

Background: Receptor guanylyl cyclase C regulates ion secretion and cytostasis in intestinal epithelial cells.
Results: Ligand-mediated activation of guanylyl cyclase C and subsequent elevation of cGMP increase levels of p21 via PKGII and p38 MAPK.
Conclusion: Guanylyl cyclase C can induce intestinal epithelial cell cytostasis and senescence via p21.
Significance: Intestinal neoplasia is controlled by cGMP and p21.
Keywords: Cyclic GMP (cGMP), Intestinal Epithelium, Microarray, Protein Kinase G (PKG), Senescence, Sp1, GCC Knock-out mice, T84 Cell, p21, Receptor Guanylyl Cyclase C
Abstract
Guanylyl cyclase C (GC-C) is expressed in intestinal epithelial cells and serves as the receptor for bacterial heat-stable enterotoxin (ST) peptides and the guanylin family of gastrointestinal hormones. Activation of GC-C elevates intracellular cGMP, which modulates intestinal fluid-ion homeostasis and differentiation of enterocytes along the crypt-villus axis. GC-C activity can regulate colonic cell proliferation by inducing cell cycle arrest, and mice lacking GC-C display increased cell proliferation in colonic crypts. Activation of GC-C by administration of ST to wild type, but not Gucy2c−/−, mice resulted in a reduction in carcinogen-induced aberrant crypt foci formation. In p53-deficient human colorectal carcinoma cells, ST led to a transcriptional up-regulation of p21, the cell cycle inhibitor, via activation of the cGMP-responsive kinase PKGII and p38 MAPK. Prolonged treatment of human colonic carcinoma cells with ST led to nuclear accumulation of p21, resulting in cellular senescence and reduced tumorigenic potential. Our results, therefore, identify downstream effectors for GC-C that contribute to regulating intestinal cell proliferation. Thus, genomic responses to a bacterial toxin can influence intestinal neoplasia and senescence.
Introduction
The high self-renewal rate of the colorectal epithelia predisposes this tissue to the development of cancers. Most colorectal cancers begin as small benign adenomatous polyps, and invariably, some of these progress into invasive carcinomas and, ultimately, metastasis (1). Both sporadic and hereditary forms of colorectal cancer develop along a well defined sequence of histopathological and genetic changes (2). Although surgery remains the first line of treatment, methods to control the proliferation of metastatic tumors are required to control later stages of the disease (3).
Higher eukaryotes have evolved multiple checkpoint mechanisms to monitor the cell cycle and halt protumorigenic activity. The tumor suppressor protein p53 mediates the DNA damage-induced checkpoint through transactivation of various growth-inhibitory or proapoptotic genes. Among these, p21 (also known as p21WAF1/Cip1) mediates a p53-dependent G1 growth arrest (4, 5). The tumor suppressor activity of p21 stems from its role in inducing growth arrest, differentiation, or senescence. Cells lacking p21 expression fail to arrest the cell cycle at the G1 checkpoint and do not senesce in response to DNA damage (6). Consequently, p21 is often found to be dysregulated in human cancers and has been identified as a risk locus associated with colorectal cancers (7). Growth inhibition of colon cancer cells mediated by p21 can occur in a p53-dependent or -independent manner (8, 9).
The guanylyl cyclase C (GC-C)3 receptor is expressed at the apical membranes of intestinal cells. Binding of its ligands guanylin and uroguanylin and the diarrhea-causing heat-stable enterotoxins (ST) results in intracellular accumulation of cyclic GMP (cGMP) and fluid-ion efflux (10). Indeed, mutations in the human GUCY2C gene have been associated with severe gastrointestinal disturbances, emphasizing the important role of this receptor in intestinal physiology and function (11, 12). In addition to maintenance of intestinal fluid-ion balance, GC-C also regulates intestinal epithelial cell proliferation. Colonic epithelia of mice deficient in GC-C are prone to colonic tumors induced by carcinogens or inherited germ line mutations and demonstrate an accelerated cell cycle, disruption of genomic stability, and activation of protumorigenic signaling pathways (13, 14). Uroguanylin and guanylin appear to play a key role in regulating the balance between proliferation and differentiation in the intestinal epithelia via cGMP and release of intracellular Ca2+ through cyclic nucleotide-gated channels (15). Guanylin knock-out mice show increased crypt depth and a higher number of proliferating cells, reiterating the role of GC-C in regulating intestinal crypt biology (16).
Uroguanylin and guanylin expression is reduced in colon carcinoma, whereas GC-C expression remains comparable with that seen in normal colonic mucosa (17–19). Thus, GC-C is a marker for metastatic colorectal carcinoma (20), and supplementation with uroguanylin has been shown to decrease tumorigenesis in mouse models of intestinal carcinogenesis (19). The role of GC-C as a moderator of cell proliferation suggests that GC-C and cGMP are vital components of a cytostatic axis, dysregulation of which promotes tumorigenesis. Here, we have delineated a signaling pathway emerging from GC-C whereby the up-regulation of p21 results in cellular cytostasis and induction of cellular senescence in intestinal epithelial cells.
EXPERIMENTAL PROCEDURES
Cell Culture
The T84 cell line was obtained from ATCC (Manassas, VA). Cells were cultured in DMEM/F-12 containing 120 mg/liter penicillin and 270 mg/liter streptomycin in the presence of 5% FBS (Invitrogen). T84 cells were transfected with the required siRNA using TransIT-TKO siRNA transfection reagent (Mirus) according to the manufacturer's protocols. Control siRNA (sc-37007) and protein kinase G II (PKGII) siRNA (sc-38974) were obtained from Santa Cruz Biotechnology. EGFP esiRNA (EHUEGFP) and p21 esiRNA (EHU003861) were obtained from Sigma-Aldrich.
Maintenance of Mice
Gucy2c−/+ mice were obtained from The Jackson Laboratory (21). They were backcrossed with C57BL/6 mice for six generations prior to use in experiments. All mice were housed under specific pathogen-free conditions and maintained in individually ventilated cages. Temperature (22 ± 2 °C) and humidity (55 ± 10%) were kept constant with a 12-h light/dark cycle, and mice had access to standard laboratory chow and water ad libitum. Litter-matched male and female Gucy2c+/+ and Gucy2c−/− mice of age 6–8 weeks were used. Animal studies and experiments were approved and carried out according to institutional guidelines for animal use and care at the Indian Institute of Science, Bangalore, India.
Colonic Crypt Isolation and Treatments
Excised colons were washed in ice-cold PBS and then cut into small pieces. The pieces were then incubated in PBS containing 3 mm EDTA and 0.5 mm DTT (PBSED) at room temperature for 60 min. The solution containing loose pieces of mesenchyme and intestine was decanted, cold PBSED was added, and the tube was shaken vigorously 10 times. The cells shed into the PBSED were discarded. The intestine pieces were again shaken vigorously 10–15 times in fresh PBSED, and the supernatant was collected. Crypts shed into the supernatant were resuspended in DMEM/F-12 containing 5% FBS and used immediately for experiments. Approximately 500 crypts were treated with STh (referred to as ST throughout the text) (22) at a concentration of 100 nm for 1 h.
Measurement of Cyclic Nucleotides
Colonic crypts from Gucy2c+/+ and Gucy2c−/− mice were prepared as described above in the presence of 3-isobutyl-1-methylxanthine (500 μm). Crypts were pelleted and lysed in 0.1 n HCl. Cyclic GMP and cAMP were estimated by radioimmunoassay as described previously (23).
Reverse Transcription-PCR
RNA was isolated using TRI Reagent (Sigma-Aldrich) according to the manufacturer's instructions. RNA was treated with DNase I (MBI, Thermo Scientific) and reverse transcribed using RevertAid reverse transcriptase (MBI, Thermo-Scientific). Real time PCR was performed using the SensiFAST SYBR kit (Bioline, India) on an ABI 7000 real time PCR machine (Applied Biosystems). 18 S RNA was used as an internal control. The sequences of primers used for PCR are available on request.
Immunoprecipitation and Western Blot Analysis
T84 cells and colonic crypts were harvested into homogenization buffer (50 mm HEPES, pH 7, 100 mm NaCl, 5 mm EDTA, 5 μg/ml leupeptin, 5 μg/ml aprotinin, 2 mm PMSF). Cells were lysed by sonication and centrifuged for 1 h at 4 °C at 13,000 rpm. The membrane fraction obtained in the form of a pellet was resuspended in resuspension buffer (50 mm HEPES, pH 7, 5 μg/ml aprotinin, 5 μg/ml leupeptin, 5 μg/ml soybean trypsin inhibitor, 20% glycerol). Protein concentration was estimated by using a modified Bradford protein assay (24).
