

Abstract
MYC is a potent oncogene that drives unrestrained cell growth and proliferation. Shortly after its discovery as an oncogene, the MYC protein was recognized as a sequence-specific transcription factor. Since that time, MYC oncogene research has focused on the mechanism of MYC-induced transcription and on the identification of MYC transcriptional target genes. Recently, MYC was shown to control protein expression through mRNA translation and to directly regulate DNA replication, thus initiating exciting new areas of oncogene research.
The MYC gene is induced by a wealth of growth factors and is essential for most normal cells to proliferate1,2. Following the deregulation of MYC expression by translocation, gene amplification or aberrant signalling, MYC becomes a potent oncoprotein that promotes unrestrained cell proliferation3. Approximately 70% of human tumours have elevated MYC expression4, and suppression of MYC expression can lead to the regression of tumours5. Therapeutic interference of MYC oncogenic activity could offer an important advance in cancer treatment6. This central role for MYC in the growth of both normal and cancer cells sustains a broad interest in MYC function. The MYC protein is a sequence-specific DNA-binding protein that functions as a transcription factor, and this role has focused MYC research on two key questions: how does MYC function biochemically to regulate transcription and what are the MYC-responsive target genes that control cell proliferation and cell transformation? Progress has been made in answering these questions (reviewed in REFS 7–9). Over the past few years, however, experimental findings have raised the intriguing possibility of transcription-independent functions of MYC.
Here, we discuss the finding that MYC regulates protein expression by a novel mechanism — that is, by regulating mRNA translation10. This mechanism can occur independently of MYC-induced transcription and is therefore a novel MYC function that is likely to have an integral role in mediating MYC-dependent functions. We also discuss a second transcription-independent function for MYC — the direct regulation of DNA replication by MYC-dependent recruitment of pre-replicative complex components11.
MYC as a transcription factor
MYC dimerizes with the basic helix-loop-helix (bHLH)/Leu-zipper protein MYC-associated factor-X (MAX) through a C-terminal HLH/Leu-zipper domain to facilitate DNA binding12. The N terminus of MYC proteins contains a transactivation domain and a number of conserved motifs known as MYC boxes (FIG. 1). MYC boxes are well conserved across species. In particular, MYC box II (MBII) is highly conserved and is the most important region of the transactivation domain. MBII is necessary for MYC binding to most cofactors, for the transactivation and repression of most MYC target genes and for the efficient execution of the biological effects of MYC9.
Figure 1. The conserved regions of MYC.
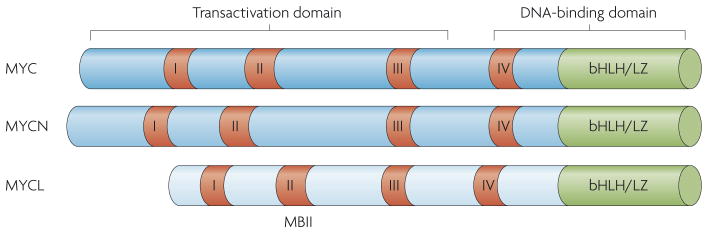
The three MYC proteins (MYC, MYCN and MYCL) are encoded by separate genes with distinct developmental regulation, but all three have been directly implicated in cancer50. The N terminus of MYC contains the transactivation domain and the C terminus contains the DNA-binding domain. The MYC boxes I, II, III and IV are indicated in red. The basic helix-loop- helix/Leu zipper (bHLH/LZ) domain is indicated in green. MYC box II (MBII) has been shown to have a crucial role in most of the biological activities of MYC. Note that MBIV is not a component of the minimal DNA-binding domain but does influence DNA binding in vivo33.
Global analyses have been performed in a comprehensive range of cellular systems and showed that MYC is a weak but expansive transcription factor that upregulates or represses the transcription of ~10% of the genome. The expression of each gene is typically regulated by less than twofold4,7,9. MYC also increases the transcription of the genes that are regulated by RNA polymerase (pol) I and RNA pol III (REFS 13–17), and regulates transcription of microRNAs18,19. Although microRNA expression might have an impact on mRNA translation, this effect is an indirect consequence of MYC-dependent transcriptional mechanisms.
