

Abstract
Short-range Notch receptor signaling is necessary for coordinating developmental activities in metazoa. To investigate this juxtacrine pathway, we mimic receptor-ligand binding within the cell-cell junction by engaging Notch1-eGFP expressing cells to a supported lipid membrane displaying Delta-like protein 4 (DLL4). DLL4-Notch1 binding, oligomerization, and transport were observed in real time, and the molecular density and stoichiometry of the complexes were determined using quantitative fluorescence imaging. A Notch transcriptional reporter readout was used to quantify how ligand lateral mobility, orientation, and density modulate receptor activation levels. These experiments demonstrate that limiting the lateral mobility of DLL4 can enhance Notch activation by 2.6-fold, thus supporting the existence of a spatio-mechanical mechanism of signal regulation.
Introduction
Cell-cell communication is essential for the development, proliferation, and survival of multicellular organisms. To coordinate complex events within multicellular organisms, certain signals need to be short-ranged and confined to a cell’s nearest neighbors. One general strategy for short-range cellular communication employs membrane-anchored ligands and receptors such that direct physical contact between adjacent cells is required for pathway activation. This type of interaction is commonly found in cell-adhesion junctions, neuronal and immunological synapses, and during cellular patterning and development (1). Despite the importance of these juxtacrine interactions, the role of receptor oligomerization, spatial organization, membrane topography, and physical forces in signal modulation remains poorly understood. The fundamental challenge pertains to the lack of methods to characterize ligand-receptor interactions in the confined and dynamic two-dimensional environment of the cell-cell junction. Therefore, new experimental strategies are needed to better understand the role of chemo-mechanical couplings in short-range, juxtacrine signaling pathways.
One important example of short-range cellular signaling is the Notch pathway, which is universally conserved and critical to development and differentiation in metazoa. The Notch receptors are a family of heterodimeric membrane proteins that bind directly to Delta/Serrate/LAG-2 ligand molecules expressed on the surface of an apposing cell (2). Ligand-induced activation of Notch receptors is characterized by a series of regulated intramembrane proteolysis events. The key proteolysis step in activation of the signaling pathway is cleavage of Notch at site 2 (S2) by a disintegrin and metalloprotease (ADAM) that results in shedding of the Notch extracellular domain (NECD). Following S2 cleavage, the Notch intracellular domain (NICD) is released from the membrane by γ-secretase proteolysis at site 3 (S3), which is located within the transmembrane domain of Notch. The NICD translocates to the nucleus where it functions as a transcription factor and upregulates gene expression of targets in the Hes and Hey families.
NMR and x-ray crystallography studies have revealed that a large conformational change is required to expose S2 for attack by ADAM (3,4). The ligand binding domain is nearly 1000 amino acid residues away from the cleavage site, and thus a mechanical force model has been proposed as a mechanism for this long-range conformational change (5–8). This mechanotransduction model suggests that the formation of ligand-receptor complexes is coupled with endocytosis to generate mechanical tension that opens up the S2 site and renders it susceptible to proteolysis. This model is supported by evidence that the receptor cannot be activated with soluble ligand molecules and that endocytosis is required for activation in both Drosophila and mammalian cells (6,7,9–12). Interestingly, one recent study found that Notch activation requires <12 pN or 0 pN of mechanical tension, but the direct observation of force exerted by a cell has yet to be observed (13).
In addition to the mechanical forces applied by cells, altering the physical properties of ligand molecules may significantly influence signaling. For example, the activity of chemically identical ligand molecules can be modulated by adjusting factors such as density, spatial organization, and lateral mobility within the membrane environment. This effect has been observed in a wide variety of signaling pathways involving immunorecognition receptors, E-cadherins, and receptor tyrosine kinases (14–17).
Current strategies for studying Delta-Notch interactions have been unable to distinguish a role for ligand density, diffusion, and oligomerization in activation of the Notch receptor, as summarized in Fig. S1 in the Supporting Material. Herein, we use a supported lipid membrane functionalized with Delta-like protein 4 (DLL4) to observe ligand-induced activation of Notch in live cells. The key advantages of this method include: i), fine control and quantification of ligand composition and concentration; ii), ability to adjust the lateral mobility of ligand molecules and the rate at which Notch and Delta molecules interact and form clusters; iii), proper orientation of the Delta ligand on the membrane through site-specific anchoring; iv), absolute number density determination of ligand and receptor molecules and clusters, and their stoichiometry within an intermembrane junction; and v), direct observation of dynamics of Delta-Notch complex formation and internalization events in real time. By frustrating the lateral motions of Notch receptors in the cell membrane, we increase activation levels by 2.6-fold compared to chemically identical ligand that is laterally mobile. Finally, we determine the stoichiometry of ligand:receptor binding to be 1:1 when approximated to the nearest integer value, in agreement with accepted structural models (3).
Material and Methods
Preparation of small unilamellar vesicles
Phospholipids (1,2-dioleoyl-sn-glycero-3-phosphocholine, DOPC; 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl], Ni2+-NTA; 1-oleoyl-2-(6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl)-sn-glycero-3-phosphocholine, NBD-PC; and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl), biotin-DPPE) were purchased from Avanti Polar Lipids (Alabaster, AL). Fluorescent lipid (N-(6-tetramethylrhodaminethiocarbamoyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine), TRITC-DHPE was from Life Technologies (Grand Island, NY). To prepare small unilamellar vesicles, lipids were combined in a round bottom flask at the desired molar ratio in chloroform and dried on a rotary evaporator to form a lipid film. The film was dried under a stream of N2 for 15 min and then resuspended with Nanopure water to achieve a lipid concentration of 2 mg/ml. The lipid solution was frozen in a dry ice-acetone bath and thawed in a 40°C water bath three times. The vesicles were passed through a 100 nm polycarbonate filter (Whatman, Florham Park, NJ) 11 times using a high-pressure extruder (Northern Lipids, Burnaby, Canada) warmed to 45°C.
Assembly of supported lipid membranes
To prepare the glass surface, a 96-well plate with #1.5 glass (Greiner Bio-One, Monroe, NC) was etched with 1 M NaOH for 1 h and rinsed with Nanopure water. A 0.5 mg/ml vesicle solution prepared in 10 mM Tris, 150 mM NaCl, pH 8.0 (for Ni2+-NTA), or phosphate buffered saline (PBS) (for biotin-DPPE) and added to the glass for 20 min to form the bilayer. The surfaces were blocked with 0.1 mg/ml bovine serum albumin for 30 min. For surfaces containing biotin-DPPE, the membranes were incubated with 36 nM streptavidin (Rockland Immunochemicals, Gilbertsville, PA) for 45 min. After rinsing unbound streptavidin, the surface was treated with 40 nM of c[RGDfK(biotin-PEG-PEG)] (Peptides International, Louisville, KY) or 50 nM of DLL4-mCherry-biotin for 1 h. For attachment of histidine-tagged proteins, the surface was treated with a 10 mM Tris, 100 mM Ni(II)Cl2, pH 8.0 solution for 5 min and then incubated overnight with either 66 nM of DLL4-568/647 or 60 nM DLL4-mCherry. Between each step of the supported membrane assembly, the wells were rinsed with 10 ml of 10 mM Tris, 300 mM NaCl, pH 8.0 (Ni2+-NTA), or PBS (biotin-DPPE).