Membrane fractions (1 mg of total protein) were solubilized in 1% Triton X-100 and 500 mm NaCl for 2 h, and GC-C was immunoprecipitated using the GCC:4D7 monoclonal antibody (mAb) overnight at 4 °C. 10 μl of equilibrated protein A/G beads (Invitrogen) was added and incubated for an additional 2 h. The beads were pelleted at 4 °C and washed thrice with wash buffer (10 mm Tris-HCl, pH 7.5, 100 mm NaCl, 0.1% Triton X-100) and twice with TBS (10 mm Tris-HCl, pH 7.5, 100 mm NaCl). The beads were then boiled in SDS sample buffer, and proteins were subsequently analyzed by SDS-PAGE and Western blotting.
For Western blot analysis, whole cell lysates were prepared from T84 cells and colonic crypts by sonicating cells in PBS containing 2% SDS. Samples were diluted, and protein was estimated by a microBCA kit (Pierce, Thermo-Fisher). 50 μg of total protein from T84 cell lysates or 100 μg of total protein from colonic crypt lysates was loaded in each lane unless otherwise specified. Sp1, cystic fibrosis transmembrane conductance regulator, and PKGII antibodies were from Santa Cruz Biotechnology. Phospho-p38 MAPK, p21, phospho-Rb, total Rb, cyclin D1, and Histone deacetylase 1–6 (HDAC1–6) antibodies were from Cell Signaling Technology. Total p38 and tubulin antibodies were from Sigma-Aldrich. Blots were detected with a horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse antibody (GE Healthcare). Blots were processed with Luminata Crescendo HRP substrate (Millipore) and visualized on a FluorChem Q MultiImage III system (Alpha Innotech). All blots were repeated at least thrice and quantitated using Alpha View software (Version 3).
N-Methyl-N-nitrosourea (MNU)-induced Colorectal Carcinogenesis Model
Litter-matched Gucy2c +/+ and Gucy2c−/− mice were divided into four groups and given the indicated treatments by oral gavage. Each group consisted of six mice of each genotype except Group 1, which had three mice of each genotype. Group 1 was treated with PBS. Group 2 was treated with MNU at 50 mg/kg. Group 3 was treated with MNU at 50 mg/kg followed by three doses (100 μl) of ST (10 nm) every 48 h for 1 week. Group 4 was treated with MNU at 50 mg/kg followed by three doses (100 μl) of 8-pCPT-cGMP (100 μm) every 48 h for 1 week. At the end of 6 weeks, the mice were sacrificed, and colons were removed, fixed in 4% paraformaldehyde, and stained with 0.2% methylene blue for evaluation of formation of aberrant crypt foci (ACF) by stereomicroscopy. To investigate the effect of long term ST administration, Gucy2c+/+ and Gucy2c−/− mice (n = 3) were given 100 μl of ST (10 nm) by oral gavage every 48 h for 10 days following which colonic crypts were prepared, lysed in 2% SDS, and analyzed by Western blotting.
Immunofluorescence
Colons were fixed with 4% paraformaldehyde, dehydrated, and embedded in paraffin. Tissue sections (5 μm) prepared from paraffin blocks were subjected to antigen retrieval in citrate buffer, pH 6. Sections were stained with anti-Ki67 (2 μg/ml) (Abcam), Alexa Fluor 488-conjugated secondary antibody (Molecular Probes, Invitrogen) and counterstained with Hoechst 33342. Images were taken on a Leica TCS SP5 II confocal microscope (Leica Microsystems, Germany).
Microarray Analysis
Microarray experiments were performed using whole human genome (4 × 44,000) oligonucleotide arrays (Agilent Technologies, Santa Clara, CA) on RNA isolated from control and 1-h ST (100 nm)-treated T84 cells using an RNeasy kit (Qiagen). Labeling of probes was performed using the low RNA input linear amplification kit (Agilent Technologies). Hybridization and washing protocols were carried out according to Agilent guidelines. The LOWESS (locally weighted scatter plot smoothing) algorithm was used to normalize the data, and -fold change was calculated from the ratio of Cy5/Cy3 (treated/untreated) intensities. For statistical analysis, Student's t test was performed using Benjamini Hochberg multiple testing correction. The Gene Expression Omnibus (GEO) accession number for the microarray data is GSE45531.
Quantitative ChIP Assay
T84 cells (∼106) were cross-linked with formaldehyde (1%) and resuspended in 1 ml of swelling buffer (25 mm HEPES, pH 7, 1.25 mm MgCl2, 10 mm KCl, 1% Nonidet P-40, 1 mm DTT, protease inhibitor mixture (Roche Applied Science)). The cells were homogenized in a Dounce homogenizer (10 strokes) followed by centrifugation at 2000 rpm for 15 min. The nuclear pellet was resuspended in sonication buffer (50 mm HEPES, pH 7, 140 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, protease inhibitor mixture). Nuclei were sonicated 10 times for 10 s each at the 80% amplitude setting with a sonicator (IKA Laborotechnik, Germany) and centrifuged at 14,000 rpm for 10 min. 50–150 μg of sonicated chromatin was diluted 10-fold in ChIP dilution buffer (50 mm HEPES, pH 7, 140 mm NaCl, 1 mm EDTA, 1% Triton X-100, 0.01% SDS, protease inhibitor mixture) and precleared for 1 h at 4 °C with normal rabbit IgG bound to 20 μl of protein A/G-agarose. The supernatant was then incubated overnight at 4 °C with specific antibody or control IgG (5 μg). Antibody-bound chromatin was recovered following a 2-h incubation at 4 °C with 20 μl of protein A/G-agarose. The beads were washed once in low salt buffer (50 mm HEPES, pH 7, 140 mm NaCl, 1 mm EDTA, 0.1% SDS, 1% Triton X-100), twice in high salt buffer (50 mm HEPES, pH 7, 500 mm NaCl, 1 mm EDTA, 0.1% SDS, 1% Triton X-100), twice in LiCl buffer (50 mm HEPES, pH 7, 1 mm EDTA, 250 mm LiCl, 1% Nonidet P-40, 1% sodium deoxycholate), and twice in Tris-EDTA, pH 8. The bound DNA was eluted by two 15-min incubations at room temperature in 50 μl of elution buffer (1% SDS, 100 mm NaHCO3). Protein-DNA cross-links were reversed by addition of 5 μl of NaCl (5 m) and incubation at 65 °C for 4 h. The chromatin was then subjected to RNase A (10 μg/ml) and proteinase K (10 μg/ml) digestion and purified by phenol-chloroform extraction. Real time PCR was performed with the eluted DNA using p21 promoter-specific primers. -Fold enrichment was calculated with reference to of input chromatin.
Senescence-associated (SA)-β-galactosidase Assay and Senescence-associated Heterochromatin Foci Formation
SA-β-galactosidase activity for detection of senescent cells was performed as described previously with minor modifications (25). Cells were washed with PBS, fixed in 0.5% glutaraldehyde, and incubated with staining solution (1 mg/ml X-gal, 5 mm potassium ferrocyanide, 5 mm potassium ferricyanide, 150 mm NaCl, 1 mm MgCl2 in PBS at pH 6.0) for 12 h at 37 °C. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) and visualized on an Axio Observer Z.1 (Carl Zeiss Microsystems, Germany). Approximately 1000 cells were counted in each experiment.
T84 cells were fixed with 4% paraformaldehyde and stained with DAPI, and senescence-associated heterochromatin foci were visualized on a Leica TCS SP5 II (Leica Microsystems). Approximately 1000 cells were manually counted in each experiment.
Soft Agar Colony Formation Assay
T84 cells (105) were treated with ST (100 nm) or 8-pCPT-cGMP (25 μm) for 10 days. ST- or 8-pCPT-cGMP-containing medium was replaced every 3 days. Subsequently, these cells were trypsinized and resuspended in 0.3% Noble agar in DMEM/F-12 supplemented with 5% FBS at a density of 2 × 104 cells/well of a 6-well plate. 3 weeks postplating, colonies were stained with 0.005% crystal violet, and colonies were quantified manually.
Statistical Analysis
All data were analyzed using Student's t test (GraphPad Prism5) unless specified otherwise.
RESULTS
Gucy2c−/− Mice Show Higher Colonic Cell Proliferation and Are Prone to Tumorigenesis
We utilized litter-matched Gucy2c-sufficient (Gucy2c+/+) and -deficient (Gucy2c−/−) mice to delineate mechanisms by which GC-C regulates intestinal cell proliferation. Crypts from Gucy2c−/− showed no GC-C expression by Western blot analysis (Fig. 1A). Equivalent expression of cystic fibrosis transmembrane conductance regulator and PKGII was seen in wild type and Gucy2c−/− mice (Fig. 1A). No changes in the levels of guanylin or uroguanylin transcripts were seen in Gucy2c−/− mice in comparison with wild type mice (Fig. 1B). The absence of GC-C in knock-out mice was correlated with a significant reduction (∼60%) in intracellular cGMP levels in the colonic epithelia of Gucy2c−/− mice, whereas cAMP levels remained unchanged (Fig. 1C).
FIGURE 1.

