

Abstract
The mammalian target of rapamycin (mTOR) pathway is an essential cellular signaling pathway involved in a number of important physiological functions, including cell growth, proliferation, metabolism, protein synthesis, and autophagy. Dysregulation of the mTOR pathway has been implicated in the pathophysiology of a number of neurological diseases. Hyperactivation of the mTOR pathway, leading to increased cell growth and proliferation, has been most convincingly shown to stimulate tumor growth in the brain and other organs in the genetic disorder, tuberous sclerosis complex (TSC). In addition, mTOR may also play a role in promoting epileptogenesis or maintaining seizures in TSC, as well as in acquired epilepsies following brain injury. Finally, the mTOR pathway may also be involved in the pathogenesis of cognitive dysfunction and other neurological deficits in developmental disorders and neurodegenerative diseases. mTOR inhibitors, such as rapamycin and its analogs, may represent novel, rational therapies for a variety of neurological disorders.
Keywords: rapamycin, epilepsy, seizure, tuberous sclerosis, neurodegenerative disease
Introduction
The mammalian target of rapamycin (mTOR) is a ubiquitous serine-threonine protein kinase that regulates a number of important physiological functions, including cell growth, proliferation, metabolism, protein synthesis, and autophagy. mTOR is also critical for a number of brain-specific mechanisms, such as synaptic plasticity, learning, and cortical development. Significant advances have been made in understanding the complicated upstream and downstream components and regulation of the mTOR pathway and have been reviewed elsewhere.(1–3) Briefly, mTOR participates in the formation of at least two functional complexes, mTORC1 and mTORC2. mTORC1 consists of mTOR itself, the regulatory protein raptor, and several other binding proteins. mTORC2 is composed of mTOR, the regulatory protein rictor, and other binding proteins. mTORC1 primarily exerts its functional effects via regulation of ribosomal biogenesis and protein synthesis through controlling the ribosomal protein S6 or the translation initiating factor, eIF4E, which secondarily can influence a variety of processes, including cell growth, proliferation, and metabolism. mTORC1 is inhibited by rapamycin and other rapamycin analogs. In contrast, mTORC2 acts primarily through regulation of a number of other protein kinases and interactions with cytoskeletal elements of cells. While mTORC2 is relatively insensitive to rapamycin inhibition, mTORC2 can be inhibited by mTOR inhibitors after prolonged exposure in certain cell types.
The mTOR pathway may respond to a variety of physiological stimuli and environmental conditions via modulation by a number of upstream signaling pathways (Fig. 1). Hamartin and tuberin, proteins encoded by the tuberous sclerosis complex 1 and 2 genes, form a complex that normally inhibits the mTOR pathway via inhibition of the intermediary GTP-binding protein Rheb (Ras homolog enriched in brain). Multiple upstream pathways activate or inhibit the mTOR pathway either directly or by interacting with the hamartin-tuberin complex. For example, in conditions of energy or nutrient surplus, growth factors, such as insulin, may stimulate the phosphoinositide-3 kinase (PI3K)/Akt pathway, which then activates the mTOR pathway and promotes increased cell growth and metabolism. In contrast, states of energy or nutrient deprivation may trigger other upstream pathways, such as the LKB1/AMPK pathway, which ultimately inhibits mTOR activity and limits cell growth and metabolism.
Fig. 1.
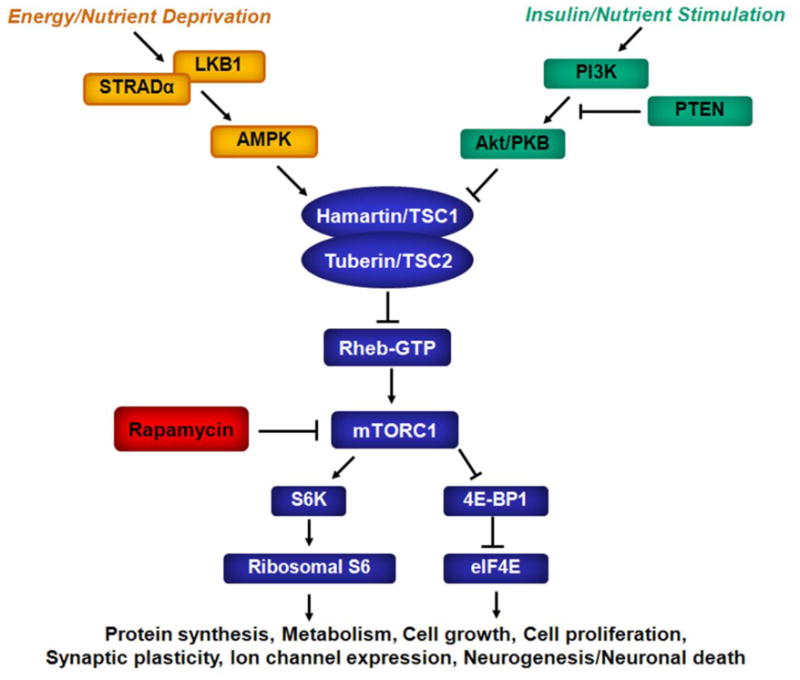
Physiological regulation of the mTOR pathway. The mammalian target of rapamycin (mTOR) is a serine-threonine protein kinase, which forms two complexes, rapamycin-sensitive mTORC1,and relatively rapamycin-insensitive mTORC2 (not shown). mTORC1 activates downstream signaling mechanisms involved in ribosomal biogenesis (S6K, Ribosomal S6) and protein translation (4E-BP1, eIF4E). Through regulation of protein synthesis and other mechanisms, the mTOR pathway modulates a number of important physiological processes, such as cell growth and proliferation, metabolism, and brain-specific functions (e.g., synaptic reorganization, ion channel expression, neuronal death). In turn, the mTOR pathway may be activated or inhibited by various physiological stimuli via various upstream signaling pathways and intermediary proteins (TSC1, TSC2, Rheb). AMPK - 5′ adenosine monophosphate-activated protein kinase; eIF4E, elongation initiation factor 4E; mTOR - mammalian target of rapamycin; PI3K - phosphoinositide-3 kinase; PTEN, phosphatase and tensin homolog on chromosome 10; Rheb - Ras homolog enriched in brain; STRADα – STE20-related kinase adapter alpha; S6 - ribosomal protein S6; S6K - ribosomal S6 kinase; TSC1 - tuberous sclerosis complex 1 protein; TSC2 - tuberous sclerosis complex 2 protein; 4E-BP1 - elongation factor 4E binding protein 1.
While physiological regulation of the mTOR pathway is essential for normal cellular function, dysregulation of the mTOR pathway may promote the development or progression of disease under pathological conditions. Given the ubiquity of mTOR in various cell types throughout the body, mTOR may be involved in the pathogenesis of a number of systemic disorders, such as diabetes, cancer, and cardiovascular disease.(3,4) In addition, abnormal mTOR signaling has also been implicated in a variety of neurological diseases (Fig. 2).(5.6) In this review, I will highlight the most recent scientific findings and clinical applications of the mTOR pathway in neurological disorders, including tuberous sclerosis complex (TSC), brain tumors, epilepsy, autism/neurodevelopmental disorders, and neurodegenerative diseases. In parallel, I will discuss the potential therapeutic utility of mTOR inhibitors for these disorders (Table 1).
Fig. 2.