Measurement of ligand diffusion coefficient
DLL4 diffusion coefficients were measured on a Nikon Ti Eclipse inverted microscope (Nikon, Melville, NY) with a 100X N.A. 1.49 CFI Apo total internal reflection fluorescence microscopy (TIRF) objective lens. A small spot (d ∼20 μm) of the membrane tethered DLL4 surface was photobleached with a 561 nm laser until the fluorescence intensity neared dark count levels. Fluorescence recovery was captured over 2 min with images taken in 2 s intervals. A series of 10 prebleach images was also collected to correct for the nonuniform illumination profile of the Intensilight excitation source. Images were acquired with an EM-CCD camera iXon DU897 (Andor, Belfast, UK) through the use of Nikon’s NIS Elements and analyzed using ImageJ (NIH, Bethesda, MD). For a detailed description of the photobleaching analysis method, please see (18).
Live cell imaging
The stably transfected Notch1-eGFP C2C12 mouse (Mus musculus) myoblast cell line (provided by G. Weinmaster, UCLA) was maintained in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum, 5% cosmic calf serum, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 2 μg/ml puromycin (Cellgro, Manassas, VA). Cells were cultured overnight on a 10 cm dish and used for experiments the following day. The cells were pipetted off of the surface using PBS and pelleted. The functionalized lipid membrane surfaces were exchanged into warm cell imaging media (Hanks’ balanced salts, 10 mM HEPES, pH 7.4). For quantitative fluorescence measurements, 25,000 cells were added into each well and incubated for 30 min at 37°C and 5% CO2. Samples were imaged at 37°C for 30 min using a Nikon Ti Eclipse microscope equipped with an Evolve EM CCD (Photometrics, Tucson, AZ) and objective warmer (Warner, Hamden, CT). The excitation sources used included an Intensilight epifluorescence source and a TIRF launcher with two laser lines: 488 nm (10 mW) and 638 nm (20 mW). Images were collected using NIS Elements software and analyzed using ImageJ. To observe live cell dynamics, ∼10,000 cells were added per membrane surface and individual cells were tracked as they attached to the membrane.
Activation of Notch reporter cell line
Following assembly and imaging of the supported membrane or physisorbed surfaces, Notch1 reporter cells were trypsinized from the cell culture flask and 10,000 cells were distributed onto each surface. Activation of the reporter was measured by epifluorescence microscopy 48 h after plating. Cells were maintained in Alpha MEM Earle’s Salts (Irvine Scientific, Santa Ana, CA) supplemented with 10% fetal bovine serum, 100 IU/ml of penicillin, 100 μg/ml streptomycin, 400 μg/ml Zeocin, 10 μg/ml Blasticidin, and 600 μg/ml Geneticin (Life Technologies, Grand Island, NY).
Calibration curves and determination of F factor
To calibrate the fluorescence intensity, TRITC-DHPE surfaces ranging from 0 to 0.08 mol % and NBD-PC surfaces from 0 to 0.75 mol % were imaged under the same conditions as the live cell images. The number of fluorophore molecules per unit area was estimated from the footprint of DOPC, which was determined to be 0.72 nm2 (19). To use the calibration curves, the intensity of the ligand molecules was compared with the lipid-fluorophore standards to obtain the F factor. The F factor is defined as , where Isol(ligand) and Isol(lipid) are the intensity of the ligand or lipid molecules in solution after being normalized for concentration. These values were measured on the fluorescence microscope by moving the focal plane (∼100 μm) into the center of the sample. It is important to note that for DLL4-568 unconjugated dye was thoroughly removed from the sample by purification with Ni-NTA magnetic agarose beads before measuring the F factor value.
Data analysis for stoichiometry measurements
To calculate the protein density, the fluorescence images were background subtracted and then quantified by dividing each image by the F factor and calibration curve slope. In the ligand channel, the unbound ligand was subtracted as this represented ligand molecules that were present on the membrane surface but not bound to receptor molecules. A correction factor was introduced to account for the 28% of ligand binding to untagged Notch homologs measured in the parental cell line, vide infra. The ligand image was then divided by the receptor image to generate the ratio image (DLL4:Notch1). A threshold was applied to the receptor image to determine areas where Notch receptors were located, and all other areas were set to zero. This mask was applied to the ratio image and each nonzero pixel value was binned into a histogram fit with a single Gaussian. The peak value for each histogram, representing an individual cell, was determined and averaged for a population of 40–80 cells.
Results and Discussion
Delta-Notch binding on fluid membranes
Supported lipid membranes were prepared as illustrated in Fig. 1 A, and two types of DLL4 ligand (residues Ser27-Pro524) were tethered to the membrane surfaces, see the Supporting Material. The first was a DLL4 tagged with 2–3 molecules of an amine-reactive Alexa Fluor 568 or 647 (DLL4-568 or DLL4-647), and the second was a DLL4-mCherry fusion, where the fluorescent protein was inserted at the C-terminus. It is important to note that the polyhistidine tag is the final element of the C-terminus for both proteins ensuring that the ligand is oriented in the correct conformation for binding with Notch (2). Fluorescence recovery after photobleaching (FRAP) shows that surface anchored DLL4 is fluid within the membrane (Fig. 1, B–D). A small area of the bilayer was photobleached (Fig. 1 B) and after ∼5 min nonphotobleached ligand molecules diffused into the area and restored fluorescence intensity to prebleached levels (Fig. 1 C). A line scan over the photobleached spot (Fig. 1 D) showed near complete recovery of the ligand fluorescence and verified the fluid nature of the supported lipid membrane tethered DLL4.
Figure 1.
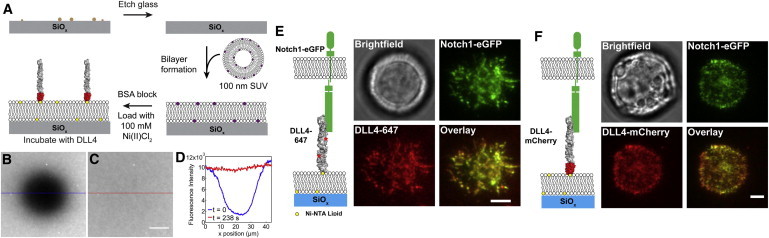
(A) Outline of the procedure used to fabricate supported lipid membranes containing tethered DLL4 molecules. Rendering of the extracellular domain of DLL4 is based upon the crystal structure of a fragment of human Jagged1, residues 187–335, which includes the DSL domain and the first 3 EGF repeats (PDB ID: 2VJ2). (B and C) Fluorescence recovery after photobleaching of the DLL4-647 bilayer surface verifying the lateral mobility of the fluorophore-labeled ligand. Scale bar = 10 μm. (D) Line scans over the indicated areas of images (B) and (C). Representative live cell images showing binding between Notch1-eGFP and membrane tethered (E) DLL4-647 and (F) DLL4-mCherry following a 30 min incubation period. Overlay images indicate a high degree of colocalization between Notch1 and DLL4. Scale bar = 5 μm. To see this figure in color, go online.