Colonic epithelia in the Gucy2c−/− mice show a higher incidence of MNU-induced ACF. A, Western blot analysis of GC-C immunoprecipitated with GCC:4D7 mAb from colonic crypts of Gucy2c+/+ and Gucy2c−/− mice and solubilized membrane fraction from T84 cells (top panel). Western blot analysis of PKGII, cystic fibrosis transmembrane conductance regulator (CFTR), and tubulin was performed with lysates prepared from colonic crypts of Gucy2c+/+ and Gucy2c−/− mice and T84 cells. B, RT-PCR analysis of uroguanylin and guanylin expression in colonic crypts from Gucy2c+/+ and Gucy2c−/− mice. C, cyclic GMP levels and cAMP levels in the colonic mucosa of Gucy2c+/+ and Gucy2c−/− mice. Values shown represent the mean ± S.E. of duplicate determinations of experiments repeated thrice (*, p < 0.05). D, formation of ACF in mice was induced by oral gavage with MNU. Mice subsequently received additional oral doses of ST or 8-pCPT-cGMP every 48 h for 1 week. Formation of ACF was analyzed 6 weeks post-MNU treatment. Statistical significance was calculated using one-way analysis of variance with Tukey's multiple comparison test: Gucy2c+/+ MNU versus Gucy2c+/+ MNU + ST, p < 0.05 (*); π = 0.76; Gucy2c+/+ MNU versus Gucy2c+/+ MNU + 8-pCPT-cGMP, p < 0.01 (**), π = 0.93; Gucy2c+/+ MNU versus Gucy2c−/− MNU, p < 0.01 (*), π = 0.86; Gucy2c−/− MNU versus Gucy2c−/− MNU + 8-pCPT-cGMP, p < 0.01 (**), π = 0.99. (error bars, S.D.; filled circles, Gucy2c+/+; open circles, Gucy2c−/−; n = 3 for PBS, n = 6 for rest). E, Ki67 staining of colonic sections from Gucy2c+/+ and Gucy2c−/− mice. Green indicates Ki67-positive nuclei. All nuclei were counterstained with Hoechst 33342 (blue).
We then looked for the presence of microscopic hyperproliferative pockets, known as ACF, in wild type and Gucy2c−/− mice following oral gavage with MNU (26, 27). ACF are considered as preneoplastic lesions and serve as surrogate biomarkers for early colorectal adenomas (28). The number of MNU-induced dysplastic ACF were higher in Gucy2c−/− mice compared with Gucy2c+/+ mice (Fig. 1D). Importantly, in vivo administration of ST after an MNU challenge reduced formation of ACF in Gucy2c+/+ mice but not in Gucy2c−/− mice. This suggested a role for cGMP in regulating intestinal cell proliferation with activation of GC-C providing a mechanism to increase intracellular cGMP levels. In agreement with this, administration of 8-pCPT-cGMP was also able to attenuate formation of ACF both in wild type and Gucy2c−/− mice (Fig. 1D).
We did not observe an appreciably higher number of actively cycling cells in the colonic crypts of Gucy2c−/− mice as assessed by Ki67 staining (Fig. 1E). Thus, we can conclude that activation of GC-C restricts tumorigenesis induced by a carcinogen in the colon in a cGMP-dependent manner.
Cyclic GMP Up-regulates p21 Expression via PKGII and p38 MAPK in Colorectal Carcinoma Cells
To elucidate the molecular mechanisms by which ST (via GC-C) regulates the proliferation of intestinal epithelial cells, we utilized the T84 human colorectal carcinoma cell line as a model. We and others have demonstrated previously that application of ST to T84 cells results in a reduced rate of cell division and G1 arrest of the cell cycle (29, 30). To identify genes whose differential regulation could mediate this cell cycle arrest, we performed global gene expression profiling in T84 cells following treatment with ST peptide. One of the up-regulated genes was CDKN1A (∼2.7-fold up-regulation of mRNA; Fig. 2A). CDKN1A encodes p21 (p21CIP1/WAF1), which is a potent cyclin-dependent kinase inhibitor and functions as a regulator of cell cycle progression at the G1-S phase of the cell cycle (5). Western blot analysis of T84 cells treated with ST or 8-pCPT-cGMP revealed that the elevation of p21 mRNA levels (Fig. 2A) was correlated with an increase in p21 protein (Fig. 2B), suggesting that p21 could be the mediator of cell cycle arrest in T84 cells (via cGMP) following ST action. Elevation of intracellular cAMP using cholera toxin or 8-pCPT-cAMP showed only a marginal increase in p21 levels (Fig. 2C), probably due to a low cross-activation of a common cyclic nucleotide-dependent downstream kinase (31, 32).
FIGURE 2.

Cyclic GMP regulates p21 expression in colorectal carcinoma cells. A, real time PCR analysis of the p21 transcript in T84 cells treated with ST (100 nm) or 8-pCPT-cGMP (25 μm) for 1 h. Values shown represent the mean ± S.E. of duplicate determinations of experiments repeated thrice. B, increase in p21 protein in response to cGMP elevation by ST (100 nm) or 8-pCPT-cGMP (25 μm). GC-C expression remained unchanged. The right panel shows quantification of Western blots performed with T84 cell lysates with experiments repeated thrice (*, p < 0.05 compared with control). C, Western blot analysis of lysates prepared from T84 cells treated with ST (100 nm), cholera toxin (100 nm), or 8-pCPT-cAMP (25 μm) for 1 h with p21 and tubulin antibodies. The right panel shows quantification of Western blots of experiments repeated thrice (*, p < 0.05 compared with control). Error bars represent S.E.
We next investigated molecular players that could transduce the signal of intracellular cGMP accumulation resulting in p21 induction. PKGII is an immediate downstream target of cGMP (33). Transfection of PKGII-specific siRNAs to T84 cells prior to ST addition abrogated p21 up-regulation (Fig. 3A). To investigate downstream effectors of PKGII, we analyzed the activation of p38, ERK1/2, and SAPK/JNK MAPKs following treatment of T84 cells with ST/8-pCPT-cGMP (Fig. 3B). No significant change in the activation of ERK1/2 or SAPK/JNK was observed, whereas p38 MAPK was distinctly activated on ST and 8-pCPT-cGMP treatment. Total levels of p38 MAPK remained unchanged (Fig. 3B). Activation of p38 MAPK following ST treatment was lost in cells lacking PKGII (Fig. 3C), indicating that activation of p38 MAPK was dependent on cGMP-mediated regulation of PKGII. Inhibition of the catalytic activity of p38 MAPK using a specific inhibitor (SB203580) prevented the up-regulation of p21 on ST treatment (Fig. 3D). Thus, these results demonstrate the role of PKGII and p38 MAPK in mediating the signal of intracellular cGMP accumulation leading to the elevation of p21 levels in the cell.
FIGURE 3.

Cyclic GMP acts via PKGII and p38 MAPK to alter p21 expression. A, Western blot analysis of p21 and PKGII expression upon ST stimulation (100 nm) in T84 cells transfected with PKGII siRNA or control siRNA. The right panel shows quantification of Western blots of experiments repeated thrice (*, p < 0.05 compared with control). B, T84 cells were serum-starved for 12 h in DMEM/F-12 containing 0.25% FBS and treated with ST (100 nm) or 8-pCPT-cGMP (25 μm) for 1 h. Western blot analysis was performed on T84 lysates with phospho-p38 antibody, p38 mAb, phospho-ERK1/2 antibody, ERK1/2 antibody, phospho-SAPK/JNK antibody, and SAPK/JNK mAb. The bottom panel shows quantification of Western blots of experiments repeated thrice (*, p < 0.05 compared with control). C, Western blot analysis of p38 MAPK activation upon ST stimulation (100 nm) in T84 cells transfected with PKGII siRNA or control siRNA. The right panel shows quantification of Western blots of experiments repeated thrice (**, p < 0.01 compared with control). D, Western blot analysis of p21 expression and p38 MAPK activation upon ST stimulation (100 nm) in T84 cells treated with SB203580 (10 μm). The right panel shows quantification of Western blots of experiments repeated thrice (*, p < 0.05 compared with control). Error bars represent S.E.
Transcriptional Regulation of p21 Is Mediated via Sp1 and HDAC
Various genetic and epigenetic mechanisms regulate p21 expression (34). Because T84 cells do not express p53, a major regulator of p21 transcription (35), we looked for additional factors that could regulate p21 transcription in a cGMP-dependent manner. Binding of the transcription factor Sp1 to the p21 promoter has been shown to induce differentiation in colorectal carcinoma cells (36) consequent to an increase in p21 transcription. Six Sp1 binding sites present in the proximal region of the p21 promoter (−1 to −117) have been described (34) (Fig. 4A). Phosphorylation of Sp1 retards its mobility on SDS-PAGE (37) and enhances its ability to bind DNA and activate transcription (38). Western blot analysis using Sp1-specific antibodies showed the presence of a slower migrating form following ST treatment that corresponds to phosphorylated Sp1 (Fig. 4B). This slower migrating form was absent in cells treated with SB203580 (Fig. 4B), indicating that p38 MAPK activation was required for Sp1 phosphorylation and that PKGII is unlikely to directly phosphorylate Sp1. Chromatin immunoprecipitation with an Sp1-specific antibody was performed on ST-treated T84 cells. Enhanced binding (∼2-fold) of Sp1 to the p21 proximal promoter was observed in ST-treated cells in comparison with untreated cells (Fig. 4C), and the increase in promoter occupancy was abrogated in the presence of SB203580. Thus, these observations suggest that p38 MAPK-mediated Sp1 phosphorylation contributed to the increase in p21 transcription.
