

Abstract
Nitric oxide (NO) mediates a substantial part of its physiologic functions via S-nitrosylation, however the cellular substrates for NO-mediated S-nitrosylation are largely unknown. Here we describe the S-nitrosoproteome using a high-density protein microarray chip containing 16,368 unique human proteins. We identified 834 potentially S-nitrosylated human proteins. Using a unique and highly specific labeling and affinity capture of S-nitrosylated proteins, 138 cysteine residues on 131 peptides in 95 proteins were determined, defining critical sites of NO's actions. Of these cysteine residues 113 are novel sites of S-nitrosylation. A consensus sequence motif from these 834 proteins for S-nitrosylation was identified, suggesting that the residues flanking the S-nitrosylated cysteine are likely to be the critical determinant of whether the cysteine is S-nitrosylated. We identify eight ubiquitin E3 ligases, RNF10, RNF11, RNF41, RNF141, RNF181, RNF208, WWP2, and UBE3A, whose activities are modulated by S-nitrosylation, providing a unique regulatory mechanism of the ubiquitin proteasome system. These results define a new and extensive set of proteins that are susceptible to NO regulation via S-nitrosylation. Similar approaches could be used to identify other post-translational modification proteomes.
It is known that NO regulates the majority of its physiologic function through S-nitrosylation (1). Protein-assisted or small molecule, S-nitrosoglutathione (GSNO)1 trans-nitrosylation, oxidative S-nitrosation, and metalloprotein-catalyzed S-nitrosylation are the prominent cellular mechanisms that are utilized to S-nitrosylate proteins (2). A number of proteins are known to be S-nitrosylated and this post-translational modification can either activate or inactivate a protein's biologic activity (1, 3). A number of attempts at probing tissue-specific S-nitrosoproteomes have been made, but the results of these are limited to proteins that are S-nitrosylated to a great degree and which are present at high concentrations (2, 4–6). Recently, to investigate determinants of S-nitrosylation, yeast and human target protein microarrays have been studied. However, these assay were limited because of the small number of proteins present on the chip (7). In addition, many proteins that are known to be S-nitrosylated have been studied through a targeted and biased approach (8). To overcome these shortcomings, we report the use of a 16,368 human protein microarray chip to better define the human S-nitrosoproteome.
Ubiquitin is a 76-amino-acid long polypeptide that can be covalently added to lysine residues on targeted proteins either as single monomers or in chains. Ubiquitination of proteins can dramatically alter their function or localization depending on the number of ubiquitin attached and the nature of their linkages. The most well characterized ubiquitin-mediated process is targeting of the protein for degradation by the 26S proteasome, which occurs via poly-ubiquitination linked together through lysine 48 on the ubiquitin monomers. Ubiquitination occurs in a three-step enzymatic process in which the third enzyme, the ubiquitin protein ligase (E3) determines protein target specificity (9). NO S-nitrosylates the RING finger E3 ligases, parkin and XIAP, modifying their function (10, 11). In the case of parkin, S-nitrosylation transiently activates its E3 ligase activity, but ultimately inhibits its activity (12). In contrast, XIAP's E3 ligase activity is unaffected by S-nitrosylation, but its anti-apoptotic function is compromised (11). Using the 16,368 human protein microarray, we identify a number of NO-regulated E3 ligases, the majority of which are activated by NO-dependent S-nitrosylation.
MATERIALS AND METHODS
Reagents
The anti-biotin antibody and anti-β-actin HRP were purchased from Sigma (St. Louis, MO) and anti-GST HRP, anti-V5, anti-V5 HRP, and anti-Cy5 (anti-mouse Alexa-fluor 647) were purchased from Invitrogen (Carlsbad,CA). Anti-ubiquitin was purchased from Dako (Glostrup, Denmark). Anti-myc, anti-myc HRP, and anti-HA HRP were purchased from Roche (Basel, Switzerland). Entry clones, which are the Invitrogen Ultimate ORF collection and 445 additional sub-cloned ORFs (The Center for High-Throughput Biology, Johns Hopkins University), for S-nitrosylated proteins were cloned to the destination GST vector (desti-15, Invitrogen). To generate mutants, site-directed mutagenesis was carried out using the quick-change site-directed mutagenesis kit (Stratagene, La Jolla, CA), and DNA sequencing analysis confirmed mutation sites. Myc-parkin and myc-XIAP were used as previously described (10, 11). Glutathione (GSH) and GSNO were purchased from Sigma.
Protein Chip
The protein microarray chip was spotted with 16,368 unique, full-length human recombinant proteins in duplicate along with several control proteins such as IgG, GST, BSA-biotin, and histones. The Invitrogen Ultimate ORF collection and 445 additional ORFs were subcloned into a yeast expression vector that allows for galactose-dependent overexpression of N-terminal GST- and 6x-His-tagged recombinant proteins as previously described (13). The quality of the purified proteins was assayed and at least 92% of the proteins were expressed correctly. The 16,368 proteins were spotted onto Fullmoon slides (Fullmoon Biosystem, Sunnyvale, CA) using the NanoPrint LM210 system (ArrayIT, Sunnyvale, CA).
Biotin Switch Method on Protein Chip
The biotin switch method was performed as previously described (14) with some modification for protein chips (Fig. 1). The protein chip was equilibrated with HEN buffer (250 mm Hepes, 1 mm EDTA, 0.1 mm Neocuproine, 0.5% Nonidet P-40) for 30 min. S-nitrosylated proteins were treated with 100 μm GSH or GSNO for 1 h on a shaking platform at 60 rpm. Three individual protein chips were treated with GSH and 3 individual protein chips were treated with GSNO. Chips were washed 3 times with HEN buffer for 10 min, and incubated with 20 mm methyl methanethiosulfonate (MMTS, Sigma) (50 rpm, 50 °C for 1 h) and excess MMTS was removed 5 times with HEN buffer for 10 min. The protein chips were incubated with 50 mm ascorbate (Sigma) and 0.4 mm N-(6-(biotinamido)hexyl)-3′-(2′-pyridyldithio)-propionamide (biotin-HPDP) (Pierce, Rockford, IL) for 12 h at RT. To remove ascorbate and biotin-HPDP, protein chips were washed 5 times with HEN buffer for 10 min. Monoclonal anti-biotin (1:1000) in 5% filtrated dry milk was applied for 2 h at RT and washed 3 times with filtrated PBS-T (137 mm NaCl, 2.7 mm KCl, 4.3 mm Na2HPO4, 1.47 mm KH2PO4, pH 7.4, 0.1% Tween 20) for 10 min. The anti-mouse Alex-fluor 647 (1:5000) in 5% filtrated dry milk was applied for 1 h and washed 3 times with PBS-T for 10 min at RT. Protein chips were dried via centrifugation at 200 g for 2 min using a 50 ml conical tube. The protein chips were scanned with GenePix 4000B (Axon Instruments, Union City, CA) for quantification and statistical analysis.