To demonstrate that the membrane-bound ligand molecules function biologically, stably transfected Notch1-eGFP C2C12 mouse myoblasts were incubated with the membrane surfaces for 30 min to bind to DLL4. Live cell images of single cells showed that both DLL4-647 and DLL4-mCherry formed ligand-receptor complexes, Fig. 1, E and F, respectively. These representative images show near complete colocalization between the DLL4 and Notch1 fluorescence signal as seen in the overlay images, thus suggesting that Notch1 is the primary binding partner for DLL4. As a control, parental C2C12 cells were incubated onto DLL4-mCherry functionalized bilayers and showed low levels of ligand-receptor binding (Fig. S2). Based on quantitative single cell intensity analysis, the amount of parental cell binding to DLL4 ligand was calculated to be 28% of Notch1-eGFP binding. It is important to note that this percentage reflects binding of all types of endogenous Notch receptors in the parental cell line compared with Notch1-eGFP and endogenous Notch binding in the transfected cell line. To verify the specificity of the DLL4-Notch1 interaction, Notch1-eGFP expressing cells were incubated with soluble DLL4/Fc (185 nM) and then placed onto the DLL4 membrane. In this case, binding of the Notch1-eGFP expressing cells to the ligand-presenting membrane was completely blocked after treatment with soluble ligand, as seen in Fig. S3. Note that the soluble DLL4/Fc-treated cells very weakly attached to the membrane; therefore, a small amount (0.1% by molar ratio) of cyclic Arg-Gly-Asp-d-Phe-Lys (cRGDfK) was incorporated into the bilayer to anchor cells and facilitate imaging (see the Supporting Material). We found that cRGDfK-integrin binding does not upregulate the downstream marker of Notch activation, Hey1 (also known as Herp2), as determined by reverse transcription-polymerase chain reaction (data not shown). Taken together, these data confirm that Notch1-eGFP specifically binds to membrane bound DLL4.
Formation and dynamics of Delta-Notch clusters
In addition to DLL4-Notch1 binding, we observed the formation of clusters of ligand-receptor complexes, in agreement with previous studies (11,20). To determine if clustering is Notch cell-driven, DLL4 supported lipid membrane surfaces were incubated with recombinant human Notch1 extracellular domain/Fc labeled with Alexa Fluor 647 (NECD-647), Fig. S4. This NECD construct is composed of the first 13 EGF repeats of the Notch receptor including the 11th and 12th repeats that are known to be necessary and sufficient for ligand binding (21). NECD-647 binds to both types of DLL4 surfaces; however, the receptor had an enhanced affinity for DLL4-mCherry compared to DLL4-568 as evidenced by a higher overall fluorescence signal in the NECD channel. One potential reason for reduced ligand potency of DLL4-568 arises as a result of fluorophore conjugation to lysine residues (K189, K190) found in the Notch receptor binding site (3). This highlights an important difference between the two ligands tested as DLL4-mCherry possesses a single fluorescent protein located at the C-terminus of the ligand, far from the Notch binding domain. Regardless of ligand affinity, when receptor ectodomain fragments were exposed to a DLL4 supported membrane no significant clustering of the ligand was observed and the membrane remained fluid as verified by FRAP (Fig. S4). These results illustrate that the Notch expressing cell is sufficient in driving ligand-induced receptor clustering.
To gain insight into the formation of ligand-receptor clusters, the dynamics of individual Notch1-eGFP cells on a DLL4 supported lipid membrane were observed using time-lapse microscopy. Interestingly, these movies revealed the depletion of Notch1-eGFP signal through two distinct routes. The first type of receptor depletion was gradual loss in Notch1-eGFP intensity on the timescale of minutes (Fig. 2, A and B, Movie S1). These clusters consisted of hundreds of receptor molecules, and the reduction in intensity was distinct from eGFP photobleaching (Fig. S5). In Fig. 2 A, two clusters labeled 1 and 2 were tracked for a total of 60 min. The eGFP intensity of cluster 1 steadily decreased over a 12 min interval, whereas the overall signal never reached zero. Cluster 2 served as a control and showed that the depletion of specific clusters was coordinated by cellular inputs and not due to fluctuations in excitation intensity or photobleaching. The second depletion behavior was represented in Fig. 2, C and D, and Movie S2 where a Notch1-eGFP cluster disappeared abruptly between two frames (t ∼ <1 min). In both types of Notch1-eGFP depletion, loss of the ligand signal was correlated with eGFP loss.
Figure 2.
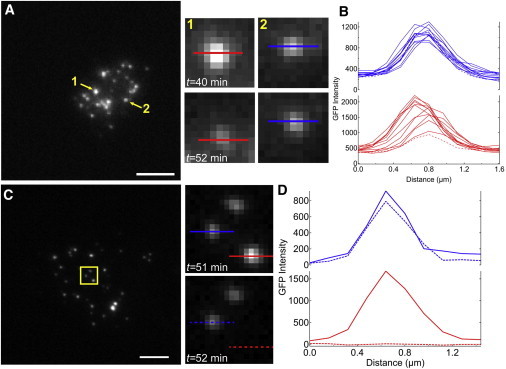
Dynamics of Notch1-eGFP cluster depletion. (A) Representative TIRFM image of Notch1-eGFP taken from a time-lapse series showing the depletion dynamics from a single cell. Two clusters are highlighted: position 1 shows attenuation of eGFP signal, whereas position 2 shows only small fluctuations in signal intensity. (B) Signal quantification using line scans taken through both clusters. Dashed line indicates intensity profile at t = 52 min. (C) Representative fluorescence image showing sudden loss of an entire Notch1-eGFP cluster and (D) line scans taken through two clusters to highlight this event. Scale bar = 5 μm. To see this figure in color, go online.
Within each set of time-lapse images, the loss of fluorescence intensity in the total internal reflection fluorescence microscopy (TIRFM) illumination field corresponds to either cleavage of the NICD from the cell membrane, activation of the Notch receptor, or internalization of the receptor. It is important to state that the ligand is rarely observed inside the cell, which suggests that the intact ligand-bound receptor complex is not internalized. To see if this coordinated signal loss is due to a mechanism independent of receptor activation, we conducted a set of live cell experiments with Notch1-eGFP cells that were DAPT (N-[N-(3,5-difluorophenacetyl-L-alanyl)]-S-phenylglycine t-butyl ester) treated. In one of these control cells, we observed simultaneous loss of DLL4 and Notch1 signal, which must be due to an internalization mechanism and not cleavage at S3 (Fig. S6). However, it is still possible that the receptor is internalized before being cleaved from the membrane and progressing to the nucleus. Therefore, the coordination between ligand and receptor depletion suggests that we are viewing either S3 cleavage, an intermediate regulatory step that takes place before S3 proteolysis, or an alternate receptor internalization process.