FIGURE 4.

Sp1 and HDAC3 are required for cGMP-dependent p21 transcription. A, transcription factor binding sites on the proximal p21 promoter (34). The Sp1 binding sites are highlighted, and the arrows indicate primers used for ChIP analysis. C/EBP, CCAAT/enhancer-binding protein; RAR, retinoic acid receptor. B, Western blot analysis of Sp1 upon ST stimulation (100 nm) in T84 cells treated with SB203580 (10 μm). * marks the lower mobility form of Sp1. C, ChIP-quantitative PCR demonstrating enhanced Sp1 binding to the p21 promoter upon treatment with ST (100 nm). Treatment with SB203580 (10 μm) abrogated this increase in Sp1 binding. IgG represents normal goat IgG, which was used as a control in ChIP analysis. Values shown represent the mean ± S.E. of experiments repeated thrice (**, p < 0.01). D, Western blot analysis of lysates of T84 cells treated with ST (100 nm) for 1 h with HDAC1, HDAC2, HDAC3, HDAC4, HDAC5, and HDAC6 antibodies. E, ChIP-quantitative PCR to estimate occupancy of the indicated HDACs on the p21 promoter upon treatment with ST (100 nm). NMI represents normal mouse IgG and NRI represents normal rabbit IgG used as controls for HDAC1–3 antibodies and HDAC4–6 antibodies, respectively, during ChIP analysis. Values shown represent the mean ± S.E. of experiments repeated thrice (**, p < 0.01). Error bars represent S.E.
HDACs have emerged as critical epigenetic regulators of cell growth and differentiation programs. HDAC3 is overexpressed in approximately half of all colorectal carcinomas, resulting in repression of p21 expression and enhanced cell proliferation (39, 40). Therefore, we analyzed the expression and binding of HDAC1, HDAC2, HDAC3, HDAC4, HDAC5, and HDAC6 to the proximal p21 promoter in T84 cells. HDACs 1, 2, 3, 4, and 6 were expressed, and no alteration in their expression levels was seen on ST treatment (Fig. 4D). However, we observed reduced HDAC3 occupancy at the p21 promoter upon elevation of cGMP (Fig. 4E). We suggest that exclusion of HDAC3 from the p21 promoter in the presence of cGMP possibly maintains transcription-permissive chromatin, which acts synergistically with Sp1 to promote p21 transcription.
GC-C Activation Leads to Induction of p21 in Murine Colonic Epithelia
We have thus far demonstrated that activation of GC-C by ST increases p21 levels in human colorectal carcinoma cells in a cGMP-dependent manner. This transcriptional regulation occurred independently of p53. Therefore, we sought to determine whether the induction of p21 transcription by cGMP occurred in intact colonic crypts, which were p53-positive.
Administration of ST to crypts isolated from wild type mice resulted in an increase in cGMP levels, whereas no elevation in cGMP was seen in Gucy2c−/− mice (Fig. 5A). In agreement with observations in T84 cells, an increase in p21 mRNA (Fig. 5B) and protein levels (Fig. 5C) following treatment with ST or 8-pCPT-cGMP was observed in crypts from Gucy2c+/+ mice. Importantly, ST was unable to up-regulate p21 in crypts from the Gucy2c−/− mice. Moreover, 8-pCPT-cGMP was able to increase p21 levels in both wild type and GC-C knock-out mouse crypts (Fig. 5C).
FIGURE 5.

The cGMP/p21 cytostatic axis is functional in the mouse colon. A, measurement of cGMP in colonic crypts obtained from Gucy2c+/+ and Gucy2c−/− mice following treatment with ST (100 nm). Values shown represent the mean ± S.E. of duplicate determinations of experiments repeated thrice. B, real time PCR analysis of p21 transcript levels in response to ST (100 nm) or 8-pCPT-cGMP (25 μm) from colonic crypts isolated from Gucy2c+/+ and Gucy2c−/− mice. Values shown represent the mean ± S.E. of duplicate determinations of experiments repeated thrice. C, p21 expression and p38 MAPK activation in response to ST (100 nm) or 8-pCPT-cGMP (25 μm) treatment of colonic crypts isolated from Gucy2c+/+ and Gucy2c−/− mice. The right panel shows quantification of Western blots of lysates prepared from colonic crypts with experiments repeated thrice (*, p < 0.05 compared with control). D, Western blot analysis of p21 expression in colonic crypt lysates upon ST treatment (100 nm) in the presence of SB203580 (10 μm) or (Rp)-8-pCPT-cGMPS (2 μm). The right panel shows quantification of Western blots of experiments repeated thrice (*, p < 0.05 compared with control). Error bars represent S.E.
We next examined p38 MAPK activation in response to elevation of cGMP. p38 MAPK was activated on ST or 8-pCPT-cGMP treatment in colonic crypts from Gucy2c+/+ mice (Fig. 5C). However, only 8-pCPT-cGMP, and not ST, was able to activate p38 MAPK in Gucy2c−/− mice (Fig. 5C). To analyze the role of PKGII and p38 MAPK in modulating p21 expression in colonic crypts, we utilized a specific inhibitor of PKGII ((Rp)-8-pCPT-cGMPS) as well as the p38 MAPK inhibitor SB203580. As seen in Fig. 5D, the cGMP-mediated increase in p21 was inhibited in the presence of either of these inhibitors. Thus, the molecular mechanisms of cGMP-mediated p21 expression delineated in a colorectal carcinoma cell line were mirrored in the murine colonic crypt, and the mechanisms underlying the regulation of p21 expression by cGMP were retained in the background of wild type p53.
Chronic ST Treatment Leads to a Cell Cycle Arrest and Induction of Cellular Senescence
In the normal intestine, GC-C is continuously stimulated by the gastrointestinal hormones guanylin and uroguanylin, and this could act as an antitumorigenic strategy. In developing countries, the frequent episodes of diarrhea mediated by ST peptides could also result in prolonged stimulation of GC-C. Therefore, we investigated the consequences of chronic stimulation of GC-C by treating T84 cells with ST peptide for 10 days. We observed a dramatic increase in p21 protein (Fig. 6A, upper panel) at the end of this treatment period. However, we did not observe a concomitant increase in the p21 mRNA at the end of 10 days (Fig. 6A, lower panel). Therefore, it appears that following the initial increase in p21 at the transcriptional level the large increase in p21 protein at 10 days may be a consequence of enhanced protein stability.
FIGURE 6.

ST-treated T84 cells show hallmarks of senescence and reduced tumorigenicity. A, p21 was up-regulated upon treatment with ST (100 nm) for 10 days at the protein (upper panel) and transcript levels (lower panel). ST-containing medium was replaced every 3 days. Values shown represent the mean ± S.E. of duplicate determinations of experiments repeated thrice. B, upper panel, quantification of SA-β-galactosidase staining of T84 cells transfected with control EGFP esiRNA or p21 esiRNA followed by treatment with ST (100 nm) for 10 days. Values shown represent the mean ± S.E. of experiments repeated thrice (**, p < 0.01). Lower panel, Western blot to confirm p21 knockdown. C, fluorescence image of DNA stained with DAPI in T84 cells treated with ST (100 nm) for 10 days. Numbers indicate the numbers of senescence-associated heterochromatin foci (SAHF)-positive cells. Scale bar, 5 μm. D, Western blot analysis of phospho-Rb, total Rb, cyclin D1, and p21 expression upon ST stimulation (100 nm) for 10 days in T84 cells transfected with EGFP esiRNA or p21 esiRNA. E, soft agar colony formation with cells transfected with EGFP esiRNA or p21 esiRNA (top panel). T84 cells were treated with ST (100 nm) or 8-pCPT-cGMP (25 μm) for 10 days and trypsinized, and colony-forming units were quantified (bottom panel). Values shown represent the mean ± S.E. of experiments repeated thrice (*, p < 0.05 compared with control EGFP esiRNA-transfected cells). Error bars represent S.E.