The prevailing model of MYC-mediated transcription postulates that MYC increases local histone acetylation at promoters7,9 (FIG. 2a). MYC binds to histone acetyltransferase complexes including TRRAP (transformation/transcription-domain-associated protein) and either general control of amino-acid-synthesis protein-5 (GCN5) or TIP60, which preferentially acetylate histones H3 or H4, respectively20–22. MYC also binds to the p300/CBP (CReB-binding protein) acetyltransferases23. Recruitment of TRRAP and associated acetylation activity is also associated with the stimulation of RNA pol I and pol III transcription14,15,17. The histone acetylation that results then opens the chromatin and provides docking sites for acetyl-histone-binding proteins, including GCN5 and the SWI/SNF chromatin-remodelling complex, both of which correlate with increased transcription24,25 (FIG. 2a). Transcription-factor-mediated recruitment of histone acetyltransferases is now recognized to be a major mechanism of transactivation, and many other transcription factors, including TCF (T-cell factor), e2F, the tumour suppressor p53 and Gal4, have been subsequently found to use this mechanism26.
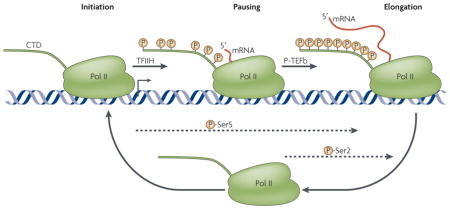
Figure 2. Mechanisms of MYC-induced transcription.

a | MYC recruits histone acetyltransferases, which promote localized modification of chromatin through acetylation of nucleosomes. MYC-associated factor-X (MAX) dimerizes with MYC to create a functional DNA-binding complex that preferentially recognizes the consensus sequence CACGTG. The MYC transactivation domain recruits acetyltransferase complexes that contain transformation/transcription-domain-associated protein (TRRAP) and either general control of amino-acid-synthesis protein-5 (GCN5) or TIP60, which preferentially acetylate histones H3 or H4, respectively. MYC can also recruit p300/CReB-binding protein (CBP). These complexes alter the acetylation of nucleosomes around MYC target genes, and these acetylation marks might also recruit other nuclear factors that influence transcription, such as the SWI/SNF chromatin-remodelling complex (not shown). b | MYC recruits basal transcription factors and promotes the clearance of promoters through RNA polymerase (pol) II. RNA pol II is frequently paused on promoters after phosphorylation of Ser5 on the RNA pol II C-terminal domain (CTD) and synthesis of a short (20–40 base) segment of mRNA28. The MYC protein can promote a paused RNA pol to continue transcription of the mRNA by recruiting the P-TeFb (positive transcription-elongation factor-b) complex, which phosphorylates the CTD on Ser2 and promotes transcriptional elongation.
Our understanding of the factors that regulate transcription has changed in light of a recent global genome analysis that reports the presence of paused RNA pol II at most cellular promoters27. This study expands on earlier work that showed paused RNA pol II at specific promoters, including those of heat shock and MYC genes28. This finding suggests that regulation of transcription also occurs at the level of transcriptional elongation and not just at transcriptional initiation. RNA pol II undergoes a cycle of phosphorylation and dephosphorylation during transcription (BOX 1) and, with its C-terminal domain (CTD) in a hypophosphorylated form, RNA pol II is recruited to promoters. Phosphorylation of the CTD occurs during transcription initiation and elongation, whereas the CTD must be dephosphorylated to allow RNA pol II to be recycled for another round of transcription. RNA pol II has been found to pause on most promoters after transcribing approximately 20–40 bases. Specific signals and cofactors then stimulate transcriptional elongation and further RNA pol II phosphorylation28,29. This model fits well with the earlier finding that MYC does not induce transcription of the target gene CAD (carbamoylphosphate synthetase-2, aspartate transcarbamylase, dihydroorotase) by driving RNA pol II recruitment, but rather stimulates the release of paused RNA pol II from the promoter and stimulates subsequent transcriptional elongation30 (FIG. 2b). This correlates with a MYC-dependent increase in RNA pol II phosphorylation at the CAD promoter, and the MYC transactivation domain was shown to bind CTD kinases31. MYC has also been found to recruit CTD kinases and basal transcription factors to a number of other promoters32. We recently expanded this finding by demonstrating that the MYC induction of RNA pol II phosphorylation occurs globally throughout the nucleus; it can be detected in the total cellular pool of RNA pol II rather than simply at MYC target-gene promoters10. This finding prompted us to question the definition of MYC as simply a traditional transcription factor and led us to investigate novel, transcription-independent roles of MYC.
