

Abstract
Adipose tissue is a complex organ that comprises a wide range of cell types with diverse energy storage, metabolic regulation, and neuroendocrine and immune functions. Because it contains various immune cells, either adaptive (B and T lymphocytes; such as regulatory T cells) or innate (mostly macrophages and, more recently identified, myeloid-derived suppressor cells), the adipose tissue is now considered as a bona fide immune organ, at the cross-road between metabolism and immunity. Adipose tissue disorders, such as those encountered in obesity and lipodystrophy, cause alterations to adipose tissue distribution and function with broad effects on cytokine, chemokine, and hormone expression, on lipid storage, and on the composition of adipose-resident immune cell populations. The resulting changes appear to induce profound consequences for basal systemic inflammation and insulin sensitivity. The purpose of this review is to synthesize the current literature on adipose cell composition remodeling in obesity, which shows how adipose-resident immune cells regulate inflammation and insulin resistance—notably through cytokine and chemokine secretion—and highlights major research questions in the field.
1. Adipose Tissue Inflammation Is Crucial in the Development of Obesity-Induced Insulin Resistance
Obesity is a growing epidemic worldwide; its prevalence has been rising tremendously over the last 30 years (WHO, 2013). Excess adiposity is an established risk factor for metabolic diseases including insulin resistance, type 2 diabetes (T2D), hypertension, nonalcoholic fatty liver disease (NAFLD), polycystic ovarian diseases, and several types of cancer [1].
Obesity is a proinflammatory condition in which hypertrophied adipocytes and adipose tissue-resident immune cells (primarily lymphocytes and macrophages) both contribute to increased circulating levels of proinflammatory cytokines. The obesity-associated state of chronic low-grade systemic inflammation, termed “metabolic inflammation,” is considered a focal point in the pathogenesis of insulin resistance and T2D in humans and rodent animal models [2–5]. Although liver and muscle show obesity-induced mild inflammatory responses, white adipose tissue (WAT) is the key site mediating systemic inflammation [6].
1.1. Adipose Tissue Promotes an Inflammatory Response in Obesity: Role of TNF-α, IL-6, Leptin, Adiponectin, and Resistin in Insulin Resistance
Adipose tissue primary function is to store excess nutrients as triacylglycerols and to release free fatty acids during fasting. A major step forward to the recognition of the major secretory and endocrine role of WAT occurred in the 1990's with the demonstration that adipocytes synthesize and secrete the proinflammatory cytokine tumor necrosis factor alpha (TNF-α) [8] and the hormone leptin which regulates appetite and energy balance [9]. Evidence shows that the adipose tissue secretes more than 50 hormones and signaling molecules, collectively called adipokines, which exert their biological roles in an autocrine, paracrine, or systemic manner and influence several physiological processes concerning energy, glucose metabolism, and immunity [10]. More specifically, adipokines can exhibit either proinflammatory or anti-inflammatory properties, thereby contributing to insulin resistance.
Adipose tissue from lean individuals preferentially secretes anti-inflammatory adipokines such as adiponectin, transforming growth factor beta (TGFβ), interleukin (IL)-10, IL-4, IL-13, IL-1 receptor antagonist (IL-1Ra), and apelin. In contrast, obese adipose tissue mainly releases proinflammatory cytokines among which are TNF-α, IL-6, leptin, visfatin, resistin, angiotensin II, and plasminogen activator inhibitor 1 [4]. In lean individuals, anti-inflammatory adipokines mediate physiological functions, whilst in states of metabolic diseases, the proinflammatory adipokines modulate insulin resistance either directly by affecting the insulin signaling pathway or indirectly via stimulation of inflammatory pathways. Indeed, serine phosphorylation of insulin receptor substrate (IRS) by various adipokines directly or via inflammatory pathways including the c-Jun N-terminal kinase (JNK) pathway and I-kappa B kinase β (IKKβ)/NFκB pathway disrupts the insulin signaling pathways, possibly giving rise to insulin resistance [11].
Adipokines enlisted in regulation of insulin resistance are adiponectin, leptin, resistin, visfatin, chemerin, TNF-α, IL-1, IL-6, IL-8, IL-10, plasminogen activator inhibitor 1, monocyte chemoattractant protein-1, and retinol binding protein-4 (Tables 1 and 2). Because this topic has been the subject of recent reviews [12, 13] it will not be discussed in detail. We will rather focus on the prototypical adipokines (TNF-α, IL-6, leptin, adiponectin, and resistin) highlighting their roles in the development of insulin resistance as well as in immunity and inflammation.
Table 1.
Adipokines increased in obesity and/or diabetes (adapted and updated from [85]).
| Adipokine | Distribution | Function | Increased in obesity |
|---|---|---|---|
| Leptin | Secreted predominantly by WAT, to a lesser degree, in hypothalamus, gastric epithelium, placenta, and gonads | Regulates energy intake, expenditure and feeding behavior. Also regulates storage of fat and insulin signaling | Increased in mouse models of obesity. Increased in human obesity and correlated with BMI and decreased with weight loss |
|
| |||
| Resistin | In rodents, secreted by adipocytes. In humans, secreted predominantly by circulating macrophages and monocytes, to a lesser degree, by WAT | Implicated in glucose metabolism, in the regulation of neoglucogenesis and insulin resistance in rodents. More proinflammatory role in humans | Increased circulating concentrations in mouse models of obesity. Increased in human obesity and correlated with insulin resistance in diabetic patients |
|
| |||
| TNF-α | Expressed by macrophages and adipocytes (visceral WAT > subcutaneous WAT) | Affects insulin and glucose metabolism. Provokes insulin resistance and stimulates lipolysis | Increased in mouse models of obesity. Increased in human obesity and correlated with BMI |
|
| |||
| IL-6 | One-third of total circulating levels are expressed predominantly by adipocytes. Also expressed in macrophages, skeletal muscle, endothelial cells, and fibroblasts | Controversial role in the development of insulin resistance. Affects glucose metabolism | Increased circulating levels in human obese subjects and correlated with adiposity and reduced with weight loss. Increased in plasma of T2D patients |
|
| |||
| IL-7 | Secreted by stromal and vascular endothelial cells | Homeostatic immune cytokine. Also regulates body weight, adipose tissue mass and function, and insulin signaling | Increased in morbidly obese subjects |
|
| |||
| IL-8 | Secreted by adipocytes (visceral WAT > subcutaneous WAT) and macrophages | Neutrophil chemotaxis | Increased in obese subjects and related to fat mass and TNF-α levels |
|
| |||
| IL-1 | Secreted mainly by adipocytes and macrophages | Role in macrophages chemotaxis and thermogenesis | Increased in obese mice. Increased in human obesity and predictive of T2D |
|
| |||
| RBP4 | Secreted by adipocytes, macrophages, and hepatocytes | Affects insulin sensitivity, hepatic glucose output, and muscle insulin signaling | Increased circulating levels in obese subjects and correlated with BMI and insulin resistance |
|
| |||
| MCP-1 | Secreted by adipose tissue | Affects insulin sensitivity and increases macrophage recruitment in adipose tissue and inflammation | Increased in mouse models of obesity. Increased in T2D subjects |
|
| |||
| PAI-1 | Expressed by WAT | Potent inhibitor of fibrinolytic pathway | Increased in human obesity and T2D subjects |
|
| |||
| CXCL5 | Secreted by macrophages within the stromal vascular fraction | Interferes with insulin signaling in muscle | Circulating levels are higher in obese insulin-resistant individuals than in obese insulin-sensitive and decreased after a 4-week period on low-calorie diet |
|
| |||
| Visfatin | Expressed in liver, muscle, WAT, bone marrow, and lymphocytes | Role in insulin sensitivity, insulin secretion and inflammatory properties | Increased in obesity and correlates with visceral adiposity in humans |
|
| |||
| Chemerin | In rodents and humans, expressed in placenta and WAT | Regulates adipocyte development and metabolic function | Increased circulating levels in obese and T2D patients and correlated with body fat, glucose, and lipid metabolism |
|
| |||
| Vaspin | Secreted by WAT, hypothalamus, pancreatic islets, and skin | Improves insulin sensitivity | Increased in obesity and T2D patients |
Table 2.