High p21 expression has been associated with irreversible exit from the cell cycle and induction of cellular senescence (41). The most widely used marker for senescence is the staining for β-galactosidase assessed at a suboptimal pH of 6.0 (known as SA-β-galactosidase) (42). The number of cells expressing the senescence marker SA-β-galactosidase following prolonged ST treatment reached a maximum of ∼20% of the total population (Fig. 6B, upper panel), whereas little or no SA-β-galactosidase activity was detected in untreated cells. A fraction of T84 cells (11%) treated with ST also displayed characteristic senescence-associated heterochromatin focus formation (Fig. 6C). To determine whether the induction of senescent cells was p21-dependent, T84 cells knocked down for p21 were treated with ST for 10 days (Fig. 6B, lower panel). A marked reduction in SA-β-galactosidase-positive cells was seen in comparison with cells treated with ST and with consistent expression of p21 (Fig. 6B, upper panel). Therefore, sustained expression of p21 following GC-C activation and cGMP accumulation was critical to induce senescence.
T84 cells treated with ST or 8-pCPT-cGMP for prolonged periods of time displayed canonical features of irreversibly arrested senescent cells, showing lower levels of phosphorylated Rb protein and a decrease in cyclin D1 levels (Fig. 6D). This is commensurate with elevated levels of p21 (41) because silencing p21 expression attenuated the ST-mediated decrease in phosphorylated Rb and cyclin D1 levels. Hence, to analyze ST-induced senescence and the role of p21 in preventing tumor formation, we depleted p21 expression using esiRNA in T84 cells and monitored colony formation in a soft agar assay. As seen in Fig. 6E, T84 cells treated with ST or 8-pCPT-cGMP for 10 days formed significantly fewer colonies than untreated cells. This compromised ability to form colonies was p21-dependent because cells in which p21 had been depleted showed no reduction in the number of colonies on ST or 8-pCPT-cGMP treatment (Fig. 6E).
We extended these studies and asked whether chronic in vivo ST treatment of mice could elevate p21 levels in colonic crypts. Indeed, 10 days of oral ST application led to markedly higher levels of p21 in colonic crypts of wild type mice. This increase in p21 levels was much reduced in the Gucy2c−/− mice (Fig. 7A). Moreover, cyclin D1 levels were significantly lowered in wild type mice treated with ST, thus underscoring the cytostatic effect of cGMP elevation in the intestinal epithelia (Fig. 7A).
FIGURE 7.
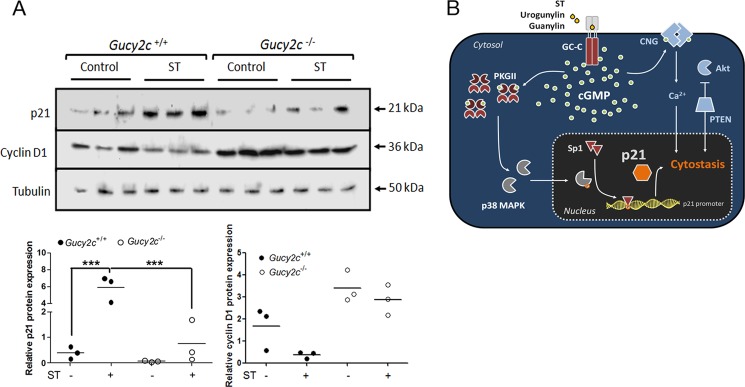
Chronic ST treatment of mice elevates p21 levels in colonic epithelia and reduces cell proliferation. A, Western blot analysis of p21 and cyclin D1 expression in colonic crypts of Gucy2c+/+ and Gucy2c−/− mice treated with ST every 48 h for 10 days. Each lane represents individual mice (n = 3 for each treatment). The bottom panels show quantification of Western blots. Statistical significance was calculated using one-way analysis of variance with Tukey's multiple comparison test (**, p < 0.01; ***, p < 0.005). B, ligand-mediated (ST/uroguanylin/guanylin) activation of GC-C in the colonic cell leads to accumulation of cGMP, which in turn activates PKGII. This results in activation of the p38 MAPK pathway and phosphorylation of the Sp1 transcription factor, which enhances transcription of the p21 mRNA. Nuclear accumulation of p21 brings about senescence and quiescence, thereby reducing colorectal tumorigenesis. Known cytostatic pathways regulated by GC-C activation are shown in blue. CNG, cyclic nucleotide-gated channel; PTEN, phosphatase and tensin homolog.
DISCUSSION
This study describes the process by which activation of GC-C initiates a transcriptional up-regulation of p21, resulting in cell cycle arrest and reduced tumorigenesis in colonic epithelia. Mice lacking the Gucy2c gene have lower homeostatic levels of cGMP in their colonic crypts (Fig. 1C), and this possibly contributes to an increase in MNU-induced ACF in these mice (Fig. 1D). Elevating cGMP by application of ST or 8-pCPT-cGMP ameliorated neoplastic growth, supporting an additional mechanism by which GC-C regulates antiproliferative signaling in the colon (43).
PKGII has emerged as an important downstream target of cGMP produced by GC-C and in a manner similar to GC-C is primarily expressed in the intestinal epithelia (33). Intriguingly, PKGII−/− mice phenocopy the GC-C knock-out animals with regard to crypt hyperplasia, indicating that PKGII mediates the homeostatic effects of GC-C/cGMP in the colon (13, 44). As seen in Fig. 3A, PKGII is vital for ST-mediated p21 induction as its depletion attenuates the effect of ST treatment. In agreement with this observation, poorly differentiated colonic adenocarcinomas show reduced PKGII expression in comparison with matched normal tissue, further highlighting the crucial role of PKGII in intestinal cell proliferation (44). In a previous study, overexpression of a constitutively activated form of PKGI has been shown to up-regulate p21 in colorectal carcinoma cells, providing additional evidence of a cGMP-p21 link (45).
The p38 MAPK acts here as a downstream signal transducer, coupling activation of PKGII with increased transcription of the p21 mRNA. Inactivation of the p38 MAPK pathway disrupts induction of cellular senescence and may render mice prone to tumor development (46). Of the three MAPK signaling pathways, p38 MAPK was significantly activated in cells with elevated levels of cGMP (Fig. 3B). There was a marginal reduction in proproliferative ERK1/2 signaling that could be a consequence of p38 MAPK or PKGII activation (47, 48). In an earlier study, enhanced p21 transcription via ERK activation was reported in rat adventitial fibroblasts in response to nitric oxide, which would elevate intracellular cGMP levels (49). Using a specific ERK inhibitor, we found that ERK1/2 activity was not essential for cGMP-dependent p21 induction in T84 cells (data not shown) as has been shown previously (50). Interestingly, the phosphorylation of p38 MAPK was critically dependent on PKGII because activation of p38 MAPK was lost in cells lacking PKGII (Fig. 3C). This observation clearly places the GC-C/cGMP/PKGII cascade upstream of p38 MAPK activation. In a peptide-based screen for substrates for PKGII, p38 MAPK was identified as a putative target (51). This raises an intriguing possibility that in colonic epithelia PKGII could directly phosphorylate and activate the p38 MAPK or the upstream mitogen-activated protein kinase kinases (MEKK3/6).
Of the various means of modulating p21 expression, transcriptional regulation is the most complex and critical for coordinating cell division (52). Consequently, deregulation of intratumoral p21 transcription is a frequent occurrence in cancer (53). Although p53 remains one of the most critical modulators of p21 transcription, results presented here demonstrate that cGMP acts in a p53-independent manner to increase p21 expression. This implies that the GC-C/p21 axis is functional even in colorectal carcinomas that have lost p53 expression.
The multiple Sp1 binding sites in the p21 promoter are required for p21 induction by diverse signaling cascades (34). In addition, PKGI has been shown to increase p21 transcription via Sp1 (54). As seen in Fig. 4B, Sp1 was found to be phosphorylated by p38 MAPK in response to cGMP elevation, and increased phosphorylation of Sp1 has been shown to enhance its DNA binding ability (55). In agreement with this, ST treatment increased the occupancy of Sp1 at the p21 promoter, an effect that was abolished in the presence of a p38 MAPK inhibitor (Fig. 4C). Thus, it appears that Sp1 plays a crucial role in transducing the signal of GC-C activation to p21 expression.
Because p38 MAPK has multiple downstream targets that also include several transcription factors, it is conceivable that Sp1 may not be the sole transcription factor responsible for the p21 induction seen in T84 cells. HDAC inhibitors have been shown to up-regulate p21 transcription in an Sp1-dependent manner (56). Here, we found that cGMP accumulation reduced HDAC3 occupancy at the proximal p21 promoter, thereby maintaining transcription-permissive chromatin (Fig. 4E). HDAC3 has been shown to inhibit p21 expression, and overexpression of HDAC3 has been frequently observed in colonic tumors (39). These observations demonstrate that the p21 promoter in the colonic epithelia is under an added level of epigenetic control that can be regulated in turn by cGMP levels.
Although the antiproliferative functions of p21 are mediated by its nuclear localization, its cytoplasmic localization has been associated with oncogenic growth (52). AKT1 phosphorylates p21 and promotes its cytoplasmic accumulation (57). AKT signaling is reduced following activation of GC-C (14), and in agreement with this, we did not observe any cytoplasmic enrichment of p21 upon ST treatment (data not shown). Thus, the proproliferative roles of cytoplasmic p21 are unlikely to play an important role in GC-C-mediated cell cycle regulation.