Pathological regulation of the mTOR pathway in neurological disease. Dysregulation of the mTOR pathway has been implicated in the pathophysiology of a variety of different neurological diseases. In some disorders (shown in black boxes), there is convincing evidence of a direct molecular or genetic link between the disease and a specific upstream component of the mTOR pathway, specifically TSC and hamartin/tuberin, polyhydramnios, megalencephaly and symptomatic epilepsy syndrome (PMSE) and STRADα, and glioblastoma and PTEN. In other disorders (shown in gray boxes), the mechanistic link to the mTOR pathway is not as well established, although there is some evidence that traumatic brain injury (TBI), other acquired epilepsies, and fragile X syndrome may involve activation of PI3K/Akt. The least is known about how neurodegenerative disorders, such as Alzheimer’s, Huntington’s, and Parkinson’s disease, affect the mTOR pathway, although downstream effects likely involve regulation of autophagy, clearance of protein aggregates, and neuronal death mechanisms. See legend to Fig. 1 for other abbreviations.
Table 1.
Effects of mTOR Inhibitors in Animal Models and Clinical Studies of Neurological Disease
| Neurological Disease | Effects on Neurological Manifestations | Proposed Mechanism(s) of Action | References |
|---|---|---|---|
| Astrocytomas and Other Tumors in TSC | |||
| Clinical studies in TSC patients | Reduction in volume of SEGAs, as well AMLs and angiofibromas; improvement in pulmonary functions (related to LAM) in TSC patients. | Inhibition of cell growth/proliferation. | 18–22 |
| Animal models of TSC | Reduction in renal tumors, brain megalencephaly, neuronal hypertrophy, and astrocyte proliferation in KO mice. | Inhibition of cell growth/proliferation. | 16,17,39–42 |
| Other (non-TSC) Brain Tumors | |||
| Clinical studies of patients with glioblastoma multiforme | No effect on primary endpoints of tumor progression and survival. | N/A | 32,33 |
| Xenoplants of human glioma in mice | Reduction in glioma cell growth | Inhibition of cell growth/proliferation, cytotoxic effects, inhibition of angiogenesis, sensitization to radiation. | 28–30 |
| Genetic Epilepsies | |||
| Clinical studies in TSC patients | Reduction in seizure frequency in TSC patients | Unknown. | 19,44–46 |
| Animal models of TSC | Prevention of epilepsy (early treatment) and reduction in seizure frequency (late treatment) in KO mice. | Inhibition of cell growth/proliferation, decreased neuronal dispersion, improved myelination and astrocyte glutamate transport. | 17,39–42 |
| Animal models with PTEN inactivation | Reduction in seizure frequency in KO mice. | Inhibition of cell growth/proliferation | 47–50 |
| Acquired Epilepsies/Seizures | |||
| Animal models of acquired epilepsy/seizures | Reduction in epilepsy following status epilepticus. Reduction in epilepsy following neonatal seizures Reduction in spasms in rats following brain injury. |
Inhibition of mossy fiber sprouting, cell death Inhibition of enhanced glutamate EPSCs Unknown |
62–64 66 67 |
| Autism/Other Neurodevelopmental Disorders | |||
| Animal models of TSC | Reversal of learning deficits in KO mice | Reversal of deficits in synaptic plasticity | 43 |
| Animal models of Fragile X and Down syndrome | Effects of mTOR inhibitors not yet reported, but mTOR pathway is abnormally activated. | N/A | 78–80 |
| Neurodegenerative Diseases | |||
| Animal models of Alzheimer’s disease | Improvement in cognitive/memory deficits in transgenic mice. | Decreased phosphorylation of tau, decreased amyloid-beta, induction of autophagy. | 82–84 |
| Animal models of Huntington’s disease | Improvement in motor deficits, decreased neuronal death in transgenic mice. | Induction of autophagy. | 85,86 |
| Animal models of Parkinson’s disease | Reduction in dopaminergic neuronal death in MPTP-treated mice. | Inhibition of pro-apoptotic protein RTP801, induction of autophagy and lysosomal biogenesis. | 87,88 |
Abbreviations: AML – angiomyolipoma, EPSC – excitatory postsynaptic currents, KO – knock-out, LAM – lymphangioleiomyomatosis, N/A – not applicable, PTEN – phosphatase and tensin homolog on chromosome 10, MPTP – 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, mTOR – mammalian target of rapamycin, SEGA – subependymal giant cell astrocytoma, TSC – tuberous sclerosis complex.
Tuberous Sclerosis Complex (TSC)
TSC probably represents the prototypical disease related to abnormal mTOR signaling. TSC is an autosomal dominant genetic disorder involving tumor or hamartoma formation in multiple organs, including the heart, kidney, lungs, eyes, skin, and brain.(7,8) While symptoms may occur as a result of tumors and other abnormal lesions in any of these organs, neurological involvement, including epilepsy, developmental delay, mental retardation, behavioral disorders, and autism, often constitutes the most common and disabling manifestations of TSC. The pathological hallmark of TSC is the cortical tuber, a focal malformation of cortical development, which is histologically identical to certain types of isolated focal cortical dysplasia. Epilepsy may be directly triggered by cortical tubers, although intracranial recordings suggest that seizures actually originate from the regions surrounding the tubers.(9) The pathophysiology of cognitive deficits and autism is even less understood, but may relate to the disruption of normal cortical circuits by tubers or to tuber-independent cellular and molecular defects of neurons and glia. In addition to tubers, subependymal nodules, consisting of small collections of dysplastic neurons and glia, commonly align the ventricular walls. While subependymal nodules are asymptomatic, subependymal giant cell astrocytomas (SEGAs) may grow progressively and constitute a substantial risk for obstructive hydrocephalus, a neurosurgical emergency. Besides the neurological manifestations of TSC, tumor growth in other organs may also cause significant clinical problems. In particular, enlarging renal angiomyolipomas (AMLs) may lead to hypertension, bleeding, and renal failure. The interstitial lung disease, lymphangioleiomyomatosis (LAM), may occur particularly in adult women with TSC and cause progressive dyspnea, pneumothoraces, and respiratory insufficiency. Until the recent emergence of mTOR inhibitors, all treatments for manifestations of TSC have involved symptomatic therapy or surgical resection of tumors.
Mutations in two different genes, TSC1 and TSC2, which encode the proteins hamartin and tuberin, respectively, have been identified to cause TSC.(10,11) A similar phenotype may result from a mutation in either of these two genes because hamartin and tuberin bind together to form a single functional complex. As the hamartin-tuberin complex normally inhibits the mTOR pathway, mutation of either TSC1 or TSC2 leads to abnormal disinhibition of the mTOR pathway. This hyperactivation of the mTOR pathway may result in increased cell growth and proliferation, which can promote tumorigenesis in TSC patients, as well as other downstream functional effects.
The relevance of the mTOR pathway in the pathophysiology of TSC was first suggested in simplified biological systems, such as drosophila and yeast, in which hamartin and tuberin were shown to inhibit mTOR signaling.(12–15) Demonstration of this basic interaction immediately indicated that mTOR inhibitors, such as rapamycin, could be of strong therapeutic value in TSC. The potential utility of mTOR inhibitors for tumors in TSC was first established for renal tumors in mouse models of TSC.(16) Subsequently, rapamycin was also shown to decrease the abnormal proliferation of astrocytes in other TSC knock-out mice, suggesting that mTOR inhibitors may represent an appropriate treatment for SEGAs.(17)
Several clinical trials of mTOR inhibitors for tumor growth in TSC patients have been conducted in recent years. First of all, rapamycin or the rapamycin analog, everolimus, has been shown to decrease SEGA growth in TSC patients.(18,19). These studies have led to the official regulatory approval of the mTOR inhibitor, everolimus, for treatment of SEGAs in TSC in the United States. Although mTOR inhibitors clearly reduce SEGA size, one important caveat is that the tumors tend to grow back if the drug is discontinued, indicating that long-term treatment may be necessary to maintain effectiveness. In addition to SEGAs, clinical trials also support the efficacy of mTOR inhibitors for renal AMLs and pulmonary LAM,(20,21) and official approval for AMLs has just occurred. Another potential use of rapamycin that is under clinical trials is topical administration for facial angiofibromas.(22) Thus, there appear to be multiple promising therapeutic applications of mTOR inhibitors for treating various tumor phenotypes in TSC.