Adipokines decreased in obesity and/or diabetes (adapted and updated from [85]).
| Adipokine | Distribution | Function | Decreased in obesity |
|---|---|---|---|
| Adiponectin | Only secreted by adipose tissue. Lower production in men | Insulin sensitizing effect. Improves insulin resistance and glucose metabolism | Decreased in mouse models of obesity. Decreased in human obesity and correlated negatively with BMI. Increased after weight loss |
|
| |||
| IL-10 | Secreted by monocytes, macrophages, dendritic cells, and B and T cells | Improves insulin sensitivity and glucose transport | Attenuated in T2D patients and increased with weight loss |
|
| |||
| Omentin | Expressed in heart, lungs, ovary, and placenta and predominantly produced by WAT | Improve glucose uptake in human adipocytes and has an anti-inflammatory effect | Decreased circulating levels in obese subjects. In impaired glucose tolerant (IGT) and subjects with T2D, circulating levels are lower those when compared with matched controls |
TNF-α is a potent proinflammatory cytokine, primarily secreted from myeloid cells via activation of MAPK and NFκB signaling pathways, resulting in the release of other inflammatory cytokines, such as IL-1β and IL-6 [14]. It was the first WAT-derived inflammatory cytokine reported to be implicated in the initiation and progression of insulin resistance [8, 15]. Although originally thought to be mainly secreted by adipocytes, it is now admitted that the majority of TNF-α is secreted by adipose tissue-resident macrophages [16]. In rodents TNF-α is overexpressed in adipose tissue from obese animals, and obese mice lacking either TNF-α or its receptor show protection against the development of insulin resistance [17]. In humans TNF-α levels are higher in plasma and adipose tissue of obese individuals, and circulating levels reduce with weight loss [18]. TNF-α levels were also found to be positively correlated with other markers of insulin resistance [19]; nonetheless, acute treatment with TNF-α inhibitor in obese subjects with type 2 diabetes reduced other systemic inflammatory markers without reducing insulin resistance [20], fueling lingering uncertainty about the biological relevance of this pathway in human insulin resistant states. More recently, the long-term assessment of anti-TNF-α inhibitor treatment to subjects diagnosed with metabolic syndrome has been shown to improve fasting blood glucose and to increase adiponectin levels, confirming a role for TNF-α in obesity-related insulin resistance in humans [21].
A key mechanism by which TNF-α induces insulin resistance involved phosphorylation of IRS-1 [22]. Beside its direct negative interference with the insulin signaling pathway, TNF-α also indirectly induces insulin resistance by altering adipocyte differentiation and adipocyte lipid metabolism. TNF-α is known to promote lipolysis and the secretion of free fatty acids, which contribute to an increase in hepatic glucose production [23]. Moreover, TNF-α inhibits the conversion of preadipocytes to mature adipocytes—notably through downregulating adipogenic genes such as peroxisome proliferator-activated receptor gamma (PPARγ) and CCAAT/enhancer binding protein (C/EBP)—allowing further recruitment of uncommitted cells and thus possible expansion of adipose tissue mass [24]. TNF-α-activated NF-κB suppressed genes involved in lipid uptake and storage [25] as well as many adipocyte-specific genes. TNF-α also downregulates the mRNA levels of adiponectin [26], an adipocyte-derived hormone which contributes to the maintenance of peripheral glucose and lipid homeostasis [27]. Nevertheless, the influence of TNF-α on immune response mostly results from its enhancing effect on the production of other cytokines, such as IL-6, rather than from a direct effect.
IL-6 is a multifaceted, pleiotropic cytokine that is a central player in the regulation of inflammation, hematopoiesis, immune responses, and host defense mechanisms [28]. IL-6 is secreted by WAT, skeletal muscle, and liver [16, 29]. Because one-third of circulating IL-6 in healthy individuals is estimated to originate from adipose tissue, IL-6 is considered an adipokine. In WAT, only a fraction of IL-6 is secreted by adipocytes, the other part being produced by other cells, particularly macrophages [16]. Similarly to TNF-α, WAT and plasma IL-6 expression correlate with increased body mass, waist circumference, and free fatty acid levels [30], with reduction in circulating IL-6 following weight loss [31]. IL-6 has been implicated as a marker for visceral adiposity because visceral adipose tissue releases more IL-6 than subcutaneous adipose tissue [32]. Nevertheless, data regarding the role of IL-6 in both obesity and insulin resistance are controversial and unresolved. While several studies indicate that increased IL-6 levels correlate with adiposity and fat mass, and not necessarily with insulin action or responsiveness [30, 33], another study has pointed to higher IL-6 levels in patients with obesity-related insulin resistance [34]. It can be inferred that relentless increase in systemic levels of IL-6 may lead to insulin resistance, whereas a transient increase in IL-6 may assist in normal glucose homeostasis. In fact, IL-6 appears to have dual functions depending on the tissue and metabolic state. During exercise, IL-6 increases glucose uptake in the skeletal muscle, leading to muscle hypertrophy and myogenesis and AMPK-mediated fatty acid oxidation, as well as having an anti-inflammatory effect [35]. In adipose tissue and liver, however, IL-6 will exert proinflammatory activities, increasing insulin resistance by upregulating SOCS3 (suppressor of cytokine signaling 3) which, in turn, impairs insulin-induced insulin receptor and IRS1 phosphorylation [36]. IL-6 may promote dysregulation of fatty acid metabolism in WAT as it enhanced mesenchymal stem cell proliferation, maintaining the cells in an undifferentiated state and inhibiting adipogenesis [37]. Additionally, IL-6 was recently shown to stimulate insulin secretion via enhanced GLP-1 (glucagon-like peptide-1) expression in pancreatic cells [38]. Thus, obesity-induced IL-6 secretion may reflect a mechanism to increase insulin production in the obese insulin resistant state. However, while elevated IL-6 secretion from WAT and liver is unfavorable, the opposite is true for skeletal muscle.
On the other hand, a number of in vitro and in vivo studies demonstrate that IL-6 is capable of inducing insulin resistance. In cultured murine adipocytes, IL-6 production is strongly increased by TNF-α and induces insulin resistance by inhibiting glucose uptake and impairing insulin signaling and action [39]. Whether or not IL-6 impairs insulin action in adipose tissue in vivo has yet to be clearly determined. Like TNF-α, IL-6 can directly affect lipid metabolism and activate pathways to promote increased energy turnover. IL-6 stimulates lipolysis in humans, increases free fatty acid (FFA) concentrations and whole body fat oxidation [40]. Several findings have shown that IL-6 can also affect other adipokines. Notably, IL-6 can decrease the expression and secretion of adiponectin in human adipocytes, as well as other markers of adipocyte differentiation [41]. Overall, IL-6 may play a pivotal role in metabolic diseases, including obesity. Therefore, understanding and clarifying its role in the regulation of metabolism is of utmost importance.
As stated above, leptin was one of the first proteins shown to be secreted from adipose tissue, through the identification and sequencing of the ob gene from the ob/ob mouse [9]. Leptin is primarily secreted by adipocytes proportionally to fat cell mass and is well known for its key contribution to energy metabolism [42]. Leptin exerts its effect on energy balance mainly by acting on the brain, either directly or indirectly by activating specific centers in the hypothalamus to decrease food intake, to increase energy expenditure, to influence glucose and lipid metabolism, or to alter neuroendocrine function. Daily injection of leptin in ob/ob mice resulted in a rapid reduction in food intake, body mass, and percentage of body fat but maintained lean muscle mass, increased energy expenditure, and restored euglycemia, confirming its important role in energy homeostasis and storage [43]. However, leptin levels are increased in obese subjects, with little or no impact to regulate energy homeostasis, which coined the well-established phrase “leptin resistance” in obesity. Indeed, preclinical and clinical experiments showed that obese rodents and humans displayed leptin resistance that may directly contribute to the reduction of lipid oxidation in insulin-sensitive organs, leading to accumulation of lipids and insulin resistance [44, 45]. Mechanisms leading to leptin resistance are still under investigation. Recently, it has been proposed that SOCS3 could be involved in negative regulation of leptin-induced intracellular signal transduction in the brain [46]. Moreover, neuronal deletion as well as whole-body knock-out of protein tyrosine phosphatase 1B (PTP1B) increased leptin and insulin sensitivity, preventing body weight gain in a diet-induced obesity animal model [47, 48], hence suggesting that, likewise SOCS3, PTP1B also orchestrates leptin resistance control. On the other hand, the role of leptin on insulin resistance is still not fully understood. Leptin is decreased in low insulin states, such as experimentally induced diabetes, and increases after insulin treatment [49]. In humans, insulin resistance is associated with elevated plasma leptin levels independently of body fat mass [50]. However, in patients with lipodystrophy, a condition characterized by almost complete lack of adipose tissue [51], leptin levels are very low and correlate significantly with markers of insulin resistance [52]. Leptin therapy in lipodystrophic patients improves their metabolic state with remarkable improvements in insulin sensitivity, suggesting that leptin acts as a signal that contributes to regulation of total body sensitivity to insulin [53].