Chronic exposure of T84 cells to ST led to the development of senescence-like features in colorectal carcinoma cells and an appreciable p21-dependent decrease in soft agar colony formation (Fig. 6E). Induction of quiescence and senescence has long been considered a barrier for tumorigenesis (58). In addition to the cytostatic nature of the senescence program, these cells also attract innate immune cells that can selectively target the tumor, reinforcing the tumor-suppressive action of senescence and facilitating tumor clearance (59). Interestingly, many of the genes found to be differentially regulated following ST treatment have been implicated in modulating cell proliferation and tumorigenesis (Table 1). For example, ETS1 (v-ets erythroblastosis virus E26 oncogene homolog 1) (60), S100P (S100 calcium-binding protein P) (61), and RAP2A (member of RAS oncogene family) (62) were all down-regulated.
TABLE 1.
List of 20 maximally regulated genes in T84 cells treated with ST for 1 h
| Up-regulated genes |
Down-regulated genes |
||
|---|---|---|---|
| Gene symbol | -Fold change | Gene symbol | -Fold change |
| GIPR | 3.12 | ETS1 | 0.02 |
| CDKN1A | 2.82 | REM2 | 0.02 |
| CLINT1 | 2.73 | USP47 | 0.03 |
| SLC6A6 | 2.71 | FNDC7 | 0.05 |
| PRKG2 | 2.58 | IQSEC1 | 0.21 |
| ISYNA1 | 2.54 | RPS29 | 0.28 |
| COL14A1 | 2.51 | ASB8 | 0.29 |
| KLF14 | 2.42 | S100P | 0.31 |
| COL18A1 | 2.42 | RND3 | 0.31 |
| ELN | 2.38 | FTH1 | 0.31 |
| TOMM20L | 2.31 | UBC | 0.35 |
| ZIC4 | 2.28 | APOL4 | 0.35 |
| ERN1 | 2.27 | TAF5L | 0.37 |
| A2ML1 | 2.25 | GPR177 | 0.4 |
| SPATA18 | 2.21 | RPS2 | 0.41 |
| CSDC2 | 2.13 | RPS27 | 0.42 |
| KRTAP4-11 | 2.12 | RPLP0 | 0.44 |
| ACRBP | 2.11 | SNX12 | 0.49 |
| MLL | 2.01 | RPL19 | 0.49 |
Worldwide, colorectal cancer is the fourth most commonly diagnosed malignant disease and the second most frequent cause of cancer-related death (63). Intriguingly, the age-adjusted occurrence of colorectal cancer in developing nations is close to the lowest rates in the world (64). Our observations linking GC-C activation and p21-dependent cytostasis may in part explain the epidemiological disparity of colorectal cancer incidence in the world as has been noted earlier (43). A common epidemiological feature of these colorectal cancer-spared regions is the prevalence of enterotoxigenic Escherichia coli infections (43). Enterotoxigenic E. coli is endemic to these regions and may be a part of the normal gut microbiota of people living in the Asian and African continents.4 Enterotoxigenic E. coli-associated diarrhea is life-threatening in infants but less severe in adults (65) and thus may provide resistance against development of colorectal cancer.
The intestinal stem cells are thought to be the “cells of origin” for colonic cancers (66). These stem cells, which lie at the bottom of the crypts, do not express GC-C and hence would not be under cGMP-mediated cytostatic control (67). However, a recent report suggests that colonic tumors can also arise from differentiated enterocytes in the context of an inflammatory stroma, supporting the “top-down” model of adenoma morphogenesis (68, 69). This means that enterocytes have the ability to dedifferentiate and form tumor-initiating cells. Therefore, the GC-C/p21 cytostatic axis, which is active in the transit-amplifying and differentiated compartments of the colonic crypt, can influence tumor development in these cells.
In conclusion, we have described in molecular detail a mechanism by which GC-C/cGMP can restrict colon carcinogenesis (Fig. 7C). The data presented raise the prospect of the use of GC-C agonists in colorectal cancer therapy, including p53-null carcinomas. The GC-C agonist linaclotide is currently a drug of choice for the treatment of chronic idiopathic constipation (70). One can envisage the use of this GC-C agonist in combination with other chemotherapeutic agents to enhance the efficacy of the current treatment regimens for colorectal cancer.
Acknowledgments
We thank Prof. P. Kondaiah and Neeraj Rana for help with the microarray and Pavithra Kumar, Vani R. Iyer, and members of the laboratory for useful discussions. We acknowledge the assistance of the late Prof. Parag P. Sadhale in early transcriptomic studies. Support from the Central Animal Facility in the Indian Institute of Science is acknowledged.
This work was supported in part by the Departments of Science and Technology and of Biotechnology, Government of India.
G. B. Nair, personal communication.
- GC-C
- guanylyl cyclase C
- ST
- heat-stable enterotoxin
- esiRNA
- endoribonuclease-prepared siRNA
- EGFP
- enhanced GFP
- Rb
- retinoblastoma protein
- HDAC
- histone deacetylase
- MNU
- N-methyl-N-nitrosourea
- 8-pCPT-cGMP
- 8-(4-chlorophenylthio)-guanosine 3′,5′-cyclic monophosphate
- ACF
- aberrant crypt foci
- SA
- senescence-associated
- (Rp)-8-pCPT-cGMPS
- (Rp)-8-[(4-chlorophenyl)thio]-guanosine-cyclic 3′,5′-hydrogen phosphorothioate triethylammonium salt.
REFERENCES
- 1. Kanthan R., Senger J. L., Kanthan S. C. (2012) Molecular events in primary and metastatic colorectal carcinoma: a review. Patholog. Res. Int. 2012, 597497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kinzler K. W., Vogelstein B. (1996) Lessons from hereditary colorectal cancer. Cell 87, 159–170 [DOI] [PubMed] [Google Scholar]
- 3. Cunningham D., Atkin W., Lenz H. J., Lynch H. T., Minsky B., Nordlinger B., Starling N. (2010) Colorectal cancer. Lancet 375, 1030–1047 [DOI] [PubMed] [Google Scholar]
- 4. Harper J. W., Adami G. R., Wei N., Keyomarsi K., Elledge S. J. (1993) The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75, 805–816 [DOI] [PubMed] [Google Scholar]
- 5. el-Deiry W. S., Harper J. W., O'Connor P. M., Velculescu V. E., Canman C. E., Jackman J., Pietenpol J. A., Burrell M., Hill D. E., Wang Y., Wiman K. G., Mercer W. E., Kastan M. B., Kohn K. W., Elledge S. J., Kinzler K. W., Vogelstein B. (1994) WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 54, 1169–1174 [PubMed] [Google Scholar]
- 6. Brown J. P., Wei W., Sedivy J. M. (1997) Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science 277, 831–834 [DOI] [PubMed] [Google Scholar]
- 7. Dunlop M. G., Dobbins S. E., Farrington S. M., Jones A. M., Palles C., Whiffin N., Tenesa A., Spain S., Broderick P., Ooi L. Y., Domingo E., Smillie C., Henrion M., Frampton M., Martin L., Grimes G., Gorman M., Semple C., Ma Y. P., Barclay E., Prendergast J., Cazier J. B., Olver B., Penegar S., Lubbe S., Chander I., Carvajal-Carmona L. G., Ballereau S., Lloyd A., Vijayakrishnan J., Zgaga L., Rudan I., Theodoratou E., Colorectal Tumour Gene Identification (CORGI) Consortium, Starr J. M., Deary I., Kirac I., Kovacevi“c D., Aaltonen L. A., Renkonen-Sinisalo L., Mecklin J. P., Matsuda K., Nakamura Y., Okada Y., Gallinger S., Duggan D. J., Conti D., Newcomb P., Hopper J., Jenkins M. A., Schumacher F., Casey G., Easton D., Shah M., Pharoah P., Lindblom A., Liu T., Swedish Low-Risk Colorectal Cancer Study Group, Smith C. G., West H., Cheadle J. P., COIN Collaborative Group, Midgley R., Kerr D. J., Campbell H., Tomlinson I. P., Houlston R. S. (2012) Common variation near CDKN1A, POLD3 and SHROOM2 influences colorectal cancer risk. Nat. Genet. 44, 770–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Archer S. Y., Meng S., Shei A., Hodin R. A. (1998) p21(WAF1) is required for butyrate-mediated growth inhibition of human colon cancer cells. Proc. Natl. Acad. Sci. U.S.A. 95, 6791–6796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Waldman T., Kinzler K. W., Vogelstein B. (1995) p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 55, 5187–5190 [PubMed] [Google Scholar]