Given that the NICD is a potent transcription factor that translocates from the membrane to the nucleus, we employed an approach developed by Toomre et al. (22) to localize the z-position of the Notch1-eGFP through tandem imaging with epifluorescence and TIRFM (23). By switching between these two excitation configurations, it was possible to determine the lateral location of the clusters and their proximity to the cell membrane (24). Live cell imaging of a Notch1-eGFP expressing cell on a DLL4-568 surface resulted in the formation of ligand-receptor clusters as evidenced by colocalization of the ligand and receptor signals (Fig. 3 A). After binding, one of the formed clusters (arrowhead, Fig. 3 A) vanished from the TIRF channel at a rate that was faster than can be expected for photobleaching (Fig. S5). There was also noticeable loss of the DLL4-568 signal indicating that the unbound ligand molecules diffused away (Fig. 3 B). Strikingly, the epifluorescence channel revealed that the receptor cluster remained near the plasma membrane and had not yet translocated to the nucleus. Our data suggest that after ligand-induced S2 cleavage occurs at the cell-supported lipid membrane junction, the NICD cluster moves at least 100 nm away from the plasma membrane.
Figure 3.
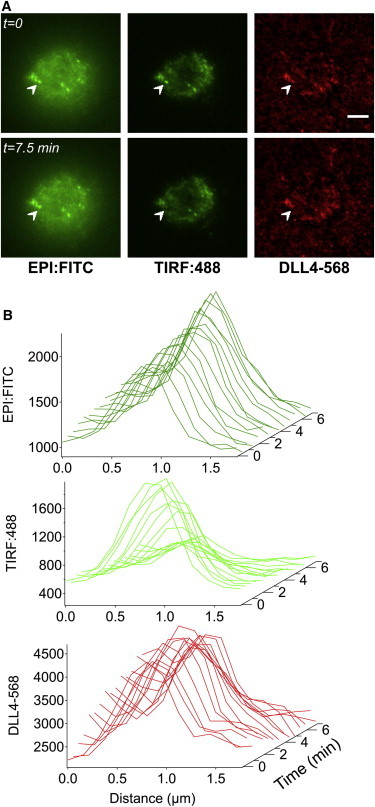
Observation of DLL4-Notch1 complexes using alternating epifluorescence-TIRFM. (A) Localization of the Notch1-eGFP at different time points is monitored with both epifluorescence and TIRFM (green), whereas DLL4 position is visualized in epifluorescence only (red). The highlighted cluster remains in the epifluorescence channel, but is diminished in TIRFM, suggesting translocation away from the membrane. Scale bar = 10 μm. (B) Waterfall plots indicate the changes in fluorescence intensity of the ligand-receptor complex (white arrow) as a function of time for each channel. To see this figure in color, go online.
To explain this series of events, it is important to emphasize that the precise cellular location of S3 cleavage is still unknown. There are studies that show that proteolysis at the S3 site occurs at the plasma membrane (25,26). In contrast, Gupta-Rossi et al. (27) show that monoubiquitination and endocytosis of the Notch receptor are necessary to trigger S3 cleavage. Alternatively, there is evidence to support that S3 cleavage may occur at both the plasma membrane and early endosomes but produce different forms of the NICD as a way to regulate Notch signaling (28). Currently, our results suggest that the cell has different ways to process the NICD and supports the hypothesis that the S3 cleavage event occurs at either the plasma membrane or early endosomes.
Determination of DLL4-Notch1 binding ratio
Given that receptor clustering is thought to play a role in signal activation (29), we were interested in determining the absolute molecular density of ligands and receptors and their stoichiometry within DLL4-Notch1 complexes of live cells. This is a general question that pertains to all juxtacrine pathways. Using a method developed by Galush et al. (30), the fluorescence intensity of the ligand and the receptor was calibrated using a spectrally similar lipid-fluorophore conjugate as a standard. In the case of the DLL4 ligand molecules, the calibration curve was constructed with TRITC-DHPE, although for the receptor molecules, NBD-PC lipid molecules were selected, as seen in Fig. 4, A–C. The second component of this analysis required that the fluorescence intensity of the ligand molecules be directly compared with the lipid-fluorophore standards. This comparison produced a unitless scaling factor, F factor, which is unique to each ligand/lipid pair, and these values are provided in Fig. 4, A–C. Based on the calibration data, the average ligand density of DLL4-568 on the membrane surface was determined to be 3700 ± 200 molecules per μm2, whereas the DLL4-mCherry density was lower at 1400 ± 300 molecules per μm2. This variation in surface packing density may be explained by the differences in protein dimension and possible interactions between the deca-histidine tag and ligand C-terminus. To calibrate the Notch1 receptor signal, a surface was prepared with dodecahistidine eGFP, and its protein density was measured at 4800 ± 300 molecules per μm2. All calculated values were consistent with previously published results for histidine-tagged proteins on membrane surfaces, thus validating the calibration method for quantifying ligand and receptor molecular densities (31).
Figure 4.
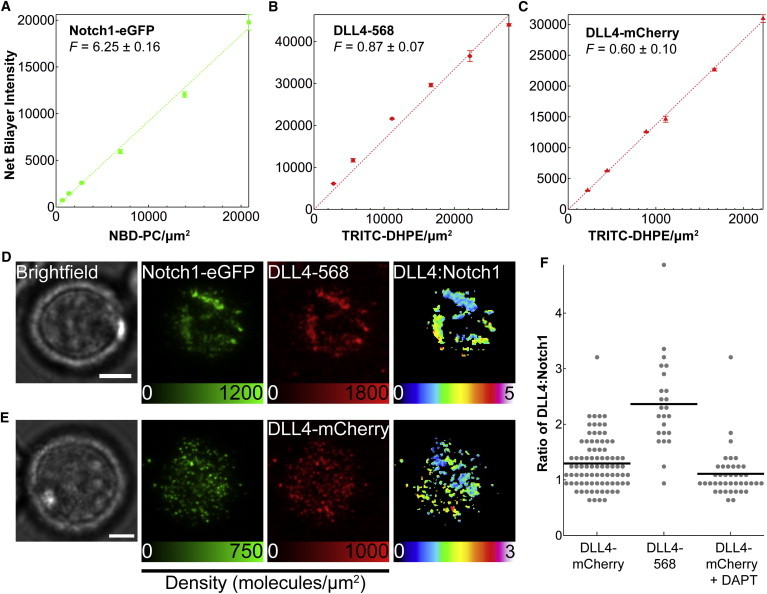
Bilayer calibration curves and calculation of binding ratio. (A) Intensity of NBD-PC bilayer at different molecular densities with the F factor comparing brightness of Notch1-eGFP and NBD-PC. (B) Calibration curve and F factor relating TRITC-DHPE and DLL4-568. (C) Calibration curve and F factor relating TRITC-DHPE and DLL4-mCherry. Representative quantitative fluorescence images of Notch1-eGFP expressing cells engaged to supported lipid membrane surfaces with (D) DLL4-mCherry and (E) DLL4-568 ligand. Each series includes images collected in brightfield, surface density maps of Notch1-eGFP and ligand (calibration bar in molecules/μm2), and a masked ratio (DLL4:Notch1) heat map. Scale bar = 5 μm. (F) Scatter plot of the ratio of DLL4:Notch1 determined from quantitative fluorescence analysis. Each dot indicates the ratio value from a single cell, and the horizontal line indicates the average value of all measurements. To see this figure in color, go online.