Box 1. Phosphorylation of the RNA polymerase II C-terminal domain during transcription.
RNA polymerase (pol) II undergoes a cycle of phosphorylation and dephosphorylation at the C-terminal domain (CTD) during transcription initiation, elongation and termination. Initial promoter recognition and pre-initiation-complex formation occurs with an unphosphorylated CTD. Following transcription initiation, the CTD is phosphorylated on Ser5 by the transcription factor TFIIH, after which the polymerase often pauses. Transcriptional elongation is stimulated by P-TEFb (positive transcription-elongation factor-b), which phosphorylates Ser2 of the CTD. The CTD is dephosphorylated during and after transcription termination to facilitate a new cycle of transcription.
Emerging novel functions of MYC
A number of experimental findings have suggested that MYC might have biologically significant, transcription-independent functions. First, MYC biological activity can be uncoupled from the regulation of transcription by mutant analysis. Mutations near the DNA-binding domain can reduce the DNA-binding activity of MYC with no effect on MYC-dependent cell proliferation and rat embryo fibroblast cell transformation33. Second, MYC mutants that cannot dimerize with MAX or lack DNA-binding activity can promote cell proliferation (albeit at half the rate that is induced by wild-type MYC)10. These findings imply that inherent DNA binding and transcriptional activation are not required for every biological activity of MYC. These observations are not completely novel. Although the neuronal PC12 cell line does not express MAX, MYC was found to inhibit differentiation of PC12 cells34,35. MAX is required for binding of MYC to DNA36,37. However, these experiments did not address whether there was another dimerization partner or rare MYC homodimer that could account for this effect.
MYC drives mRNA cap methylation
How can the expression of MYC mutants that lack DNA-binding activity (such as expression of the transactivation domain alone) promote cell proliferation? We recently found that MYC can increase protein abundance by directly regulating the translation of individual mRNAs. The first hint towards this novel MYC mechanism came from the observation that the protein levels of several cyclin and cyclin-dependent kinases (CDKs), which are required for cell-cycle progression and transcription, abundantly increased in response to MYC expression without any change in their mRNA levels or in their requirement for the DNA-binding domain of MYC10. Conversely, reducing the level of MYC in normal fibroblasts by small interfering RNA led to a suppression of cyclin and CDK protein levels without causing a suppression of mRNA levels, demonstrating that endogenous MYC protein has an activity that is comparable to the MYC mutants that lack direct DNA-binding activity.
Although there were other possible explanations that could have accounted for this, such as increased protein stability, the mechanism by which MYC increases expression of cyclins and CDKs proved to be surprising. MYC was found to increase the translation of specific mRNAs by promoting the methylation of the 5′ mRNA guanine or ‘cap’ (REF. 10), which is an essential step for protein-coding gene expression38,39. Genes that are subject to MYC-dependent cap methylation, for example, cyclin T1 and CDK9 (REF. 10), represent a novel set of MYC-responsive genes. As native levels of MYC regulate the expression of these proteins without changes in mRNA abundance, this transcription-independent activity has the potential to influence all aspects of MYC biology in both normal and tumour cells.