Non-TSC-related Brain Tumors
In addition to SEGAs in TSC, the mTOR pathway has been implicated in the pathophysiology of other brain tumors unrelated to TSC, particularly other types of gliomas. Active expression of upstream and downstream mTOR pathway markers, such as Akt, S6K, and S6, occurs in human gliomas and correlates with the malignancy grade.(23,24) Furthermore, specific genetic mutations have been found in gliomas that could lead to downstream activation of the PI3K/Akt/mTOR signaling, such as in the epidermal growth factor receptor (EGFR) and phosphatase and tensin homolog on chromosome 10 (PTEN) genes.(25–27) mTOR inhibitors have been reported to inhibit growth of tumor cells in xenografts of human gliomas implanted into mouse models.(28–30) The mechanism of the antitumor effects of rapamycin against gliomas is still being investigated, but may involve direct cytotoxic and antiproliferative effects, inhibition of vascular endothelial growth factor and angiogenesis, decreased invasive propensity, and increased sensitivity to radiation.(30,31)
Based on the encouraging basic science and preclinical findings, a couple of clinical trials have been conducted testing the effect of mTOR inhibitors on patients with gliomas. Phase II trials of the mTOR inhibitor, temsirolimus (CCI-779), in patients with glioblastoma multiforme have reported good tolerability and modest effects on radiographic or other clinical criteria in a subset of patients, but overall did not demonstrate efficacy in primary endpoints of time to tumor progression and survival.(32,33) Possible reasons for poor efficacy include pharmacokinetic issues and blood-brain barrier penetration. However, much attention is now focused on the complex role of parallel and feedback signaling mechanisms in tumorigenesis. In particular, primary or feedback activation of Akt with mTOR inhibition may lead to alternative pathway activation that may cause resistance to mTOR inhibitors and persistent tumor growth. Newer research strategies have focused on inhibiting multiple mTOR-related signaling mechanisms, such as using dual PI3K-mTOR inhibitors, mTORC1/mTORC2 inhibitors, and other combination therapies.(34,35) However, unlike the established efficacy of rapamycin in treating SEGAs in TSC patients, targeting the mTOR pathway as anti-tumor treatments for non-TSC-related gliomas is clearly more complicated and awaits further investigations before establishment in clinical practice.
Epilepsy
While the role of the mTOR pathway in tumorigenesis and the associated utility of mTOR inhibitors for treating tumors in TSC is firmly documented, the importance of mTOR in the common, disabling neurological symptoms of TSC, in particular epilepsy, autism, and cognitive deficits, is not as well established. These neurological manifestations of TSC typically are not directly related to tumor growth per se. Even if epilepsy is caused by cortical tubers, tubers are developmental malformations that do not grow progressively like tumors. Other cellular and molecular abnormalities, not directly related to cell growth and proliferation, likely promote epileptogenesis, such as aberrant circuit formation and dysregulated neurotransmitter receptors or ion channels.(36) Given the known role of the mTOR pathway in regulating such functions as protein synthesis and synaptic plasticity, it is reasonable to hypothesize that mTOR is also involved in mechanisms of epileptogenesis in TSC. However, whether mTOR inhibitors represent a rational treatment for seizures and neuropsychiatric symptoms in TSC patients has not been definitively established at this point.
Although the specific downstream mechanisms of epileptogenesis are still uncertain, animal models of TSC do indicate that the mTOR pathway is important for the pathophysiology of epilepsy in TSC and that mTOR inhibitors may represent appropriate treatments to either prevent epilepsy (antiepileptogenic) or inhibit existing seizures (antiseizure).(37,38) Several mouse models of TSC, involving conditional inactivation of Tsc1 or Tsc2 in neurons, glia, or brain progenitor cells, have been documented to have epilepsy. Early treatment with rapamycin prior to the onset of seizures can prevent the development of epilepsy, as well as many of the pathological and molecular changes in the brain that likely promote epileptogenesis in these mice.(17,39–42) In addition, later treatment of mice that already have epilepsy can decrease seizure frequency. mTOR inhibitors may also improve cognitive deficits in mouse models of TSC.(43) Similar to the reversible effects on tumor growth in TSC patients, unfortunately the beneficial effects of rapamycin on seizures do not persist if rapamycin is stopped.
Data on the utility of mTOR inhibitors for epilepsy in TSC patients are more limited. A couple of case reports suggest that mTOR inhibitors may decrease seizure frequency in TSC patients being treated for SEGAs.(44,45) In larger clinical trials primarily testing the effect of everolimus on SEGA growth, seizures were examined as a secondary measure and a significant decrease in seizure frequency was reported with everolimus.(19) However, these previous reports could have been confounded by an indirect effect of everolimus on seizures related to decreased SEGA growth and hydrocephalus. Another clinical trial is currently ongoing, examining the effect of everolimus on seizure frequency as the primary outcome measure, independent of SEGAs. Preliminary results from this uncontrolled study also support that mTOR inhibitors may decrease seizure frequency in TSC patients with established epilepsy.(46) However, larger, placebo-controlled trials are still needed to definitively establish whether mTOR inhibitors are an effective treatment for epilepsy in TSC.
Beyond symptomatic treatment of ongoing epilepsy, perhaps a much more significant question is whether mTOR inhibitors can actually prevent epilepsy when started prior to the first seizure.(37,38) While current seizure medications simply suppress seizures as symptomatic therapy, it has been recognized for a while that novel treatments are needed that have disease-modifying or “antiepileptogenic” properties to prevent the development or progression of epilepsy. TSC patients may represent a very appropriate population to develop and test a preventative/antiepileptogenic therapy for a couple of reasons. While many patients with epilepsy present with seizures unpredictably without any previously known risk factors, some TSC patients can be identified at a young age before the onset of epilepsy due to non-neurological findings of TSC. Thus, it is feasible to identify and start a preventative treatment on this subset of TSC patients. Furthermore, TSC patients are at high risk (~80–90%) of developing epilepsy. Therefore, it can be justified to start a neurologically-asymptomatic TSC patient on antiepileptogenic therapy as a preventative measure. However, despite the evidence for antiepileptogenic effects of mTOR inhibitors in animal models of TSC, designing and conducting antiepileptogenic clinical trials are extremely difficult and at this point, there are no human data indicating whether mTOR inhibitors have antiepileptogenic properties to prevent epilepsy in TSC patients.
Similar to TSC, several other, relatively rare genetic disorders involve dysregulation of the mTOR pathway and an increased risk for tumors and epilepsy. Pten is a tumor suppressor gene that regulates growth via modulation of the phosphatidylinositol-3 kinase (PI3K)/AKT pathway which is an upstream activator of the mTOR pathway (Fig. 1). Pten knock-out mice exhibit neuronal hypertrophy, megalencephaly, and seizures, and treatment with mTOR inhibitors decreases the pathological abnormalities and suppresses seizures.(47–49) In one Pten mouse model, rapamycin not only decreased seizures, but also reversed deficits in social behavior suggestive of autism.(50) Similar to the TSC models, seizures return within weeks of cessation of rapamycin therapy, although subsequent intermittent rapamycin treatment is able to maintain a long-term antiseizure effect.(49) The link between mTOR and epilepsy is further supported in patients with a rare disorder known as polyhydramnios, megalencephaly, and symptomatic epilepsy syndrome (PMSE), in which deletions in the STRADα gene cause dysregulation of mTOR signaling via a reduction in the LKB1/AMPK pathway.(51) Finally, genetic polymorphisms and biochemical markers of mTOR activation have been identified in patients with isolated focal cortical dysplasia, a common etiology of intractable epilepsy.(52–60) While mTOR inhibitors may represent rational therapies for seizures in these other types of epilepsy, such clinical use has not yet been reported.