Fig. 1.
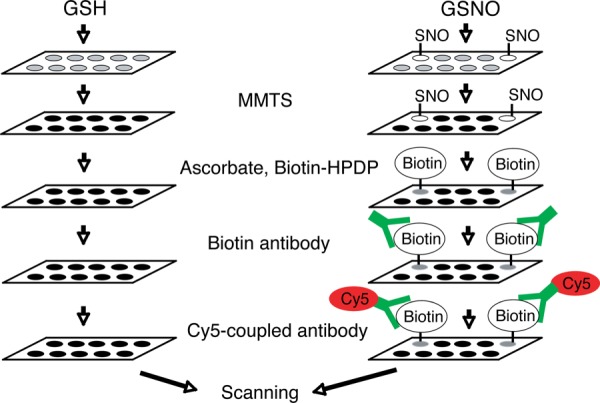
Diagram of the modified biotin-switch method for use on the protein microarray chip. In vitro S-nitrosylation of proteins on the chip. The protein chips were incubated with 100 μm S-nitrosoglutathione (GSNO), which is a NO donor and 100 μm glutathione (GSH) as the control compound. The non-nitrosylated thiol of cysteine residues are blocked with methyl methanethiosulfonate (MMTS) via methylthiolation, whereas nitrosothiol and disulfide bonds cannot react with MMTS. Nitrosothiols bonds are then decomposed with ascorbate, which results in the reduction of nitrosothiols to thiols, which are capable of being labeled with the thiol-specific biotinylation reagent (Biotin-HPDP). S-nitrosylated proteins are then identified by anti-biotin and cy5-coupled (anti-mouse Alex-fluor 647) antibody and scanned for quantification and statistical analysis.
Gene Ontology and Functional Protein Analysis
The list of 834 identified S-nitrosylated proteins was submitted to the online DAVID functional annotation resource (v6.7) (15, 16) and overrepresented proteins were identified and compared with the human proteome using the SP_PIR_KEYWORDS category. The molecular class, biological process, and compartment localization of all 834 proteins was downloaded from the human protein reference database (HPRD) (17).
Preparation of Human Protein Mixtures for Site-specific Identification of S-Nitrosylated Proteins
GST-human recombinant proteins were pooled and exposed to 100 μm GSNO in HEN buffer (250 mm Hepes, pH 7.7, containing 1 mm EDTA, 0.1 mm neocuproine, 1% Triton X-100) and protease inhibitors for 1 h at RT. Proteins were then precipitated with 3 volumes of acetone for 20 min at −20 °C followed by centrifugation at 3000 × g for 10 min at 4 °C. Pellets were then washed twice with 1 ml of cold acetone, solubilized in 10 ml blocking buffer (HEN, 2.5% SDS, and 20 mm methyl methanethiosulfonate (MMTS), and incubated for 30 min at 50 °C with frequent vortexing. Following blocking, excess MMTS was removed by protein precipitation with 3 volumes of acetone for 30 min at −20 °C followed by centrifugation at 4500 × g for 10 min at 4 °C. Protein pellets were suspended into 5 ml of blocking buffer (without MMTS) and precipitated as described above. Protein pellets were then suspended in 3 ml of loading buffer (250 mm MES, 1 mm DTPA, pH 6.0, 1% SDS) and analyzed by organomercury resin as described below.
Capture of S-Nitrosylated Proteins Using Organomercury Resin (MRC) and On-column Digestion
MRC solid-phase capture columns preparation as well as protein capture was performed according to established protocols (2) with minor modifications described below. MMTS-blocked protein lysates (3 ml), prepared as described above, were applied to activated MRC columns and incubated under stationary conditions for 1 h at RT. Next, the columns were washed with 25 bed volumes of 300 mm NaCl, 0.05% SDS, followed by 25 bed volumes 300 mm NaCl, 1% Triton X-100. Columns were then washed with 200 bed volumes of deionized water, followed by 10 bed volumes of 0.1 m ammonium bicarbonate. The captured proteins were digested on-column with trypsin (5 μg) in 0.1 m ammonium bicarbonate in the dark for 16 h at RT. The resin was washed with 25 volumes of 1 m ammonium bicarbonate, pH 7.4, containing 300 mm NaCl, followed by 25 volumes of the same buffer without NaCl. Columns were then washed with 25 volumes of 0.1 m ammonium bicarbonate followed by 250 volumes of deionized water. To elute the bound peptides, the resin was incubated with 1 bed volume of performic acid in water (generated by reacting 1 m formic acid with 0.3 m H2O2 for 30 min in the dark) for 30 min at room temperature. Eluted peptides were recovered by washing the resin with 1 bed volume of deionized water. The sample was then lyophilized and resuspended in 200 μl of 0.1% formic acid. The volume was then reduced by Speedvac to less than 5 μl and adjusted to 10 μl with 0.1% formic acid. Eluted peptides (5 μl) were analyzed by LC-MS/MS as described below and previously (2, 18).
LC-MS/MS Analysis, Generation, and Evaluation of SEQUEST Peptide Assignments
Liquid chromatography-tandem mass spectrometry analysis of tryptic peptide digests, engine-assisted search of RAW files and database search were performed as previously described (2, 18). DTA files were generated from MS/MS spectra extracted from the RAW data file (intensity threshold of; minimum ion count of 50) and processed by the ZSA and Correction algorithms of the SEQUEST Browser program. DTA files were submitted to Sorcerer-SEQUEST (ver. 4.0.3, rev 11; Sagen Research) using the following parameters: Database searching was performed against a Uniprot database (Release v3.57; 3/24/2009) containing Homo sapiens sequences from Swiss-Prot plus common contaminants, which were then reversed and appended to the forward sequences (total 32344 entries). The database was indexed with the following parameters: mass range of 600–3500, tryptic cleavages with a maximum of two missed cleavage and static modifications of cysteine by sulfonic acid (+48 amu) and methionine sulfone (+32 amu). The DTA files were searched with a 10 ppm peptide mass tolerance and 1.0 amu fragment ion mass tolerance.