- 10. Basu N., Arshad N., Visweswariah S. S. (2010) Receptor guanylyl cyclase C (GC-C): regulation and signal transduction. Mol. Cell. Biochem. 334, 67–80 [DOI] [PubMed] [Google Scholar]
- 11. Fiskerstrand T., Arshad N., Haukanes B. I., Tronstad R. R., Pham K. D., Johansson S., Håvik B., Tønder S. L., Levy S. E., Brackman D., Boman H., Biswas K. H., Apold J., Hovdenak N., Visweswariah S. S., Knappskog P. M. (2012) Familial diarrhea syndrome caused by an activating GUCY2C mutation. N. Engl. J. Med. 366, 1586–1595 [DOI] [PubMed] [Google Scholar]
- 12. Romi H., Cohen I., Landau D., Alkrinawi S., Yerushalmi B., Hershkovitz R., Newman-Heiman N., Cutting G. R., Ofir R., Sivan S., Birk O. S. (2012) Meconium ileus caused by mutations in GUCY2C, encoding the CFTR-activating guanylate cyclase 2C. Am. J. Hum. Genet. 90, 893–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li P., Schulz S., Bombonati A., Palazzo J. P., Hyslop T. M., Xu Y., Baran A. A., Siracusa L. D., Pitari G. M., Waldman S. A. (2007) Guanylyl cyclase C suppresses intestinal tumorigenesis by restricting proliferation and maintaining genomic integrity. Gastroenterology 133, 599–607 [DOI] [PubMed] [Google Scholar]
- 14. Lin J. E., Li P., Snook A. E., Schulz S., Dasgupta A., Hyslop T. M., Gibbons A. V., Marszlowicz G., Pitari G. M., Waldman S. A. (2010) The hormone receptor GUCY2C suppresses intestinal tumor formation by inhibiting AKT signaling. Gastroenterology 138, 241–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kazerounian S., Pitari G. M., Shah F. J., Frick G. S., Madesh M., Ruiz-Stewart I., Schulz S., Hajnóczky G., Waldman S. A. (2005) Proliferative signaling by store-operated calcium channels opposes colon cancer cell cytostasis induced by bacterial enterotoxins. J. Pharmacol. Exp. Ther. 314, 1013–1022 [DOI] [PubMed] [Google Scholar]
- 16. Currie M. G., Fok K. F., Kato J., Moore R. J., Hamra F. K., Duffin K. L., Smith C. E. (1992) Guanylin: an endogenous activator of intestinal guanylate cyclase. Proc. Natl. Acad. Sci. U.S.A. 89, 947–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Steinbrecher K. A., Tuohy T. M., Heppner Goss K., Scott M. C., Witte D. P., Groden J., Cohen M. B. (2000) Expression of guanylin is downregulated in mouse and human intestinal adenomas. Biochem. Biophys. Res. Commun. 273, 225–230 [DOI] [PubMed] [Google Scholar]
- 18. Camici M. (2008) Guanylin peptides and colorectal cancer (CRC). Biomed. Pharmacother. 62, 70–76 [DOI] [PubMed] [Google Scholar]
- 19. Shailubhai K., Yu H. H., Karunanandaa K., Wang J. Y., Eber S. L., Wang Y., Joo N. S., Kim H. D., Miedema B. W., Abbas S. Z., Boddupalli S. S., Currie M. G., Forte L. R. (2000) Uroguanylin treatment suppresses polyp formation in the ApcMin/+ mouse and induces apoptosis in human colon adenocarcinoma cells via cyclic GMP. Cancer Res. 60, 5151–5157 [PubMed] [Google Scholar]
- 20. Carrithers S. L., Barber M. T., Biswas S., Parkinson S. J., Park P. K., Goldstein S. D., Waldman S. A. (1996) Guanylyl cyclase C is a selective marker for metastatic colorectal tumors in human extraintestinal tissues. Proc. Natl. Acad. Sci. U.S.A. 93, 14827–14832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schulz S., Lopez M. J., Kuhn M., Garbers D. L. (1997) Disruption of the guanylyl cyclase-C gene leads to a paradoxical phenotype of viable but heat-stable enterotoxin-resistant mice. J. Clin. Investig. 100, 1590–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dwarakanath P., Visweswariah S. S., Subrahmanyam Y. V., Shanthi G., Jagannatha H. M., Balganesh T. S. (1989) Cloning and hyperexpression of a gene encoding the heat-stable toxin of Escherichia coli. Gene 81, 219–226 [DOI] [PubMed] [Google Scholar]
- 23. Brooker G., Harper J. F., Terasaki W. L., Moylan R. D. (1979) Radioimmunoassay of cyclic AMP and cyclic GMP. Adv. Cyclic Nucleotide Res. 10, 1–33 [PubMed] [Google Scholar]
- 24. Zor T., Selinger Z. (1996) Linearization of the Bradford protein assay increases its sensitivity: theoretical and experimental studies. Anal. Biochem. 236, 302–308 [DOI] [PubMed] [Google Scholar]
- 25. Dimri G. P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E. E., Linskens M., Rubelj I., Pereira-Smith O., Peacocke M, Campisi J. (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. U.S.A. 92, 9363–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pretlow T. P., Barrow B. J., Ashton W. S., O'Riordan M. A., Pretlow T. G., Jurcisek J. A., Stellato T. A. (1991) Aberrant crypts: putative preneoplastic foci in human colonic mucosa. Cancer Res. 51, 1564–1567 [PubMed] [Google Scholar]
- 27. Hosono K., Endo H., Takahashi H., Sugiyama M., Uchiyama T., Suzuki K., Nozaki Y., Yoneda K., Fujita K., Yoneda M., Inamori M., Tomatsu A., Chihara T., Shimpo K., Nakagama H., Nakajima A. (2010) Metformin suppresses azoxymethane-induced colorectal aberrant crypt foci by activating AMP-activated protein kinase. Mol. Carcinog. 49, 662–671 [DOI] [PubMed] [Google Scholar]
- 28. Takayama T., Katsuki S., Takahashi Y., Ohi M., Nojiri S., Sakamaki S., Kato J., Kogawa K., Miyake H., Niitsu Y. (1998) Aberrant crypt foci of the colon as precursors of adenoma and cancer. N. Engl. J. Med. 339, 1277–1284 [DOI] [PubMed] [Google Scholar]
- 29. Basu N., Bhandari R., Natarajan V. T., Visweswariah S. S. (2009) Cross talk between receptor guanylyl cyclase C and c-src tyrosine kinase regulates colon cancer cell cytostasis. Mol. Cell. Biol. 29, 5277–5289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pitari G. M., Di Guglielmo M. D., Park J., Schulz S., Waldman S. A. (2001) Guanylyl cyclase C agonists regulate progression through the cell cycle of human colon carcinoma cells. Proc. Natl. Acad. Sci. U.S.A. 98, 7846–7851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Forte L. R., Thorne P. K., Eber S. L., Krause W. J., Freeman R. H., Francis S. H., Corbin J. D. (1992) Stimulation of intestinal Cl- transport by heat-stable enterotoxin: activation of cAMP-dependent protein kinase by cGMP. Am. J. Physiol. Cell Physiol. 263, C607–C615 [DOI] [PubMed] [Google Scholar]
- 32. Miller J. P., Boswell K. H., Muneyama K., Simon L. N., Robins R. K., Shuman D. A. (1973) Synthesis and biochemical studies of various 8-substituted derivatives of guanosine 3′,5′-cyclic phosphate, inosine 3′,5′-cyclic phosphate, and xanthosine 3′,5′-cyclic phosphate. Biochemistry 12, 5310–5319 [DOI] [PubMed] [Google Scholar]
- 33. Lohmann S. M., Vaandrager A. B., Smolenski A., Walter U., De Jonge H. R. (1997) Distinct and specific functions of cGMP-dependent protein kinases. Trends Biochem. Sci. 22, 307–312 [DOI] [PubMed] [Google Scholar]
- 34. Gartel A. L., Tyner A. L. (1999) Transcriptional regulation of the p21(WAF1/CIP1) gene. Exp. Cell Res. 246, 280–289 [DOI] [PubMed] [Google Scholar]
- 35. Liu Y., Bodmer W. F. (2006) Analysis of P53 mutations and their expression in 56 colorectal cancer cell lines. Proc. Natl. Acad. Sci. U.S.A. 103, 976–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gartel A. L., Goufman E., Najmabadi F., Tyner A. L. (2000) Sp1 and Sp3 activate p21 (WAF1/CIP1) gene transcription in the Caco-2 colon adenocarcinoma cell line. Oncogene 19, 5182–5188 [DOI] [PubMed] [Google Scholar]
- 37. Olofsson B. A., Kelly C. M., Kim J., Hornsby S. M., Azizkhan-Clifford J. (2007) Phosphorylation of Sp1 in response to DNA damage by ataxia telangiectasia-mutated kinase. Mol. Cancer Res. 5, 1319–1330 [DOI] [PubMed] [Google Scholar]