While genetic epilepsies affecting the mTOR pathway are relatively rare, there is increasing interest as to whether the mTOR pathway may be involved in other, more common types of epilepsy, such as following acquired brain injury. In animal models, mTOR signaling is abnormally increased following traumatic brain injury or status epilepticus that leads to chronic epilepsy.(61–65) The mTOR pathway is also activated by hypoxia or toxin-related insults that acutely induce neonatal seizures or infantile spasms.(66,67) mTOR inhibitors can decrease pathological abnormalities that are associated with epileptogenesis, in particular mossy fiber sprouting.(61–64) Rapamycin may decrease behavioral and cognitive deficits in animal models of traumatic brain injury.(68) There is some evidence that mTOR inhibitors can inhibit existing seizures or prevent epilepsy in some animal models of acquired epilepsy,(62–64,66,67) although other studies have found negative results.(69–71) Variability among different studies in effectiveness for epilepsy may depend on a number factors, such as dose, timing, and animal model. Even when found to be effective, whether mTOR inhibitors primarily have antiepileptogenic/preventative effects or traditional seizure-suppressing effects is still debated. With a number of issues still to be resolved on a basic science and animal model level, mTOR inhibitors have not been tested in patients with acquired brain injury or post-injury epilepsy.
Other Neurodevelopmental Disorders and Autism
In addition to TSC, there are a number of other genetic disorders characterized primarily by neurodevelopmental disabilities, such as autism, mental retardation, and other cognitive deficits, as well as epilepsy. Interestingly, abnormal mTOR signaling has also been implicated in many of these diseases, including Fragile X syndrome, Down syndrome, and Rett syndrome.(72) The potential involvement of dysregulated mTOR signaling in multiple neurological disorders characterized by cognitive deficits should not be surprising, as the mTOR pathway has been implicated in mediating physiological mechanisms of learning and memory. In particular, experience-dependent forms of synaptic plasticity, such as long-term potentiation and depression, largely rely on protein synthesis activated by the mTOR pathway.(73,74) Furthermore, changes in dendritic morphology may represent a structural substrate for memory and can be regulated by the mTOR pathway,(75,76) whereas abnormalities in dendritic morphology have been demonstrated in these neurogenetic syndromes, including TSC, Fragile X, Down, and Rett syndromes.(77)
Fragile X syndrome is one of the most common causes of inherited mental retardation and autism. In this disease, abnormal trinucleotide repeat expansion of the Fragile X mental retardation 1 (FMR1) gene on the X chromosome leads to loss of expression of the corresponding protein (FMRP). FMRP is an RNA binding protein that normally prevents protein synthesis via binding of messenger RNA. In addition, the mTOR pathway has been found to have increased activation in a Fmr1 KO mouse and in pathological brain specimens from Fragile X patients.(78,79) Although the biochemical link between FMRP and mTOR is not entirely clear, there is some evidence that FMRP may normally inhibit the PI3K/Akt pathway and that loss of FMRP function would then lead to hyperactivation of PI3K/Akt and subsequently the mTOR pathway.(78)
Similarly, in a mouse model of Down syndrome, the most common genetic cause of mental retardation, there is also evidence for increased mTOR pathway activity, which may relate to increased levels of brain-derived neurotrophic factor.(80) In contrast, in a mouse model of Rett syndrome, another common genetic cause of mental retardation and autism, the mTOR pathway shows signs of decreased activation.(81)
Overall, these findings suggest the possibility of using mTOR modulators to treat many of these neurogenetic causes of mental retardation and autism associated with mTOR hyperactivation. In a mouse model of TSC, rapamycin reversed deficits in spatial learning and long-term potentiation.(43) Effects of rapamycin on cognitive deficits in mouse models of Fragile X and Down syndrome are currently under investigation.(72) While no clinical reports of the effects of mTOR inhibitors on autism or cognitive measures presently exist in people, there is currently at least one ongoing clinical trial investigating effects of mTOR inhibitors on cognitive outcomes in TSC patients (NCT01289912, clinicaltrials.gov).
Neurodegenerative Diseases
On the opposite end of the developmental spectrum, classic neurodegenerative diseases, such as Alzheimer’s, Parkinson’s, and Huntington’s disease, are characterized by accumulation of abnormal misfolded or toxic proteins and associated neuronal death. As the mTOR pathway is involved in regulation of cell death and survival mechanisms, it makes sense that mTOR has also been implicated in the pathophysiology of these neurodegenerative disorders.(6) In particular, mTOR signaling may modulate mechanisms of apoptotic cell death. Furthermore, the mTOR pathway normally inhibits autophagic mechanisms, which help degrade and clear aggregated or accumulated proteins. Thus, mTOR inhibitors may represent a rational therapy for this group of diseases by inducing autophagy or directly regulating neuronal death mechanisms.
Alzheimer’s disease involves progressive cognitive dysfunction and memory loss associated with development of both extracellular plaques containing amyloid-beta aggregates and intracellular neurofibrillary tangles composed of hyperphosphorylated microtubular protein tau. Increased mTOR activity is seen in mouse models of Alzheimer’s disease and correlates with the progression of abnormal amyloid-beta levels and tau pathology.(82) Furthermore, mTOR and its downstream signaling kinases promoting phosphorylation are abnormally increased in neurofibrillary tangles from Alzheimer’s brains and may contribute directly to hyperphosphorylation of tau.(83) The relationship between amyloid-beta and mTOR is more complex, as amyloid-beta itself can increase mTOR activity,(82) but conversely rapamycin can reduce amyloid-beta levels.(84) Thus, mTOR inhibitors may represent rational therapy for Alzheimer’s disease by reducing the hyperphosphorylation of tau, decreasing amyloid-beta levels, and inducing autophagy. In fact, mTOR inhibitors have been reported to reverse cognitive deficits in mouse models of Alzheimer’s disease.(82,84)
Huntington’s disease, an autosomal dominant disorder, involves relatively selective neurodegeneration in the basal ganglia and cortex, related to trinucleotide repeat expansion of polyglutamine on the huntingtin protein. Mutant huntingtin protein is not effectively cleared from neurons, leading to accumulation of toxic intracellular aggregates and associated neuronal death. In mouse models and human brains of Huntington’s disease, mTOR has been shown to be sequestered in polyglutamine aggregates. mTOR inhibitors can enhance autophagy and consequently reduce huntingtin accumulation and associated neuronal death in cellular and animal models of Huntington’s disease.(85,86) Furthermore, rapamycin can improve motor deficits in mouse models of Huntington’s disease.(85)
Finally, Parkinson’s disease is characterized by a progressive movement disorder related to loss of specific populations of central and peripheral neurons, including dopaminergic cells in the substantia nigra. The mTOR pathway may regulate cell death mechanisms in Parkinson’s disease via interaction with a stress response protein RTP801, which is activated by oxidative stress and induces apoptotic cell death. Rapamycin has been reported to inhibit expression of RTP801 and correspondingly protect against neuronal death in in vitro and in vivo models of Parkinson’s disease.(87) Alternatively, dopaminergic neuronal death in Parkinson’s disease has been hypothesized to be related to oxidative stress-induced lysosomal depletion and a resulting accumulation of undegraded autophagosomes. By inducing autophagy, rapamycin was shown to restore normal lysosomal levels and attenuate dopaminergic neuronal death in a cellular model of Parkinson’s disease.(88) Although the mechanisms may vary, cumulative evidence supports that mTOR inhibitors are neuroprotective in animal models of a variety of neurodegenerative diseases. At this point, however, no clinical trials have been conducted testing the effect of mTOR inhibitors on patients with these neurodegenerative disorders.
