

Abstract
Pollen development in angiosperms is one of the most important processes controlling plant reproduction and thus productivity. At the same time, pollen development is highly sensitive to environmental fluctuations, including temperature, drought, and nutrition. Therefore, pollen biology is a major focus in applied studies and breeding approaches for improving plant productivity in a globally changing climate. The most accessible developmental stages of pollen are the mature pollen and the pollen tubes, and these are thus most frequently analyzed. To reveal a complete quantitative proteome map, we additionally addressed the very early stages, analyzing eight stages of tobacco pollen development: diploid microsporocytes, meiosis, tetrads, microspores, polarized microspores, bipolar pollen, desiccated pollen, and pollen tubes. A protocol for the isolation of the early stages was established. Proteins were extracted and analyzed by means of a new gel LC-MS fractionation protocol. In total, 3817 protein groups were identified. Quantitative analysis was performed based on peptide count. Exceedingly stage-specific differential protein regulation was observed during the conversion from the sporophytic to the gametophytic proteome. A map of highly specialized functionality for the different stages could be revealed from the metabolic activity and pronounced differentiation of proteasomal and ribosomal protein complex composition up to protective mechanisms such as high levels of heat shock proteins in the very early stages of development.
Plants contain numerous specialized cell types, each of them expressing a specific set of proteins. In recent studies much effort has been put into isolating and analyzing proteins of these individual cell types (1) rather than whole organs. New emerging methods have led to the in-depth analysis of different plant cell types, including guard cells (2, 3), mesophyll cells (4), trichomes (5–9), root hair cells (10–12), and egg cells (13). Additionally, because of their easy availability, mature pollen and in vitro–grown pollen tubes are among the most frequently studied cell types. Pollen and pollen tube proteomes have been analyzed, for example, from Arabidopsis (14–16), lily (17, 18), tomato (19, 20), rice (21, 22), quercus and pine trees (23–27), and tobacco (28).
As pollen represents the severely reduced male gametophyte of higher plants, it expresses a very unique set of genes (29) required for the fast and energy-consuming polar outgrowth of the pollen tube during the fertilization process (30). Enzymes required for metabolism and energy generation are overrepresented, but there are also components of the exocytotic machinery, including signaling proteins (14) required for the deposition of pectin compounds at the tip of the growing pollen tube.
Although mature pollen and in vitro–grown pollen tubes have been the focus of research because of the ease of harvesting procedures and are widely used for cell biological studies (31–36), this is not the case for earlier stages of pollen development.
In angiosperms, mature pollen develops from microsporocytes in the anthers of the flower in a series of distinct stages (37). After the microsporocytes have completed meiosis, they form tetrads that release microspores with one central haploid nucleus. These microspores undergo polarization and asymmetric mitosis. The bigger vegetative cell internalizes the smaller cell, which later divides again and forms the two sperm cells. Finally, the pollen desiccates. When the pollen falls on the stigma, it rehydrates, and the vegetative cell forms a pollen tube that delivers the two sperm cells through the transmitting tract to the ovule (38).
Even though pollen development studies using electron microscopy date back to the 1960s (39) and many mutants are described that are disrupted in this process (14), information on the proteome of developing pollen remains relatively sparse and is mostly restricted to whole anthers (40, 41). Only very recently, a work on tomato pollen was conducted covering five developmental stages (42).
The transcriptome of Arabidopsis pollen has been analyzed from the microspore stage on (43), but the earlier stages of microsporocytes, meiosis, and tetrads were not studied, most likely because of a limitation of available material. However, this study was able to show dramatic changes in the transcriptome during the development from microspores to the mature pollen. Similar studies have been performed with Brassica napus (44) and rice pollen (45).
A comparative analysis of the proteome from these stages, as presented in our study, can have special relevance, because in pollen the proteome can greatly differ from the transcriptome not only quantitatively, but also qualitatively, as has been shown for Arabidopsis pollen (14). It seems that often the mRNA is degraded while the protein persists or mRNA is stored in desiccated pollen to be transcribed after rehydration (14).
Additionally, in our proteomic study, we extended the analysis to even earlier stages, including the stage of meiosis. We were able to compare, for the first time, the proteome of a total of eight stages: the diploid microsporocytes, cells undergoing meiosis, tetrads, microspores, polarized microspores (undergoing mitosis I), bipolar pollen, desiccated pollen, and finally pollen tubes. We found that the proteome underwent great changes during development, especially during the polarized microspore stage.
EXPERIMENTAL PROCEDURES
Plant Growth and Pollen Collection
Tobacco was grown under greenhouse conditions (12 h of light, 120 μmol m−2 s−1, 23 °C during the day, 20 °C at night, 60% humidity). Flowers of different sizes were collected, and the anthers of individual flowers were sampled in 200 μl of 10% mannitol. Anthers were gently squeezed open and vortexed, and the supernatant including the released pollen was transferred to a new tube. Pollen was spun down at 100 × g for 1 min and washed twice with 10% mannitol. A subfraction of the pollen of each individual flower was analyzed under a microscope to determine the developmental stage. Samples not representing a stage with at least 90% of their pollen were discarded.
Pollen tubes were grown for 5 h in pollen tube medium (10% sucrose, 15 mm MES-KOH pH 5.9, 1 mm CaCl2, 1 mm KCl, 0.8 mm MgSO4, 1.6 mm H3BO3, 30 μm CuSO4) slightly modified according to Read et al. (46).
Young leaves and roots were ground in liquid nitrogen, and proteins were extracted accordingly.
Microscopy
Pollen samples were fixed in 10% mannitol and 4% formaldehyde overnight, collected via centrifugation, and resuspended in 1 μg/ml DAPI and 1% triton X-100 5 min prior to microscopy.
Images were recorded with an upright point laser scanning confocal microscope (LSM780, Zeiss, Oberkochen, Germany) using a 405-nm diode laser for excitation and a band-pass filter ranging from 450–550 nm. Acquired images were processed using Fiji software.
Quantitative Proteome Analysis (GeLC-LTQ-Orbitrap MS)
For each sample, pollen from between 5 and 30 flowers (depending on the stage) was pooled, freeze-dried, cooled in liquid nitrogen, and ground for 3 min in a shaking mill using three 2-mm steel balls per tube. The pollen fragments were resuspended in 200 μl of protein extraction buffer (62.5 mm Tris-HCl pH 6.5, 5% SDS (w/v), 10% glycerol (v/v), 10 mm DTT, 1.2% (v/v) plant protease inhibitor mixture (Sigma P9599)) and incubated for 5 min at room temperature. After this time, the samples were mixed again by pipetting, incubated for 3 min at 90 °C, and then centrifuged at 21,000 × g for 5 min at room temperature. Supernatants were carefully transferred to a new tube. After the addition of an equal volume of 1.4 m sucrose, proteins were extracted twice with Tris-EDTA buffer–equilibrated phenol. The combined phenolic phases were counter-extracted with 0.7 m sucrose and subsequently mixed with five volumes of 0.1 m ammonium-acetate in methanol to precipitate the proteins. After 16 h of incubation at −20 °C, samples were centrifuged for 5 min at 5000 × g at 5 °C. The pellet was washed twice with 0.1 m ammonium-acetate and once with acetone and then air-dried. Pellets were redissolved in 6 m urea, 5% SDS, and protein concentrations were estimated via bicinchoninic acid assay (47).
Proteins were analyzed via a new gel-LC-MS protocol (48). 40 μg of protein were loaded into a mini-protean cell and run for 1.5 cm. Gels were fixed and stained with methanol:acetic acid:water:Coomassie Brilliant Blue R-250 (40:10:50:0.001). Gels were destained in methanol:water (40:60), and then each lane was divided into two fractions. Gel pieces were destained, equilibrated, and digested with trypsin as previously described (49). Peptides were then desalted with the use of Bond-Elute C-18 stage tips (50) and concentrated in a SpeedVac. Prior to mass spectrometric measurement, protein digest pellets were dissolved in 4% (v/v) acetonitrile, 0.1% (v/v) formic acid. 10 μg of digested peptides were loaded per injection into a one-dimensional nano-flow LC-MS/MS system equipped with a pre-column (Eksigent, Redwood City, CA, USA). Peptides were eluted using a monolithic C18 column Chromolith RP-18r (Merck, Darmstadt, Germany) of 15-cm length and 0.1-mm internal diameter during an 80-min gradient from 5% to 50% (v/v) acetonitrile/0.1% (v/v) formic acid with a controlled flow rate of 500 nl/min.