During the early stages of transcription mRNA is capped and methylated (BOX 2). Cap methylation is necessary for the binding of translation factors to the mRNA and thus is required for translation38–40. As the nascent mRNA emerges from RNA pol II, capping enzyme (Ce) adds an inverted guanylyl group or ‘cap’ to the 5′ end. All mRNA that can be isolated from eukaryotic cells is probably capped (guanylylated) because uncapped mRNA is rapidly degraded41. However, mRNA cannot be translated efficiently unless the cap is subsequently methylated by an RNA methyl transferase (RNMT)39 (BOX 2). Both Ce and RNMT are recruited to mRNA after transcription initiation by binding specifically to the RNA pol II CTD, which has been phosphorylated on Ser5 by the kinase TFIIH38. This phosphorylation occurs during the early stages of transcription, thus coordinating the recruitment of Ce and RNMT with the emergence of the mRNA 5′ end. The finding that MYC induces mRNA cap methylation reveals for the first time that these processes are not constitutive for every mRNA and that this can be an important regulatory mechanism for some genes10.
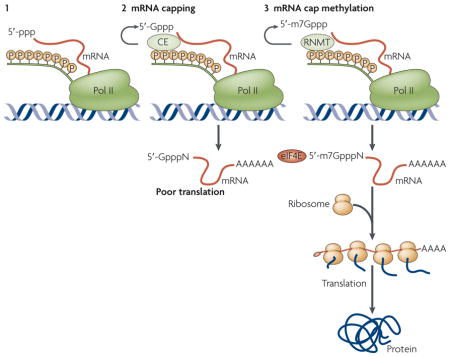
Box 2. Mechanism of mRNA cap methylation.
The primary mRNA transcript contains a triphosphate at its 5′ end, 5′-ppp (step 1). Capping enzyme (CE) is recruited to the phosphorylated C-terminal domain (CTD) of RNA polymerase (pol) II during the early stages of transcription and adds an inverted guanine ‘cap’ at the 5′ end, thereby producing 5′-Gppp (step 2). CE is a bifunctional enzyme that has both triphosphatase and guanylyltransferase activities. The 5′-Gppp cap is then methylated on N7 by a second enzyme, the cap RNA methyltransferase (RNMT), to produce 5′-m7Gppp (step 3). 5′-Gppp-capped mRNAs are stable but are weakly translated. Methylation of this cap is required for eukaryotic translation initiation factor-4E (eIF4E) binding and recruitment onto ribosomes for translation.
Previously, mRNA cap methylation was not widely recognized to be a regulated process because both biochemical and genetic studies have assessed the modification of the total mRNA pool39. However, the potential for differential regulation of mRNA capping and cap methylation was known to exist. Biochemical studies had shown that mRNA cap addition and cap methylation are distinct biochemical steps in mRNA processing42. Furthermore, yeast experiments have shown that the RNMT moves with RNA pol II further into the gene body than the guanylyltransferase43,44, where it might have a role in transcription elongation that is independent of its methyltransferase activity45.
RNA pol II Ser5 is phosphorylated predominantly by TFIIH, and MYC has been found to promote TFIIH recruitment and therefore promote subsequent RNA pol II phosphorylation10,32. TFIIH recruitment and RNA pol II phosphorylation both correlate with increased mRNA cap methyl ation10. However, MYC can stimulate the methylation of specific mRNA, which prompts us to propose a model whereby methyltransferase recruitment requires a higher or qualitatively different level of RNA pol II phosphorylation than the guanylyltransferase. An unexpected finding was that TFIIH recruitment, RNA pol II phosphorylation and mRNA cap methylation on a subset of genes could be driven by the expression of the MYC transactivation domain alone — independently of the DNA-binding domain of MYC and of direct binding of MYC to DNA10 (FIG. 3).
Figure 3. Mechanism of MYC-induced mrNa cap methylation.