- 38. Tan N. Y., Khachigian L. M. (2009) Sp1 phosphorylation and its regulation of gene transcription. Mol. Cell. Biol. 29, 2483–2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wilson A. J., Byun D. S., Popova N., Murray L. B., L'Italien K., Sowa Y., Arango D., Velcich A., Augenlicht L. H., Mariadason J. M. (2006) Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J. Biol. Chem. 281, 13548–13558 [DOI] [PubMed] [Google Scholar]
- 40. Spurling C. C., Godman C. A., Noonan E. J., Rasmussen T. P., Rosenberg D. W., Giardina C. (2008) HDAC3 overexpression and colon cancer cell proliferation and differentiation. Mol. Carcinog. 47, 137–147 [DOI] [PubMed] [Google Scholar]
- 41. Kagawa S., Fujiwara T., Kadowaki Y., Fukazawa T., Sok-Joo R., Roth J. A., Tanaka N. (1999) Overexpression of the p21 sdi1 gene induces senescence-like state in human cancer cells: implication for senescence-directed molecular therapy for cancer. Cell Death Differ. 6, 765–772 [DOI] [PubMed] [Google Scholar]
- 42. Debacq-Chainiaux F., Erusalimsky J. D., Campisi J., Toussaint O. (2009) Protocols to detect senescence-associated β-galactosidase (SA-βgal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 4, 1798–1806 [DOI] [PubMed] [Google Scholar]
- 43. Pitari G. M., Zingman L. V., Hodgson D. M., Alekseev A. E., Kazerounian S., Bienengraeber M., Hajnóczky G., Terzic A., Waldman S. A. (2003) Bacterial enterotoxins are associated with resistance to colon cancer. Proc. Natl. Acad. Sci. U.S.A. 100, 2695–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang R., Kwon I. K., Thangaraju M., Singh N., Liu K., Jay P., Hofmann F., Ganapathy V., Browning D. D. (2012) Type 2 cGMP-dependent protein kinase regulates proliferation and differentiation in the colonic mucosa. Am. J. Physiol. Gastrointest. Liver Physiol. 303, G209–G219 [DOI] [PubMed] [Google Scholar]
- 45. Deguchi A., Thompson W. J., Weinstein I. B. (2004) Activation of protein kinase G is sufficient to induce apoptosis and inhibit cell migration in colon cancer cells. Cancer Res. 64, 3966–3973 [DOI] [PubMed] [Google Scholar]
- 46. Han J., Sun P. (2007) The pathways to tumor suppression via route p38. Trends Biochem. Sci. 32, 364–371 [DOI] [PubMed] [Google Scholar]
- 47. Wu Y., Chen Y., Qu R., Lan T., Sang J. (2012) Type II cGMP-dependent protein kinase inhibits EGF-triggered signal transduction of the MAPK/ERK-mediated pathway in gastric cancer cells. Oncol. Rep. 27, 553–558 [DOI] [PubMed] [Google Scholar]
- 48. Junttila M. R., Li S. P., Westermarck J. (2008) Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB J. 22, 954–965 [DOI] [PubMed] [Google Scholar]
- 49. Gu M., Lynch J., Brecher P. (2000) Nitric oxide increases p21Waf1/Cip1 expression by a cGMP-dependent pathway that includes activation of extracellular signal-regulated kinase and p70S6k. J. Biol. Chem. 275, 11389–11396 [DOI] [PubMed] [Google Scholar]
- 50. Saha S., Chowdhury P., Pal A., Chakrabarti M. K. (2008) Downregulation of human colon carcinoma cell (COLO-205) proliferation through PKG-MAP kinase mediated signaling cascade by E. coli heat stable enterotoxin (STa), a potent anti-angiogenic and anti-metastatic molecule. J. Appl. Toxicol. 28, 475–483 [DOI] [PubMed] [Google Scholar]
- 51. Kawasaki Y., Kugimiya F., Chikuda H., Kamekura S., Ikeda T., Kawamura N., Saito T., Shinoda Y., Higashikawa A., Yano F., Ogasawara T., Ogata N., Hoshi K., Hofmann F., Woodgett J. R., Nakamura K., Chung U. I., Kawaguchi H. (2008) Phosphorylation of GSK-3β by cGMP-dependent protein kinase II promotes hypertrophic differentiation of murine chondrocytes. J. Clin. Investig. 118, 2506–2515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Abbas T., Dutta A. (2009) p21 in cancer: intricate networks and multiple activities. Nat. Rev. Cancer 9, 400–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pasz-Walczak G., Kordek R., Faflik M. (2001) P21 (WAF1) expression in colorectal cancer: correlation with P53 and cyclin D1 expression, clinicopathological parameters and prognosis. Pathol. Res. Pract. 197, 683–689 [DOI] [PubMed] [Google Scholar]
- 54. Cen B., Deguchi A., Weinstein I. B. (2008) Activation of protein kinase G Increases the expression of p21CIP1, p27KIP1, and histidine triad protein 1 through Sp1. Cancer Res. 68, 5355–5362 [DOI] [PubMed] [Google Scholar]
- 55. D'Addario M., Arora P. D., McCulloch C. A. (2006) Role of p38 in stress activation of Sp1. Gene 379, 51–61 [DOI] [PubMed] [Google Scholar]
- 56. Sowa Y., Orita T., Hiranabe-Minamikawa S., Nakano K., Mizuno T., Nomura H., Sakai T. (1999) Histone deacetylase inhibitor activates the p21/WAF1/Cip1 gene promoter through the Sp1 sites. Ann. N.Y. Acad. Sci. 886, 195–199 [DOI] [PubMed] [Google Scholar]
- 57. Li Y., Dowbenko D., Lasky L. A. (2002) AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival. J. Biol. Chem. 277, 11352–11361 [DOI] [PubMed] [Google Scholar]
- 58. Campisi J., d'Adda di Fagagna F. (2007) Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8, 729–740 [DOI] [PubMed] [Google Scholar]
- 59. Xue W., Zender L., Miething C., Dickins R. A., Hernando E., Krizhanovsky V., Cordon-Cardo C., Lowe S. W. (2007) Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dittmer J. (2003) The biology of the Ets1 proto-oncogene. Mol. Cancer 2, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Arumugam T., Simeone D. M., Van Golen K., Logsdon C. D. (2005) S100P promotes pancreatic cancer growth, survival, and invasion. Clin. Cancer Res. 11, 5356–5364 [DOI] [PubMed] [Google Scholar]
- 62. Bigler D., Gioeli D., Conaway M. R., Weber M. J., Theodorescu D. (2007) Rap2 regulates androgen sensitivity in human prostate cancer cells. Prostate 67, 1590–1599 [DOI] [PubMed] [Google Scholar]
- 63. Ferlay J., Shin H. R., Bray F., Forman D., Mathers C., Parkin D. M. (2010) Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 127, 2893–2917 [DOI] [PubMed] [Google Scholar]
- 64. Mohandas K. M. (2011) Colorectal cancer in India: controversies, enigmas and primary prevention. Indian J. Gastroenterol. 30, 3–6 [DOI] [PubMed] [Google Scholar]
- 65. Ghosh A. R., Koley H., De D., Paul M., Nair G. B., Sen D. (1996) Enterotoxigenic Escherichia coli associated diarrhoea among infants aged less than six months in Calcutta, India. Eur. J. Epidemiol. 12, 81–84 [DOI] [PubMed] [Google Scholar]
- 66. Barker N., Ridgway R. A., van Es J. H., van de Wetering M., Begthel H., van den Born M., Danenberg E., Clarke A. R., Sansom O. J., Clevers H. (2009) Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457, 608–611 [DOI] [PubMed] [Google Scholar]
- 67. Nandi A., Bhandari R., Visweswariah S. S. (1997) Epitope conservation and immunohistochemical localization of the guanylin/stable toxin peptide receptor, guanylyl cyclase C. J. Cell Biochem. 66, 500–511 [DOI] [PubMed] [Google Scholar]
- 68. Schwitalla S., Fingerle A. A., Cammareri P., Nebelsiek T., Göktuna S. I., Ziegler P. K., Canli O., Heijmans J., Huels D. J., Moreaux G., Rupec R. A., Gerhard M., Schmid R., Barker N., Clevers H., Lang R., Neumann J., Kirchner T., Taketo M. M., van den Brink G. R., Sansom O. J., Arkan M. C., Greten F. R. (2013) Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 152, 25–38 [DOI] [PubMed] [Google Scholar]
- 69. Shih I. M., Wang T. L., Traverso G., Romans K., Hamilton S. R., Ben-Sasson S., Kinzler K. W., Vogelstein B. (2001) Top-down morphogenesis of colorectal tumors. Proc. Natl. Acad. Sci. U.S.A. 98, 2640–2645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lembo A. J., Schneier H. A., Shiff S. J., Kurtz C. B., MacDougall J. E., Jia X. D., Shao J. Z., Lavins B. J., Currie M. G., Fitch D. A., Jeglinski B. I., Eng P., Fox S. M., Johnston J. M. (2011) Two randomized trials of linaclotide for chronic constipation. N. Engl. J. Med. 365, 527–536 [DOI] [PubMed] [Google Scholar]