- Knowles LL. Estimating species trees: methods of phylogenetic analysis when there is incongruence across genes. Syst Biol. 2009;58:463–467. doi: 10.1093/sysbio/syp061. [DOI] [PubMed] [Google Scholar]
- Koch M. Monophyly and phylogenetic position of the Diplura (Hexapoda) Pedobiologia. 1997;41:9–12. [Google Scholar]
- Kristensen NP. Phylogeny of insect orders. Annu Rev Entomol. 1981;26:135–157. [Google Scholar]
- Kristensen NP. The ground plan and basal diversification of the hexapods. In: Fortey RA, Thomas RH, editors. Arthropod relationships, systematic association. Special volume series 55. London: Chapman & Hall; 1997. pp. 281–293. [Google Scholar]
- Kück P. ALICUT: a Perlscript which cuts ALISCORE identified RSS. 2009. Version 2.0 ed. Bonn (Germany): Department of Bioinformatics, Zoologisches Forschungs Museum A. Koenig (ZFMK). [cited 2013 Oct 30]. Available from: http://www.zfmk.utilities.de. [Google Scholar]
- Kück P, Meusemann K. FASconCAT: convenient handling of data matrices. Mol Phylogenet Evol. 2010;56:1115–1118. doi: 10.1016/j.ympev.2010.04.024. [DOI] [PubMed] [Google Scholar]
- Kück P, Meusemann K, Dambach J, Thormann B, von Reumont BM, Waegele JW, Misof B. Parametric and non-parametric masking of randomness in sequence alignments can be improved and leads to better resolved trees. Front Zool. 2010;7:10. doi: 10.1186/1742-9994-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukalová-Peck J. New Carboniferous Diplura, Monura and Thysanura, the hexapod ground plan, and the role of thoracic side lobes in the origin of wings (Insecta) Can J Zool. 1987;65:2327–2345. [Google Scholar]
- Kukalová-Peck J. Fossil history and the evolution of hexapod structures. In: Naumann ID, editor. Insects of Australia: a textbook for students and research workers. Melbourne (Australia): CSIRO, Melbourne University Press; 1991. pp. 141–179. [Google Scholar]
- Le SQ, Gascuel O. An improved general amino acid replacement matrix. Mol Biol Evol. 2008;25:1307–1320. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- Lehtonen S. Can sensitivity analysis help to detect long-branch attraction? Mol Phylogenet Evol. 2011;61:899–903. doi: 10.1016/j.ympev.2011.08.007. [DOI] [PubMed] [Google Scholar]
- Luan Y, Mallatt JM, Xie R, Yang Y, Yin W. The phylogenetic positions of three basal-hexapod groups (Protura, Diplura, and Collembola) based on ribosomal RNA gene sequences. Mol Biol Evol. 2005;22:1579–1592. doi: 10.1093/molbev/msi148. [DOI] [PubMed] [Google Scholar]
- Machida R. Evidence from embryology for reconstructing the relationships of hexapod basal clades. Arthropod Syst Phylogeny. 2006;64:95–104. [Google Scholar]
- Mallatt J, Giribet G. Further use of nearly complete 28S and 18S rRNA genes to classify Ecdysozoa: 37 more arthropods and a kinorhynch. Mol Phylogenet Evol. 2006;40:772–794. doi: 10.1016/j.ympev.2006.04.021. [DOI] [PubMed] [Google Scholar]
- Mallatt JM, Craig CW, Yoder MJ. Nearly complete rRNA genes assembled from across the metazoan animals: effects of more taxa, a structure-based alignment, and paired-sites evolutionary models on phylogeny reconstruction. Mol Phylogenet Evol. 2010;55:1–17. doi: 10.1016/j.ympev.2009.09.028. [DOI] [PubMed] [Google Scholar]
- Meusemann K, von Reumont BM, Simon S, et al. (16 co-authors) A phylogenomic approach to resolve the arthropod tree of life. Mol Biol Evol. 2010;27:2451–2464. doi: 10.1093/molbev/msq130. [DOI] [PubMed] [Google Scholar]
- Meyer B, Misof B. MARE: matrix reduction—a tool to select optimized data subsets from supermatrices for phylogenetic inference. Bonn (Germany): Zentrum für Molekulare Biodiversitätsforschung (zmb) am ZFMK; 2010. [cited 2013 Oct 30]. Available from: http://mare.zfmk.de (current version) [Google Scholar]
- Minh BQ, Nguyen MAT, von Haeseler A. Ultrafast approximation for phylogenetic bootstrap. Mol Biol Evol. 2013;30:1188–1195. doi: 10.1093/molbev/mst024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misof B, Niehuis O, Bischoff I, Rickert A, Erpenbeck D, Staniczek A. Towards an 18S phylogeny of hexapods: accounting for group-specific character covariance in optimized mixed nucleotide/doublet models. Zoology. 2007;110:409–429. doi: 10.1016/j.zool.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Misof B, Misof K. A Monte Carlo approach successfully identifies randomness in multiple sequence alignments: a more objective means of data exclusion. Syst Biol. 2009;58:21–34. doi: 10.1093/sysbio/syp006. [DOI] [PubMed] [Google Scholar]
- Ott M, Zola J, Stamatakis A, Aluru S. Large-scale maximum likelihood-based phylogenetic analysis on the IBM BlueGene/L. Proceedings of the 2007 ACM/IEEE conference on Supercomputing. 2007. IEEE/ACM Supercomputing conference 2007 (SC2007); November 2007. Reno (NV): ACM. [Google Scholar]