MS analysis was performed on an Orbitrap LTQ XL mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). Specific tune settings for the MS were as follows: the spray voltage was set to 1.8 kV using a needle with a 30-μm inner diameter (PicoTip Emitter, New Objective, Woburn, MA), and the temperature of the heated transfer capillary was set at 180 °C. Fourier transform MS was operated as follows: full scan mode, centroid, resolution of 30,000, covering the range of 300–1800 m/z, and cyclomethicone used as a lock mass. Each full MS scan was followed by 10 dependent MS/MS scans performed in the ion trap, in which the 10 most abundant peptide molecular ions were dynamically selected with a dynamic exclusion window set to 90 s and an exclusion list set to 500. Dependent fragmentations were performed in collision-induced dissociation mode with a normalized collision energy of 35, an isolation width of 2.0, an activation Q of 0.250, and an activation time of 30 ms. Ions with an unassigned charge or a charge of +1 were excluded for fragmentation. The minimum signal threshold was set at 1000.
Raw data were searched with the SEQUEST algorithm present in Proteome Discoverer version 1.3 (Thermo, Germany) as described elsewhere (51). In brief, identification confidence was set at a 5% false discovery rate, and the variable modifications were set as acetylation of the N terminus, oxidation of methionine, and carbamidomethyl cysteine formation, with mass tolerances of 10 ppm for the parent ion and 0.8 Da for the fragment ion. Up to two missed cleavage sites were permitted. Three different databases were employed (tobacco 7.0, a CDNA library from the gene index project with 120,122 entries; a tobacco protein database from UniProt 09.2011 with 4826 entries; and a genomic sequence database from the Tobacco Genome Initiative 11.2008 with 349,877 entries, resulting in 2,099,262 entries after six-frame translation). Databases were translated with an in-house tool, taking into consideration only the longest open reading frame of all reading frames. In the case of the genomic sequences, the longest open reading frames of all reading frames were considered.
When the database from the gene index project was used, additional variable modifications were allowed: phosphorylation of threonine, serine, and tyrosine; methylation and dimethylation of lysine and arginine; and acetylation and trimethylation of lysine.
Peptides were matched against these databases plus decoys, with a significant hit considered as one in which the peptide confidence was at least medium or high, and the xcorr score threshold was established at 2.5 for +2 ions and 3.5 for charge states of +3 or greater. The high thresholds were chosen to minimize false identifications based on the incomplete databases used.
The identified proteins were quantitated via a label-free approach based on peptide count followed by a normalized spectral abundance factor (NSAF)1 normalization strategy (52),
in which the total number of spectra counts for the matching peptides from protein k (PSM) was divided by the protein length (L) and then divided by the sum of PSM/L for all N proteins.
Multivariate Statistical and Bioinformatic Data Analysis
Multivariate statistical analyses such as principal components analysis (PCA) and k-means clustering were performed with the statistical toolbox COVAIN (53). The software and parameter settings can be accessed online. Missing values were estimated from the dataset, and data were log transformed before the PCA. For cluster analysis, the mean NSAF value of each developmental stage was calculated and normalized for each protein, setting the total amount throughout the stages to 1.
All proteins in the three used databases were blasted for the closest Arabidopsis (TAIR10) homologue using an unpublished Python script in conjunction with stand-alone BLAST v2.2.26+ using the default matrix, and entries in the TAIR Arabidopsis MapMan mapping file (Ath_AGI_LOCUS_TAIR10_Aug2012) were replaced as previously described (54). This way, most tobacco protein accessions could be assigned to a functional bin and an Arabidopsis homologue.
Tobacco and Arabidopsis microarray results were binned according to the MapMan mapping files Ntob_AGILENT44K_mapping and Ath_AGI_LOCUS_TAIR10_Aug2012, respectively. Tobacco bin numbers were slightly adjusted to fit the tobacco protein bins.
Further blasting of the tobacco protein sequences versus the list of Arabidopsis proteins found in Arabidopsis pollen (14) and a list of pollen-affected Arabidopsis mutants (extended list from Ref. 14) was performed using the same Python script.
RESULTS AND DISCUSSION
Isolation and Proteomic Analysis of Early and Late Pollen Developmental Stages
Pollen from a total of eight developmental stages was harvested for proteomic analysis (Figs. 1 and 2).
Fig. 1.

Confocal images of pollen purifications as were used for proteomic analysis. A–D, microsporocytes; E–H, meiotic cells; I–L, tetrads; M–P, microspores. Pollen was stained with DAPI and images were obtained at 250× and 630× magnification.
Fig. 2.

Confocal images of pollen purifications as used for proteomic analysis. A–D, polarized microspores; E–H, binuclear pollen; I–L, desiccated and rehydrated pollen; M–P, in vitro grown pollen tubes. Pollen was stained with DAPI and images were obtained at 250× and 630× magnification.
These distinct stages were diploid microsporocytes (also referred to as stage A), meiosis (stage B), tetrad stage (stage C), microspores (stage D), polarized microspores (stage E), bipolar pollen (stage F), desiccated pollen (stage G), and pollen tubes (stage H).
Immature pollen was obtained by gently opening the anther buds of individual flowers and vortexing in 10% mannitol. In this way, pollen in the supernatant could be easily separated from larger cell debris via simple pipetting. Smaller cell debris and soluble proteins could be removed with the supernatant after low-speed centrifugation.
Although pollen from the microsporocyte and meiosis stages was obtained as large aggregates, which were associated with cell debris (Fig. 1), it was possible to isolate individual cells (or tetrads) from later stages (Figs. 1 and 2).
As the stage of pollen development cannot be easily determined by the size of the flower or anthers, especially in early stages, the developmental stage of the pollen of each individual flower was determined via microscopy, and a sufficient amount of pollen was pooled for protein extraction.
Desiccated pollen was harvested after anthesis, and pollen tubes were grown in vitro for 6 h.
For comparison, proteins from young tobacco leaves and roots were analyzed.
Three biological replicates of each stage (or tissue) were analyzed and separated into two fractions via SDS-PAGE prior to tryptic digestion and LC-MS/MS analysis.
The spectra of all identified peptides (supplemental Table S1) from the different stages can be reviewed online in the proteomics database PROMEX (http://promex.pph.univie.ac.at/promex/Experiment; Nic taba002 for pollen and Nic taba003 for roots and leaves). Additionally, the mass spectrometry proteomics data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository (55) with the dataset identifier PXD000469.
In total, 3817 protein groups were identified from all pollen stages (Table I, supplemental Table S2), with stages A–D and F–H showing the most overlap (Fig. 3A). When the results were compared with data on extracts from tobacco roots and leaves, a total of 4262 protein groups were identified: 1217 from leaves, 1285 from roots, and 3888 from pollen (Fig. 3B, Table I, supplemental Table S3; the increased number of pollen protein groups is due to different groupings of the proteins). The high number of identified pollen proteins was in part caused by the large tobacco genome (4.5 billion bp), leading to the finding of many homologue isoforms, but it is also attributable to the great changes that took place in the proteome during the development.
Table I. Number of identified protein groups by stage/tissue. Values given in parentheses are the numbers of protein groups when only pollen proteins are considered, which leads to a smaller number of protein groups from the same number of proteins (due to different grouping).
| Stage | Label | n proteins |
|---|---|---|
| Microsporocyte | A | 1775 (1741) |
| Meiosis | B | 1596 (1573) |
| Tetrad | C | 1748 (1719) |
| Microspore | D | 1288 (1264) |
| Polarized microspore | E | 2004 (1956) |
| Binuclear pollen | F | 1770 (1740) |
| Dessicated pollen | G | 1604 (1580) |
| Pollen tubes | H | 2526 (2485) |
| Pollen total | 3888 (3817) | |
| Leaves | 1217 | |
| Roots | 1285 | |
Fig. 3.
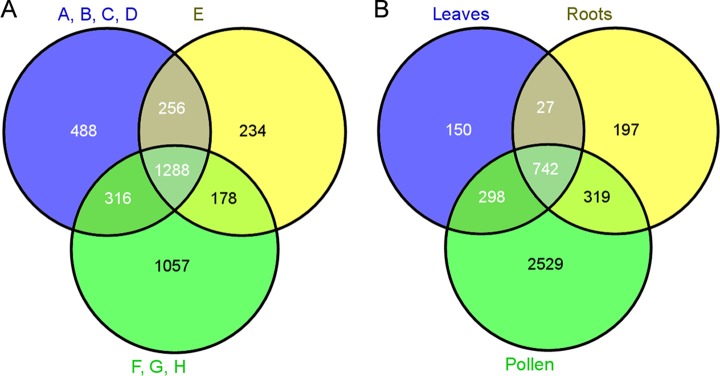
Venn diagrams. A, number of proteins identified throughout development. A–D, microsporocytes, meiotic cells, tetrads, and microspores, respectively; E, polarized microspores; F–H, binuclear pollen, desiccated pollen, and pollen tubes, respectively. B, number of identified proteins in all pollen stages, leaves, and roots.
The protein groups represent a total of 12,728 putative protein accessions in pollen (supplemental Table S2) and 14,323 proteins in all the samples (supplemental Table S3). For easier reading, the protein groups are referred to as proteins hereinafter.