Potential sequence-to-spectrum peptide assignments generated by Sorcerer-SEQUEST were loaded into Scaffold (version 2.6.02; Proteome Software) to validate protein identifications and perform manual inspection of MS/MS. Empirically defined SEQUEST Xcorr thresholds were applied to filter peptides IDs with at least one peptide per protein. Xcorr thresholds were applied to sulfonic acid-containing and nonmodified peptides independently so peptide FDR was ≤5%. Because one unique peptide per protein was permitted, manual inspection of all MS/MS spectra was performed. The criteria for manual inspection were the following: (1) assignment of the majority of fragment ion abundance, (2) sulfonic acid (+48 amu) modification supported by either y- or b- ions series (≥ 4 consecutive fragments), and (3) assigned charge state and diagnostic markers, such as N-terminal proline, C-terminal aliphatic amino acids, and loss of H2O/ammonia consistent with amino acid sequence (2, 18).
Motif Discovery
The short amino acid sequences (i.e. 15mers) containing the S-nitrosylated cysteine sites were aligned with the cysteines at center. Based on the alignment, we constructed for the 131 MS/MS determined sites an initial foreground positional weight matrix (PWM) Mf (i, a), which denotes the frequency of amino acid type a at position i. Similarly, we also constructed a background set for all cysteine sites in human proteome and used it to construct background PWM Mb (i, a). We then scanned all the S-nitrosylation sites according to a relative entropy formula
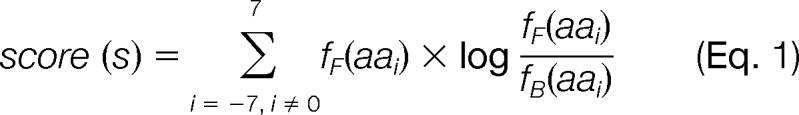 |
Here, aai represents the amino acid at position i of the site of interest. The fF(aai) and fB(aai) represent the values of Mf(i, aai) and Mb(i, aai). The top ten nitrosylation sites with the highest scores were selected. These sites represent the most important characteristics of the nitrosylation sites. Therefore, we used them to rebuild the foreground PWM and rescan the remaining sites. We will include a site if its score is above a threshold. The threshold is determined by the top 5% of the matching scores for all sites in the background. In this process, we will extract at most two sites from each protein. The iterative procedure will stop if either (1) no more site can be added to the foreground set, or (2) the number of sites in the foreground is equal N, which is the number of proteins containing the MS/MS verified S-nitrosylated cysteine sites.
In Vitro Ubiquitination Assay
GST-purified E3 ligases with 0.4 μg of E1, 1 μg of ubiquitin, 0.5 μg of UbcH5c, deionized water, and 10× reaction buffer (50 mm Tris-HCl (pH 7.5), 2 mm ATP, 2.5 mm MgCl2) in a total sample volume of 50 μl were incubated for 1 h at 37 °C. Laemml sample buffer (with 2-ME) was added and samples were boiled for 5 min and subjected to immunoblot analysis with anti-ubiquitin.
In Vivo Ubiquitination Assay
HEK293 cells were transiently transfected with V5-tagged E3 ligase as well as HA-tagged ubiquitin using the lipofectamine with PLUS reagent (Invitrogen). Cells were then treated with 100 μm GSNO at various time-points. Cells were washed with PBS and then lysed in IP lysis buffer (25 mm Hepes (pH 7.4), 1 mm EDTA, 1.5 mm NaCl, 1% Triton X-100, proteasome inhibitor mixture) after detaching cells using a cell scraper. Protein concentration was assayed using the Bicinchoninic acid method and protein levels were equalized. Samples were immunoprecipitated with anti-V5 antibody using protein G Sepharose beads (GE Healthcare Life Science, Pittsburgh, PA) overnight in a rotating incubator at 4 °C. Beads were washed with IP washing buffer (IP lysis buffer modified to 100 mm NaCl) three times via centrifugation at 4000 × g for 1 min. Proteins were eluted in Laemmli sample buffer (with 2-ME) and boiled for 5 min. Samples were subjected to immunoblot analysis using anti-HA-HRP, anti-V5-HRP, or anti-myc antibodies.
Statistical Analysis
Following scanning, signal intensity value for each spot was obtained as the ratio of foreground to background signals and normalized with GST signal intensity. The mean and standard deviation of signal intensity of all proteins on the chip were calculated. The Student's t test was used for comparisons between GSH and GSNO means with three data sets, and p ≤ 0.05 was considered to be statistically significant.
RESULTS
Identification of the Human S-nitrosoproteome
The biotin-switch method (BSM) (19), which converts an S-nitrosocysteine into an S-biotinylated cysteine, was adapted for use on the human protein microarray chip, which contains 16,368 unique GST-tagged full-length human proteins (Fig. 1) (19). The protein chip was treated in parallel with either 100 μm Glutathione (GSH) or 100 μm S-nitrosoglutathione (GSNO) for 1 h at room temperature (2, 10–12, 14), followed by the BSM. The S-biotinylated cysteine was detected with an anti-biotin antibody using a Cy5-coupled secondary antibody. The chip was then scanned using a GenePix 4000B scanner. To normalize the Cy5 signal, the protein chip was then probed with an anti-GST antibody, followed by scanning. Two representative protein blocks, each containing 400 proteins in duplicate, are shown (Fig. 2A and supplemental Fig. S1). A high-powered view of a random region containing seven, two, two and four proteins in duplicate, to illustrate the Cy5 and GST signal, is shown (Fig. 2B). To determine the S-nitrosylation (SNO) signal on potential S-nitrosylated proteins, a SNO/GSH normalization ratio was calculated by first normalizing the SNO signal to the corresponding GST signal divided by the normalized GSH/GST signal (Data Set S1). The data distribution of each of the chips followed a normal distribution (data not shown). 834 proteins (transcriptional variants included) were found to be significantly S-nitrosylated over the course of three replicates (Data Set S2). Forty-six of these proteins contain only one cysteine in their sequence, the site of S-nitrosylation (Data Set S1). 23 of these proteins do not have a cysteine and thus represent likely false positives. These 23 proteins were excluded from further analysis. 55% of the 834 S-nitrosylated proteins contain one or more cysteines that are part of a known functional domain or motif (17). A representative column graph of the p values for the 15 random proteins shown in Fig. 2B is presented in Fig. 2C.
Fig. 2.

Detection of S-nitrosylated proteins on the human proteome chip. A, Cy5 fluorescence of two representative blocks (#26 and #27 of 48) of the protein chip treated with GSH (100 μm, 1 h) or GSNO (100 μm, 1 h). IgG and biotin labeled bovine serum albumin (BSA-biotin) serve as positive controls. A gradient of GST at the bottom right of chip serves as a GST control. White rectangles indicate magnification area for (B). B, Magnified images of proteins identified as S-nitrosylated are shown in red. C, The p values of the 15 proteins shown in (B). All experiments were repeated three times.