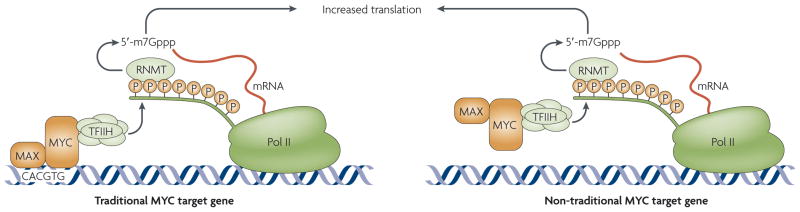
MYC promotes recruitment of the transcription factor TFIIH kinase to promoters and RNA polymerase (pol) II phosphorylation. Increased RNA pol II phosphorylation increases cap RNA methyltransferase (RNMT) recruitment and/or activity, which correlates with MYC-dependent mRNA cap methylation. At direct MYC target genes (left), TFIIH enhances the recruitment or activity of the cap RNMT to increase the fraction of cap methylated mRNA. Direct binding of MYC also stimulates a modest increase in transcription through TFIIH or acetyltransferases (see FIG. 2). At other promoters (right), MYC stimulates mRNA cap methylation through TFIIH stimulation by the MYC transactivation domain and through the subsequent recruitment or activation of RNMT by C-terminal domain phosphorylation. In doing so, MYC functions as a transcription-independent factor. Activation of direct targets is MYC-associated factor-X (MAX)-dependent (left), whereas activation of transcription-independent targets is MAX-independent (right).
Insight into this mechanism came from the finding that the transactivation domain could be recruited to chromatin-packaged promoters in cells, probably via a protein–protein interaction10. We propose a model in which the MYC transactivation domain can be recruited to promoters by MYC cofactors, provided these cofactors have already been recruited by other transcription factors. If the MYC transactivation domain binds to cofactors that are recruited to promoters through some other DNA-binding protein, then MYC will colocalize to the same promoters. This model could explain earlier observations, which showed that MYC binds to a large fraction of promoters with no apparent requirement for a consensus binding site46. However, we found that MYC binding correlated with changes in RNA pol II CTD phosphorylation and cap methylation. This correlation implies that the MYC transactivation domain is not simply engaged in cofactor recruitment but might also modulate the activity of cofactors. This mechanism is similar to the mechanism proposed for the stimulation of RNA pol III transcription, in which MYC recruits TRRAP and promotes histone H3 acetylation, which in turn recruits TFIIIB and RNA pol III. However, in contrast to RNA pol II, RNA pol III stimulation involves increased RNA pol III recruitment to the promoter and requires both the N terminus and C terminus of MYC13,14.
MYC controls DNA replication
Along with transcription, the most important nuclear function is DNA replication. The genome must be faithfully replicated each cell cycle and the chromosomes must be segregated to the daughter cells. Disruption of any step in this process, such as a stalled replication fork or DNA damage incurred during S phase, activates a checkpoint that halts the cell cycle until the lesion can be repaired47. Failure to correct this damage leads to a mutation and/or genomic instability.
Previous studies have provided a link between MYC and genomic instability48,49, but it was postulated that this was an indirect consequence of transcriptional activity. A recent study describes a direct, non-transcriptional role for MYC in the initiation of DNA replication11. MYC was found to bind to numerous components of the pre-replicative complex, including MCM proteins, ORC2, CDC6 and CDT1, and localize to early sites of DNA replication. These observations suggested that MYC might directly control the initiation of S phase and that the MYC effects on genomic instability might not depend on the transcriptional induction of S-phase-promoting genes. Taking advantage of Xenopus extracts (which support cell-cycle-regulated DNA replication in the absence of transcription and new protein synthesis), it was found that depletion of MYC decreased DNA synthesis and addition of recombinant MYC protein rescued DNA synthesis. This activity required intact MYC protein because neither the N terminus nor C terminus alone could rescue the defects observed. Levels of MYC protein seem to govern the number of active replication origins in both Xenopus and mammalian cells, suggesting that MYC functions to control origin selection. Aberrant firing of replication origins can be a source of replicative stress and can elicit a DNA-damage response. DNA damage stimulated the accumulation of H2A.X Ser139 phosphorylation, which was also stimulated by MYC induction. Phospho-H2A.X accumulation was dependent on MYC overexpression in S phase. This argues that the DNA-damage response was due to a defect in DNA replication.