Protein abundances were quantified by peptide count and an NSAF normalization strategy (52). For further analysis, only proteins that were detected in all three biological replicates of at least one of the developmental stages (or tissues) were considered, leading to datasets of 1869 proteins when only pollen proteins were considered and 2135 proteins when leaves and roots were included. Proteins were classified by identifying the closest Arabidopsis homologue and assigning a function according to functional Arabidopsis mapping for MapMan (supplemental Tables S2, S3, and S7).
Of the 2135 proteins used for quantification, 837 were not detected in any of the root or leaf samples (supplemental Table S3). It cannot be ruled out that these proteins are also present in minor amounts in these organs or in other non-analyzed tissues. However, the proteins showing high expression levels in one of the pollen stages (Table II) can be considered as especially strong candidates for being specifically expressed in developing pollen, or at least for serving specific purposes in these cell types. One example is the highly abundant ethanol dehydrogenase, which serves a specific function in the primary metabolism of pollen tubes (56) and is not needed in roots and leaves, at least under normal conditions, as well as cell wall degrading enzymes. Another protein that was also not found in roots and leaves is the Rab-GDP dissociation inhibitor, which is crucial for G-protein signaling and, thus, maintaining cell polarity during polar tip growth.
Table II. Proteins with the highest NSAF scores (maximum of eight stages) that were not detected in roots and leaves.
| Accession number | Proposed function | Closest Arabidopsis homologue | A | B | C | D | E | F | G | H |
|---|---|---|---|---|---|---|---|---|---|---|
| CN824898 | Unknown | — | 8.73 | |||||||
| AM795719 | Unknown | — | 3.29 | 0.44 | 5.27 | 7.58 | ||||
| Q42953 | Alcohol dehydrogenase | AT1G77120.1 | 0.06 | 3.46 | 4.98 | 2.94 | ||||
| TC125213 | Globulin-like protein, storage | AT1G07750.1 | 0.06 | 0.32 | 0.06 | 0.25 | 4.73 | 3.28 | 4.27 | 2.38 |
| 191520275 | Unknown | — | 4.63 | |||||||
| AM837330 | Fructokinase | AT4G10260.1 | 0.22 | 3.28 | 3.06 | 4.39 | 2.52 | |||
| TC131738 | Globulin-like protein, storage | AT1G07750.1 | 0.04 | 0.33 | 0.04 | 0.20 | 4.32 | 2.63 | 4.07 | 1.85 |
| 191442209 | Polygalacturonase inhibitor protein precursor | AT5G06860.1 | 2.00 | 4.19 | 2.91 | 2.19 | 2.96 | 0.07 | 0.22 | |
| TC127895 | Polygalacturonase inhibitor protein precursor | AT5G06860.1 | 2.10 | 3.98 | 3.32 | 2.70 | 2.80 | 0.20 | 0.15 | |
| 191443577 | Polygalacturonase inhibitor protein precursor | AT5G06860.1 | 2.11 | 3.85 | 3.02 | 2.74 | 2.75 | 0.21 | 0.16 | |
| TC156020 | Glyceraldehyde 3-phosphate dehydrogenase | AT1G13440.2 | 3.74 | 1.86 | 2.38 | |||||
| O24625 | Chalcone/stilbene synthase | AT1G02050.1 | 3.66 | 0.43 | 0.12 | 2.87 | 0.06 | |||
| D4I601 | Alcohol dehydrogenase | AT1G77120.1 | 0.12 | 1.65 | 2.86 | 3.45 | ||||
| TC129072 | Alcohol dehydrogenase | AT1G77120.1 | 1.48 | 3.37 | 2.72 | |||||
| TC132846 | Late embryogenesis abundant protein | AT3G15670.1 | 3.27 | |||||||
| TC150130 | Rab-GDP dissociation inhibitor | AT5G09550.1 | 0.66 | 3.11 | 0.71 | |||||
| 190837846 | Unknown | AT3G59510.1 | 1.40 | 3.09 | 1.63 | 1.22 | 2.29 | 0.60 | ||
| 191572052 | Unknown | AT1G12570.1 | 3.08 | 2.10 | ||||||
| CV021106 | Ribosomal 60S subunit.L32 | AT4G18100.1 | 1.35 | 1.41 | 1.05 | 2.95 | 0.17 | 0.28 | 0.26 | |
| AM806415 | Membrane transporter | AT3G08580.2 | 0.16 | 0.37 | 2.70 | 1.33 | 1.06 |
Notes: Average NSAF scores over three biological replicates multiplied by 1000. A, microsporocytes; B, meiotic cells; C, tetrads and microspores; E, polarized microspores; F, binuclear pollen; G, desiccated pollen; H, pollen tubes.
Multivariate Statistical Data Mining
A PCA of the pollen proteins alone revealed that, on the proteome level, tobacco pollen development could be separated into three major phases (Fig. 4A), with the first one including the stages from microsporocytes to microspores (A–D), the second one including only the polarized microspores (E), and the third one including the binuclear pollen stage to the pollen tubes (F–H).
Fig. 4.

Multivariant statistic analysis. A, PCA plot of the eight stages of pollen development. B, sum of the NSAF scores of the 30 highest and 30 most negative loadings of PC1 and PC2, respectively. C, PCA plot of the eight stages of pollen development including leaves and roots. PCA was based on log values of the NSAF scores. D, correlation coefficient according to calculated average NSAF values of three replicates. A, microsporocytes; B, meiotic cells; C, tetrads; D, microspores; E, polarized microspores; F, binuclear pollen; G, desiccated pollen; H, pollen tubes; L, leaves; R, roots.
This separation in the PCA and the stage specificity of the proteomes are clearly based on different cell functionalities. In the first four stages (A–D), the principal function of the pollen is its own transformation from diploid microsporocytes to microspores, whereas the obviously very different function of the rehydrated pollen is to produce and elongate a pollen tube. To facilitate a quick outgrowth, many proteins that support this function are apparently already synthesized prior to desiccation, which leads to the observed similarity of the last three stages (F–H). The polarized microspore stage (E) could be a transition stage. However, this stage also contains a unique set of proteins not present in any of the other stages.
The distinct composition of the proteome of this stage was also apparent in the individual principal components (supplemental Table S4). PC1 separated the samples according to their ongoing development, and PC2 separated stage E from all other stages (Fig. 4A).
The proteins with the highest loadings in PC2 showed comparatively high abundance in stage E (Fig. 4B), but they were also present in stages A, F, and H. Among these proteins were several subunits of the 26S proteasome.
In comparison, the proteins with the most negative loadings in PC2 showed an inverse expression pattern (Fig. 4B). The proteins with the highest PC2 loadings were ribosomal proteins, hinting at a severe rearrangement of the ribosomal complex during stage E: from a total of 155 detected ribosomal proteins, 71 were missing in stage E, and 44 of these were detected in all other stages. A specific set of 38 ribosomal proteins had higher loadings in stage E than the average over all samples.
Thus, we observed a pronounced reprogramming of the protein synthesis machinery that might prepare the ribosomal complex machinery for the high demands of protein synthesis during pollen tube growth.
As PC1 differentiated the samples according to development, the negative loadings represented proteins with high abundance in early stages that declined during development, whereas positive loadings represented proteins with high expression levels in the last stages (Fig. 4B).
Among the negative loadings, histones were especially well represented, probably because of their lower cell-volume-to-nucleus ratios in early stages relative to mature pollen.
The highest positive loadings included proteins required for pollen tube growth such as enzymes of the primary metabolism, ethanolic fermentation, and cell wall synthesis.
A PCA additionally including roots and leaves revealed a clear separation of the different tissues (Fig. 4C, supplemental Table S4). Whereas PC1 discriminated especially between the different pollen stages and the sporophytic tissues, PC2 discriminated between roots and leaves, with the earlier developmental pollen stages being more closely related to leaves. This closer connection of leaves and the early male gametophytes was also apparent from the correlation coefficients (Pearson's R, Fig. 4C).
In order to further group the pollen proteins according to their presence in the different stages, the NSAF scores were normalized for each protein and the proteins were clustered using the k means algorithm (supplemental Table S6, supplemental Fig. S1). 12 of the 35 clusters showed proteins that were almost exclusively expressed in one of the stages (Table III).
Table III. Overview of stage-specific clusters identified via k means clustering.
| Stage | Cluster number | N proteins |
|---|---|---|
| A | 10 | 22 |
| B | 27 | 5 |
| C | 33 | 15 |
| D | 2 | 7 |
| E | 5, 6, 21 | 59, 51, 29 |
| F | 30 | 10 |
| G | 13, 24 | 17, 33 |
| H | 11, 15 | 39, 47 |
| B + E | 23 | 33 |
| F + H | 22 | 37 |
Again, stage E stood out in terms of the number of specifically expressed proteins (clusters 5, 6, and 21; Fig. 5A). Among them were three proteins similar to Skp1 (BP531238, TC168823, and 191216821), a core component of the E3 ubiquitin ligase that targets protein for degradation by the 26S proteasome. The isoform expressed in stage E might interact with specific F-Box proteins, which could target a distinctive set of proteins for ubiquitination and breakdown, in order to adjust the proteome as required for the change in cellular function.