Importantly, leptin also plays a key role in controlling immunity and inflammation [54]. Leptin has proinflammatory functions: it stimulates T-cell proliferative responses, polarized naïve CD4+ T-cell proliferation towards the Th1 phenotype, promotes a marked increase in Th1-type cytokine production, induces the expression of proinflammatory cytokines by macrophages and monocytes, and acts directly on hepatocytes to promote C-reactive protein expression [55]. The proinflammatory nature of leptin has been noted in several studies, with intravenous injection of endotoxin inducing a sudden rise in leptin levels [56], as well as endotoxin-induced fever and anorexia in rats, again inducing an increase in leptin levels as part of the inflammatory response [57]. The importance of leptin in immunity was confirmed in obese mice with homozygous mutation in leptin (ob/ob mice) or leptin receptor (db/db mice), in which high levels of lymphocyte atrophy and significant reduced thymus cortex were evidenced [58]. Replacement of leptin in the ob/ob mice or in congenital leptin-deficient children is able to restore normal thymic function, to increase the number of CD4+/CD8+ T-cells, to promote Th1 differentiation, and to reduce thymic apoptosis. We also reported impaired functionality of T-lymphocytes, dendritic cells, and macrophages in ob/ob and high-fat (HF) diet-fed mice [59, 60]. More recently, leptin has also been shown to activate human B lymphocytes to secrete TNF-α, IL-6, and IL-10 via the JAK2, STAT3, p38MAPK, and ERK signaling pathways [61]. Besides acting on adaptive immunity, leptin also regulates innate immune cells such as polymorphonuclear neutrophils, monocytes, and natural killer (NK) cells [62]. Leptin can induce chemotaxis of neutrophils, is involved in the development and maintenance of a functional NK (natural killer) pool, and induces the production of IL-6 and TNF-α from macrophages [55].
Unlike leptin, the circulating levels of adiponectin, a hormone produced predominantly by adipocytes, are decreased in obesity [63]. Adiponectin has important insulin-sensitizing effect: adiponectin-deficient transgenic mouse showed improved insulin sensitivity [64] and association studies have consistently linked plasma adiponectin levels to insulin sensitivity in rodent models and in humans [65]. Among the three major adiponectin isoforms, high-molecular weight (HMW) adiponectin is the most biologically active form and best reflective of the reduction in total adiponectin levels associated with obesity. Indeed, HMW adiponectin levels have been identified as an independent risk factor for insulin resistance [66]. In addition to improving insulin sensitivity, adiponectin exerts anti-inflammatory activity. Adiponectin can suppress the production of TNF-α and IFNγ (interferon gamma) and is a negative regulator of T cells, notably through its effect on the T-cell presenting function of dendritic cells [67]. Adiponectin maintains a mutual antagonistic action to TNF-α: as mentioned above TNF-α inhibits the expression of adiponectin [26], and conversely adiponectin suppresses lipopolysaccharide- (LPS-) induced TNF-α production [68].
Resistin is another unique adipocyte-derived signaling cysteine-rich molecule that was first identified in obese mice, deriving its name because of its resistance to the action of insulin. In rodents, resistin is secreted primarily from adipose tissue, whereas in humans resistin can be detected in other tissues like placenta, skeletal muscle, small intestine, spleen, stomach, thymus, thyroid gland, and uterus, being predominantly expressed in macrophages [69]. In rodents, initial studies reported increased resistin levels in various models of obesity and insulin resistance [70]. Rajala et al. [71] demonstrated that circulating resistin levels are elevated and positively concordant with rising levels of insulin, glucose, and lipids in ob/ob mice and that leptin administration improved insulin sensitivity associated with a decrease in resistin gene expression. Moreover, transgenic mice overexpressing a dominant negative form of resistin showed increased adiposity, possibly owing to enhanced adipose tissue differentiation and adipocyte hypertrophy [72]. Resistin appears to interfere with normal insulin signaling by decreasing insulin receptor and insulin receptor substrate (IRS1 and 2) protein expression and phosphorylation level in preadipose 3T3-L1 cells [73]. In addition, resistin has been showed to decrease AMPK activation which is known to be implicated as a potential insulin sensitizing molecule [74].
However, the role of resistin in the development of insulin resistance in humans is not as clear as in rodents. Since resistin is preferentially expressed by macrophages in humans, it suggests a proinflammatory role of resistin rather than a role in regulating glucose metabolism. Resistin mRNA expression level is higher in obese subjects, likely resulting from increased infiltration of macrophages in the adipose tissue. Several studies have reported positive correlations between resistin levels and insulin resistance in vivo and in vitro [70]. Moreover, genetic studies showed that two single nucleotide polymorphisms (SNPs: −537A > C and −420C > G) were associated with increased resistin levels in diabetic patients, but not in control subjects [75]. Recently, associations have been reported between resistin and metabolic syndrome components on one hand and early atherosclerosis in obese children on the other hand [76]. Finally, resistin has been demonstrated to stimulate the secretion of several inflammatory factors (e.g., TNF-α, IL-6, IL-8, and MCP-1) known to play a role in the induction of insulin resistance [77]. Therefore, resistin may have an indirect effect on insulin resistance in humans through exacerbating inflammation, which has been shown to disturb insulin sensitivity.
1.2. Interleukin-7 Regulates Adipose Tissue Mass and Function
During the past decades, IL-7 has been identified as the major homeostatic cytokine supporting the survival of αβ and γδ T cells, NKT cells, innate lymphoid cells, and regulatory T cells (Tregs) [78]. IL-7 is predominantly produced by stromal and vascular endothelial cells, with very low levels of IL7 transcripts detectable in adult animals, consistent with the concept that under basal states there are limited amounts of IL-7 available for lymphocytes in vivo. In a homeostatic animal, IL-7 amount is thought to be constant yet stroma-derived IL-7 production can be induced by overt inflammation. IL-7 receptor (IL-7R) is composed of the private IL-7Rα chain (CD127) combined with the common gamma (γ c; CD132) chain and is expressed mainly by T lymphocytes but also by NK cells, macrophages, dendritic cells, lymphoid tissue inducer cells, and certain subsets of B cells. One central characteristic of IL-7R expression is its dynamic regulation by cytokines and by the overall metabolic and differentiation state of the cells. For example, TNF-α has been reported to upregulate IL-7Rα expression [79] and IL-6 to be a critical effector of IL-7R signaling [80].
Without IL-7 the lymphoid system cannot be built and maintained. Interestingly, the role of IL-7 on lymphocyte homeostasis was shown to partly rely on its control of basal lymphocyte glucose metabolism through the expression of the glucose transporter GLUT-1, which promotes glucose uptake and increases metabolic activity as well as cell size [81].
Recently, we and others identified IL-7 as a new secretory product of the adipose tissue, mostly produced by cells of the stromal vascular fraction [82, 83]. Furthermore, we reported that IL-7 also contributes to body weight regulation via both hypothalamic [84] and adipose tissue [82] control. Regarding the latter, we showed that IL-7 modulates the adipose tissue through acting on its mass and function. In fact, a single administration of IL-7 was sufficient to decrease adipose tissue inflammation and to protect mice from obesity in three different models of experimentally induced obesity (i.e., monosodium glutamate-induced hypothalamic obesity [84], gold thioglucose-induced hypothalamic obesity (Wolowczuk I, unpublished data), and HF diet- (HFD-) induced obesity [82]).
Strikingly, we showed that IL-7 overexpressing mice presented a lipodystrophy-like phenotype: reduced WAT mass is associated with impaired adipocyte differentiation and intolerance to glucose and insulin resistance, these traits being commonly associated with lipodystrophy in both animals and humans [51].
2. Adipose Tissue Cellular Remodeling in Obesity
The first part of our review showed that the apparent metabolic simplicity of the adipose tissue is illusory; this is also true regarding its cellular composition. Besides lipid-filled mature adipocytes, the tissue is also composed of various stromal cells, including preadipocytes, endothelial cells, fibroblasts, and immune cells [13]. During the progression of obesity, both the adipocyte and the stroma vascular fractions are changed: adipocytes grow larger, secrete predominantly proinflammatory cytokines, and are insulin resistant; coincidently, the nature of WAT immune cells is also modified.