Ligand and receptor densities in live cell experiments were determined after allowing the Notch1-eGFP cells to incubate on DLL4 functionalized membranes for 30 min. Representative brightfield, Notch1-eGFP, and DLL4 images are shown in Fig. 4, D–E. The fluorescence intensity in each channel was converted into a surface density map, which displays the density of ligand and receptor molecules within individual clusters. A summary of the image analysis procedure is provided in Fig. S7. To determine the binding ratio, the DLL4 image was divided by the Notch1-eGFP image to generate a heat map of ratio values (DLL4:Notch1). To identify individual clusters, a mask was applied to the ratio image by selecting the regions of the cell expressing Notch1-eGFP. Analysis of these images showed an observed ratio value that varied from 0 to 5 under each cell, and the overall DLL4:Notch1 ratio was generally lower for DLL4-mCherry than for DLL4-568 ligand.
To compare ratio values across a statistically meaningful population of cells, a total of 40 to 85 cells were analyzed for each type of ligand. A summary of ratio values is provided in Fig. 4 F and Table S1. These values were obtained from histograms of ratio distributions fit with a Gaussian function (Fig. S8). The binding stoichiometry was calculated to be 1.3 ± 0.5 for DLL4-mCherry:Notch1 and 2.4 ± 0.8 for DLL4-568:Notch1. To explain the higher DLL4-568:Notch1 value, we hypothesized that a fraction of labeled, but unbound ligand was present within ligand-receptor clusters due to the higher initial density on the membrane surface. We tested this idea by tuning the ligand concentration on the membrane by lowering the percentage of Ni2+-NTA lipid molecules within the bilayer and observing the effect on the DLL4:Notch1 ratio, as shown in Fig. S9. As the density of DLL4-568 was decreased from 3700 to 300 molecules per μm2, the stoichiometry of DLL4-568:Notch1 decreased from 2.4 ± 0.8 to 1.6 ± 0.4, nearing the value obtained with DLL4-mCherry. The minimum amount of ligand required for cell adhesion was determined to be near 0.25 mol % or 20 molecules per μm2 (Fig. S10).
Another possible explanation for the slightly elevated amount of DLL4 is because the eGFP is fused to the NICD on the cytoplasmic portion of the receptor. Therefore, the ratio values reported more accurately relate NICD density to DLL4, and it is possible that S2 cleaved DLL4-Notch1 complexes remain trapped within the cell-supported membrane junction, whereas the S3 cleaved NICD diffuses away from the plasma membrane. Diffusion of membrane proteins surrounded by other proteins within the bilayer is highly hindered and would support this hypothesis (32). To test this idea, cells were treated with DAPT to prevent S3 cleavage, and the stoichiometry of DLL4:Notch1 was determined to be 1.1 ± 0.4, Fig. 4 F. The similarity of the values for DAPT treated and untreated populations suggest that the presence of the inhibitor does not affect binding between the ligand and receptor. Although this measurement establishes the ratio of ligand/receptor molecules, it is not sufficiently sensitive to be used as an assay of Notch activation by reporting changes in the ratio value.
Biological activity of surface tethered DLL4
To quantify the biological activity of different forms of the membrane-bound ligand, two independent methods were used to measure Notch activity levels. The first strategy employed a reporter cell line engineered by Sprinzak et al. (33) to monitor Notch1 activation through the expression of a citrine (YFP) reporter, which was controlled by the 12XCSL promoter. Supported lipid membranes were prepared with histidine-tagged, DLL4, DLL4-mCherry or DLL4-568, and the fluorescence intensity of the YFP reporter was measured after 48 h of cell incubation to maximize reporter expression. Reporter fluorescence was normalized to cells treated with the γ-secretase inhibitor, DAPT. For Notch reporter cells engaged with the DLL4-functionalized membrane, the reporter signal intensity within the nucleus was 2.5 ± 0.5 times greater than the control (Fig. 5 A, DLL4). In addition, DLL4-568 and DLL4-mCherry membranes showed slightly lower activation levels when compared to decahistidine DLL4 that was not fluorescently tagged (Fig. 5 A, DLL4-mC and DLL4-568). Variation in activation levels for different ligand types is attributed to fluctuations in the ligand surface density as well as ligand efficacy. The average surface density was calculated to be ∼3700 DLL4-568 per μm2 compared with ∼1400 DLL4-mCherry per μm2 (Table S1). However, as mentioned previously, it is likely that chemical modification of critical lysine residues in the receptor-binding domain of the DLL4-568 molecule alters its binding affinity.
Figure 5.
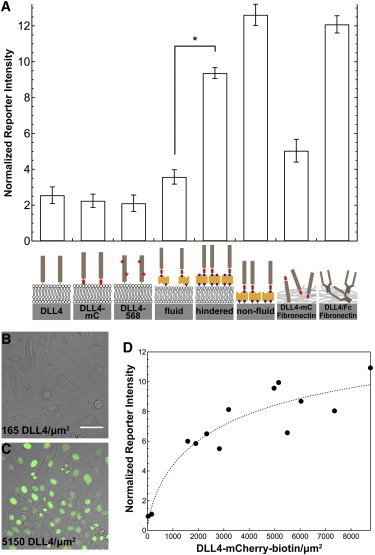
Role of ligand tether and lateral mobility on Notch activation levels. A reporter cell line engineered by Sprinzak et al. containing full length Notch1 and a YFP reporter controlled by a synthetic 12XCSL promoter was used to measure Notch activation levels. (A) Mean fluorescence intensity of reporter cells was measured after 48 h for the following surface types (left to right): supported membrane with histidine-tagged variants of DLL4, supported membrane with laterally mobile DLL4-mCherry-biotin (fluid), supported membrane with hindered DLL4-mCherry-biotin (hindered), functionalized glass with nonfluid DLL4-mCherry-biotin (nonfluid), DLL4-mCherry physisorbed with fibronectin, and DLL4/Fc physisorbed with fibronectin, ∗P < 0.001. Error bars represent SE for experiments performed in triplicate. Orange rectangles represent streptavidin and purple circles represent biotin moieties. Abbreviations for mCherry = mC; biotin = bio. Representative images of Notch reporter cells on supported membranes with (B) 165 molecules/μm2 of nonfluid DLL4-mCherry-biotin and (C) 5150 molecules/μm2 of nonfluid DLL4-mCherry-biotin. Scale bar = 10 μm. (D) Calibration of reporter cell line response to different surface densities of nonfluid DLL4-mCherry-biotin. The data were fit to a Hill function, which is indicated by the dashed line. To see this figure in color, go online.