Fig. 5.

Cluster analysis. Relative abundance of proteins in a selection of groups obtained via k means clustering. NSAF scores were averaged over three biological replicates and normalized for each protein to represent their proportion of the total abundance over all eight stages. A, microsporocytes; B, meiotic cells; C, tetrads; D, microspores; E, polarized microspores; F, binuclear pollen; G, desiccated pollen; H, pollen tubes.
It is also possible that the skp1 proteins are directly involved in mitosis. The 26S proteasome is a key factor in the degradation of cell cycle proteins (57). Also, a skp1-like 1 (ASK1) of Arabidopsis is essential for meiosis in pollen (58), where it is essential for nuclear reorganization and homolog juxtapositioning (59). It was also found to be involved in mitosis (60).
Several proteases identified in stage E could also be in part responsible for the major rearrangement of the pollen proteome during in this transition stage.
Many potential cellulases, glucosidases, and mannosidases were expressed during stage E (supplemental Table S6). They have previously been proposed to support the loosening of the cell wall required for cell expansion taking place between stages D and F (61).
A subset of proteins grouped in cluster 23 (Fig. 5B) was expressed predominantly during meiosis (B) and mitosis (E), and these proteins might take part in the regulation of mitosis and meiosis, as they are specifically expressed in these stages (supplemental Table S6). One example is annexins (TC137724, BP533244, Q56D09), which might act in targeted secretion (62), required for cytokinesis. Another example is a set of potential subtilases (TC132351, TC133164, TC133288, 191501021) that could take part in signaling or specific protein cleavage and degradation.
Other proteins expressed during these two stages are predicted to play a role in secondary metabolism, which makes them unlikely to take direct part in the cell cycle. Three of them (191361943, CN949712, and O24625) show homologies to the anther expressed proteins less adhesive pollen 5 and 6, which show similarities to chalcone synthases and are essential for exine formation (63).
Before pollination, the pollen desiccates and has to drastically adjust its physiology to protect its membranes from breaking and its proteins from denaturation. In the cluster analysis, a set of proteins was grouped (clusters 13 and 24; Fig. 5C) that was almost exclusively expressed in the desiccated stage (G) and disappeared after rehydration and pollen tube growth (H). Among these proteins were late early abundant proteins (TC132846, TC146808, TC165472, 191501982) that also play a role in the desiccation of seeds (64) and are proposed to protect pollen during dehydration (65). Also, a homologue of an Arabidopsis tonoplast monosaccharide transporter (TC129132) and potential signaling proteins that could play a role in adaption to desiccation were grouped in this cluster.
Additionally, a set of proteins (cluster 22; Fig. 5D) was identified showing that some proteins might be degraded prior to desiccation and resynthesized after rehydration. Proteins in this cluster included enzymes of β-oxidation and of other primary metabolic pathways (supplemental Table S4). We can only speculate about the reason for this temporary degradation. The proteins might have a negative effect on the adaption to desiccation or be unstable under this condition.
Functional Remodeling of the Proteome During Pollen Development
During development, the pollen cells have to adjust their metabolism to suit their functions. In order to get a better overview of the functionality, proteins were matched against their closest Arabidopsis homologues and grouped according to their predicted functions (supplemental Table S7). The total NSAF scores were added (Fig. 6).
Fig. 6.

Functional analysis. Total NSAF scores of proteins grouped in functional groups. NSAF scores were averaged over three biological replicates and multiplied by 1000. Red indicates high abundance in relation to the other stages. Standard deviation can be assessed from supplemental Table S7. L, leaves; R, roots; A, microsporocytes; B, meiotic cells; C, tetrads; D, microspores; E, polarized microspores; F, binuclear pollen; G, desiccated pollen; H, pollen tubes.
Some functional groups were predominantly present in specific stages, such as the already mentioned late early abundant proteins in desiccated pollen (stage G); the gluco-, galacto-, and mannosidases, factors of protein degradation in polarized microspores (stage E); and the enzymes of secondary metabolism during meiosis and mitosis (stages B and E).
Starch synthesis seems to occur in microsporocytes and binuclear pollen prior to desiccation (stages A and F), most likely to store energy for cell division and pollen tube growth (66), respectively. The synthesis of storage proteins starts during polarization of the microspore.
Many enzymes required for energy-consuming pollen tube growth are synthesized starting from the polarized microspore or binuclear pollen stage (stages E and F). These include enzymes required for ethanolic fermentation, which is performed by pollen tubes due to anoxia caused by rapid oxygen consumption during their growth (56). Furthermore, enzymes of sucrose, lipid, and amino acid degradation and proteins involved in cell wall metabolism and vesicle trafficking follow a similar expression pattern. These proteins are required to support pollen tube growth with sufficient energy (30) and the machinery to deposit large amounts of cell wall and membrane at the tip of the growing pollen tube (67).
Interestingly, the abundance of proteins involved in anabolic pathways like gluconeogenesis and sucrose synthesis is also increased in the later stages, which is somewhat surprising, as sucrose can be taken up by the pollen tube from the surrounding tissue (68) and was also present in large amounts in the medium used for cultivation in this study.
Proteins associated with cell division showed increased abundance during the polarized microspore stage, but their levels were also increased in desiccated pollen, probably in preparation for the mitosis of the generative cell, which takes place after pollen tube germination. One subgroup of proteins (C5MQG8, FG636560, Q1G0Z1, TC141620) in this functional group, the cell cycle controlling CDC48 (69), has numerous functions (70), including spindle disassembly at the end of mitosis (71). The heterozygous Arabidopsis mutant cdc48a (72) displayed incomplete pollen germination of the mutated pollen. Our data suggest an additional role of CDC48 in pollen mitosis, which was not affected in the described mutant, the reason being most likely the presence of two other isoforms of CDC48a in the Arabidopsis genome (73).
Hot and cold temperature stresses can be detrimental to all phases of pollen development and have major effects on sexual reproduction. Heat shock proteins have been described as strongly abundant in pollen during the later stages of development in comparison to other tissues (74). The analysis of earlier stages including microsporocytes, meiotic cells, and tetrads in our study revealed an even greater abundance of proteins associated with heat stress.
A total of 67 heat-stress-associated proteins were identified in our study, including isoforms of the chaperones HSP 70 and HSP 90 and luminal binding proteins (supplemental Table S7). Taken together, these proteins constituted up to 10% of the total protein abundance according to our NSAF calculation (Fig. 6), highlighting the importance of these groups of proteins for pollen development, especially in early stages.
The functional comparison of the pollen stages with roots and leaves displayed a number of functional groups including ethanolic fermentation, polyamine metabolism, and late early abundant and storage proteins, which were almost not found in the sporophytic tissues and which highlight the highly specialized functionality of the developing pollen (Fig. 6). The comparison also displayed the previously described high rate of anabolic metabolism, especially in the late pollen stages. Also, the synthesis of fatty acids seemed to be much higher in pollen tubes than in leaves and roots, as the abundance of the related proteins was much higher. This shows that pollen tubes do not rely solely on previously synthesized and oil-body stored fatty acids and also require de novo synthesis to cope with the rapidly expanding membranes.
The rate of protein synthesis, in contrast, did not seem to be strongly enhanced in developing pollen or, especially, pollen tubes when taking into account the abundance of proteins associated with protein synthesis (ribosomal and nonribosomal). This observation once more supports the idea that growing pollen tubes rely strongly on presynthesized proteins.
Analysis with Respect to Previous Transcriptomics, Proteomics, and Genetic Studies
In order to find out to what extent protein and transcript levels differed, the data in this study were compared with expression data from a previous study (75). The microarrays of the latter study were based on transcripts obtained from mature tobacco pollen grains and pollen tubes.
Because different accessions were used in this and the previous study and in order to simplify the comparison, the transcripts and proteins were grouped according to their MapMan bins, and the individual values were added (supplemental Table S8).
From the comparison, it is apparent that the transcripts of proteins involved in signaling were much higher than the protein levels, maybe because of high turnover rates (Table IV). In contrast, protein levels of enzymes of the primary metabolism were much higher relative to their transcript levels (Table IV). This could be because the translation rate of these transcripts is much higher or the turnover rate of the proteins is lower. Another possibility is that the proteins are synthesized in earlier stages of pollen development and persist while the mRNA is degraded. Unfortunately, no expression data on developing tobacco pollen are available to date.