2.1. Changes in Immune Cell Composition in the Obese Adipose Tissue: A Focus on MCP-1 and CCR5 Chemokines
Obesity is characterized by the accumulation of diverse immune cells in the adipose tissue. Notably, proinflammatory macrophage infiltration and inflammation-related gene expression precede the development of insulin resistance and appear to be a cardinal feature of obesity in rodents and humans [16].
Adipose tissue macrophages (ATMs) accumulate in both the subcutaneous and visceral expanding fat depots, even though macrophage infiltration appears to be more prominent in the latter [86]. Apart from increasing in numbers, adipose tissue macrophages are also phenotypically changed during obesity: while anti-inflammatory M2 macrophages reside in WAT of lean mice, obese WAT predominantly contains proinflammatory M1 macrophages [87]. Activated M1 ATMs are a prominent source of proinflammatory cytokines such as TNF-α and IL-6, which can block insulin action in adipocytes via autocrine/paracrine signaling causing systemic insulin resistance via endocrine signaling (cf. Section 1). Thus, both recruitment and proinflammatory polarization of ATMs are required for the development of insulin resistance. In both humans and rodents, ATMs content positively correlates with inflammation and insulin resistance [16]. Despite its importance in adipose tissue inflammatory responses and systemic insulin sensitivity, the mechanisms underlying M1 versus M2 macrophage polarization still remain poorly understood. The recent discovery of microRNAs (miRNAs) provides a new opportunity to understand this complicated but crucial network for macrophage activation and adipose tissue function. miRNAs, which correspond to a group of highly conserved, small (i.e., approximately 22 nucleotides in length) noncoding RNAs, can trigger either a block in translation and/or mRNA degradation [88, 89]. Numerous studies have provided compelling evidence that miRNAs are key regulators of cell fate determination and significantly contribute to the pathogenesis of complex diseases, including obesity-associated metabolic diseases [90–92]. Zhuang et al. [93] recently identified miRNA-223 (miR-223) as a potent regulator of macrophage polarization and provided strong evidence supporting the functional significance of this new pathway in metabolic homeostasis. The authors showed a suppressive effect of miR-223 on macrophage proinflammatory activation (M1) and a stimulatory effect on anti-inflammatory activation (M2): high-fat diet-fed miR-223-deficient mice displayed increased adipose tissue inflammation and were more insulin resistant. At the molecular level, a major target of miR-223 in macrophages is Pknox1, which itself favors the proinflammatory activation pathway. However, a key question still unanswered by now is how the miR-223/Pknox1 pathway interacts with known regulatory pathways that control macrophage activation. The identification of mechanisms underlying functional polarization of macrophages into M1 or M2 might provide new insights into a basis for macrophage-centered therapeutic strategies for metabolic diseases.
Similarly to any immune and inflammatory response, macrophage infiltration in the obese adipose tissue results from blood monocyte influx, mainly attracted by the chemokine monocyte chemoattractant protein-1 (MCP-1) which is secreted by hypertrophic adipocytes. It has been reported that MCP-1 secretion is markedly enhanced locally and in plasma of obese rodents and humans [94]. Overexpression, deficiency, or mutation-induced dysfunction of MCP-1 in different mouse models were shown to interfere with ATMs accumulation, along with insulin-resistance development [95, 96]. However, the role of MCP-1 in promoting ATM recruitment and insulin resistance has recently been challenged by the absence of noticeable impact on macrophage accumulation and glucose intolerance resulting from MCP-1 genetic disruption [97]. Furthermore, HFD-fed MCP-1 receptor i.e., CCR2-deficient mice (namely, ccr2 −/− mice) do not normalize ATM content and insulin resistance to the levels of lean animals [96], suggesting that ATM recruitment and insulin resistance are also regulated by MCP-1/CCR2 independent signaling pathways.
Kitade et al. recently identified and characterized a critical role for CCR5, another C-C motif chemokine receptor, in the regulation of obesity-induced WAT inflammatory response and insulin resistance [98]. These authors reported accumulation of CCR5 expressing ATMs in HFD-fed mice. Importantly, Ccr5 −/− mice were protected from insulin resistance induced by HF feeding through both reduction in ATM accumulation and induction of anti-inflammatory M2 shift in those cells [98]. Additionally, a bone marrow transplantation study revealed that lack of CCR5 expression in macrophages alone could protect mice from the HFD-induced insulin resistance, this being associated with a significant reduction in ATM infiltration. In humans, recent studies have also shown upregulation of CCR5 in the visceral fat of morbidly obese individuals in whom macrophage infiltration has been confirmed [99]. However, further studies are needed to evaluate whether CCR5 inhibitor treatment (e.g., maraviroc) affects macrophage activation and other aspects of adipose tissue biology in obese patients. Also, it remains to be established whether the two C-C chemokine receptors, CCR2 and CCR5, play common or unique roles in obesity-induced adipose tissue inflammation and insulin resistance.
Alterations in ATM content and polarization state occur fairly late in the progression of obesity and probably are not initiating events of inflammation and development of sustained insulin resistance. Evidence has accumulated showing that other changes in adipose-resident immune cells may precede these events. Under this scenario, ATMs will be effectors of a coordinated inflammatory response that includes the accumulation of proinflammatory T cells (CD8+ and Th1 CD4+ T cells) and the loss of anti-inflammatory regulatory T cells (Tregs), as well as the appearance of B cells, NK cells, NKT cells, eosinophils, neutrophils, and mast cells [100].
Adipocytes in lean adipose tissue produce factors such as IL-4 and IL-13 that induce M2 activation of macrophages and Th2 activation of CD4+ T cells and maintain Treg cell and eosinophil numbers. In obesity, the progressive accumulation of adipose tissue is accompanied by early increased infiltration of proinflammatory CD8+ T cells and a shift towards a higher CD8+/CD4+ ratio. CD8+ infiltration appears to be a key event preceding the depletion of adipose Tregs and the increased CD4+ Th1 cell activation observed in murine models of diet-induced obesity [101, 102]. Increased adipose-resident CD8+ T-cell activation also potentiates adipocyte expression of IL-6 and TNF-α in mice, while CD8+ T-cell depletion reverses this effect.
Neutrophils are known to play a role in the early stages of inflammatory responses, and it has been recently reported a sustained increased in adipose tissue neutrophil content in HFD-induced obesity with neutrophil secreted elastase being a key effector in this process [103]. The enhanced release of IL-8, a factor involved in neutrophil chemotaxis, by hypertrophic adipocytes, may partly explain neutrophil recruitment. Adipose tissue neutrophils produce chemokines and cytokines, facilitating macrophage infiltration, which could contribute to development of insulin resistance.
2.2. Myeloid-Derived Suppressor Cells: A Novel Actor in the Control of Insulin Sensitivity
In the 1980s, a new cell population known as “natural suppressor cells,” distinct from T and NK cells, was described in the bone marrow and spleen of tumor-bearing mice [104, 105]. Later on, these cells were defined as “myeloid-derived suppressor cells” (MDSC) because of their myeloid origin and their ability to suppress immune responses [106]. In fact, MDSCs represent a heterogeneous and metabolically plastic population of immature myeloid cells in different stages of differentiation, having in common the capacity to inhibit effector immune responses and to accumulate under conditions of inflammation [107]. The term plasticity here refers to the ability of MDSCs to change both their expression of various mediators of suppression (e.g., iNOS, arginase I) in response to environmental influences (e.g., local IL-4/13 or IFNγ concentration) and also their differentiation state (e.g., becoming more/less neutrophil or myeloid cells). These cells are also defined by their immature state of macrocytic/monocytic, granulocytic/neutrophilic, and dendritic cell precursors and are characterized by the increased production of extracellular degradative enzymes, cytokines, and reactive oxygen and nitrogen species [108].
In mice, MDSC are commonly identified as coexpressing the cell surface markers CD11b and Gr-1. Since there are several subpopulations within Gr-1+CD11b+ cells, several groups further subcategorized MDSC into “monocytic” MDSC (CD11b+Ly6G−Ly6Chigh) and “granulocytic/neutrophil-like” MDSC (CD11b+Ly6G+Ly6Clow), based on the expression of Ly6C and Gr-1/Ly6G [109]. There is no human marker equivalent to mouse Gr-1, human MDSC being typically defined as CD11b+CD33+CD34+CD14−HLA-DR− cells [110]. In addition to heterogeneity, discrepancies exist in cell surface expression of certain activation/maturation markers, such as MHC II and costimulatory molecules, and of lineage markers (e.g., F4/80) between MDSC. This heterogeneity supports the notion that MDSC include multiple subpopulations of myeloid-derived cells that are at various stages of maturity.