These findings provide an unexpected link between MYC and a non-transcriptional process, DNA replication. Because this activity is dependent on the integrity of both the N-terminal and C-terminal domains of MYC, it suggests that MYC directly binds to DNA to recruit factors that govern the firing of replication origins. The selection of origins of replication in metazoan cells remains a puzzle47, but MYC was found to localize to a known origin of replication of the MYC gene itself11. If MYC is stimulating the firing of replication origins in normal cells, then continual overexpression of MYC in tumours could affect tumorigenesis by causing increased DNA damage. Continual accumulation of DNA damage from replicative stress and genomic instability could accelerate tumorigenesis in any tumour with abnormally high levels of MYC.
Conclusions and future perspectives
The sequence-specific transcription-factor field is built on the general assumption that all biological activity is dependent on conventional DNA-binding mechanisms, predominantly through cofactor recruitment to promoters. We consider the MYC oncogenic transcription factor in a new light: that the modular domains containing DNA-binding and transactivation activities might have independent functions with significant biological impact. The transactivation domain alone can stimulate both cell proliferation and global RNA pol II phosphorylation at physiological levels. By separating the functions of the transactivation and DNA-binding domains, a novel mechanism of gene regulation was revealed, namely the modulation of mRNA cap methylation. Changes in mRNA cap methylation can enhance or suppress protein expression in the absence of any changes in mRNA levels and hence alter gene expression through a transcription-independent mechanism. Beyond the transcriptional and transcription-independent functions of MYC in controlling gene expression, MYC might have an important role in setting the number of origins in DNA replication. The potent oncogenic activity of MYC might be dependent on both widespread activation and repression of target genes and on a transcription-independent capacity to regulate the translation and replication apparatus. These findings add to the complexity of understanding the function of MYC in cancer cells and might provide novel activities for therapeutic targets.
Acknowledgments
This work was funded by National Institutes of Health research grants to M.D.C. and a Medical Research Council Career Development Award to V.H.C.
Footnotes
DATABASES
Entrez Gene: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene
CAD | c-Myc
UniProtKB: http://www.uniprot.org
CDC6 | CDK9 | CDT1 | cyclin T1 | GCN5 | MAX | ORC2 | RNMT | TIP60 | TRRAP
FURTHER INFORMATION
Michael D. Cole’s homepage: http://www.dartmouth.edu/~colelab/Cole.html
Victoria H. Cowling’s homepage: http://www.lifesci.dundee.ac.uk/people/victoria_cowling
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
Contributor Information
Michael D. Cole, Departments of Pharmacology and Genetics, Dartmouth Medical School, Norris Cotton Cancer Center, One Medical Center Drive, Lebanon, New Hampshire 03756, USA
Victoria H. Cowling, Departments of Pharmacology and Genetics, Dartmouth Medical School, Norris Cotton Cancer Center, One Medical Center Drive, Lebanon, New Hampshire 03756, USA. Division of Cell Biology and Immunology, College of Life Sciences, University of Dundee, Dow Street, Dundee, DD1 5EH, UK
References
- 1.Oster SK, Ho CS, Soucie EL, Penn LZ. The myc oncogene: MarvelouslY Complex. Adv Cancer Res. 2002;84:81–154. doi: 10.1016/s0065-230x(02)84004-0. [DOI] [PubMed] [Google Scholar]
- 2.Pirity M, Blanck JK, Schreiber-Agus N. Lessons learned from Myc/Max/Mad knockout mice. Curr Top Microbiol Immunol. 2006;302:205–234. doi: 10.1007/3-540-32952-8_8. [DOI] [PubMed] [Google Scholar]