Future Directions
A number of issues need to be addressed in order to optimize the current use of mTOR inhibitors and to expand the clinical indications in various neurological diseases. With regard to the present indication for SEGAs in TSC patients, while everolimus has proven efficacy in decreasing tumor growth, a potential limitation is the ill-defined duration of therapy. Cessation of treatment seems to result in a regrowth of tumors, so mTOR inhibitors are not a cure for tumors in TSC and long-term treatment is likely necessary to maintain efficacy.(18–20) Since SEGAs often present in early childhood, this potentially would require treatment for decades or longer. Significant side effects that may occur with mTOR inhibitors, including chronic immunosuppression and associated opportunistic infections, make this prospect of long-term treatment less attractive. One potential option to alleviate this concern would be to use “drug holidays” or treatment paradigms with intermittent application of mTOR inhibitors. Animal model data suggest that intermittent use of mTOR inhibitors can maintain efficacy, but reduce risk of side effects, such as immunosuppression.(49,89–91)
To expand the clinical indications of mTOR inhibitors beyond TSC-related tumors, additional basic and clinical data are needed. For epilepsy, if the encouraging preliminary data from ongoing uncontrolled clinical trials in TSC are confirmed, a placebo-controlled trial of an mTOR inhibitor for intractable epilepsy in TSC patients would be a reasonable, feasible next step. In contrast, testing the potential antiepileptogenic efficacy of mTOR inhibitors for preventing epilepsy is a much more complicated endeavor, involving a number of practical barriers and ethical issues. Some TSC patients can be identified at an early age prior to the onset of seizures due to non-neurological findings and may be appropriate candidates for starting a potential antiepileptogenic therapy. However, while a large proportion of TSC patients will develop epilepsy, a subset will not, thus exposing such patients to an unnecessary treatment with side effects. Ideally, biomarkers could first identify those patients that are at highest risk for epilepsy and the best candidates for an antiepileptogenic therapy. Although such biomarkers for epilepsy have not been definitely established, future studies might establish specific EEG or brain MRI abnormalities as biomarkers for epilepsy in TSC. Beyond TSC, other high-risk populations, such as patients with traumatic brain injury, could be candidates for antiepileptogenic treatment. Furthermore, cognitive deficits and autism from other neurogenetic syndromes with mTOR hyperactivation (e.g., Fragile X, Down syndrome) could be targeted, and neurodegenerative disorders could be considered for neuroprotective trials with mTOR inhibitors. However, expansion to these non-TSC neurological disorders should await more definitive data from animal models.
Summary/Conclusions
There is accumulating evidence that the mTOR pathway may be involved in the pathophysiology of a number of neurological diseases. At this point, this evidence is predominantly derived from basic science and animal model studies. However, these findings are starting to be translated into the clinical arena. TSC is the one disease in which the mTOR pathway has been established as a central component of its pathogenesis and mTOR inhibitors have proven benefit. The rapamycin analog, everolimus, has recently been approved for treatment of SEGA and AML growth in TSC patients, and similar approval for other TSC-related tumors, such as LAM, and angiofibromas, may soon follow. In contrast, for non-TSC-related gliomas, such as glioblastoma multiforme, initial clinical trials have not demonstrated clear efficacy of mTOR inhibitors, and focus is now shifted to dual pathway inhibitors and combination therapies. Preliminary clinical studies suggest that mTOR inhibitors may be beneficial for treating seizures in TSC patients with established epilepsy, but await definitive confirmation from controlled trials. While several studies with TSC mouse models suggest that mTOR inhibitors have antiepileptogenic properties for preventing epilepsy, clinical antiepileptogenic drug trials are difficult to conduct and have not yet been attempted with mTOR inhibitors in TSC patients. Similarly, there is a variety of evidence that mTOR inhibition may also have disease-modifying effects in animal models of acquired epilepsy and various common neurodegenerative diseases, such as Alzheimer’s, Huntington’s, and Parkinson’s disease, but use of mTOR inhibitors have not been tested in patients with these diseases. Overall, considerable progress has been made in demonstrating the potential utility of mTOR inhibitors in a variety of neurological disorders. With ongoing progress in basic and clinical research, there are reasons to be optimistic that the clinical indications of mTOR inhibitors for neurological disease will continue to expand in the future.
Acknowledgments
This article is a summary of the Professor Yu-Zen Shen Memorial Lecture presented at the 16th Annual Meeting of the Taiwan Child Neurology Society. MW receives funding from the National Institutes of Health (R01 NS056872), the US Department of Defense Congressionally Directed Medical Research Programs, and the Washington University/Pfizer Biomedical Research Collaboration.
References
- 1.Weber JD, Gutmann DH. Deconvoluting mTOR biology. Cell Cycle. 2012;11:236–248. doi: 10.4161/cc.11.2.19022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cho CH. Frontier of epilepsy research - mTOR signaling pathway. Exp Mol Med. 2011;43:231–274. doi: 10.3858/emm.2011.43.5.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zoncu R, Efevan A, Sabitini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dazert E, Hall MN. mTOR signaling in disease. Current Opin Cell Biol. 2011;23:744–755. doi: 10.1016/j.ceb.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 5.Chong ZZ, Shang YC, Zhang L, Wang S, Maiese K. Mammalian target of rapamycin: Hitting the bull’s-eye for neurological disorders. Oxid Med Cell Longev. 2010;3:374–391. doi: 10.4161/oxim.3.6.14787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bove J, Martinez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci. 2011;12:437–452. doi: 10.1038/nrn3068. [DOI] [PubMed] [Google Scholar]
- 7.Orlova KA, Crino PB. The tuberous sclerosis complex. Ann NY Acad Sci. 2010;1184:87–105. doi: 10.1111/j.1749-6632.2009.05117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 9.Major P, Rakowski S, Simon MV, Cheng ML, Eskandar E, Baron J, Leeman BA, Frosch MP, Thiele EA. Are cortical tubers epileptogenic? Evidence from electrocorticography. Epilepsia. 2009;50:147–154. doi: 10.1111/j.1528-1167.2008.01814.x. [DOI] [PubMed] [Google Scholar]
- 10.European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 1993;75:1305–1315. doi: 10.1016/0092-8674(93)90618-z. [DOI] [PubMed] [Google Scholar]