Proteomic Analysis and Functional Analysis of the S-nitrosoproteome
The 834 S-nitrosylated proteins were matched to Gene Ontology annotation including transcriptional variants drawn from the Human Protein Reference Database (17) and were analyzed for functional classification (Data Set S2). Diverse classes of proteins are found in the S-nitrosoproteome. For example, 211 proteins are involved in metabolism, 190 proteins are involved in signal transduction, 111 are involved in DNA/RNA regulation, 59 are involved in transport, 42 are involved in cell growth and maintenance, and 21 are involved in apoptosis. Seventeen are involved in a variety of processes including cell adhesion, cell motility, cytoskeletal organization and biogenesis, protein folding, and muscle function. One hundred eighty-three do not fall into any known signaling category (Fig. 3A). Functional annotation analysis was conducted using the DAVID database to determine which functional categories are overrepresented in the list of S-nitrosylated proteins compared with the background human proteome (15, 16). Interestingly, proteins known to be acetylated show 1.9-fold enrichment in our data set, (p = 1.2e−18). Proteins localized to the cytoplasm are overrepresented in the human S-nitrosoproteome, exhibiting 2.3-fold enrichment (p = 9.2e−21). Of the S-nitrosylated proteins, 11.7% are group transfer enzymes (1.7-fold enrichment, p = 1.2e−6) and include phosphotransferases (2.9-fold enrichment, p = 1.5e−5).
Fig. 3.
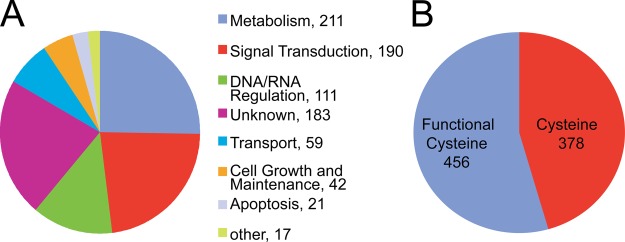
Analysis of S-nitrosylated protein candidates. A, Gene Ontology analysis of the 834 S-nitrosylated proteins. B, Schematic representation of the number of proteins with cysteines in a functional domain or motif.
Of the 834 proteins, 456 were found to contain one or more cysteines in their sequence that are part of a functional domain or motif, whereas 378 were found to contain cysteines that are not part of a known functional domain or motif (Fig. 3B). 29 proteins were selected for validation by BSM. Among them, 19 are confirmed to be strongly S-nitrosylated using BSM assay, five of them are weakly S-nitrosylated (Fig. 4A). Only five of them appear not to be S-nitrosylated. Thus, 82.8% of the proteins chosen for validation are indeed S-nitrosylated. Interestingly, eight E3 ubiquitin ligases, RNF10, RNF11, RNF41, RNF141, RNF181, RNF208, WWP2, UBE3A variant 3 are robustly S-nitrosylated (Fig. 4A).
Fig. 4.
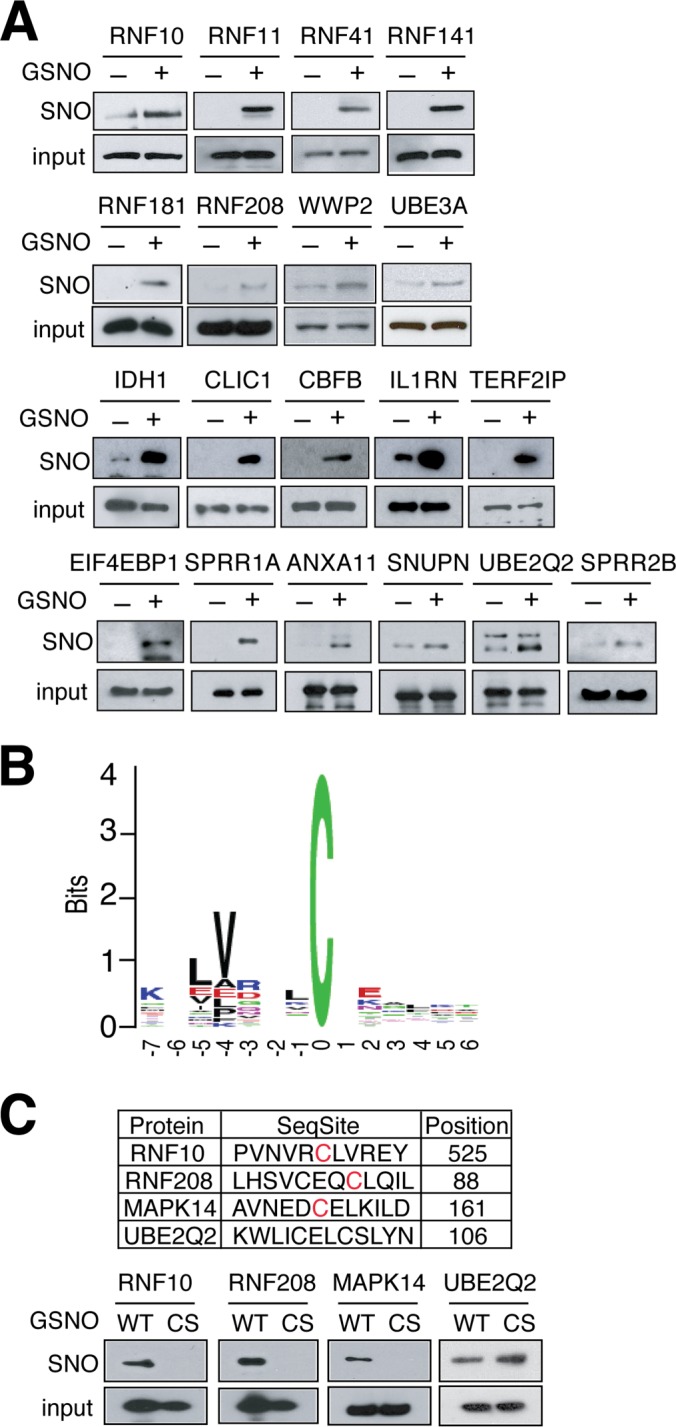
Validation of S-nitrosylated protein candidates. A, For confirmation of human protein microarray screening, GST fused-recombinant proteins were purified and treated with GSH or GSNO (100 μm, 1h), and analyzed by the BSM and followed by Western blotting with an anti-GST antibody. B, Predicted S-nitrosylation motif. Sequence motifs for residues flanking S-nitrosylated cysteine residue from 138 cysteines identified by mass spectrometry. C, Validation of predicted S-nitrosylation motif (red color) by the BSM in presence GSNO (100 μm, 1h). WT, wild type; CS, cysteine to serine substitution. Data were reproduced two times with similar results.