Table IV. Comparison of tobacco pollen transcript and protein data.
| Bin | Function | Transcript |
Protein NSAF |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| G | H | A | B | C | D | E | F | G | H | ||
| 1.3.3 + 4.1.11 | 3-phosphoglycerate kinase | 0.2 | 0.2 | 12.1 | 8.9 | 10.4 | 13.0 | 11.7 | 18.2 | 20.8 | 16.9 |
| 4.1.8 | Glyceraldehyde 3-phosphate dehydrogenase | 0.9 | 0.5 | 27.8 | 17.9 | 31.0 | 31.3 | 20.4 | 40.8 | 33.2 | 23.9 |
| 5.2 | Pyruvate decarboxylase | 0.2 | 0.1 | 0.1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.3 | 2.9 | 2.1 |
| 5.3 | Alcohol dehydrogenase | 0.5 | 0.2 | 0.0 | 0.4 | 0.0 | 0.0 | 0.0 | 13.4 | 22.6 | 17.7 |
| 30.3 | Calcium signaling | 30.7 | 31.4 | 8.5 | 11.5 | 11.5 | 7.6 | 5.8 | 2.6 | 5.8 | 3.5 |
| 30.5 | G-proteins | 20.2 | 23.6 | 8.4 | 5.9 | 7.5 | 6.1 | 9.0 | 10.3 | 5.0 | 11.8 |
| 20.2.1 | Stress abiotic heat | 6.4 | 7.1 | 142.7 | 158.8 | 174.4 | 160.5 | 132.6 | 96.5 | 38.1 | 45.7 |
Notes: Transcript data were previously published by Hafidh et al. (75). Transcripts and proteins were binned according to MapMan. The complete dataset can be surveyed in supplemental Table S8. Normalized values were multiplied by 1000. A, microsporocytes; B, meiotic cells; C, tetrads; D, microspores; E, polarized microspores; F, binuclear pollen; G, desiccated pollen; H, pollen tubes.
Transcript data on Arabidopsis stages ranging from unicellular microspores to pollen tubes have been previously generated (43, 76), and we compared our dataset to these transcript levels. Again, the transcripts and proteins were grouped according to their MapMan bins (supplemental Table S9). Once more it was apparent that the abundance of proteins of the primary metabolism was greater than the corresponding transcript levels. Additionally, they followed a different pattern. The enzymes phosphoglycerate kinase and pyruvate decarboxylase, for example (Fig. 7), showed their greatest abundance in desiccated pollen, whereas the corresponding Arabidopsis transcripts peaked much earlier and were completely abolished in mature pollen and pollen tubes, respectively. It could be speculated that this difference is simply due to the different analyzed species. The detection of both proteins in substantial amounts in a proteomic survey of mature Arabidopsis pollen (14), however, makes a strong case that proteins are synthesized in earlier stages and persist though desiccation, rehydration, and pollen tube growth, by which time their transcripts are already degraded. This seems to be true for many enzymes of glycolysis, as the total protein and transcript abundance of this pathway follows a similar pattern. Another group that shows high protein levels in desiccated tobacco pollen and pollen tubes contains proteins associated with cell wall metabolism. Here, however, the Arabidopsis transcripts show a similar dynamic, maybe because the proteins in this group show a higher turnover rate and have to be resynthesized.
Fig. 7.

Protein and transcript comparison. Tobacco protein abundances (NSAF scores over three replicates) and Arabidopsis transcript levels were grouped according to MapMan. All transcript data excluding pollen tubes were derived from Honys et al. (43). Pollen tube transcript data were derived from Wang et al. (76). Arabidopsis stages: UNM, unicellular microspores; BCP, bicellular pollen; TCP, tricellular pollen; MPG, mature pollen grains; PT, pollen tubes. Tobacco stages: D, microspores; E, polarized microspores; F, binuclear pollen; G, desiccated pollen; H, pollen tubes.
In order to be able to study the different dynamics of transcripts and proteins in better detail, it should be a future goal to generate either protein data from earlier Arabidopsis stages or transcript data throughout tobacco pollen development.
It must be concluded that the transcript levels in mature pollen can be very misleading when considering the importance of a specific gene for pollen tube growth or, even worse, pollen development, especially when only the transcript levels in mature pollen are considered, which is often the case (as, for example, in the commonly used open access version of GENEVESTIGATOR).
To find out how tobacco pollen might differ from Arabidopsis pollen, we compared the proteins in our study to the proteins found in the already mentioned proteomic survey of Arabidopsis (14), blasting all the sequences of the identified proteins from tobacco against the protein sequences from Arabidopsis pollen. All matches with an E-value equal to or less than 10−10 were considered as homologues (supplemental Table S10).
Of the 3817 proteins in this study, only 1055 did not have a homologue in Arabidopsis pollen. Of the 1869 proteins considered for quantification, an even lower proportion (320 proteins) had no homologues in Arabidopsis pollen. Even though this indicates high similarity of the proteomes, there are some distinct differences.
The ortholog of Arabidopsis alcohol dehydrogenase (At1g77120), catalyzing the conversion from acetaldehyde to ethanol, was one of the enzymes with the greatest abundance in tobacco pollen tubes but was not found itself in Arabidopsis pollen. On the transcript level, this gene also showed only very weak expression in microspores and bicellular pollen and no expression in later stages. As the production of ethanol is one of the hallmarks of pollen tube metabolism in many species such as lily (77), tobacco, and petunia (56), this indicates a strong difference in primary metabolism between Arabidopsis and tobacco, probably based on the much shorter growing distance of Arabidopsis pollen tubes, which decreases the problem of anoxia. It is also possible that another enzyme takes this role in Arabidopsis pollen tubes, as several other proteins in Arabidopsis pollen showed a high similarity to the tobacco alcohol dehydrogenase. However, the protein with the highest similarity, ADH2, is already described as a glutathione reductase. It remains to be investigated whether Arabidopsis pollen tubes produce ethanol during growth.
Another possibility is that the acetaldehyde produced by pyruvate decarboxylase (which was found in Arabidopsis pollen) is directly converted to acetate and, later, acetyl-CoA, a pathway termed the pyruvate dehydrogenase bypass. An enzyme that is a strong candidate to perform the oxidation of acetaldehyde, aldehyde dehydrogenase (78), was detected in substantial amounts in Arabidopsis.
Another example of how the primary metabolism might differ is in the conversion of fructose-6-phosphate to fructose-1,6-bisphosphate. Whereas in Arabidopsis pollen only the phosphofructokinase was found, tobacco pollen and pollen tubes additionally contained several pyrophosphate-fructose-6-P phosphotransferase isoforms with a total abundance that was more than 8-fold greater than that of the phosphofructokinase (supplemental Table S9). This way, pyrophosphate can be used instead of ATP for the second activation step of glycolysis, increasing the ATP yield per hexose by one. Although this increase might not be so significant under aerobic conditions, it does make a big difference when ATP is generated via fermentation, which is the case in tobacco but might not be in Arabidopsis.
In Arabidopsis, many mutants are described that are affected in pollen development and pollen tube growth. After a survey of the literature, we updated a previously published list (14) of affected genes from 127 to 215 (supplemental Fig. S11). From these, we found 135 to have homologues (E-value equal to or less than 10−10) in tobacco pollen. This supports the theory that most proteins with important functions in pollen development could be detected in tobacco pollen. However, from the 3817 proteins identified, only 320 homologues have been described so far in Arabidopsis mutant studies according to our literature survey, leaving tremendous room for future pollen research.
Post-translational Modifications
The identification of post-translational modifications was not the focus of this study, and the available material from early developmental stages was too limited for the enrichment of modified peptides. However, 655 potentially modified peptides were detected, including methylation of lysine and arginine; acetylation of lysine; and phosphorylation of serine, threonine, and tyrosine (supplemental Table S12). The protein with the greatest number of modifications was a homologue of elongation factor 1-α (Fig. 8). This protein is a member of the family of small G-proteins and serves a multitude of functions (79) including elongation of protein translation (80), regulation of the cytoskeleton (81, 82), and signaling (83–85). It has been previously shown to be methylated (86, 87) and acetylated (88). Multiple potential methylation sites were found; however, they must be considered with caution, as the mass of the peptide can be identical to a nonmodified peptide of a homologue protein, leading to ambiguous identifications (supplemental Table S12). This is an even bigger problem when the organism used in the study is not entirely sequenced, as potential ambiguous identifications might be missed because of the incomplete database. Also, methylated lysine and arginine residues lie at the N terminus of peptides cleaved by tryptic digest, making a confirmation of the modification based on the MS2 spectrum harder, as y-ions are too small to be detected. Therefore, only methylation sites can be considered as strong candidates (Fig. 8) if the modified amino acid lies in the middle of a miscleaved peptide and the MS2 spectrum shows the correct b- and y-ions of the modified amino acid.
Fig. 8.

Post-translational modifications observed in a homologue of translation elongation factor 1-á (TC 141641). Sequences highlighted in green were identified via LC-MS/MS analysis. Modification sites are marked above the sequence. M, methylation; D, dimethylation; T, trimethylation.