- 11.Van Slegtenhorst MM, de Hoogt R, Hermans C, Nellist M, Janssen B, Verhoef S, Lindhout D, van den Ouweland A, Halley D, Young J, Burley M, Jeremiah S, Woodward K, Nahmias J, Fox M, Ekong R, Osborne J, Wolfe J, Povey S, Snell RG, Cheadle JP, Jones AC, Tachataki M, Ravine D, Sampson JR, Reeve MP, Richardson P, Wilmer F, Munro C, Hawkins TL, Sepp T, Ali JB, Ward S, Green AJ, Yates JR, Kwiatkowska J, Henske EP, Short MP, Haines JH, Jozwiak S, Kwiatkowski DJ. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277:805–808. doi: 10.1126/science.277.5327.805. [DOI] [PubMed] [Google Scholar]
- 12.Gao X, Zhang Y, Arrazola P, Hino O, Kobayashi T, Yeung RS, Ru B, Pan D. Tsc tumour suppressor proteins antagonize amino acid-TOR signalling. Nat Cell Biol. 2002;4:699–704. doi: 10.1038/ncb847. [DOI] [PubMed] [Google Scholar]
- 13.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signaling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 14.Tee AR, Fingar DC, Manning BD, Kwaitkowski DJ, Cantley LC, Blenis J. Tuberous sclerosis complex-1 and –2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci USA. 2002;99:13571–13576. doi: 10.1073/pnas.202476899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El-Hashemite N, Zhang H, Henske EP, Kwiatkowski DJ. Mutation in TSC2 and activation of mammalian target of rapamycin signaling pathway in renal angiomyolipoma. Lancet. 2003;361:1348–1349. doi: 10.1016/S0140-6736(03)13044-9. [DOI] [PubMed] [Google Scholar]
- 16.Lee L, Sudentas P, Donohue B, Asrican K, Worku A, Walker V, Sun Y, Schmidt K, Albert MS, El-Hashemite N, Lader AS, Onda H, Zhang H, Kwiatokowski DJ, Dabora SL. Efficacy of a rapamycin analog (CCI-779) and IFN-gamma in tuberous sclerosis mouse models. Genes Chromosomes Cancer. 2005;42:213–227. doi: 10.1002/gcc.20118. [DOI] [PubMed] [Google Scholar]
- 17.Zeng L, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of Tuberous Sclerosis Complex. Ann Neurol. 2008;63:444–453. doi: 10.1002/ana.21331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franz DN, Leonard J, Tudor C, Chuck G, Care M, Sethuraman G, Dinopoulos A, Thomas G, Crone KR. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59:490–498. doi: 10.1002/ana.20784. [DOI] [PubMed] [Google Scholar]
- 19.Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, Wilson KA, Byars A, Sahmoud T, Franz DN. Everolimus for subependymal giant-cell astrocytomas in Tuberous Sclerosis. N Engl J Med. 2010;363:1801–11. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- 20.Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, Schmithorst VJ, Laor T, Brody AS, Bean J, Salibury S, Franz DN. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358:140–151. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dabora SL, Franz DN, Ashwal S, Sagalowsky A, DiMario FJ, Jr, Miles D, Cutler D, Krueger D, Uppot RN, Rabenou R, Camposano S, Paolini J, Fennessy F, Lee N, Woodrum C, Manola J, Garber J, Thiele EA. Multicenter phase 2 trial of sirolimus for tuberous sclerosis: kidney angiomyolipomas and other tumors regress and VEGF-D levels decrease. PLoS One. 2011;6:e23379. doi: 10.1371/journal.pone.0023379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haemel AK, O’Brian AL, Teng JM. Topical rapamycin: a novel approach to facial angiofibromas in tuberous sclerosis. Arch Dermatol. 2010;146:715–718. doi: 10.1001/archdermatol.2010.125. [DOI] [PubMed] [Google Scholar]
- 23.Li XY, Zhang LQ, Zhang XG, Li X, Ren YB, Ma XY, Li XG, Wang LX. Association between AKT/mTOR signaling and malignancy grade of human gliomas. J Neurooncol. 2011;103:453–458. doi: 10.1007/s11060-010-0424-1. [DOI] [PubMed] [Google Scholar]
- 24.Riemenschneider MJ, Betensky RA, Pasedag SM, Louis DN. AKT activation in human glioblastomas enhances proliferation via TSC2 and S6 kinase signaling. Cancer Res. 2006;66:5618–5623. doi: 10.1158/0008-5472.CAN-06-0364. [DOI] [PubMed] [Google Scholar]
- 25.Wong AJ, Ruppert JM, Bigner SH, Gzeschik CH, Humphrey PA, Bigner DS, Vogelstein B. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci USA. 1992;89:2965–2969. doi: 10.1073/pnas.89.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang SI, Puc J, Li J, Bruce JN, Cairns P, Sidransky D, Parsons R. Somatic mutations of PTEN in glioblastoma multiforme. Cancer Res. 1997;57:4183–4186. [PubMed] [Google Scholar]
- 27.Duerr EM, Rollbrocker B, Hayashi Y, Peters N, Meyer-Puttlitz B, Louis DN, Schramm J, Wiestler OD, Parsons R, Eng C, von Deimling A. PTEN mutations in gliomas and glioneuronal tumors. Oncogene. 1998;16:2259–2264. doi: 10.1038/sj.onc.1201756. [DOI] [PubMed] [Google Scholar]
- 28.Houchens DP, Ovejera AA, Riblet SM, Slagel DE. Human brain tumor xenografts in nuce mice as a chemotherapy model. Eur J Cancer Clin Oncol. 1983;19:799–805. doi: 10.1016/0277-5379(83)90012-3. [DOI] [PubMed] [Google Scholar]
- 29.Eshleman JS, Carlson BL, Mladek AC, Kastner BD, Shide KL, Sarkaria JN. Inhibition of the mammalian target of rapamycin sensitizes U87 xenografts to fractionated radiation therapy. Cancer Res. 2002;62:7291–7297. [PubMed] [Google Scholar]
- 30.Heimberger AB, Wang E, McGary EC, Hess KR, Henry VK, Shono T, Cohen Z, Gumin J, Sawaya R, Conrad CA, Lang FF. Mechanisms of action of rapamycin in gliomas. Neuro-oncol. 2005;7:1–11. doi: 10.1215/S1152851704000420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anandharaj A, Cinghu S, Park WY. Rapamycin-mediated mTOR inhibition attenuates surviving and sensitizes glioblastoma cells to radiation. Acta Biochim Biophys Sin. 2011;43:292–300. doi: 10.1093/abbs/gmr012. [DOI] [PubMed] [Google Scholar]
- 32.Chang SM, Wen P, Cloughesy T, Greenberg H, Schiff D, Conrad C, Fink K, Robins HI, De Angelis L, Raizer J, Hess K, Aldape K, Lamborn KR, Kuhn J, Dancey J, Prados MD North American Brain Tumor Consortium and the National Cancer Institute. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Invest New Drugs. 2005;23:357–361. doi: 10.1007/s10637-005-1444-0. [DOI] [PubMed] [Google Scholar]
- 33.Galanis E, Buckner JC, Maurer MJ, Kreisberg JI, Ballman K, Boni J, Peralba JM, Jenkins RB, Dakhil SR, Morton RF, Jaeckle KA, Scheithauer BW, Dancey J, Hidalgo M, Walsh DJ North Central Cancer Treatment Group. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. J Clin Oncol. 2005;23:5294–5304. doi: 10.1200/JCO.2005.23.622. [DOI] [PubMed] [Google Scholar]