Identification of S-nitrosylation Sites by Mass Spectrometry
The top 300 most significantly S-nitrosylated proteins (p ≤ 0.03) were shuttled to the pDEST15 bacterial expression system for GST purification. Of these proteins 120 had sufficient expression to enable determination of the cysteines that are S-nitrosylated via mass spectrometry. The 120 proteins (greater than or equal to 10 μg each) were pooled and treated with 100 μm GSNO for 1 h. Proteins were precipitated and applied to activated organomercury resin columns. The captured proteins were digested on-column and peptides were eluted and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Protein identification was accomplished by a Uniprot database search and validated by the program Scaffold using empirically defined SEQUEST thresholds (2, 18). Mass spectrometry analysis identified 138 S-nitrosylated cysteines in 131 peptides from 95 proteins (Data Set S3). Peptide sequences, the mass of the monoisotopic peptide as well as the peptide identification scores are provided in Data Set S4. MS/MS spectra are provided in Data Set S5. We recovered many previously known S-nitrosylated cysteines sites (e.g. Cys118 and Cys80 in p21 Ras, Cys94 in interleukin 1 receptor antagonist, Cys27 and Cys97 in actin-related protein 10) confirming the specificity of the identification method (Data Set S3) (2–6, 17, 20). We also identified 113 S-nitrosylated cysteines that have not been previously described, representing novel S-nitrosylated cysteines sites. Of the S-nitrosylated cysteines identified, 43 cysteines fall within a known functional domain or motif (16).
To gain insight into the specificity of S-nitrosylation process, we examined whether there exist consensus sequences for the sites identified by MS/MS. The flanking sequences were analyzed using a motif discovery program (see Materials and Methods in Supporting Information) and a motif (consensus sequence) was predicted as -H-H-X-X-X-C- (H-hydrophobic) (Fig. 4B). The positions of −4 and −5 of motif are mainly hydrophobic amino acids (such as Alanine, Valine, Leucine, and Isoleucine). In fact, among the 131 peptides, 58% of them have such amino acids at positions −4 and −5. We then scanned the predicted motif against all the S-nitrosylated proteins identified by protein microarray. The 834 proteins contain 5933 cysteines. Among these sites, 366 matched with our predicted motif (366/5933 = 6.2%). In contrast, we only expect 5.4% of cysteine sites in the human proteome (all proteins on the protein microarray) matched with the motif. The enrichment of the motif in the identified substrates is statistically significant (p < 0.0038). To validate the S-nitrosylation motif the BSM was conducted on WT RNF10, RNF208, MAPK14, and UBE2Q2 and a CS substitution mutant for each in which the predicted cysteine was replaced with a serine. S-nitrosylation is prevented by mutation of the predicted cysteine to serine in RNF10, RNF208, and MAPK14 confirming the predictive value of the S-nitrosylation motif. However mutation of the predicted cysteine in UBE2Q2 to serine did not eliminate S-nitrosylation (Fig. 4C). Cysteine to serine site mutants that are not predicted by the motif in RNF10, RNF208, MAPK14 did not eliminate S-nitrosylation confirming the predictive value of the motif (supplementary Fig. S2).
E3 Ligase Activity is Modulated by S-nitrosylation at Specific Cysteine Residues
It is known that NO regulates the ubiquitin E3 ligase parkin via S-nitrosylation, inhibiting its E3 ligase activity and protective function (10). The E3 ligase XIAP is also regulated by NO by inhibiting its anti-apoptotic function without affecting its E3 ligase activity (11). Based on the fact that NO can regulate E3 ligases, the effect of NO on E3 ligases identified in this study that are S-nitrosylated were subjected to further analysis to validate and confirm the utility of the human proteome chip to identify functional sites of S-nitrosylation. The E3 ligase auto-ubiquitination activity of RNF10, RNF11, RNF41, RNF141, RNF181, RNF208, WWP2, and UBE3A was monitored in the presence and absence of NO at 2, 4, 6, and 12 h after HEK293 cells were transfected with V5-tagged cDNA constructs (Fig. 5). GSNO potently increases the auto-ubiquitination activity of RNF10 (Fig. 5A), RNF41 (Fig. 5B), and UBE3A (Fig. 5C). GSNO biphasically increases RNF141 (Fig. 5D). GSNO transiently increases the auto-ubiquitination activity of RNF11 (Fig. 5E) and WWP2 (Fig. 5F). The auto-ubiquitination activity of RNF181 (Fig. 5G) and RNF208 (Fig. 5H) are decreased. As positive controls, the auto-ubiquitination activity of parkin and XIAP were examined (10, 11). As previously shown, GSNO initially activates parkin at 2 h and then inactivates parkin's E3 ligase activity (supplemental Fig. S3A), whereas GSNO has no effect on XIAP's E3 ligase activity (supplementary Fig. S3B).
Fig. 5.

Ubiquitin E3 ligase activity of A, RNF10, B, RNF41, C, UBE3A, D, RNF141, E, RNF11, F, WWP2, G, RNF181, and H, RNF208 was analyzed by an in vivo ubiquitination activity assay with or without GSNO treatment for 0, 2, 4, 6, and 12 h.
From the mass spectrometry analysis, Cys4 is S-nitrosylated in RNF11, Cys106, Cys134, and Cys238 are S-nitrosylated in RNF41, and Cys82 and Cys175 are S-nitrosylated in RNF141 (Fig. 6A and Data Set S3). A serine substitution for each cysteine that is S-nitrosylated was made to determine the functional consequences of S-nitrosylation in each RNF protein. The C4S RNF11 protein is not S-nitrosylated (Fig. 6B). The C106S RNF41 protein S-nitrosylation signal is dramatically reduced compared with WT RNF41, whereas C134S and C238S mutants have minimal effect on the S-nitrosylation of RNF41 (Fig. 6B). The C82S and C175S RNF141 proteins both show reduced S-nitrosylation compared with WT RNF141, suggesting that both cysteines can be S-nitrosylated (Fig. 6B).
Fig. 6.