To reaffirm the biological activity of these ligands, we used a second readout by immunostaining for the NICD to visualize proteolytically cleaved receptor within the nucleus. Surfaces were prepared in a similar manner as described previously and Notch1-eGFP cells were incubated on the surfaces for 3 h. The cells were fixed, permeabilized, and treated with a primary antibody specific for cleaved NICD (Val1744). Representative fluorescence images of either DAPT-treated or untreated Notch1-eGFP expressing cells with DAPI stained nuclei and antibody stained NICD on an unlabeled DLL4 functionalized membrane are shown in Fig. S11. To quantify activation levels, the nucleus was identified using the DAPI signal, and the mean NICD intensity was measured within each nucleus (Fig. S11). The trends in Notch activation levels were consistent with those observed with the reporter cell line.
Another key factor that may influence activation levels of the Notch receptor is the stability of the ligand tether to the supported lipid membrane. For this reason, we designed a biotin ligase-modified DLL4-mCherry. The biotin-streptavidin association (Kd ∼4 × 10−14 M) (34) provides a more stable anchor to the membrane surface when compared with histidine-NTA binding (Kd ∼10 × 10−6 M) (35). Although the overall DLL4 structure remains unchanged, use of a biotin tether increased the Notch activation levels to 3.6 ± 0.4-fold over the control (Fig. 5 A, fluid). This result indicates that if the ligand remains firmly associated with the membrane surface, then Notch activation levels increase.
It is unlikely that the observed increase in activation is due to formation of ligand dimers on tetrameric streptavidin. First, streptavidin is attached to the membrane surface through head-modified biotin lipid molecules; therefore, two of the binding sites are unavailable to the ligand (Fig. 5 A, fluid) (36). In addition, the density of fluorophore-labeled streptavidin on a fluid biotin-DPPE surface was measured to be ∼730 molecules per μm2. The density of DLL4-mCherry-biotin was calculated to be ∼770 molecules per μm2 suggesting that there is close to one ligand molecule per streptavidin. In addition, it is sterically unfavorable for two ligand molecules to be bound to the same streptavidin molecule. The two binding sites of streptavidin (on the same face) are separated by ∼2.0 nm (37,38), and the diameter of the mCherry beta-barrel is ∼3.0 nm (39) leaving little room for both ligand molecules to bind to the same side of the streptavidin.
Role of lateral ligand mobility on Notch activation
An important advantage of the supported membrane platform is that lateral mobility of the DLL4 ligand is dictated by the fluidity of the supported lipid membrane. Therefore, synthetic membrane surfaces can be tuned to present chemically identical DLL4 ligands that differ only by their lateral diffusion coefficient (D). To alter the lateral mobility of ligand molecules, the percentage of biotin-DPPE lipids doped into DOPC vesicles was varied from 0.1 to 4 mol %. At 0.1 mol % biotin-DPPE, membrane-tethered ligand molecules remain fluid, but at 4 mol % biotin-DPPE the ligand displays hindered long-range diffusion (Fig. S12) as the surface nears the packing density limit of streptavidin (40,41). Line scans over the photobleached areas qualitatively indicate the difference in lateral mobility of DLL4-mCherry-biotin (Fig. S12). The ligand density of the 4 mol % biotin-DPPE membrane was reduced to match the ligand density of the 0.1 mol % biotin-DPPE surface by titration with free D-biotin (Fig. S13). When Notch reporter cells were cultured on these surfaces and the activation levels quantified, the hindered DLL4-mCherry-biotin ligand displayed significant enhancement in activation (2.6-fold) over the fully fluid ligands (P < 0.001), Fig. 5 A (compare fluid to hindered).
Given that the 4 mol % biotin-DPPE membrane presented a small fraction of lipid molecules that remained mobile and some lipid dissociation from the membrane occurs over the 48 h incubation time, we next sought to test the activity of covalently immobilized ligand. Accordingly, DLL4-mCherry was fully immobilized using two different strategies (see the Supporting Material). First, a mixture of fibronectin and DLL4-mCherry or DLL4/Fc was adsorbed onto a glass substrate. This resulted in a normalized reporter response of 5.0 ± 0.6 and 12.1 ± 0.5 (Fig. 5 A, DLL4-mC and DLL4/Fc Fibronectin), respectively. This approach has been reported in the literature and results in randomly oriented ligand molecules within a thin film of fibronectin. In the second strategy, the DLL4-mCherry-biotin ligand was directly anchored to the glass slide through biotin groups that were covalently attached to the glass slide. Following incubation with the reporter cells, Notch activation levels were 12.6 ± 0.6 times greater than the DAPT control, which was the highest level measured on any type of surface tested (Fig. 5 A, nonfluid). This result confirms that proper ligand orientation coupled with nonlabile surface immobilization lead to the most potent ligand activity. Taken together, these data show that limiting lateral membrane fluidity can enhance receptor activation and lends support to the proposed mechanotransduction model of Notch receptor activation (10). Moreover, the difference in activation between fluid and nonfluid ligand also indicates that there is a lateral component to Notch tension-driven activation, which may provide an additional level of spatio-mechanical regulation of receptor activation.
Given that the Notch transcriptional reporter generates a nonlinear response to ligand density, we next aimed to develop a quantitative calibration relating the absolute molecular ligand density (input) with reporter YFP intensity (output). We quantified ligand densities using the fluorescence-based lipid membrane calibration and recorded the corresponding reporter response using a series of nonfluid DLL4-mCherry-biotin functionalized surfaces, Fig. 5, B–D. We found that the response of the reporter cell line to immobilized ligand molecules was steeply graded (Fig. 5 D) as captured in the representative images shown in Fig. 5, B–C. The shape of this plot is in agreement with prior literature precedent (33), and indicates that the reporter response rapidly saturates when cells engage surfaces presenting densities >∼2000 DLL4-mCherry-biotin molecules per μm2.
To better understand the interplay between DLL4 ligand density and D, and their role in modulating Notch activation, we performed a more detailed analysis using a set of membrane surfaces with D ranging from ∼0 to 0.6 μm2/s and densities ranging from 120 to 26,000 molecules/μm2. Although such parameters may exceed the range of physiological values, the absolute lateral mobility and surface density of ligands and receptors within living systems is not known. We quantified D of membrane tethered DLL4-mCherry-biotin by employing an established FRAP-based analysis method (18). Interestingly, for surfaces with ligand densities of 1500 molecules per μm2 or lower, activation of the Notch reporter showed a nonlinear and sharp increase as D approached 0 (Fig. 6 A). Although ligand density and D are not completely independent parameters, especially in densely packed membranes, high levels of reporter activation (>fivefold over DAPT control) were observed as D approached 0 over a wide range of ligand densities (Fig. 6 B). Both plots indicate that restricting the lateral mobility of DLL4 drastically increases Notch activation, whereas ligand density appears to be a less sensitive parameter for tuning activation. These results confirm that the mechanical properties of the ligand (lateral mobility) alter its biochemical activity in a highly nonlinear fashion, which is suggestive of the mechanoregulatory model of Notch activation.
Figure 6.