In the steady state, MDSCs are predominantly present in the bone marrow and participate in the normal process of myelopoiesis. However, under various pathological inflammatory conditions such as cancer, infection, sepsis, graft-versus-host disease, and bone marrow transplantation, a variety of cytokines and soluble factors released induce rapid expansion of MDSC that will accumulate in peripheral lymphoid organs and blood, as well as in tumors, where they have been described to block CD4+ and CD8+ T-cell responses thus favoring cancer development [111]. In cancer, one key factor controlling MDSC expansion and tumor progression is PPARγ [112]. Vascular endothelial growth factor (VEGF), macrophage colony-stimulating factor (M-CSF), or IL-6 is also required for MDSC expansion. It has been suggested that MDSCs contribute to tumor progression by both facilitating neoangiogenesis and metastasis [113] and by inhibiting antitumor responses [111]. In addition to their recognized role in tumor tolerance, MDSCs may also be involved in the induction and maintenance of transplant tolerance [114].
Recently, an exciting observation has been described by Xia et al., showing that MDSCs and M2 macrophage induction may be a physiological response to promotion of insulin sensitivity [115]. These authors showed that obese ob/ob mice, as well as wild-type mice fed on high-fat diet, have marked accumulation of anti-inflammatory MDSCs and M2 macrophages in adipose tissue. Furthermore, the increase in MDSCs and M2 macrophage number was associated with higher response to insulin. Adoptive transfer of MDSCs (e.g., Gr-1+ cells) into high-fat diet-fed mice improved the response of the recipient mice to insulin while, in contrast, MDSCs depletion (after treatment with anti-Gr-1 antibody) increased their susceptibility to obesity and further worsened their resistance to insulin. Yin et al. have also described the ability of MDSCs to delay onset of type 1 diabetes and insulin resistance [116], through inducing expansion of antigen-specific Tregs and suppressing T-cell proliferation. The mechanisms by which obesity-associated chronic inflammation induces expansion/accumulation of MDSCs are arguably stepwise. However, the initial proinflammatory state created in early obesity may induce the accumulation of MDSC in an attempt to curtail overt inflammation, as described in other well-described models of inflammation [107], and may improve insulin sensitivity. In addition, we recently showed that the mechanistic target of rapamycin (mTOR) signaling pathway might be involved in the expansion of MDSCs (Makki, MS submitted), as well as in myelopoiesis [117]. Although mechanisms by which MDSCs enhance insulin sensitivity are unknown, it has been proposed that upregulation of insulin growth factor-1 (IGF-1) in the setting of insulin resistance may lead to the accumulation of MDSCs or M2 macrophages [118]. Suggestions have been made that not only does insulin resistance induce physiological response for MDSC and M2 macrophage expansion, but insulin may also modulate direct gene transcriptional control of these cells. Therefore, pharmacological enhancement of insulin sensitivity in obese individuals may preemptively hinder the development of MDSCs.
3. Concluding Remarks
The complex alterations in adipose tissue secretion of cytokines, adipokines, and chemokines and immune cell composition observed in adipose tissue-related pathologies such as obesity (Figure 1) have been, and still are, an active research area. As the proportion of overweight and obese (even among the youngest) continues to rise worldwide, understanding the role of adipose tissue in the pathogenesis of obesity and its metabolic and immune-based complications will be critical to optimize long-term health outcomes. As summarized in the present review, there might be a potential therapeutic value of targeting certain immune resident cells (such as M2, Tregs, or MDSC) and/or certain cytokines, adipokines, or chemokines (such as MCP-1/CCR2 or CCR5) to improve insulin resistance and restrain organ damage in type 2 diabetic obese patients by limiting the proinflammatory milieu.
Figure 1.
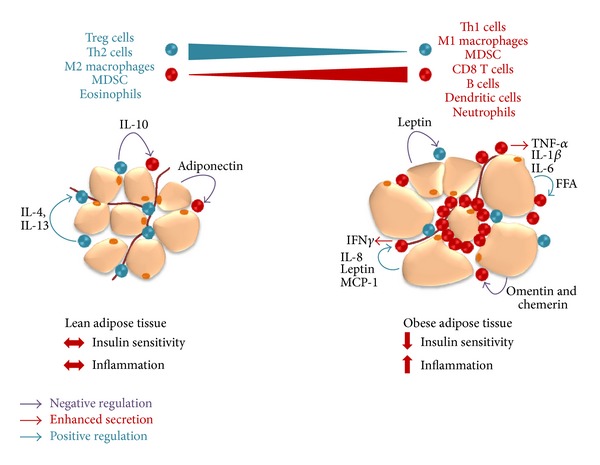
Adipose tissue-resident cells, cytokines, and hormones: role in insulin sensitivity (adapted and updated from [7]).
Conflict of Interests
The authors do not report any conflict of interests.
Acknowledgments
This work was supported by the Centre National de la Recherche Scientifique (CNRS). The authors thank Dr. G. Rocheleau for his careful reading of the paper. Kassem Makki was funded by a PhD fellowship from Lille II University.
References
- 1.Hotamisligil GS, Erbay E. Nutrient sensing and inflammation in metabolic diseases. Nature Reviews Immunology. 2008;8(12):923–934. doi: 10.1038/nri2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annual Review of Immunology. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 3.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 4.Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nature Reviews Immunology. 2011;11(2):85–97. doi: 10.1038/nri2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. The Journal of Clinical Investigation. 2006;116(7):1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Odegaard JI, Chawla A. Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science. 2013;339:172–177. doi: 10.1126/science.1230721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han JM, Levings MK. Immune regulation in obesity-associated adipose inflammation. The Journal of Immunology. 2013;191:527–532. doi: 10.4049/jimmunol.1301035. [DOI] [PubMed] [Google Scholar]
- 8.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-α: direct role in obesity-linked insulin resistance. Science. 1993;259(5091):87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 10.Waki H, Tontonoz P. Endocrine functions of adipose tissue. Annual Review of Pathology. 2007;2:31–56. doi: 10.1146/annurev.pathol.2.010506.091859. [DOI] [PubMed] [Google Scholar]
- 11.Tilg H, Moschen AR. Inflammatory mechanisms in the regulation of insulin resistance. Molecular Medicine. 2008;14(3-4):222–231. doi: 10.2119/2007-00119.Tilg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Havel PJ. Update on adipocyte hormones: regulation of energy balance and carbohydrate/lipid metabolism. Diabetes. 2004;53(supplement 1):S143–S151. doi: 10.2337/diabetes.53.2007.s143. [DOI] [PubMed] [Google Scholar]
- 13.Lucas S, Verwaerde C, Wolowczuk I. Is the adipose tissue the key road to inflammation? Immunology and Immunogenetics Insights. 2009;1:3–14. [Google Scholar]
- 14.Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. 2002;296(5573):1634–1635. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- 15.Hotamisligil GS, Spiegelman BM. Tumor necrosis factor α: a key component of the obesity-diabetes link. Diabetes. 1994;43(11):1271–1278. doi: 10.2337/diab.43.11.1271. [DOI] [PubMed] [Google Scholar]
- 16.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr. Obesity is associated with macrophage accumulation in adipose tissue. The Journal of Clinical Investigation. 2003;112(12):1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-α function. Nature. 1997;389(6651):610–614. doi: 10.1038/39335. [DOI] [PubMed] [Google Scholar]
- 18.Kern PA, Saghizadeh M, Ong JM, Bosch RJ, Deem R, Simsolo RB. The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. The Journal of Clinical Investigation. 1995;95(5):2111–2119. doi: 10.1172/JCI117899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hivert M-F, Sullivan LM, Fox CS, et al. Associations of adiponectin, resistin, and tumor necrosis factor-α with insulin resistance. Journal of Clinical Endocrinology and Metabolism. 2008;93(8):3165–3172. doi: 10.1210/jc.2008-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dominguez H, Storgaard H, Rask-Madsen C, et al. Metabolic and vascular effects of tumor necrosis factor-α blockade with etanercept in obese patients with type 2 diabetes. Journal of Vascular Research. 2005;42(6):517–525. doi: 10.1159/000088261. [DOI] [PubMed] [Google Scholar]