The identification of additional modification sites and the study of their dynamics should be goals for the future. At least, the material available from the microspore stage on should be sufficient for metal oxide affinity chromatography enrichment of phosphopeptides, as has been performed recently for desiccated and activated pollen (28).
CONCLUSION
The comparative proteomic analysis of pollen development was, for the first time, extended to eight stages ranging from diploid microsporocytes to pollen tubes. In order to compare the data to results for sporophytic tissues, leaves and roots were also investigated, leading to the identification of a total of 4262 proteins.
Based on these data, pollen development can be divided into three phases (Fig. 9). The early phase that is still more closely related to leaves ranges from the microsporocytes to meiosis, extends to the formation of tetrads, and ends with the release of the microspores. The proteome of this phase is relatively static, with a high abundance of heat shock proteins to protect the cells in the process of meiosis and cell division. It appears that the “sporophytic proteome” synthesized in the microsporocytes is sustained throughout the development to early microspores.
Fig. 9.

Key features of the tobacco pollen proteome through development. A, microsporocytes; B, meiotic cells; C, tetrads; D, microspores; E, polarized microspores; F, binuclear pollen; G, desiccated pollen; H, pollen tubes.
The late phase ranges from binuclear pollen via desiccated pollen to pollen tubes and presents a “gametophytic proteome.” Many proteins required for pollen tube growth, such as enzymes of the primary metabolism, and for cell wall synthesis are already produced prior to desiccation, so as to later allow a rapid outgrowth of the pollen tube without a bulk protein synthesis, as has also been previously observed (18, 24).
From the comparison of our protein data to Arabidopsis transcript data, it could also be concluded that many proteins, especially from the primary metabolism, do not seem to be further synthesized during pollen tube growth.
In between the early and the late phase, which are clearly very different in their cellular functionality, the pollen undergoes an intermediate phase. During this polarized microspore stage, the “sporophytic proteome” is partially degraded, accompanied by ribosomal rearrangement and a strong increase in the abundance of proteins associated with protein degradation. However, this phase not only appears to be a turning point between the sporophytic and the gametophytic phase, but also seems to represent a phase on its own, because many proteins identified in this work were exclusively found during this stage.
Reasons for this could be the strong expansion in cell size, which is unique to this cell stage in pollen development, the performed polarization and asymmetric mitosis, and also the degradation of the sporophytic proteome, which would need distinct protein degradation machinery.
The great changes in the proteome observed during the three phases underlie the complexity of the protein networks required for male gametogenesis, which are just starting to get unraveled. As this work represents a first thorough proteomic map of pollen development, it could lay the base for a better understanding of these networks, especially of the early stages.
Acknowledgments
We thank EMBO for supporting Till Ischebeck with a long-term fellowship (EMBO-ALTF 299-2010). We thank Luis Recuenco-Munoz, Thomas Naegele, Stefanie Wienkoop, and Christiana Staudinger for helpful discussions. We are very grateful for the expert plant care by Thomas Joch and Andreas Schröfl. We also thank the PRIDE team for their great support.
Supplementary Material
Footnotes
* T.I. was supported by a long-term EMBO fellowship (EMBO-ALTF 299-2010)., L.V. was supported by the European Union through a Marie Curie IEF-grant (FP7-PEOPLE-2009-IEF-255109), and D.L. was funded by FWF Project Number P23441-B20.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- NSAF
- normalized spectral abundance factor
- PCA
- principal components analysis.
REFERENCES
- 1. Dai S., Chen S. (2012) Single-cell-type proteomics: toward a holistic understanding of plant function. Mol. Cell. Proteomics 11, 1622–1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhao Z., Zhang W., Stanley B. A., Assmann S. M. (2008) Functional proteomics of Arabidopsis thaliana guard cells uncovers new stomatal signaling pathways. Plant Cell 20, 3210–3226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhu M., Simons B., Zhu N., Oppenheimer D. G., Chen S. (2010) Analysis of abscisic acid responsive proteins in Brassica napus guard cells by multiplexed isobaric tagging. J. Proteomics 73, 790–805 [DOI] [PubMed] [Google Scholar]
- 4. Zhu M., Dai S., McClung S., Yan X., Chen S. (2009) Functional differentiation of Brassica napus guard cells and mesophyll cells revealed by comparative proteomics. Mol. Cell. Proteomics 8, 752–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wienkoop S., Zoeller D., Ebert B., Simon-Rosin U., Fisahn J., Glinski M., Weckwerth W. (2004) Cell-specific protein profiling in Arabidopsis thaliana trichomes: identification of trichome-located proteins involved in sulfur metabolism and detoxification. Phytochemistry 65, 1641–1649 [DOI] [PubMed] [Google Scholar]
- 6. Van Cutsem E., Simonart G., Degand H., Faber A. M., Morsomme P., Boutry M. (2011) Gel-based and gel-free proteomic analysis of Nicotiana tabacum trichomes identifies proteins involved in secondary metabolism and in the (a)biotic stress response. Proteomics 11, 440–454 [DOI] [PubMed] [Google Scholar]
- 7. Amme S., Rutten T., Melzer M., Sonsmann G., Vissers J. P., Schlesier B., Mock H. P. (2005) A proteome approach defines protective functions of tobacco leaf trichomes. Proteomics 5, 2508–2518 [DOI] [PubMed] [Google Scholar]
- 8. Xie Z., Kapteyn J., Gang D. R. (2008) A systems biology investigation of the MEP/terpenoid and shikimate/phenylpropanoid pathways points to multiple levels of metabolic control in sweet basil glandular trichomes. Plant J. 54, 349–361 [DOI] [PubMed] [Google Scholar]
- 9. Schilmiller A. L., Miner D. P., Larson M., McDowell E., Gang D. R., Wilkerson C., Last R. L. (2010) Studies of a biochemical factory: tomato trichome deep expressed sequence tag sequencing and proteomics. Plant Physiol. 153, 1212–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nestler J., Schutz W., Hochholdinger F. (2011) Conserved and unique features of the maize (Zea mays L.) root hair proteome. J. Proteome Res. 10, 2525–2537 [DOI] [PubMed] [Google Scholar]
- 11. Brechenmacher L., Lee J., Sachdev S., Song Z., Nguyen T. H., Joshi T., Oehrle N., Libault M., Mooney B., Xu D., Cooper B., Stacey G. (2009) Establishment of a protein reference map for soybean root hair cells. Plant Physiol. 149, 670–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wan J., Torres M., Ganapathy A., Thelen J., DaGue B. B., Mooney B., Xu D., Stacey G. (2005) Proteomic analysis of soybean root hairs after infection by Bradyrhizobium japonicum. Mol. Plant Microbe Interact. 18, 458–467 [DOI] [PubMed] [Google Scholar]
- 13. Okamoto T., Higuchi K., Shinkawa T., Isobe T., Lorz H., Koshiba T., Kranz E. (2004) Identification of major proteins in maize egg cells. Plant Cell Physiol. 45, 1406–1412 [DOI] [PubMed] [Google Scholar]
- 14. Grobei M. A., Qeli E., Brunner E., Rehrauer H., Zhang R., Roschitzki B., Basler K., Ahrens C. H., Grossniklaus U. (2009) Deterministic protein inference for shotgun proteomics data provides new insights into Arabidopsis pollen development and function. Genome Res. 19, 1786–1800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zou J., Song L., Zhang W., Wang Y., Ruan S., Wu W. H. (2009) Comparative proteomic analysis of Arabidopsis mature pollen and germinated pollen. J. Integr. Plant Biol. 51, 438–455 [DOI] [PubMed] [Google Scholar]
- 16. Holmes-Davis R., Tanaka C. K., Vensel W. H., Hurkman W. J., McCormick S. (2005) Proteome mapping of mature pollen of Arabidopsis thaliana. Proteomics 5, 4864–4884 [DOI] [PubMed] [Google Scholar]