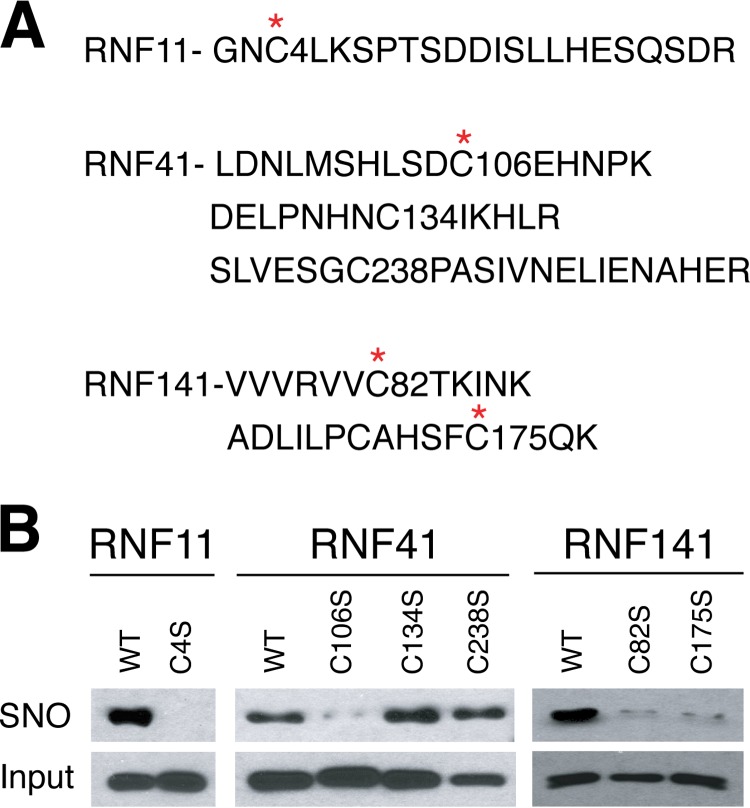
S-nitrosylation of RNF11, RNF41 and RNF141. A, S-nitrosylated cysteines identified by liquid chromatography-tandem mass spectrometry (LC-MS/MS) are indicated via an asterisk (*) in the amino acid sequence of RNF11, RNF41 and RNF141 detected using LC-MS/MS. B, S-nitrosylation of WT and serine substitution for cysteine mutants of RNF11 (C4S), RNF41 (C106S, C134S, C238S) and RNF141 (C82S, C175S) as analyzed by the BSM after GSNO (100 μm) treatment followed by immunoblot with an anti-GST antibody.
An in vitro ubiquitination assay was utilized to determine the E2 that each RING finger protein uses in an auto-ubiquitination reaction (supplemental Fig. S4A–4D). RNF11 uses UbcH5c and UbcH6 and RNF41 and RNF141 use UbcH5c (supplemental Fig. S4A–4D). The effect of these cysteines to serine substitution mutants on the auto-ubiquitination activity of RNF11, RNF41, and RNF141 was analyzed in the presence of ubiquitin, E1 and E2 enzymes. GSNO activation is dramatically attenuated in C4S RNF11, C106S RNF41, and C82S RNF141 in vitro (Fig. 7A). To confirm these results, V5-tagged RNF constructs were transfected into HEK293 cells and auto-ubiquitination for the three RNF proteins was monitored in presence and absence of GSNO. GSNO potently increases the auto-ubiquitination of RNF11, RNF41, and RNF141, but has no effect on C4S RNF11, C106S RNF41, and C82S RNF141 (Fig. 7B). C175S RNF141 displays GSNO activation in vitro (supplemental Fig. S5A) and in vivo (supplemental Fig. S5B), suggesting that Cys175 is not the functionally relevant S-nitrosylated cysteine in RNF141.
Fig. 7.

Increased ubiquitin E3 ligase activity after S-nitrosylation of RNF11, RNF41 and RNF141. A, In vitro ubiquitination activity assay with or without GSNO treatment for 1 h in presence of ubiquitin, E1 and E2 (UbcH5c) enzymes analyzed by immunoblot with anti-ubiquitin and anti-GST antibodies. B, In vivo ubiquitination activity assay with or without GSNO treatment for 6 h analyzed by immunoblot with anti-HA and anti-V5 antibodies. Actin and HA-ubiquitin expression were monitored as loading controls. All experiments were repeated three times.
DISCUSSION
The method described here provides an unbiased means of probing nearly the entire human proteome for S-nitrosylated proteins and the identification of the specific cysteines that are modified in these proteins. Previous attempts to identify the human S-nitrosoproteome have been limited to a small number of yeast and human proteins (less than 4000) in a protein microarray-based analysis and to those proteins that exhibit relatively high expression in MS-based experimental systems. Protein chip microarray methods are especially helpful in discovering previously unknown S-nitrosoproteins that are low in concentration in the cell or whose modulation by NO may be subtle (7). These and related studies are essential for understanding the physiological roles of S-nitrosoproteins. Through the use of a 16,368 human protein microarray chip and subsequent generation of GST expression vectors from the identified S-nitrosylated proteins, coupled with MS-based identification of S-nitrosylated cysteines, this limitation is surmounted. The results of our experiment yield proteins that are S-nitrosylated in an in vitro system and represent possible targets for future study. The functional annotation analysis of the 834 identified S-nitrosylated proteins showed that the S-nitrosoproteome regulates a wide swath of the human proteome, confirming previous reports (1, 2, 7). The identification of an overrepresentation of acetylated proteins, as well as phosphotransferases, may signify that there is widespread cross talk between these post-translational signaling mechanisms.
Motif search analysis was conducted using the 138 S-nitrosylated cysteines identified by mass spectrometry. A significant linear motif was identified and reinforces the current theory that the hydrophobicity of flanking residues in a protein plays the most important part in determining which cysteine is available for S-nitrosylation. Although this motif was obtained from a limited pool of S-nitrosylated cysteines it seems to have some predictive value. A variety of mechanisms exist for the S-nitrosylation of proteins. Although the production of nitric oxide in vivo is primarily derived from the enzymatic function of nitric oxide synthases. S-nitrosylation can be accomplished by at least three different mechanism: (1) Oxidative mechanisms that include metal catalysis (21–23), (2) exchange reactions with S-nitrosoglutathione (GSNO) and (3) trans-nitrosylation reactions between proteins. Despite the presence of many proteins with reduced cysteine residues, only a few appear to be modified by S-nitrosylation (24, 25). To explain the apparent selectivity it has been proposed that positioning of reduced cysteine residues between acidic and basic residues promotes S-nitrosylation reactions particularly those mediated by GSNO (26). It has been also suggested that the strict requirement is for proton donation and receiving groups to be within close proximity of the sulfur atom within the three-dimensional structure of the protein (27). Alternatively, the presence of charged residues located distally to the modified cysteine residues could provide sites for GSNO docking that may facilitate site specific S-nitrosylation (25, 28). Most likely, different sequence properties are relevant for the different means of S-nitrosylation, with solvent accessibility and pKa being the most important for low-mass S-nitrosylating species targeting, whereas charged residues near the target cysteine may be critical for the necessary protein–protein interactions involved in trans-nitrosylation.