- 21.Stanley TL, Zanni MV, Johnsen S, et al. TNF-α antagonism with etanercept decreases glucose and increases the proportion of high molecular weight adiponectin in obese subjects with features of the metabolic syndrome. Journal of Clinical Endocrinology and Metabolism. 2011;96(1):E146–E150. doi: 10.1210/jc.2010-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanety H, Feinstein R, Papa MZ, Hemi R, Karasik A. Tumor necrosis factor α-induced phosphorylation of insulin receptor substrate-1 (IRS-1). Possible mechanism for suppression of insulin-stimulated tyrosine phosphorylation of IRS-1. The Journal of Biological Chemistry. 1995;270(40):23780–23784. doi: 10.1074/jbc.270.40.23780. [DOI] [PubMed] [Google Scholar]
- 23.Fève B, Bastard J-P. The role of interleukins in insulin resistance and type 2 diabetes mellitus. Nature Reviews Endocrinology. 2009;5(6):305–311. doi: 10.1038/nrendo.2009.62. [DOI] [PubMed] [Google Scholar]
- 24.Xu H, Sethi JK, Hotamisligil GS. Transmembrane tumor necrosis factor (TNF)-α inhibits adipocyte differentiation by selectively activating TNF receptor 1. The Journal of Biological Chemistry. 1999;274(37):26287–26295. doi: 10.1074/jbc.274.37.26287. [DOI] [PubMed] [Google Scholar]
- 25.Ruan H, Miles PDG, Ladd CM, et al. Profiling gene transcription in vivo reveals adipose tissue as an immediate target of tumor necrosis factor-α: implications for insulin resistance. Diabetes. 2002;51(11):3176–3188. doi: 10.2337/diabetes.51.11.3176. [DOI] [PubMed] [Google Scholar]
- 26.Hector J, Schwarzloh B, Goehring J, et al. TNF-α alters visfatin and adiponectin levels in human fat. Hormone and Metabolic Research. 2007;39(4):250–255. doi: 10.1055/s-2007-973075. [DOI] [PubMed] [Google Scholar]
- 27.Whitehead JP, Richards AA, Hickman IJ, Macdonald GA, Prins JB. Adiponectin—a key adipokine in the metabolic syndrome. Diabetes, Obesity and Metabolism. 2006;8(3):264–280. doi: 10.1111/j.1463-1326.2005.00510.x. [DOI] [PubMed] [Google Scholar]
- 28.Eder K, Baffy N, Falus A, Fulop AK. The major inflammatory mediator interleukin-6 and obesity. Inflammation Research. 2009;58(11):727–736. doi: 10.1007/s00011-009-0060-4. [DOI] [PubMed] [Google Scholar]
- 29.Wieckowska A, Papouchado BG, Li Z, Lopez R, Zein NN, Feldstein AE. Increased hepatic and circulating interleukin-6 levels in human nonalcoholic steatohepatitis. American Journal of Gastroenterology. 2008;103(6):1372–1379. doi: 10.1111/j.1572-0241.2007.01774.x. [DOI] [PubMed] [Google Scholar]
- 30.Vozarova B, Weyer C, Hanson K, Tataranni PA, Bogardus C, Pratley RE. Circulating interleukin-6 in relation to adiposity, insulin action, and insulin secretion. Obesity Research. 2001;9(7):414–417. doi: 10.1038/oby.2001.54. [DOI] [PubMed] [Google Scholar]
- 31.Bastard J-P, Jardel C, Bruckert E, et al. Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. Journal of Clinical Endocrinology and Metabolism. 2000;85(9):3338–3342. doi: 10.1210/jcem.85.9.6839. [DOI] [PubMed] [Google Scholar]
- 32.Fried SK, Bunkin DA, Greenberg AS. Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: depot difference and regulation by glucocorticoid. Journal of Clinical Endocrinology and Metabolism. 1998;83(3):847–850. doi: 10.1210/jcem.83.3.4660. [DOI] [PubMed] [Google Scholar]
- 33.Hansen D, Dendale P, Beelen M, et al. Plasma adipokine and inflammatory marker concentrations are altered in obese, as opposed to non-obese, type 2 diabetes patients. European Journal of Applied Physiology. 2010;109(3):397–404. doi: 10.1007/s00421-010-1362-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kern PA, Ranganathan S, Li C, Wood L, Ranganathan G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. American Journal of Physiology—Endocrinology and Metabolism. 2001;280(5):E745–E751. doi: 10.1152/ajpendo.2001.280.5.E745. [DOI] [PubMed] [Google Scholar]
- 35.Starkie R, Ostrowski SR, Jauffred S, Febbraio M, Pedersen BK. Exercise and IL-6 infusion inhibit endotoxin-induced TNF-alpha production in humans. The FASEB Journal. 2003;17(8):884–886. doi: 10.1096/fj.02-0670fje. [DOI] [PubMed] [Google Scholar]
- 36.Senn JJ, Klover PJ, Nowak IA, et al. Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. The Journal of Biological Chemistry. 2003;278(16):13740–13746. doi: 10.1074/jbc.M210689200. [DOI] [PubMed] [Google Scholar]
- 37.Pricola KL, Kuhn NZ, Haleem-Smith H, Song Y, Tuan RS. Interleukin-6 maintains bone marrow-derived mesenchymal stem cell stemness by an ERK1/2-dependent mechanism. Journal of Cellular Biochemistry. 2009;108(3):577–588. doi: 10.1002/jcb.22289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ellingsgaard H, Hauselmann I, Schuler B, et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nature Medicine. 2011;17(11):1481–1489. doi: 10.1038/nm.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rotter V, Nagaev I, Smith U. Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. The Journal of Biological Chemistry. 2003;278(46):45777–45784. doi: 10.1074/jbc.M301977200. [DOI] [PubMed] [Google Scholar]
- 40.van Hall G, Steensberg A, Sacchetti M, et al. Interleukin-6 stimulates lipolysis and fat oxidation in humans. Journal of Clinical Endocrinology and Metabolism. 2003;88(7):3005–3010. doi: 10.1210/jc.2002-021687. [DOI] [PubMed] [Google Scholar]
- 41.Sopasakis VR, Sandqvist M, Gustafson B, et al. High local concentrations and effects on differentiation implicate interleukin-6 as a paracrine regulator. Obesity Research. 2004;12(3):454–460. doi: 10.1038/oby.2004.51. [DOI] [PubMed] [Google Scholar]
- 42.Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395(6704):763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 43.Campfield LA, Smith FJ, Guisez Y, Devos R, Burn P. Recombinant mouse OB protein: evidence for a peripheral signal linking adiposity and central neural networks. Science. 1995;269(5223):546–549. doi: 10.1126/science.7624778. [DOI] [PubMed] [Google Scholar]
- 44.van den Hoek AM, Teusink B, Voshol PJ, Havekes LM, Romijn JA, Pijl H. Leptin deficiency per se dictates body composition and insulin action in ob/ob mice. Journal of Neuroendocrinology. 2008;20(1):120–127. doi: 10.1111/j.1365-2826.2007.01626.x. [DOI] [PubMed] [Google Scholar]
- 45.Zhang H, Xie H, Zhao Q, et al. Relationships between serum adiponectin, apelin, leptin, resistin, visfatin levels and bone mineral density, and bone biochemical markers in post-menopausal Chinese women. Journal of Endocrinological Investigation. 2010;33(10):707–711. doi: 10.1007/BF03346674. [DOI] [PubMed] [Google Scholar]
- 46.Mori H, Hanada R, Hanada T, et al. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nature Medicine. 2004;10(7):739–743. doi: 10.1038/nm1071. [DOI] [PubMed] [Google Scholar]
- 47.Bence KK, Delibegovic M, Xue B, et al. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nature Medicine. 2006;12(8):917–924. doi: 10.1038/nm1435. [DOI] [PubMed] [Google Scholar]
- 48.Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, et al. PTP1B regulates leptin signal transduction in vivo. Developmental Cell. 2002;2(4):489–495. doi: 10.1016/s1534-5807(02)00148-x. [DOI] [PubMed] [Google Scholar]
- 49.Macdougald OA, Hwang C-S, Fan H, Lane MD. Regulated expression of the obese gene product (leptin) in white adipose tissue and 3T3-L1 adipocytes. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(20):9034–9037. doi: 10.1073/pnas.92.20.9034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Segal KR, Landt M, Klein S. Relationship between insulin sensitivity and plasma leptin concentration in lean and obese men. Diabetes. 1996;45(3):988–991. doi: 10.2337/diab.45.7.988. [DOI] [PubMed] [Google Scholar]