- 17. Han B., Chen S., Dai S., Yang N., Wang T. (2010) Isobaric tags for relative and absolute quantification-based comparative proteomics reveals the features of plasma membrane-associated proteomes of pollen grains and pollen tubes from Lilium davidii. J. Integr. Plant Biol. 52, 1043–1058 [DOI] [PubMed] [Google Scholar]
- 18. Pertl H., Schulze W. X., Obermeyer G. (2009) The pollen organelle membrane proteome reveals highly spatial-temporal dynamics during germination and tube growth of lily pollen. J. Proteome Res. 8, 5142–5152 [DOI] [PubMed] [Google Scholar]
- 19. Sheoran I. S., Ross A. R., Olson D. J., Sawhney V. K. (2007) Proteomic analysis of tomato (Lycopersicon esculentum) pollen. J. Exp. Bot. 58, 3525–3535 [DOI] [PubMed] [Google Scholar]
- 20. Lopez-Casado G., Covey P. A., Bedinger P. A., Mueller L. A., Thannhauser T. W., Zhang S., Fei Z., Giovannoni J. J., Rose J. K. (2012) Enabling proteomic studies with RNA-Seq: the proteome of tomato pollen as a test case. Proteomics 12, 761–774 [DOI] [PubMed] [Google Scholar]
- 21. Dai S., Li L., Chen T., Chong K., Xue Y., Wang T. (2006) Proteomic analyses of Oryza sativa mature pollen reveal novel proteins associated with pollen germination and tube growth. Proteomics 6, 2504–2529 [DOI] [PubMed] [Google Scholar]
- 22. Dai S., Chen T., Chong K., Xue Y., Liu S., Wang T. (2007) Proteomics identification of differentially expressed proteins associated with pollen germination and tube growth reveals characteristics of germinated Oryza sativa pollen. Mol. Cell. Proteomics 6, 207–230 [DOI] [PubMed] [Google Scholar]
- 23. Wu X., Chen T., Zheng M., Chen Y., Teng N., Samaj J., Baluska F., Lin J. (2008) Integrative proteomic and cytological analysis of the effects of extracellular Ca(2+) influx on Pinus bungeana pollen tube development. J. Proteome Res. 7, 4299–4312 [DOI] [PubMed] [Google Scholar]
- 24. Fernando D. D. (2005) Characterization of pollen tube development in Pinus strobus (Eastern white pine) through proteomic analysis of differentially expressed proteins. Proteomics 5, 4917–4926 [DOI] [PubMed] [Google Scholar]
- 25. Chen T., Wu X., Chen Y., Li X., Huang M., Zheng M., Baluska F., Samaj J., Lin J. (2009) Combined proteomic and cytological analysis of Ca2+-calmodulin regulation in Picea meyeri pollen tube growth. Plant Physiol. 149, 1111–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen Y., Chen T., Shen S., Zheng M., Guo Y., Lin J., Baluska F., Samaj J. (2006) Differential display proteomic analysis of Picea meyeri pollen germination and pollen-tube growth after inhibition of actin polymerization by latrunculin B. Plant J. 47, 174–195 [DOI] [PubMed] [Google Scholar]
- 27. Valero Galvan J., Valledor L., Gonzalez Fernandez R., Navarro Cerrillo R. M., Jorrin-Novo J. V. (2012) Proteomic analysis of Holm oak (Quercus ilex subsp. ballota [Desf.] Samp.) pollen. J. Proteomics 75, 2736–2744 [DOI] [PubMed] [Google Scholar]
- 28. Fila J., Matros A., Radau S., Zahedi R. P., Capkova V., Mock H. P., Honys D. (2012) Revealing phosphoproteins playing role in tobacco pollen activated in vitro. Proteomics 12, 3229–3250 [DOI] [PubMed] [Google Scholar]
- 29. Becker J. D., Boavida L. C., Carneiro J., Haury M., Feijo J. A. (2003) Transcriptional profiling of Arabidopsis tissues reveals the unique characteristics of the pollen transcriptome. Plant Physiol. 133, 713–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tadege M., Kuhlemeier C. (1997) Aerobic fermentation during tobacco pollen development. Plant Mol. Biol. 35, 343–354 [DOI] [PubMed] [Google Scholar]
- 31. Helling D., Possart A., Cottier S., Klahre U., Kost B. (2006) Pollen tube tip growth depends on plasma membrane polarization mediated by tobacco PLC3 activity and endocytic membrane recycling. Plant Cell 18, 3519–3534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ischebeck T., Stenzel I., Heilmann I. (2008) Type B phosphatidylinositol-4-phosphate 5-kinases mediate Arabidopsis and Nicotiana tabacum pollen tube growth by regulating apical pectin secretion. Plant Cell 20, 3312–3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ischebeck T., Stenzel I., Hempel F., Jin X., Mosblech A., Heilmann I. (2011) Phosphatidylinositol-4,5-bisphosphate influences Nt-Rac5-mediated cell expansion in pollen tubes of Nicotiana tabacum. Plant J. 65, 453–468 [DOI] [PubMed] [Google Scholar]
- 34. Ischebeck T., Vu L. H., Jin X., Stenzel I., Lofke C., Heilmann I. (2010) Functional cooperativity of enzymes of phosphoinositide conversion according to synergistic effects on pectin secretion in tobacco pollen tubes. Mol. Plant 3, 870–881 [DOI] [PubMed] [Google Scholar]
- 35. Kost B. (2008) Spatial control of Rho (Rac-Rop) signaling in tip-growing plant cells. Trends Cell Biol. 18, 119–127 [DOI] [PubMed] [Google Scholar]
- 36. Dowd P. E., Coursol S., Skirpan A. L., Kao T. H., Gilroy S. (2006) Petunia phospholipase c1 is involved in pollen tube growth. Plant Cell 18, 1438–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Berger F., Twell D. (2011) Germline specification and function in plants. Annu. Rev. Plant Biol. 62, 461–484 [DOI] [PubMed] [Google Scholar]
- 38. Boavida L. C., Becker J. D., Feijo J. A. (2005) The making of gametes in higher plants. Int. J. Dev. Biol. 49, 595–614 [DOI] [PubMed] [Google Scholar]
- 39. Echlin P., Godwin H. (1968) The ultrastructure and ontogeny of pollen in Helleborus foetidus L. II. Pollen grain development through the callose special wall stage. J. Cell Sci. 3, 175–186 [DOI] [PubMed] [Google Scholar]
- 40. Kerim T., Imin N., Weinman J. J., Rolfe B. G. (2003) Proteome analysis of male gametophyte development in rice anthers. Proteomics 3, 738–751 [DOI] [PubMed] [Google Scholar]
- 41. Imin N., Kerim T., Weinman J. J., Rolfe B. G. (2001) Characterisation of rice anther proteins expressed at the young microspore stage. Proteomics 1, 1149–1161 [DOI] [PubMed] [Google Scholar]
- 42. Chaturvedi P., Ischebeck T., Egelhofer V., Lichtscheidl I., Weckwerth W. (2013) Cell-specific analysis of the tomato pollen proteome from pollen mother cell to mature pollen provides evidence for developmental priming. J. Proteome Res. 12, 4892–4903 [DOI] [PubMed] [Google Scholar]
- 43. Honys D., Twell D. (2004) Transcriptome analysis of haploid male gametophyte development in Arabidopsis. Genome Biol. 5, R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Whittle C. A., Malik M. R., Li R., Krochko J. E. (2010) Comparative transcript analyses of the ovule, microspore, and mature pollen in Brassica napus. Plant Mol. Biol. 72, 279–299 [DOI] [PubMed] [Google Scholar]
- 45. Wei L. Q., Xu W. Y., Deng Z. Y., Su Z., Xue Y., Wang T. (2010) Genome-scale analysis and comparison of gene expression profiles in developing and germinated pollen in Oryza sativa. BMC Genomics 11, 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Read S. M., Clarke A. E., Bacic A. (1993) Stimulation of growth of cultured Nicotiana-Tabacum W-38 pollen tubes by poly(ethylene glycol) and Cu-(Ii) salts. Protoplasma 177, 1–14 [Google Scholar]
- 47. Smith P. K., Krohn R. I., Hermanson G. T., Mallia A. K., Gartner F. H., Provenzano M. D., Fujimoto E. K., Goeke N. M., Olson B. J., Klenk D. C. (1985) Measurement of protein using bicinchoninic acid. Anal. Biochem. 150, 76–85 [DOI] [PubMed] [Google Scholar]
- 48. Valledor L., Weckwerth W. (2013) An improved detergent-compatible gel-fractionation LC-LTQ-Orbitrap workflow for plant and microbial proteomics. Methods Mol. Biol. 1072, 347–358 [DOI] [PubMed] [Google Scholar]
- 49. Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. (2007) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 50. Ishihama Y., Rappsilber J., Mann M. (2006) Modular stop and go extraction tips with stacked disks for parallel and multidimensional peptide fractionation in proteomics. J. Proteome Res. 5, 988–994 [DOI] [PubMed] [Google Scholar]
- 51. Valledor L., Recuenco-Munoz L., Egelhofer V., Wienkoop S., Weckwerth W. (2012) The different proteomes of Chlamydomonas reinhardtii. J. Proteomics 75, 5883–5887 [DOI] [PubMed] [Google Scholar]
- 52. Paoletti A. C., Parmely T. J., Tomomori-Sato C., Sato S., Zhu D., Conaway R. C., Conaway J. W., Florens L., Washburn M. P. (2006) Quantitative proteomic analysis of distinct mammalian Mediator complexes using normalized spectral abundance factors. Proc. Natl. Acad. Sci. U.S.A. 103, 18928–18933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sun X., Weckwerth W. (2012) COVAIN: a toolbox for uni- and multivariate statistics, time-series and correlation network analysis and inverse estimation of the differential Jacobian from metabolomics covariance data. Metabolomics 8, 81–93 [Google Scholar]