- 3.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- 4.Dang CV, et al. The c-Myc target gene network. Semin Cancer Biol. 2006;16:253–264. doi: 10.1016/j.semcancer.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 5.Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4:199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- 6.Vita M, Henriksson M. The Myc oncoprotein as a therapeutic target for human cancer. Semin Cancer Biol. 2006;16:318–330. doi: 10.1016/j.semcancer.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 7.Patel JH, Loboda AP, Showe MK, Showe LC, McMahon SB. Analysis of genomic targets reveals complex functions of MYC. Nature Rev Cancer. 2004;4:562–568. doi: 10.1038/nrc1393. [DOI] [PubMed] [Google Scholar]
- 8.Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nature Rev Mol Cell Biol. 2005;6:635–645. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- 9.Cowling VH, Cole MD. Mechanism of transcriptional activation by the Myc oncoproteins. Semin Cancer Biol. 2006;16:242–252. doi: 10.1016/j.semcancer.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 10.Cowling VH, Cole MD. The Myc transactivation domain promotes global phosphorylation of the RNA polymerase II carboxy-terminal domain independently of direct DNA binding. Mol Cell Biol. 2007;27:2059–2073. doi: 10.1128/MCB.01828-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dominguez-Sola D, et al. Non-transcriptional control of DNA replication by c-Myc. Nature. 2007;448:445–451. doi: 10.1038/nature05953. [DOI] [PubMed] [Google Scholar]
- 12.Hurlin PJ, Huang J. The MAX-interacting transcription factor network. Semin Cancer Biol. 2006;16:265–274. doi: 10.1016/j.semcancer.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 13.Gomez-Roman N, Grandori C, Eisenman RN, White RJ. Direct activation of RNA polymerase III transcription by c-Myc. Nature. 2003;421:290–294. doi: 10.1038/nature01327. [DOI] [PubMed] [Google Scholar]
- 14.Kenneth NS, et al. TRRAP and GCN5 are used by c-Myc to activate RNA polymerase III transcription. Proc Natl Acad Sci USA. 2007;104:14917–14922. doi: 10.1073/pnas.0702909104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grandori C, et al. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nature Cell Biol. 2005;7:311–318. doi: 10.1038/ncb1224. [DOI] [PubMed] [Google Scholar]
- 16.Grewal SS, Li L, Orian A, Eisenman RN, Edgar BA. Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nature Cell Biol. 2005;7:295–302. doi: 10.1038/ncb1223. [DOI] [PubMed] [Google Scholar]
- 17.Arabi A, et al. c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nature Cell Biol. 2005;7:303–310. doi: 10.1038/ncb1225. [DOI] [PubMed] [Google Scholar]
- 18.O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 19.Chang TC, et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nature Genet. 2008;40:43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McMahon SB, Van Buskirk HA, Dugan KA, Copeland TD, Cole MD. The novel ATM-related protein TRRAP is an essential cofactor for the c-Myc and E2F oncoproteins. Cell. 1998;94:363–374. doi: 10.1016/s0092-8674(00)81479-8. [DOI] [PubMed] [Google Scholar]
- 21.McMahon SB, Wood MA, Cole MD. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol Cell Biol. 2000;20:556–562. doi: 10.1128/mcb.20.2.556-562.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frank SR, Schroeder M, Fernandez P, Taubert S, Amati B. Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes Dev. 2001;15:2069–2082. doi: 10.1101/gad.906601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vervoorts J, et al. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep. 2003;4:484–490. doi: 10.1038/sj.embor.embor821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn’t fit all. Nature Rev Mol Cell Biol. 2007;8:284–295. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- 25.Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 26.Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 27.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saunders A, Core LJ, Lis JT. Breaking barriers to transcription elongation. Nature Rev Mol Cell Biol. 2006;7:557–567. doi: 10.1038/nrm1981. [DOI] [PubMed] [Google Scholar]
- 29.Price DH. Poised polymerases: on your mark…get set…go! Mol Cell. 2008;30:7–10. doi: 10.1016/j.molcel.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 30.Eberhardy SR, Farnham PJ. c-Myc mediates activation of the cad promoter via a post-RNA polymerase II recruitment mechanism. J Biol Chem. 2001;276:48562–48571. doi: 10.1074/jbc.M109014200. [DOI] [PubMed] [Google Scholar]