- 51.Vantyghem MC, Balavoine AS, Douillard C, et al. How to diagnose a lipodystrophy syndrome. Annales d'Endocrinologie . 2012;73:170–189. doi: 10.1016/j.ando.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 52.Moon HS, Dalamaga M, Kim SY, et al. Leptin's role in lipodystrophic and nonlipodystrophic insulin-resistant and diabetic individuals. Endocrine Reviews. 2013;34:377–412. doi: 10.1210/er.2012-1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oral EA, Simha V, Ruiz E, et al. Leptin-replacement therapy for lipodystrophy. The New England Journal of Medicine. 2002;346(8):570–578. doi: 10.1056/NEJMoa012437. [DOI] [PubMed] [Google Scholar]
- 54.Matarese G, Moschos S, Mantzoros CS. Leptin in immunology. The Journal of Immunology. 2005;174(6):3137–3142. doi: 10.4049/jimmunol.174.6.3137. [DOI] [PubMed] [Google Scholar]
- 55.Loffreda S, Yang SQ, Lin HZ, et al. Leptin regulates proinflammatory immune responses. The FASEB Journal. 1998;12(1):57–65. [PubMed] [Google Scholar]
- 56.Landman RE, Puder JJ, Xiao E, Freda PU, Ferin M, Wardlaw SL. Endotoxin stimulates leptin in the human and nonhuman primate. Journal of Clinical Endocrinology and Metabolism. 2003;88(3):1285–1291. doi: 10.1210/jc.2002-021393. [DOI] [PubMed] [Google Scholar]
- 57.Sachot C, Poole S, Luheshi GN. Circulating leptin mediates lipopolysaccharide-induced anorexia and fever in rats. Journal of Physiology. 2004;561(1):263–272. doi: 10.1113/jphysiol.2004.074351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dardenne M, Savino W, Gastinel LN, Nabarra B, Bach JF. Thymic dysfunction in the mutant diabetic (db/db) mouse. The Journal of Immunology. 1983;130(3):1195–1199. [PubMed] [Google Scholar]
- 59.Macia L, Belacre M, Abboud G, et al. Impairment of dendritic cell functionality and steady-state number in obese mice. The Journal of Immunology. 2006;177(9):5997–6006. doi: 10.4049/jimmunol.177.9.5997. [DOI] [PubMed] [Google Scholar]
- 60.Verwaerde C, Delanoye A, Macia L, Tailleux A, Wolowczuk I. Influence of high-fat feeding on both naive and antigen-experienced T-cell immune response in DO10.11 mice. Scandinavian Journal of Immunology. 2006;64(5):457–466. doi: 10.1111/j.1365-3083.2006.01791.x. [DOI] [PubMed] [Google Scholar]
- 61.Agrawal S, Gollapudi S, Su H, Gupta S. Leptin activates human B cells to secrete TNF-α, IL-6, and IL-10 via JAK2/STAT3 and p38MAPK/ERK1/2 signaling pathway. Journal of Clinical Immunology. 2011;31(3):472–478. doi: 10.1007/s10875-010-9507-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zarkesh-Esfahani H, Pockley G, Metcalfe RA, et al. High-dose leptin activates human leukocytes via receptor expression on monocytes. The Journal of Immunology. 2001;167(8):4593–4599. doi: 10.4049/jimmunol.167.8.4593. [DOI] [PubMed] [Google Scholar]
- 63.Arita Y, Kihara S, Ouchi N, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochemical and Biophysical Research Communications. 1999;257(1):79–83. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- 64.Combs TP, Pajvani UB, Berg AH, et al. A transgenic mouse with a deletion in the collagenous domain of adiponectin displays elevated circulating adiponectin and improved insulin sensitivity. Endocrinology. 2004;145(1):367–383. doi: 10.1210/en.2003-1068. [DOI] [PubMed] [Google Scholar]
- 65.Berg AH, Combs TP, Scherer PE. ACRP30/adiponectin: an adipokine regulating glucose and lipid metabolism. Trends in Endocrinology and Metabolism. 2002;13(2):84–89. doi: 10.1016/s1043-2760(01)00524-0. [DOI] [PubMed] [Google Scholar]
- 66.Almeda-Valdes P, Cuevas-Ramos D, Mehta R, et al. Total and high molecular weight adiponectin have similar utility for the identification of insulin resistance. Cardiovascular Diabetology. 2010;9, article 26 doi: 10.1186/1475-2840-9-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tsang JYS, Li D, Ho D, et al. Novel immunomodulatory effects of adiponectin on dendritic cell functions. International Immunopharmacology. 2011;11(5):604–609. doi: 10.1016/j.intimp.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 68.Park P-H, Huang H, McMullen MR, Mandal P, Sun L, Nagy LE. Suppression of lipopolysaccharide-stimulated tumor necrosis factor-α production by adiponectin is mediated by transcriptional and post-transcriptional mechanisms. The Journal of Biological Chemistry. 2008;283(40):26850–26858. doi: 10.1074/jbc.M802787200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schwartz DR, Lazar MA. Human resistin: found in translation from mouse to man. Trends in Endocrinology and Metabolism. 2011;22(7):259–265. doi: 10.1016/j.tem.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Steppan CM, Bailey ST, Bhat S, et al. The hormone resistin links obesity to diabetes. Nature. 2001;409(6818):307–312. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- 71.Rajala MW, Qi Y, Patel HR, et al. Regulation of resistin expression and circulating levels in obesity, diabetes, and fasting. Diabetes. 2004;53(7):1671–1679. doi: 10.2337/diabetes.53.7.1671. [DOI] [PubMed] [Google Scholar]
- 72.Kim K-H, Lee K, Moon YS, Sul HS. A cysteine-rich adipose tissue-specific secretory factor inhibits adipocyte differentiation. The Journal of Biological Chemistry. 2001;276(14):11252–11256. doi: 10.1074/jbc.C100028200. [DOI] [PubMed] [Google Scholar]
- 73.Palanivel R, Maida A, Liu Y, Sweeney G. Regulation of insulin signalling, glucose uptake and metabolism in rat skeletal muscle cells upon prolonged exposure to resistin. Diabetologia. 2006;49(1):183–190. doi: 10.1007/s00125-005-0060-z. [DOI] [PubMed] [Google Scholar]
- 74.Fisher JS. Potential role of the AMP-activated protein kinase in regulation of insulin action. Cell Science. 2006;2:68–81. [PMC free article] [PubMed] [Google Scholar]
- 75.Cho YM, Youn B-S, Chung SS, et al. Common genetic polymorphisms in the promoter of resistin gene are major determinants of plasma resistin concentrations in humans. Diabetologia. 2004;47(3):559–565. doi: 10.1007/s00125-003-1319-x. [DOI] [PubMed] [Google Scholar]
- 76.Chen XY, Zhang JH, Liu F, Liu HM, Song YY, Liu YL. Association of serum resistin levels with metabolic syndrome and early atherosclerosis in obese Chinese children. Journal of Pediatric Endocrinology and Metabolism. 2013;17:1–6. doi: 10.1515/jpem-2012-0326. [DOI] [PubMed] [Google Scholar]
- 77.Bokarewa M, Nagaev I, Dahlberg L, Smith U, Tarkowski A. Resistin, an adipokine with potent proinflammatory properties. The Journal of Immunology. 2005;174(9):5789–5795. doi: 10.4049/jimmunol.174.9.5789. [DOI] [PubMed] [Google Scholar]
- 78.Kang J, Coles M. IL-7: the global builder of the innate lymphoid network and beyond, one niche at a time. Seminars in Immunology. 2012;24:190–197. doi: 10.1016/j.smim.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tian B, Nowak DE, Jamaluddin M, Wang S, Brasier AR. Identification of direct genomic targets downstream of the nuclear factor-κB transcription factor mediating tumor necrosis factor signaling. The Journal of Biological Chemistry. 2005;280(17):17435–17448. doi: 10.1074/jbc.M500437200. [DOI] [PubMed] [Google Scholar]
- 80.Pellegrini M, Calzascia T, Toe JG, et al. IL-7 engages multiple mechanisms to overcome chronic viral infection and limit organ pathology. Cell. 2011;144(4):601–613. doi: 10.1016/j.cell.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 81.Rathmell JC, Farkash EA, Gao W, Thompson CB. IL-7 enhances the survival and maintains the size of naive T cells. The Journal of Immunology. 2001;167(12):6869–6876. doi: 10.4049/jimmunol.167.12.6869. [DOI] [PubMed] [Google Scholar]
- 82.Lucas S, Taront S, Magnan C, et al. Interleukin-7 regulates adipose tissue mass and insulin sensitivity in high-fat diet-fed mice through lymphocyte-dependent and independent mechanisms. PLoS One. 2012;7 doi: 10.1371/journal.pone.0040351.e40351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maury E, Ehala-Aleksejev K, Guiot Y, Detry R, Vandenhooft A, Brichard SM. Adipokines oversecreted by omental adipose tissue in human obesity. American Journal of Physiology—Endocrinology and Metabolism. 2007;293(3):E656–E665. doi: 10.1152/ajpendo.00127.2007. [DOI] [PubMed] [Google Scholar]