- 54. Staudinger C., Mehmeti V., Turetschek R., Lyon D., Egelhofer V., Wienkoop S. (2012) Possible role of nutritional priming for early salt and drought stress responses in Medicago truncatula. Front. Plant Sci. 3, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vizcaino J. A., Cote R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Perez-Riverol Y., Reisinger F., Rios D., Wang R., Hermjakob H. (2013) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–D1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bucher M., Brander K. A., Sbicego S., Mandel T., Kuhlemeier C. (1995) Aerobic fermentation in tobacco pollen. Plant Mol. Biol. 28, 739–750 [DOI] [PubMed] [Google Scholar]
- 57. Peters J. M. (2002) The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol. Cell 9, 931–943 [DOI] [PubMed] [Google Scholar]
- 58. Yang M., Hu Y., Lodhi M., McCombie W. R., Ma H. (1999) The Arabidopsis SKP1-LIKE1 gene is essential for male meiosis and may control homologue separation. Proc. Natl. Acad. Sci. U.S.A. 96, 11416–11421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhao D. Z., Yang X. H., Quan L., Timofejeva L., Rigel N. W., Ma H., Makaroff C. A. (2006) ASK1, a SKP1 homolog, is required for nuclear reorganization, presynaptic homolog juxtaposition and the proper distribution of cohesin during meiosis in Arabidopsis. Plant Mol. Biol. 62, 99–110 [DOI] [PubMed] [Google Scholar]
- 60. del Pozo J. C., Boniotti M. B., Gutierrez C. (2002) Arabidopsis E2Fc functions in cell division and is degraded by the ubiquitin-SCFAtSKP2 pathway in response to light. Plant Cell 14, 3057–3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hruba P., Honys D., Twell D., Capkova V., Tupy J. (2005) Expression of beta-galactosidase and beta-xylosidase genes during microspore and pollen development. Planta 220, 931–940 [DOI] [PubMed] [Google Scholar]
- 62. Konopka-Postupolska D., Clark G., Hofmann A. (2011) Structure, function and membrane interactions of plant annexins: an update. Plant Sci. 181, 230–241 [DOI] [PubMed] [Google Scholar]
- 63. Dobritsa A. A., Lei Z., Nishikawa S., Urbanczyk-Wochniak E., Huhman D. V., Preuss D., Sumner L. W. (2010) LAP5 and LAP6 encode anther-specific proteins with similarity to chalcone synthase essential for pollen exine development in Arabidopsis. Plant Physiol. 153, 937–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hanin M., Brini F., Ebel C., Toda Y., Takeda S., Masmoudi K. (2011) Plant dehydrins and stress tolerance: versatile proteins for complex mechanisms. Plant Signal. Behav. 6, 1503–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wolkers W. F., McCready S., Brandt W. F., Lindsey G. G., Hoekstra F. A. (2001) Isolation and characterization of a D-7 LEA protein from pollen that stabilizes glasses in vitro. Biochim. Biophys. Acta 1544, 196–206 [DOI] [PubMed] [Google Scholar]
- 66. Pacini E., Guarnieri M., Nepi M. (2006) Pollen carbohydrates and water content during development, presentation, and dispersal: a short review. Protoplasma 228, 73–77 [DOI] [PubMed] [Google Scholar]
- 67. Franklin-Tong V. E. (1999) Signaling and the modulation of pollen tube growth. Plant Cell 11, 727–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jansen M. A. K., Sessa G., Malkin S., Fluhr R. (1992) Pepc-mediated carbon fixation in transmitting tract cells reflects style pollen-tube interactions. Plant J. 2, 507–515 [Google Scholar]
- 69. Moir D., Stewart S. E., Osmond B. C., Botstein D. (1982) Cold-sensitive cell-division-cycle mutants of yeast: isolation, properties, and pseudoreversion studies. Genetics 100, 547–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yamanaka K., Sasagawa Y., Ogura T. (2012) Recent advances in p97/VCP/Cdc48 cellular functions. Biochim. Biophys. Acta 1823, 130–137 [DOI] [PubMed] [Google Scholar]
- 71. Cao K., Nakajima R., Meyer H. H., Zheng Y. X. (2003) The AAA-ATPase Cdc48/p97 regulates spindle disassembly at the end of mitosis. Cell 115, 355–367 [DOI] [PubMed] [Google Scholar]
- 72. Park S., Rancour D. M., Bednarek S. Y. (2008) In planta analysis of the cell cycle-dependent localization of AtCDC48A and its critical roles in cell division, expansion, and differentiation. Plant Physiol. 148, 246–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rancour D. M., Dickey C. E., Park S., Bednarek S. Y. (2002) Characterization of AtCDC48. Evidence for multiple membrane fusion mechanisms at the plane of cell division in plants. Plant Physiol. 130, 1241–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mascarenhas J. P., Crone D. F. (1996) Pollen and the heat shock response. Sex. Plant Reprod. 9, 370–374 [Google Scholar]
- 75. Hafidh S., Breznenova K., Ruzicka P., Fecikova J., Capkova V., Honys D. (2012) Comprehensive analysis of tobacco pollen transcriptome unveils common pathways in polar cell expansion and underlying heterochronic shift during spermatogenesis. BMC Plant Biol. 12, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang Y., Zhang W. Z., Song L. F., Zou J. J., Su Z., Wu W. H. (2008) Transcriptome analyses show changes in gene expression to accompany pollen germination and tube growth in Arabidopsis. Plant Physiol. 148, 1201–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Obermeyer G., Fragner L., Lang V., Weckwerth W. (2013) Dynamic adaption of metabolic pathways during germination and growth of lily pollen tubes after inhibition of the electron transport chain. Plant Physiol. 162, 1822–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wei Y., Lin M., Oliver D. J., Schnable P. S. (2009) The roles of aldehyde dehydrogenases (ALDHs) in the PDH bypass of Arabidopsis. BMC Biochem. 10, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ransom-Hodgkins W. D. (2009) The application of expression analysis in elucidating the eukaryotic elongation factor one alpha gene family in Arabidopsis thaliana. Mol. Genet. Genomics 281, 391–405 [DOI] [PubMed] [Google Scholar]
- 80. Andersen G. R., Nissen P., Nyborg J. (2003) Elongation factors in protein biosynthesis. Trends Biochem. Sci. 28, 434–441 [DOI] [PubMed] [Google Scholar]
- 81. Demma M., Warren V., Hock R., Dharmawardhane S., Condeelis J. (1990) Isolation of an abundant 50,000-dalton actin filament bundling protein from Dictyostelium amoebae. J. Biol. Chem. 265, 2286–2291 [PubMed] [Google Scholar]
- 82. Ohta K., Toriyama M., Miyazaki M., Murofushi H., Hosoda S., Endo S., Sakai H. (1990) The mitotic apparatus-associated 51-kDa protein from sea urchin eggs is a GTP-binding protein and is immunologically related to yeast polypeptide elongation factor 1 alpha. J. Biol. Chem. 265, 3240–3247 [PubMed] [Google Scholar]
- 83. Yang W., Boss W. F. (1994) Regulation of phosphatidylinositol 4-kinase by the protein activator PIK-A49. Activation requires phosphorylation of PIK-A49. J. Biol. Chem. 269, 3852–3857 [PubMed] [Google Scholar]
- 84. Chang Y. W., Traugh J. A. (1998) Insulin stimulation of phosphorylation of elongation factor 1 (eEF-1) enhances elongation activity. Eur. J. Biochem. 251, 201–207 [DOI] [PubMed] [Google Scholar]
- 85. Umikawa M., Tanaka K., Kamei T., Shimizu K., Imamura H., Sasaki T., Takai Y. (1998) Interaction of Rho1p target Bni1p with F-actin-binding elongation factor 1alpha: implication in Rho1p-regulated reorganization of the actin cytoskeleton in Saccharomyces cerevisiae. Oncogene 16, 2011–2016 [DOI] [PubMed] [Google Scholar]
- 86. Zobel-Thropp P., Yang M. C., Machado L., Clarke S. (2000) A novel post-translational modification of yeast elongation factor 1A—methylesterification at the C terminus. J. Biol. Chem. 275, 37150–37158 [DOI] [PubMed] [Google Scholar]
- 87. Coppard N. J., Clark B. F. C., Cramer F. (1983) Methylation of elongation-factor 1-alpha in mouse 3t3b and 3t3b Sv40 cells. FEBS Lett. 164, 330–334 [DOI] [PubMed] [Google Scholar]
- 88. Wu X., Oh M. H., Schwarz E. M., Larue C. T., Sivaguru M., Imai B. S., Yau P. M., Ort D. R., Huber S. C. (2011) Lysine acetylation is a widespread protein modification for diverse proteins in Arabidopsis. Plant Physiol. 155, 1769–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.