- 34.Fan QW, Knight ZA, Goldenberg DD, Yu W, Mostov KE, Stokoe D, Shokat KM, Weiss WA. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell. 2006;9:341–349. doi: 10.1016/j.ccr.2006.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fan QW, Weiss WA. Inhibition of PI3K-Akt-mTOR signaling in glioblastoma by mTORC1/2 inhibitors. Methods Mol Biol. 2012;821:349–359. doi: 10.1007/978-1-61779-430-8_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong M. Mechanisms of epileptogenesis in tuberous sclerosis complex and related malformations of cortical development. Epilepsia. 2008;49:8–21.23. doi: 10.1111/j.1528-1167.2007.01270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McDaniel SS, Wong M. Therapeutic role of mammalian target of rapamycin (mTOR) inhibition in preventing epileptogenesis. Neurosci Lett. 2011;497:231–239. doi: 10.1016/j.neulet.2011.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wong M. Mammalian target of rapamycin (mTOR) inhibition as a potential antiepileptogenic therapy: From tuberous sclerosis to common acquired epilepsies. Epilepsia. 2010;51:27–36. doi: 10.1111/j.1528-1167.2009.02341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, Kwiatkowski Response of a neuronal model of Tuberous Sclerosis to mammalian target of Rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci. 2008;28:5422–5432. doi: 10.1523/JNEUROSCI.0955-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goto J, Talos DM, Klein P, Qin W, Chekaluk YI, Anderl S, Malinowska IA, Di Nardo A, Bronson RT, Chan JA, Vinters HV, Kernie SG, Jensen FE, Sahin M, Kwiatkowski DJ. Regulable neural progenitor-specific TSC1 loss yields giant cells with organellar dysfunction in a model of tuberous sclerosis complex. PNAS. 2011;108:1070–1079. doi: 10.1073/pnas.1106454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zeng L, Rensing NR, Zhang B, Gutmann DH, Gambello MJ, Wong M. TSC2 gene inactivation causes a more severe epilepsy phenotype than TSC1 inactivation in a mouse model of Tuberous Sclerosis Complex. Hum Mol Genet. 2011;20:445–454. doi: 10.1093/hmg/ddq491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carson RP, Van Nielen DL, Winzenburger PA, Ess KC. Neuronal and glia abnormalities in TSC1-deficient forebrain and partial rescue by rapamycin. Neurobiol Dis. 2012;45:369–380. doi: 10.1016/j.nbd.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ. Reversal of learning deficits in a TSC2+/− mouse model of tuberous sclerosis. Nat Med. 2008;14:843–848. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muncy J, Butler IJ, Koenig M. Rapamycin reduces seizure frequency in Tuberous Sclerosis Complex. J Child Neurol. 2009;24:477. doi: 10.1177/0883073808324535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perek-Poinik M, Jozwiak S, Jurkiewicz E, Perek D, Kotulska K. Effective everolimus treatment of inoperable, life-threatening subependymal giant cell astrocytoma and intractable epilepsy in a patient with tuberous sclerosis complex. Eur J Paediatr Neurol. 2012;16:83–85. doi: 10.1016/j.ejpn.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 46.Wilfong A, Krueger DA, Holland-Bouley K, Anderson AE, Agricola K, Schultz RJ, Tudor C, Mayws M, Lopez CM, Franz DN. Everolimus improves seizure control in tuberous sclerosis complex. American Epilepsy Society Meeting Abstracts. 2011 Late-breaking Abstract #3.329. [Google Scholar]
- 47.Kwon C, Zhu X, Zhang J, Baker SJ. mTor is required for hypertrophy of Pten-deficient neuronal soma in vivo. Proc Natl Acad Sci USA. 2003;100:12923–12928. doi: 10.1073/pnas.2132711100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ljungberg MC, Sunnen CN, Lugo JN, Anderson AE, D’Arcangelo G. Rapamycin suppresses seizures and neuronal hypertrophy in a mouse model of cortical dysplasia. Dis Model Mech. 2009;2:389–398. doi: 10.1242/dmm.002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sunnen CN, Brewster AL, Lugo JN, Vanegas F, Turclos E, Mukhi S, Parghi D, D’Arcangelo G, Anderson AE. Inhibition of the mammalian target of rapamycin blocks epilepsy progression in NS-Pten conditional knockout mice. Epilepsia. 2011;52:2065–2075. doi: 10.1111/j.1528-1167.2011.03280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou J, Blundell J, Ogawa S, Kwon C, Zhang W, Sinton C, Powell CM, Parada LF. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci. 2009;29:1773–1783. doi: 10.1523/JNEUROSCI.5685-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Orlova KA, Parker WE, Heuer GG, Tsai V, Yoon J, Baybis M, Fenning RS, Strauss K, Crino PB. STRADα deficiency results in aberrant mTORC1 signaling during corticogenesis in humans and mice. J Clin Invest. 2010;120:1591–1602. doi: 10.1172/JCI41592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baybis M, Yu J, Lee A, Golden JA, Weiner H, McKhann G, Aronica E, Crino PB. mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann Neurol. 2004;56:478–487. doi: 10.1002/ana.20211. [DOI] [PubMed] [Google Scholar]
- 53.Schick V, Majores M, Engels G, Hartmann W, Elger CE, Schramm J, Schoch S, Becker AJ. Differential Pi3K-pathway activation in cortical tubers and focal cortical dysplasias with balloon cells. Brain Pathol. 2007;17:165–173. doi: 10.1111/j.1750-3639.2007.00059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Becker AJ, Urbach H, Scheffler BJ, Baden T, Normann S, Lahl R, Pennek HW, Tuxhorn I, Elger CE, Schramm J, Wiestler OD, Blumcke I. Focal cortical dysplasia of Taylor’s balloon cell type: mutational analysis of the TSC1 gene indicates a pathogenic relationship to Tuberous Sclerosis. Ann Neurol. 2002;52:29–37. doi: 10.1002/ana.10251. [DOI] [PubMed] [Google Scholar]
- 55.Schonberger A, Niehusmann P, Urbach H, Majores M, Grote A, Holthausen H, Blumcke I, Deckert M, Becker AJ. Increased frequency of distinct TSC2 alleleic variants in focal cortical dysplasias with balloon cells and mineralization. Neuropathology. 2009;29:559–565. doi: 10.1111/j.1440-1789.2009.01018.x. [DOI] [PubMed] [Google Scholar]
- 56.Gumbinger C, Rohsbach CB, Schulze-Bonhage A, Korinthenberg R, Zentner J, Haffner M, Fauser S. Focal cortical dysplasia: a genotype-phenotype analysis of polymorphisms and mutations in the TSC genes. Epilepsia. 2009;50:1396–1408. doi: 10.1111/j.1528-1167.2008.01979.x. [DOI] [PubMed] [Google Scholar]
- 57.Grajkowski W, Kotulska K, Matyja E, Larysz-Brysz M, Mandera M, Roszkowski M, Domanska-Pakilela D, Lewik-Kowalik J, Jozwiak S. Expression of tuberin and hamartin in tuberous sclerosis complex-associated and sporadic cortical dysplasia of Taylor’s balloon cell type. Folia Neuropathol. 2008;46:43–48. [PubMed] [Google Scholar]
- 58.Lugnier C, Majores M, Fassunke J, Pernhorst K, Niehusmann P, Simon M, Nellist M, Schoch S, Becker A. Hamartin variants that are frequent in focal dysplasias and cortical tubers have reduced tuberin binding and aberrant subcellular distribution in vitro. J Neuropathol Exp Neurol. 2009;68:1136–1146. doi: 10.1097/NEN.0b013e3181b9a699. [DOI] [PubMed] [Google Scholar]
- 59.Ljungberg MC, Bhattacharjee MB, Lu Y, Armstrong DL, Yoshor D, Swann JW, Sheldon M, D’Arcangelo G. Activation of mammalian target of rapamycin in cytomegalic neurons of human cortical dysplasia. Ann Neurol. 2006;60:420–429. doi: 10.1002/ana.20949. [DOI] [PubMed] [Google Scholar]
- 60.Miyata H, Chiang ACY, Vinters HV. Insulin signaling pathways in cortical dysplasia and TSC-tubers: tissue microarray analysis. Ann Neurol. 2004;56:510–519. doi: 10.1002/ana.20234. [DOI] [PubMed] [Google Scholar]