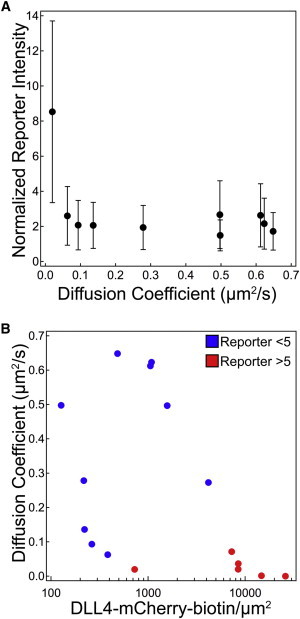
The effect of DLL4 lateral mobility, as characterized by diffusion coefficient (D), and density on Notch activation levels. (A) For membrane surfaces with <1500 DLL4-mCherry-biotin molecules per μm2, D was varied from ∼0 to 0.6 μm2/s. Notch activation sharply increased as D approached 0. Error bars indicate the standard deviation in reporter intensity as measured from 1000 cells (or more) 48 h after plating. The average D for each surface was determined using a previously published FRAP assay (18). (B) When the D and density were both varied, high levels of Notch activation (normalized reporter intensities >5, red marker) were observed when D neared 0 and when ligand density was increased. To see this figure in color, go online.
Conclusions
To study the molecular mechanisms of the Notch receptor pathway, where chemo-mechanical coupling within cellular junctions is increasingly appreciated, it is clear that new experimental strategies must be developed. The use of supported proteolipid membranes overcomes some of the limitations of other conventional methods for studying the Notch signaling pathway. We have shown that DLL4-Notch1 binding can be recaptured at the interface between a cell membrane and supported lipid membrane surface. Using quantitative fluorescence microscopy, it was observed that ligand and receptors actively formed clusters on fluid membranes containing, on average, hundreds of molecules per μm2. The stoichiometry of DLL4 to Notch1 within these clusters was ∼1.3 ± 0.5, near the expected 1:1 value. Through live cell imaging, the dynamic nature of ligand-receptor binding and clustering was captured including observation of coordinated loss of ligand and receptor molecules at the intermembrane junction. Using epi-TIRFM, the NICD was observed to leave the cell-supported membrane interface but remain near the plasma membrane. Finally, the mechanical properties and lateral mobility of the ligand molecules play a significant role in modulating activation of the Notch receptor. More specifically, ligand activity increased drastically as it became hindered or nonmobile. Despite the limited evidence showing that cells modulate the lateral mobility of Notch ligands or receptors within the plasma membrane, this work points toward an unprecedented mechanism of physical regulation of Notch receptor activation by simply modulating lateral mobility. This work opens the door to investigating Notch mechanotransduction mechanisms (42,43), and for screening of targets that may alter Notch ligand-receptor mobility and internalization dynamics.
Acknowledgments
The authors thank Prof. Gerry Weinmaster for helpful discussions and for providing the stable Notch1-eGFP C2C12 cell line used in this study. We thank Dr. Oskar Laur from the Emory Custom Cloning Core Facility for constructing the pcDNA3-DLL4-mCherry plasmid. Samples of deca-histidine eGFP were generously provided by the lab of Prof. Jay T. Groves.
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award No. R01GM097399, and in part by the Emory University Integrated Cellular Imaging Microscopy Core of the Winship Cancer Institute comprehensive cancer center grant, P30CA138292.
Supporting Material
References
- 1.Fagotto F., Gumbiner B.M. Cell contact-dependent signaling. Dev. Biol. 1996;180:445–454. doi: 10.1006/dbio.1996.0318. [DOI] [PubMed] [Google Scholar]
- 2.Gordon W.R., Arnett K.L., Blacklow S.C. The molecular logic of Notch signaling—a structural and biochemical perspective. J. Cell Sci. 2008;121:3109–3119. doi: 10.1242/jcs.035683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cordle J., Johnson S., Handford P.A. A conserved face of the Jagged/Serrate DSL domain is involved in Notch trans-activation and cis-inhibition. Nat. Struct. Mol. Biol. 2008;15:849–857. doi: 10.1038/nsmb.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gordon W.R., Roy M., Blacklow S.C. Structure of the Notch1-negative regulatory region: implications for normal activation and pathogenic signaling in T-ALL. Blood. 2009;113:4381–4390. doi: 10.1182/blood-2008-08-174748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stephenson N.L., Avis J.M. Direct observation of proteolytic cleavage at the S2 site upon forced unfolding of the Notch negative regulatory region. Proc. Natl. Acad. Sci. USA. 2012;109:E2757–E2765. doi: 10.1073/pnas.1205788109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meloty-Kapella L., Shergill B., Weinmaster G. Notch ligand endocytosis generates mechanical pulling force dependent on dynamin, epsins, and actin. Dev. Cell. 2012;22:1299–1312. doi: 10.1016/j.devcel.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shergill B., Meloty-Kapella L., Botvinick E. Optical tweezers studies on Notch: single-molecule interaction strength is independent of ligand endocytosis. Dev. Cell. 2012;22:1313–1320. doi: 10.1016/j.devcel.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen J., Zolkiewska A. Force-induced unfolding simulations of the human Notch1 negative regulatory region: possible roles of the heterodimerization domain in mechanosensing. PLoS ONE. 2011;6:e22837. doi: 10.1371/journal.pone.0022837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Varnum-Finney B., Wu L., Bernstein I.D. Immobilization of Notch ligand, Delta-1, is required for induction of notch signaling. J. Cell Sci. 2000;113:4313–4318. doi: 10.1242/jcs.113.23.4313. [DOI] [PubMed] [Google Scholar]
- 10.Parks A.L., Klueg K.M., Muskavitch M.A. Ligand endocytosis drives receptor dissociation and activation in the Notch pathway. Development. 2000;127:1373–1385. doi: 10.1242/dev.127.7.1373. [DOI] [PubMed] [Google Scholar]
- 11.Nichols J.T., Miyamoto A., Weinmaster G. DSL ligand endocytosis physically dissociates Notch1 heterodimers before activating proteolysis can occur. J. Cell Biol. 2007;176:445–458. doi: 10.1083/jcb.200609014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tiyanont K., Wales T.E., Blacklow S.C. Evidence for increased exposure of the Notch1 metalloprotease cleavage site upon conversion to an activated conformation. Structure. 2011;19:546–554. doi: 10.1016/j.str.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X., Ha T. Defining single molecular forces required to activate integrin and notch signaling. Science. 2013;340:991–994. doi: 10.1126/science.1231041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carroll-Portillo A., Spendier K., Timlin J.A. Formation of a mast cell synapse: Fc epsilon RI membrane dynamics upon binding mobile or immobilized ligands on surfaces. J. Immunol. 2010;184:1328–1338. doi: 10.4049/jimmunol.0903071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu C.-J., Hsieh W.-T., Baumgart T. Ligand mobility modulates immunological synapse formation and T cell activation. PLoS ONE. 2012;7:e32398. doi: 10.1371/journal.pone.0032398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsai J., Kam L.C. Lateral mobility of E-cadherin enhances Rac1 response in epithelial cells. Cell. Mol. Bioeng. 