We focused on the E3 ligases RNF10, RNF11, RNF41, RNF141, RNF181, RNF208, WWP2, and UBE3A. The mutational and functional analysis of NO regulation of E3 ligases represents an expanded view that NO may regulate to a great extent the ubiquitin proteasome pathway. NO can activate and inhibit E3 ligase activity or modify the function of the E3 ligase without modifying its activity. S-nitrosylation of cysteines that are not within the active site RING domains of these proteins results in an increase in the E3 ligase activity of RNF11, RNF41, and RNF141. The other E3 ligases that are activated by NO may be S-nitrosylated on nonactive site cysteines as well. The mechanism by which S-nitrosylation of these nonactive site cysteines affects E3 ligase activity requires further investigation, but it is possible that the S-nitrosylated cysteines change the protein folding dynamics, resulting in a three-dimensional configuration that enhances ubiquitination activity. It is likely that for E3 ligases that are inhibited by NO, the S-nitrosylation is at an active site, but this requires confirmation. Most of the E3 ligases reported here have a limited repertoire of substrates and RNF10, RNF11, RNF141, RNF181, and RNF208 have no known E3 ligase substrate. The 16,368 human protein microarray chip could be utilized to identify the S-nitrosylation-dependent E3 ligase substrate(s) of these E3 ligases by carrying out a modified ubiquitination activity assay on the microarray chip. Thus, the technology described herein can be used not only to probe the human proteome for S-nitrosylated proteins, but may also be co-opted for use in the functional exploration of E3 ligase substrates.
The E3 ligases we found to be S-nitrosylated are mainly involved in the regulation of apoptosis and thus might be critical players in cancer and neurodegenerative and neurodevelopmental diseases. Thus functional exploration of the physiologic consequences of NO regulation of these E3 ligases may provide heretofore unknown mechanisms that regulate these biological processes. For instance, RNF11 and RNF41 have known roles in regulation of NF-κB signaling (29, 30). Although the substrate(s) for RNF11 are not known, RNF41 is known to ubiquitinate MyD88, a component of Toll-like receptor mediated NF-κB signaling (30). RNF181 plays a role in platelet function and in the suppression of hepatocellular carcinoma growth (31, 32). It will be important in future studies to determine the impact of NO regulation of the activity of these and the other NO regulated E3 ligases.
In summary, the experimental design described here demonstrates the use of high-throughput screening of the human proteome to probe for the human S-nitrosoproteome, followed by MS identification of the specific cysteines that are modified. This represents an unbiased approach to the challenge of identifying new members of the human S-nitrosoproteome and has yielded a large number of previously unknown targets of NO as well as a putative S-nitrosylation motif that will undoubtedly provide numerous avenues for the future exploration of the S-nitrosoproteome and its importance in cellular physiology and pathophysiology. This general strategy of post-translational modification-proteome identification via an in vitro experiment designed for use on the protein microarray chip followed by the LC-MS/MS-based identification of PTM sites, can be applied to a number of other post-translation modification systems, such as phosphorylation, acetylation, ubiquitination, SUMOylation, palmitoylation, poly(ADP-ribosyl)ation and others.
Supplementary Material
Footnotes
Author Contributions: T.M.D. and V.L.D. conceived and supervised the project. Y.-I.L., D.G., H.C.K., Y.L., and M.G. performed the experiments. Y.-I.L., and D.G. assembled the figure and tables. P.-T.D. and H.I. conducted the mass spectrometry analysis. J.S.J., H.Z. and S.B. contributed new reagents. Z.X., J.H., and J.Q. contributed the analysis of the chip data. T.M.D, V.L.D. Y.-I.L., and D.G. wrote and finalized the manuscript. All authors contributed to the final edits of the manuscript.
* This work was supported by grants from the NIH/NINDS NS38377, NS67525, NIH/NIDA DA000266, NIH/NIA AG13966, NIH/NHLBI HL054926 and NIH/NIEHS ES013508. This work was also supported in part by the National Research Foundation of Korea(NRF) grant funded by the Korea government(MSIP) [Science Research Center (SRC) program No. 2011-0030830]. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases.
 This article contains supplemental Figs. S1 to S5 and Data Sets S1 to S5.
This article contains supplemental Figs. S1 to S5 and Data Sets S1 to S5.
Competing Financial Interests: The authors declare no competing financial interests.
1 The abbreviations used are:
- GSNO
- S-nitrosoglutathione
- BSM
- biotin switch method
- HPRD
- human protein reference database.