- 61.Buckmaster PS, Ingram EA, Wen X. Inhibition of the mammalian target of rapamycin signaling pathway suppresses dentate granule cell axon sprouting in a rodent model of temporal lobe epilepsy. J Neurosci. 2009;29:8259–69. doi: 10.1523/JNEUROSCI.4179-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zeng LH, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci. 2009;29:6964–72. doi: 10.1523/JNEUROSCI.0066-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang X, Zhang H, Yang J, Wu J, McMahon J, Lin Y, Cao Z, Gruenthal M, Huang Y. Pharmacological inhibition of the mammalian target of rapamycin pathway suppresses acquired epilepsy. Neurobiol Dis. 2010;40:193–9. doi: 10.1016/j.nbd.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.van Vliet EA, Forte G, Holtman L, den Burger JCG, Sinjewel A, de Vries HE, Aronica E, Gorter JA. Inhibition of mammalian target of rapamycin reduces epileptogenesis and blood-brain barrier leakage but not microglia activation. Epilepsia. 2012 doi: 10.1111/j.1528-1167.2012.03513.x. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 65.Chen S, Atkins CM, Liu CL, Alonso OF, Dietrich WD, Hu BR. Alterations in mammalian target of rapamycin signaling pathways after traumatic brain injury. J Cereb Blood Flow Metab. 2007;27:939–49. doi: 10.1038/sj.jcbfm.9600393. [DOI] [PubMed] [Google Scholar]
- 66.Talos DM, Sun H, Zhou X, Fitzgerald EC, Jackson MC, Klein PM, Lan VJ, Joseph A, Jensen F. The interaction between early life epilepsy and autistic-like behavioral consequences: a role for the mammalian target of rapamycin (mTORC1) pathway. PLoS ONE. 2012;7:e35885. doi: 10.1371/journal.pone.0035885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Raffo E, Coppola A, Ono T, Briggs Sw, Galanopoulo AS. A pulse rapamycin therapy for infantile spasms and associated cognitive decline. Neurobiol Dis. 2011;43:322–8. doi: 10.1016/j.nbd.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Erlich S, Alexandrovich A, Shohami E, Pinkas-Kramarski R. Rapamycin is a neuroprotective treatment for traumatic brain injury. Neurobiol Dis. 2007;26:86–93. doi: 10.1016/j.nbd.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 69.Buckmaster PS, Lew FH. Rapamycin suppresses mossy fiber sprouting but not seizure frequency in a mouse model of temporal lobe epilepsy. J Neurosci. 2011;31:2337–47. doi: 10.1523/JNEUROSCI.4852-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sliwa A, Plucinska G, Bednarczyk J, Lukasiuk K. Post-treatment with rapamycin does not prevent epileptogenesis in the amygdala stimulation model of temporal lobe epilepsy. Neurosci Lett. 2012 doi: 10.1016/j.neulet.2011.12.051. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 71.Chachua T, Yum MS, Veliskova J, Velisek L. Validation of the rat model of cryptogenic infantile spasms. Epilepsia. 2011;52:1666–77. doi: 10.1111/j.1528-1167.2011.03220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Troca-Marin JA, Alves-Sampaio A, Montesinos ML. Deregulated mTOR-mediated translation in intellectual disability. Prog Neurobiol. 2012;96:268–272. doi: 10.1016/j.pneurobio.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 73.Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci USA. 2002;99:467–472. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stoica L, Zhu PJ, Huang W, Zhou H, Kozma SC, Costa-Mattioli M. Selective pharmacogenetic inhibition of mammalian target of rapamycin complex 1 (mTORC1) blocks long-term synaptic plasticity and memory storage. Proc Natl Acad Sci USA. 2011;108:3791–3796. doi: 10.1073/pnas.1014715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M. Control of dendritic arborization by the phosphoinositide-3′-kinase-Akt-mammalian target of rapamycin pathway. J Neurosci. 2005;25:11300–11312. doi: 10.1523/JNEUROSCI.2270-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kumar V, Zhang MX, Swank MW, Kung J, Wu GY. Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J Neurosci. 2005;25:11288–11299. doi: 10.1523/JNEUROSCI.2284-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dierssen M, Ramakers GJA. Dendritic pathology in mental retardation: from molecular genetics to neurobiology. Genes Brain Behav. 2006;5(Suppl 2):48–60. doi: 10.1111/j.1601-183X.2006.00224.x. [DOI] [PubMed] [Google Scholar]
- 78.Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, Zukin RS. Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci. 2010;30:694–702. doi: 10.1523/JNEUROSCI.3696-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hoeffer CA, Sanchez E, Hagerman RJ, Mu Y, Nguyen DV, Wong H, Whelan AM, Zukin RS, Klann E, Tassone F. Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Gene Brain Behav. 2012;11:332–341. doi: 10.1111/j.1601-183X.2012.00768.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Troca-Marin JA, Alves-Sampaio A, Montesinos ML. An increase in basal BDNF provokes hyperactivation of the Akt-mammalian target of rapamycin pathway and deregulation of local dendritic translation in a mouse model of Down’s syndrome. J Neurosci. 2011;31:9445–9455. doi: 10.1523/JNEUROSCI.0011-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ricciardi S, Boggio EM, Grosso S, Lonetti G, Forlani G, Stefanelli G, Calcagno E, Morello N, Landsberger N, Biffo S, Pizzorusso T, Giustetto M, Broccoli V. Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum Mol Genet. 2011;20:1182–1196. doi: 10.1093/hmg/ddq563. [DOI] [PubMed] [Google Scholar]
- 82.Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107–13120. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li X, Alafuzoff I, Soininen H, Winblad B, Pei JJ. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J. 2005;272:4211–4220. doi: 10.1111/j.1742-4658.2005.04833.x. [DOI] [PubMed] [Google Scholar]
- 84.Spilman P, Podluskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PloS One. 5:e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 86.Floto RA, Sarkar S, Perlstein EO, Kampmann B, Shreiber SL, Rubinsztein DC. Small molecule enhancers of rapamycin-induced TOR inhibition promotes autophagy, reduce toxicity in Huntington’s Disease models and enhance killing of mycobacteria by macrophages. Autophagy. 3:620–622. doi: 10.4161/auto.4898. [DOI] [PubMed] [Google Scholar]
- 87.Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J Neurosci. 2010;30:1166–1175. doi: 10.1523/JNEUROSCI.3944-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dehav B, Bove J, Rodriguez-Muela N, Perier C, Recasens A, Bova P, Vila M. Pathogenic lysosomal depletion in Parkinson’s disease. J Neurosci. 2010;30:12535–12544. doi: 10.1523/JNEUROSCI.1920-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Boulay A, Zumstein-Mecker S, Stephan C, Beuvink I, Zilbermann F, Haller R, Tobler S, Heusser C, O’Reilly T, Stolz B, Marti A, Thomas G, Lane HA. Antitumor efficacy of intermittent treatment schedules with the rapamycin derivative RAD001 correlates with prolonged inactivation of ribosomal protein S6 kinase 1 in peripheral blood mononuclear cells. Cancer Res. 2004;64:252–261. doi: 10.1158/0008-5472.can-3554-2. [DOI] [PubMed] [Google Scholar]
- 90.Woodrum C, Nobil A, Dabora SL. Comparison of three rapamycin dosing schedules in A/J Tsc2+/− mice and improved survival with angiogenesis inhibitor or asparaginase treatment in mice with subcutaneous tuberous sclerosis related tumors. J Transl Med. 2010;8:14. doi: 10.1186/1479-5876-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rivera VM, Squillace RM, Miller D, Berk L, Wardwell SD, Ning Y, Pollock R, Narasimhan NI, Iuliucci JD, Wang F, Clackson T. Ridaforolimus (AP23573; MK-8669), a potent mTOR inhibitor, has broad antitumor activity and can be optimally administered using intermittent dosing regimens. Mol Cancer Ther. 2011;10:1059–1071. doi: 10.1158/1535-7163.MCT-10-0792. [DOI] [PubMed] [Google Scholar]