- 31.Eberhardy SR, Farnham PJ. Myc recruits P-TEFb to mediate the final step in the transcriptional activation of the cad promoter. J Biol Chem. 2002;277:40156–40162. doi: 10.1074/jbc.M207441200. [DOI] [PubMed] [Google Scholar]
- 32.Bouchard C, Marquardt J, Bras A, Medema RH, Eilers M. Myc-induced proliferation and transformation require Akt-mediated phosphorylation of FoxO proteins. EMBO J. 2004;23:2830–2840. doi: 10.1038/sj.emboj.7600279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cowling VH, Chandriani S, Whitfield ML, Cole MD. A conserved Myc protein domain, MBIV, regulates DNA binding, apoptosis, transformation, and G2 arrest. Mol Cell Biol. 2006;26:4226–4239. doi: 10.1128/MCB.01959-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maruyama K, Schiavi SC, Huse W, Johnson GL, Ruley HE. myc and E1A oncogenes alter the responses of PC12 cells to nerve growth factor and block differentiation. Oncogene. 1987;1:361–367. [PubMed] [Google Scholar]
- 35.Spandidos DA. The effect of exogenous human ras and myc oncogenes in morphological differentiation of the rat pheochromocytoma PC12 cells. Int J Dev Neurosci. 1989;7:1–4. doi: 10.1016/0736-5748(89)90039-7. [DOI] [PubMed] [Google Scholar]
- 36.Hopewell R, Ziff EB. The nerve growth factor-responsive PC12 cell line does not express the Myc dimerization partner Max. Mol Cell Biol. 1995;15:3470–3478. doi: 10.1128/mcb.15.7.3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ribon V, Leff T, Saltiel AR. c-Myc does not require max for transcriptional activity in PC-12 cells. Mol Cell Neurosci. 1994;5:277–282. doi: 10.1006/mcne.1994.1032. [DOI] [PubMed] [Google Scholar]
- 38.Bentley DL. Rules of engagement: co-transcriptional recruitment of pre-mRNA processing factors. Curr Opin Cell Biol. 2005;17:251–256. doi: 10.1016/j.ceb.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 39.Shatkin AJ. Capping of eucaryotic mRNAs. Cell. 1976;9:645–653. doi: 10.1016/0092-8674(76)90128-8. [DOI] [PubMed] [Google Scholar]
- 40.Shuman S. What messenger RNA capping tells us about eukaryotic evolution. Nature Rev Mol Cell Biol. 2002;3:619–625. doi: 10.1038/nrm880. [DOI] [PubMed] [Google Scholar]
- 41.Schwer B, Mao X, Shuman S. Accelerated mRNA decay in conditional mutants of yeast mRNA capping enzyme. Nucleic Acids Res. 1998;26:2050–2057. doi: 10.1093/nar/26.9.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moteki S, Price D. Functional coupling of capping and transcription of mRNA. Mol Cell. 2002;10:599–609. doi: 10.1016/s1097-2765(02)00660-3. [DOI] [PubMed] [Google Scholar]
- 43.Komarnitsky P, Cho EJ, Buratowski S. Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 2000;14:2452–2460. doi: 10.1101/gad.824700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schroeder SC, Schwer B, Shuman S, Bentley D. Dynamic association of capping enzymes with transcribing RNA polymerase II. Genes Dev. 2000;14:2435–2440. doi: 10.1101/gad.836300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schroeder SC, Zorio DA, Schwer B, Shuman S, Bentley D. A function of yeast mRNA cap methyltransferase, Abd1, in transcription by RNA polymerase II. Mol Cell. 2004;13:377–387. doi: 10.1016/s1097-2765(04)00007-3. [DOI] [PubMed] [Google Scholar]
- 46.Li Z, et al. A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc Natl Acad Sci USA. 2003;100:8164–8169. doi: 10.1073/pnas.1332764100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Machida YJ, Hamlin JL, Dutta A. Right place, right time, and only once: replication initiation in metazoans. Cell. 2005;123:13–24. doi: 10.1016/j.cell.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 48.Felsher DW, Bishop JM. Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc Natl Acad Sci USA. 1999;96:3940–3944. doi: 10.1073/pnas.96.7.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mai S, et al. Chromosomal and extrachromosomal instability of the cyclin D2 gene is induced by Myc overexpression. Neoplasia. 1999;1:241–252. doi: 10.1038/sj.neo.7900030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cole MD. The myc oncogene: its role in transformation and differentiation. Ann Rev Gen. 1986;20:361–385. doi: 10.1146/annurev.ge.20.120186.002045. [DOI] [PubMed] [Google Scholar]