2010;3:84–90. doi: 10.1007/s12195-010-0104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salaita K.S., Nair P.M., Groves J.T. Restriction of receptor movement alters cellular response: physical force sensing by EphA2. Science. 2010;327:1380–1385. doi: 10.1126/science.1181729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jönsson P., Jonsson M.P., Höök F. A method improving the accuracy of fluorescence recovery after photobleaching analysis. Biophys. J. 2008;95:5334–5348. doi: 10.1529/biophysj.108.134874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vacklin H.P., Tiberg F., Thomas R.K. Formation of supported phospholipid bilayers via co-adsorption with beta-D-dodecyl maltoside. Biochim. Biophys. Acta. 2005;1668:17–24. doi: 10.1016/j.bbamem.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 20.Luty W.H., Rodeberg D., Vyas Y.M. Antiparallel segregation of notch components in the immunological synapse directs reciprocal signaling in allogeneic Th:DC conjugates. J. Immunol. 2007;179:819–829. doi: 10.4049/jimmunol.179.2.819. [DOI] [PubMed] [Google Scholar]
- 21.Rebay I., Fleming R.J., Artavanis-Tsakonas S. Specific EGF repeats of Notch mediate interactions with Delta and Serrate: implications for Notch as a multifunctional receptor. Cell. 1991;67:687–699. doi: 10.1016/0092-8674(91)90064-6. [DOI] [PubMed] [Google Scholar]
- 22.Toomre D., Steyer J.A., Simons K. Fusion of constitutive membrane traffic with the cell surface observed by evanescent wave microscopy. J. Cell Biol. 2000;149:33–40. doi: 10.1083/jcb.149.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merrifield C.J., Feldman M.E., Almers W. Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nat. Cell Biol. 2002;4:691–698. doi: 10.1038/ncb837. [DOI] [PubMed] [Google Scholar]
- 24.Mattheyses A.L., Simon S.M., Rappoport J.Z. Imaging with total internal reflection fluorescence microscopy for the cell biologist. J. Cell Sci. 2010;123:3621–3628. doi: 10.1242/jcs.056218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tarassishin L., Yin Y.I., Li Y.M. Processing of Notch and amyloid precursor protein by gamma-secretase is spatially distinct. Proc. Natl. Acad. Sci. USA. 2004;101:17050–17055. doi: 10.1073/pnas.0408007101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sorensen E.B., Conner S.D. γ-secretase-dependent cleavage initiates notch signaling from the plasma membrane. Traffic. 2010;11:1234–1245. doi: 10.1111/j.1600-0854.2010.01090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gupta-Rossi N., Six E.M., Brou C. Monoubiquitination and endocytosis direct gamma-secretase cleavage of activated Notch receptor. J. Cell Biol. 2004;166:73–83. doi: 10.1083/jcb.200310098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tagami S., Okochi M., Takeda M. Regulation of Notch signaling by dynamic changes in the precision of S3 cleavage of Notch-1. Mol. Cell. Biol. 2008;28:165–176. doi: 10.1128/MCB.00863-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hicks C., Ladi E., Weinmaster G. A secreted Delta1-Fc fusion protein functions both as an activator and inhibitor of Notch1 signaling. J. Neurosci. Res. 2002;68:655–667. doi: 10.1002/jnr.10263. [DOI] [PubMed] [Google Scholar]
- 30.Galush W.J., Nye J.A., Groves J.T. Quantitative fluorescence microscopy using supported lipid bilayer standards. Biophys. J. 2008;95:2512–2519. doi: 10.1529/biophysj.108.131540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nye J.A., Groves J.T. Kinetic control of histidine-tagged protein surface density on supported lipid bilayers. Langmuir. 2008;24:4145–4149. doi: 10.1021/la703788h. [DOI] [PubMed] [Google Scholar]
- 32.Lippincott-Schwartz J., Snapp E., Kenworthy A.K. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2001;2:444–456. doi: 10.1038/35073068. [DOI] [PubMed] [Google Scholar]
- 33.Sprinzak D., Lakhanpal A., Elowitz M.B. Cis-interactions between Notch and Delta generate mutually exclusive signalling states. Nature. 2010;465:86–90. doi: 10.1038/nature08959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Green N.M. Avidin and streptavidin. Methods Enzymol. 1990;184:51–67. doi: 10.1016/0076-6879(90)84259-j. [DOI] [PubMed] [Google Scholar]
- 35.Lata S., Gavutis M., Piehler J. Specific and stable fluorescence labeling of histidine-tagged proteins for dissecting multi-protein complex formation. J. Am. Chem. Soc. 2006;128:2365–2372. doi: 10.1021/ja0563105. [DOI] [PubMed] [Google Scholar]
- 36.Lou C., Wang Z., Wang S.-W. Two-dimensional protein crystals on a solid substrate: effect of surface ligand concentration. Langmuir. 2007;23:9752–9759. doi: 10.1021/la701399s. [DOI] [PubMed] [Google Scholar]
- 37.Hendrickson W.A., Pähler A., Phizackerley R.P. Crystal structure of core streptavidin determined from multiwavelength anomalous diffraction of synchrotron radiation. Proc. Natl. Acad. Sci. USA. 1989;86:2190–2194. doi: 10.1073/pnas.86.7.2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ren C.-L., Carvajal D., Szleifer I. Streptavidin-biotin binding in the presence of a polymer spacer. A theoretical description. Langmuir. 2009;25:12283–12292. doi: 10.1021/la901735d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang F., Moss L.G., Phillips G.N., Jr. The molecular structure of green fluorescent protein. Nat. Biotechnol. 1996;14:1246–1251. doi: 10.1038/nbt1096-1246. [DOI] [PubMed] [Google Scholar]
- 40.Mao H., Yang T., Cremer P.S. Design and characterization of immobilized enzymes in microfluidic systems. Anal. Chem. 2002;74:379–385. doi: 10.1021/ac010822u. [DOI] [PubMed] [Google Scholar]
- 41.Nguyen T.T., Sly K.L., Conboy J.C. Comparison of the energetics of avidin, streptavidin, neutrAvidin, and anti-biotin antibody binding to biotinylated lipid bilayer examined by second-harmonic generation. Anal. Chem. 2012;84:201–208. doi: 10.1021/ac202375n. [DOI] [PubMed] [Google Scholar]
- 42.Stabley D.R., Jurchenko C., Salaita K.S. Visualizing mechanical tension across membrane receptors with a fluorescent sensor. Nat. Methods. 2012;9:64–67. doi: 10.1038/nmeth.1747. [DOI] [PubMed] [Google Scholar]
- 43.Liu Y., Yehl K., Salaita K.S. Tension sensing nanoparticles for mechano-imaging at the living/nonliving interface. J. Am. Chem. Soc. 2013;135:5320–5323. doi: 10.1021/ja401494e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beckett D., Kovaleva E., Schatz P.J. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 1999;8:921–929. doi: 10.1110/ps.8.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen I., Howarth M., Ting A.Y. Site-specific labeling of cell surface proteins with biophysical probes using biotin ligase. Nat. Methods. 2005;2:99–104. doi: 10.1038/nmeth735. [DOI] [PubMed] [Google Scholar]
- 46.Muralidharan V., Muir T.W. Protein ligation: an enabling technology for the biophysical analysis of proteins. Nat. Methods. 2006;3:429–438. doi: 10.1038/nmeth886. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.