REFERENCES
- 1. Hess D. T., Matsumoto A., Kim S. O., Marshall H. E., Stamler J. S. (2005) Protein S-nitrosylation: purview and parameters. Nature Rev. 6, 150–166 [DOI] [PubMed] [Google Scholar]
- 2. Doulias P. T., Greene J. L., Greco T. M., Tenopoulou M., Seeholzer S. H., Dunbrack R. L., Ischiropoulos H. (2010) Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc. Natl. Acad. Sci. U. S. A. 107, 16958–16963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xue Y., Liu Z., Gao X., Jin C., Wen L., Yao X., Ren J. (2010) GPS-SNO: computational prediction of protein S-nitrosylation sites with a modified GPS algorithm. PloS One 5, e11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Camerini S., Polci M. L., Restuccia U., Usuelli V., Malgaroli A., Bachi A. (2007) A novel approach to identify proteins modified by nitric oxide: the HIS-TAG switch method. J. Proteome Res. 6, 3224–3231 [DOI] [PubMed] [Google Scholar]
- 5. Hao G., Derakhshan B., Shi L., Campagne F., Gross S. S. (2006) SNOSID, a proteomic method for identification of cysteine S-nitrosylation sites in complex protein mixtures. Proc. Natl. Acad. Sci. U. S. A. 103, 1012–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu M., Hou J., Huang L., Huang X., Heibeck T. H., Zhao R., Pasa-Tolic L., Smith R. D., Li Y., Fu K., Zhang Z., Hinrichs S. H., Ding S. J. (2010) Site-specific proteomics approach for study protein S-nitrosylation. Anal. Chem. 82, 7160–7168 [DOI] [PubMed] [Google Scholar]
- 7. Foster M. W., Forrester M. T., Stamler J. S. (2009) A protein microarray-based analysis of S-nitrosylation. Proc. Natl. Acad. Sci. U. S. A. 106, 18948–18953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Uehara T., Nishiya T. (2011) Screening systems for the identification of S-nitrosylated proteins. Nitric Oxide 25, 108–111 [DOI] [PubMed] [Google Scholar]
- 9. Fang S., Weissman A. M. (2004) A field guide to ubiquitylation. Cell Mol. Life Sci. 61, 1546–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chung K. K., Thomas B., Li X., Pletnikova O., Troncoso J. C., Marsh L., Dawson V. L., Dawson T. M. (2004) S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science 304, 1328–1331 [DOI] [PubMed] [Google Scholar]
- 11. Tsang A. H., Lee Y. I., Ko H. S., Savitt J. M., Pletnikova O., Troncoso J. C., Dawson V. L., Dawson T. M., Chung K. K. (2009) S-nitrosylation of XIAP compromises neuronal survival in Parkinson's disease. Proc. Natl. Acad. Sci. U. S. A. 106, 4900–4905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lipton S. A., Nakamura T., Yao D., Shi Z. Q., Uehara T., Gu Z. (2005) Comment on “S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function”. Science 308, 1870; author reply 1870 [DOI] [PubMed] [Google Scholar]
- 13. Hu S., Xie Z., Onishi A., Yu X., Jiang L., Lin J., Rho H. S., Woodard C., Wang H., Jeong J. S., Long S., He X., Wade H., Blackshaw S., Qian J., Zhu H. (2009) Profiling the human protein-DNA interactome reveals ERK2 as a transcriptional repressor of interferon signaling. Cell 139, 610–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jaffrey S. R., Snyder S. H. (2001) The biotin switch method for the detection of S-nitrosylated proteins. Science Signaling, pl1, DOI:10.1126/scisignal.862011pl1 [DOI] [PubMed] [Google Scholar]
- 15. Huang da W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protocols 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 16. Huang da W., Sherman B. T., Lempicki R. A. (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Prasad T. S. K., Goel R., Kandasamy K., Keerthikumar S., Kumar S., Mathivanan S., Telikicherla D., Raju R., Shafreen B., Venugopal A., Balakrishnan L., Marimuthu A., Banerjee S., Somanathan D. S., Sebastian A., Rani S., Ray S., Kishore C. J. H., Kanth S., Ahmed M., Kashyap M., Mohmood R., Ramachandra Y. L., Krishna V., Rahiman A. B., Mohan S., Ranganathan P., Ramabadran S., Chaerkady R., Pandey A. (2009) Human Protein Reference Database - 2009 update. Nucleic Acids Res. 37, D767–D772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Greco T. M., Hodara R., Parastatidis I., Heijnen H. F., Dennehy M. K., Liebler D. C., Ischiropoulos H. (2006) Identification of S-nitrosylation motifs by site-specific mapping of the S-nitrosocysteine proteome in human vascular smooth muscle cells. Proc. Natl. Acad. Sci. U. S. A. 103, 7420–7425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jeong J. S., Jiang L., Albino E., Marrero J., Rho H. S., Hu J., Hu S., Vera C., Bayron-Poueymiroy D., Rivera-Pacheco Z. A., Ramos L., Torres-Castro C., Qian J., Bonaventura J., Boeke J. D., Yap W. Y., Pino I., Eichinger D. J., Zhu H., Blackshaw S. (2012) Rapid identification of monospecific monoclonal antibodies using a human proteome microarray. Mol. Cell. Proteomics 11, O111 016253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marino S. M., Gladyshev V. N. (2010) Structural analysis of cysteine S-nitrosylation: a modified acid-based motif and the emerging role of trans-nitrosylation. J. Mol. Biol. 395, 844–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang Y., Hogg N. (2005) S-Nitrosothiols: cellular formation and transport. Free Radic. Biol. Med. 38, 831–838 [DOI] [PubMed] [Google Scholar]
- 22. Bosworth C. A., Toledo J. C., Jr., Zmijewski J. W., Li Q., Lancaster J. R., Jr. (2009) Dinitrosyliron complexes and the mechanism(s) of cellular protein nitrosothiol formation from nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 106, 4671–4676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Weichsel A., Maes E. M., Andersen J. F., Valenzuela J. G., Shokhireva T., Walker F. A., Montfort W. R. (2005) Heme-assisted S-nitrosation of a proximal thiolate in a nitric oxide transport protein. Proc. Natl. Acad. Sci. U.S.A. 102, 594–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Doulias P. T., Tenopoulou M., Greene J. L., Raju K., Ischiropoulos H. (2013) Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci. Signal. 6, rs1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Doulias P. T., Greene J. L., Greco T. M., Tenopoulou M., Seeholzer S. H., Dunbrack R. L., Ischiropoulos H. (2010) Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc. Natl. Acad. Sci. U.S.A. 107, 16958–16963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hess D. T., Matsumoto A., Kim S. O., Marshall H. E., Stamler J. S. (2005) Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol. 6, 150–166 [DOI] [PubMed] [Google Scholar]
- 27. Liu L., Yan Y., Zeng M., Zhang J., Hanes M. A., Ahearn G., McMahon T. J., Dickfeld T., Marshall H. E., Que L. G., Stamler J. S. (2004) Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell 116, 617–628 [DOI] [PubMed] [Google Scholar]
- 28. Marino S. M., Gladyshev V. N. (2010) Structural analysis of cysteine S-nitrosylation: a modified acid-based motif and the emerging role of trans-nitrosylation. J. Mol. Biol. 395, 844–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shembade N., Parvatiyar K., Harhaj N. S., Harhaj E. W. (2009) The ubiquitin-editing enzyme A20 requires RNF11 to downregulate NF-kappaB signalling. EMBO J. 28, 513–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang C., Chen T., Zhang J., Yang M., Li N., Xu X., Cao X. (2009) The E3 ubiquitin ligase Nrdp1 ‘preferentially’ promotes TLR-mediated production of type I interferon. Nat. Immunol. 10, 744–752 [DOI] [PubMed] [Google Scholar]
- 31. Wang S., Huang X., Li Y., Lao H., Zhang Y., Dong H., Xu W., Li J. L., Li M. (2011) RN181 suppresses hepatocellular carcinoma growth by inhibition of the ERK/MAPK pathway. Hepatology 53, 1932–1942 [DOI] [PubMed] [Google Scholar]
- 32. Brophy T. M., Raab M., Daxecker H., Culligan K. G., Lehmann I., Chubb A. J., Treumann A., Moran N. (2008) RN181, a novel ubiquitin E3 ligase that interacts with the KVGFFKR motif of platelet integrin alpha(IIb)beta3. Biochem. Biophys. Res. Commun. 369, 1088–1093 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.