- Pattengale ND, Alipour M, Bininda-Emonds OR, Moret BM, Stamatakis A. How many bootstrap replicates are necessary? J Comput Biol. 2010;17:337–354. doi: 10.1089/cmb.2009.0179. [DOI] [PubMed] [Google Scholar]
- Pfeiffer W, Stamatakis A. 2010. Hybrid MPI/Pthreads parallelization of the RAxML phylogenetics code. Paper presented at HICOMB workshop, held in conjunction with IPDPS 2010; April 2010; Atlanta, GA. [Google Scholar]
- Philippe H, Brinkmann H, Lavrov DV, Littlewood DT, Manuel M, Worheide G, Baurain D. Resolving difficult phylogenetic questions: why more sequences are not enough. PLoS Biol. 2011;9:e1000602. doi: 10.1371/journal.pbio.1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regier JC, Shultz JW, Ganley AR, et al. (11 co-authors) Resolving arthropod phylogeny: exploring phylogenetic signal within 41 kb of protein-coding nuclear gene sequence. Syst Biol. 2008;57:920–938. doi: 10.1080/10635150802570791. [DOI] [PubMed] [Google Scholar]
- Regier J, Shultz J, Zwick A, Hussey A, Ball B, Wetzer R, Martin JW, Cunningham CW. Arthropod relationships revealed by phylogenomic analysis of nuclear protein-coding sequences. Nature. 2010;463:1079–1083. doi: 10.1038/nature08742. [DOI] [PubMed] [Google Scholar]
- Salichos L, Rokas A. Inferring ancient divergences requires genes with strong phylogenetic signals. Nature. 2013;497: 327–331. doi: 10.1038/nature12130. [DOI] [PubMed] [Google Scholar]
- Sanderson MJ, McMahon MM, Steel M. Phylogenomics with incomplete taxon coverage: the limits to inference. BMC Evol Biol. 2010;10:155. doi: 10.1186/1471-2148-10-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt HA, Strimmer K, Vingron M, von Haeseler A. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18:502–504. doi: 10.1093/bioinformatics/18.3.502. [DOI] [PubMed] [Google Scholar]
- Shao HG, Zhang YP, Xie RD, Yin WY. Mitochondria cytochrome b sequences variation of Protura and molecular systematics of Apterygota. Chin Sci Bull. 1999;44:2031–2036. [Google Scholar]
- Simmons MP, Freudenstein JV. Spurious 99% bootstrap and jackknife support for unsupported clades. Mol Phylogenet Evol. 2011;61:177–191. doi: 10.1016/j.ympev.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Simon S, Narechania A, DeSalle R, Hadrys H. Insect phylogenomics: exploring the source of incongruence using new transcriptomic data. Genome Biol Evol. 2012;4:1295–1309. doi: 10.1093/gbe/evs104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Steel M, Sanderson MJ. Characterizing phylogenetically decisive taxon coverage. Appl Math Lett. 2010;23:82–86. [Google Scholar]
- Strimmer K, von Haeseler A. Likelihood-mapping: a simple method to visualize phylogenetic content of a sequence alignment. Proc Natl Acad Sci U S A. 1997;94:6815–6819. doi: 10.1073/pnas.94.13.6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöver BC, Müller KF. TreeGraph 2: combining and visualizing evidence from different phylogenetic analyses. BMC Bioinformatics. 2010;11:7. doi: 10.1186/1471-2105-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szucsich NU, Pass G. Incongruent phylogenetic hypotheses and character conflicts in morphology: the root and early branches of the hexapodan tree. Mitt Dtsch Ges Allg Angew Ent. 2008;16:415–430. [Google Scholar]
- Trautwein MD, Wiegmann BM, Beutel R, Kjer KM, Yeates DK. Advances in insect phylogeny at the dawn of the postgenomic era. Annu Rev Entomol. 2012;57:449–468. doi: 10.1146/annurev-ento-120710-100538. [DOI] [PubMed] [Google Scholar]
- von Reumont BM, Meusemann K, Szucsich N, et al. (14 co-authors) Can comprehensive background knowledge be incorporated into substitution models to improve phylogenetic analyses? A case study on major arthropod relationships. BMC Evol Biol. 2009;9:119. doi: 10.1186/1471-2148-9-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Reumont MB, Jenner RA, Wills MA, et al. (13 co-authors) Pancrustacean phylogeny in the light of new phylogenomic data: support for Remipedia as the possible sister group of Hexapoda. Mol Biol Evol. 2012;29:1031–1045. doi: 10.1093/molbev/msr270. [DOI] [PubMed] [Google Scholar]
- Wiens JJ. Missing data and the design of phylogenetic analyses. J Biomed Inform. 2006;39:34–42. doi: 10.1016/j.jbi.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Yang Z. Among-site rate variation and its impact on phylogenetic analyses. Trends Ecol Evol. 1996;11(9):367–372. doi: 10.1016/0169-5347(96)10041-0. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhang Y, Luan Y, Chen Y, Yin W. Phylogeny of higher taxa of Hexapoda according to 12sRNA sequences. Chin Sci Bull. 2001;46:840–842. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.