- 84.Macia L, Viltart O, Delacre M, et al. Interleukin-7, a new cytokine targeting the mouse hypothalamic Arcuate nucleus: role in body weight and food intake regulation. PLoS ONE. 2010;5(4) doi: 10.1371/journal.pone.0009953.e9953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Piya MK, McTernan PG, Kumar S. Adipokine inflammation and insulin resistance: the role of glucose, lipids and endotoxin. Journal of Endocrinology. 2013;216:T1–T15. doi: 10.1530/JOE-12-0498. [DOI] [PubMed] [Google Scholar]
- 86.Bruun JM, Lihn AS, Pedersen SB, Richelsen B. Monocyte chemoattractant protein-1 release is higher in visceral than subcutaneous human adipose tissue (AT): implication of macrophages resident in the AT. Journal of Clinical Endocrinology and Metabolism. 2005;90(4):2282–2289. doi: 10.1210/jc.2004-1696. [DOI] [PubMed] [Google Scholar]
- 87.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annual Review of Physiology. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 88.Lim LP, Lau NC, Garrett-Engele P, et al. Microarray analysis shows that some microRNAs downregulate large numbers of-target mRNAs. Nature. 2005;433(7027):769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- 89.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gauthier BR, Wollheim CB. MicroRNAs: “Ribo-regulators” of glucose homeostasis. Nature Medicine. 2006;12(1):36–38. doi: 10.1038/nm0106-36. [DOI] [PubMed] [Google Scholar]
- 91.Xie H, Lim B, Lodish HF. MicroRNAs induced during adipogenesis that accelerate fat cell development are downregulated in obesity. Diabetes. 2009;58(5):1050–1057. doi: 10.2337/db08-1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lodish HF, Zhou B, Liu G, Chen C-Z. Micromanagement of the immune system by microRNAs. Nature Reviews Immunology. 2008;8(2):120–130. doi: 10.1038/nri2252. [DOI] [PubMed] [Google Scholar]
- 93.Zhuang G, Meng C, Guo X, et al. A novel regulator of macrophage activation: miR-223 in obesity-associated adipose tissue inflammation. Circulation. 2012;125:2892–2903. doi: 10.1161/CIRCULATIONAHA.111.087817. [DOI] [PubMed] [Google Scholar]
- 94.Kim C-S, Park H-S, Kawada T, et al. Circulating levels of MCP-1 and IL-8 are elevated in human obese subjects and associated with obesity-related parameters. International Journal of Obesity. 2006;30(9):1347–1355. doi: 10.1038/sj.ijo.0803259. [DOI] [PubMed] [Google Scholar]
- 95.Kanda H, Tateya S, Tamori Y, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. The Journal of Clinical Investigation. 2006;116(6):1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Weisberg SP, Hunter D, Huber R, et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. The Journal of Clinical Investigation. 2006;116(1):115–124. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Inouye KE, Shi H, Howard JK, et al. Absence of CC chemokine ligand 2 does not limit obesity-associated infiltration of macrophages into adipose tissue. Diabetes. 2007;56(9):2242–2250. doi: 10.2337/db07-0425. [DOI] [PubMed] [Google Scholar]
- 98.Kitade H, Sawamoto K, Nagashimada M, et al. CCR5 plays a critical role in obesity-induced adipose tissue inflammation and insulin resistance by regulating both macrophage recruitment and M1/M2 status. Diabetes. 2012;61:1680–1690. doi: 10.2337/db11-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Huber J, Kiefer FW, Zeyda M, et al. CC chemokine and CC chemokine receptor profiles in visceral and subcutaneous adipose tissue are altered in human obesity. Journal of Clinical Endocrinology and Metabolism. 2008;93(8):3215–3221. doi: 10.1210/jc.2007-2630. [DOI] [PubMed] [Google Scholar]
- 100.Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. The Journal of Clinical Investigation. 2011;121(6):2111–2117. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Deiuliis J, Shah Z, Shah N, et al. Visceral adipose inflammation in obesity is associated with critical alterations in tregulatory cell numbers. PLoS ONE. 2011;6(1) doi: 10.1371/journal.pone.0016376.e16376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nishimura S, Manabe I, Nagasaki M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nature Medicine. 2009;15(8):914–920. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- 103.Talukdar S, Oh da Y, Bandyopadhyay G, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nature Medicine. 2012;18:1407–1412. doi: 10.1038/nm.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Maier T, Holda JH, Claman HN. Natural suppressor cells. Progress in Clinical and Biological Research. 1989;288:235–244. [PubMed] [Google Scholar]
- 105.Strober S. Natural suppressor (NS) cells, neonatal tolerance, and total lymphoid irradiation: exploring obscure relationships. Annual Review of Immunology. 1984;2:219–237. doi: 10.1146/annurev.iy.02.040184.001251. [DOI] [PubMed] [Google Scholar]
- 106.Gabrilovich DI, Bronte V, Chen S-H, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Research. 2007;67(1):p. 425. doi: 10.1158/0008-5472.CAN-06-3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nagaraj S, Gabrilovich DI. Myeloid-derived suppressor cells in human cancer. Cancer Journal. 2010;16(4):348–353. doi: 10.1097/PPO.0b013e3181eb3358. [DOI] [PubMed] [Google Scholar]
- 108.Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. The Journal of Immunology. 2001;166(9):5398–5406. doi: 10.4049/jimmunol.166.9.5398. [DOI] [PubMed] [Google Scholar]
- 109.Zhou Z, French DL, Ma G, et al. Development and function of myeloid-derived suppressor cells generated from mouse embryonic and hematopoietic stem cells. Stem Cells. 2010;28(3):620–632. doi: 10.1002/stem.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nature Reviews Immunology. 2009;9(3):162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Khaled YS, Ammori BJ, Elkord E. Myeloid-derived suppressor cells in cancer: recent progress and prospects. Immunology and Cell Biology. 2013;91:493–502. doi: 10.1038/icb.2013.29. [DOI] [PubMed] [Google Scholar]
- 112.Wu L, Yan C, Czader M, et al. Inhibition of PPARγ in myeloid-lineage cells induces systemic inflammation, immunosuppression, and tumorigenesis. Blood. 2012;119(1):115–126. doi: 10.1182/blood-2011-06-363093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shojaei F, Ferrara N. Role of the microenvironment in tumor growth and in refractoriness/resistance to anti-angiogenic therapies. Drug Resistance Updates. 2008;11(6):219–230. doi: 10.1016/j.drup.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 114.Hock BD, MacKenzie KA, Cross NB, et al. Renal transplant recipients have elevated frequencies of circulating myeloid-derived suppressor cells. Nephrology Dialysis Transplantation. 2012;27(1):402–410. doi: 10.1093/ndt/gfr264. [DOI] [PubMed] [Google Scholar]
- 115.Xia S, Sha H, Yang L, Ji Y, Ostrand-Rosenberg S, Qi L. Gr-1+ CD11b+ myeloid-derived suppressor cells suppress inflammation and promote insulin sensitivity in obesity. The Journal of Biological Chemistry. 2011;286(26):23591–23599. doi: 10.1074/jbc.M111.237123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yin B, Ma G, Yen C-Y, et al. Myeloid-derived suppressor cells prevent type 1 diabetes in murine models. The Journal of Immunology. 2010;185(10):5828–5834. doi: 10.4049/jimmunol.0903636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Martelli AM, Chiarini F, Evangelisti C, et al. The phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin signaling network and the control of normal myelopoiesis. Histology and Histopathology. 2010;25(5):669–680. doi: 10.14670/HH-25.669. [DOI] [PubMed] [Google Scholar]
- 118.Lu H, Huang D, Saederup N, Charo IF, Ransohoff RM, Zhou L. Macrophages recruited via CCR2 produce insulin-like growth factor-1 to repair acute skeletal muscle injury. The FASEB Journal. 2011;25(1):358–369. doi: 10.1096/fj.10-171579. [DOI] [PMC free article] [PubMed] [Google Scholar]