

Main Text
The spondylometaphyseal dysplasias (SMDs) are a group of about a dozen rare disorders characterized by short stature, irregular, flat vertebrae, and metaphyseal abnormalities. Aside from spondylometaphyseal dysplasia Kozlowski type (MIM 184252) caused by mutations in TRPV4 (MIM 605427) and spondyloenchondrodysplasia (MIM 607944) resulting from mutations in ACP5 (MIM 171640), the genetic etiologies of SMDs are unknown.1 Two of these unexplained SMDs have ophthalmologic manifestations: SMD with cone-rod dystrophy (SMD-CRD [MIM 608940]) and axial SMD with retinal degeneration (MIM 602271).
Delineated clinically a decade ago, SMD-CRD is a presumed autosomal-recessive disorder with postnatal growth deficiency leading to profound short stature; rhizomelia with bowing of the lower extremities; platyspondyly with anterior vertebral protrusions; progressive metaphyseal irregularity and cupping with shortened tubular bones; and early-onset, progressive visual impairment associated with a pigmentary maculopathy and electroretinographic evidence of cone-rod dysfunction.2–5 In contrast to retinitis pigmentosa, the CRDs have early involvement of cone photoreceptors.6
Here, we report loss-of-function mutations in PCYT1A (MIM 123695) as the cause of SMD-CRD. PYCT1A encodes CTP:phosphocholine cytidylyltransferase (CCTα),7,8 a key enzyme in the CDP-choline or Kennedy pathway for de novo phosphatidylcholine biosynthesis.
We used whole-exome and targeted sequencing of members of six unrelated families with eight individuals with SMD-CRD (Figure 1). Three families were submitted to the Baylor-Hopkins Center for Mendelian Genomics (BHCMG) through the online submission portal PhenoDB9 and, to confirm our observations in the first three families, we recruited three additional families for targeted candidate gene sequencing. Local approval for this study was provided by the Johns Hopkins Institutional Review Board, and all participants signed an informed consent. The clinical features of these individuals are summarized in Table 1 and briefly reviewed here. Six of the subjects have been described in previous publications.2,5
Figure 1.
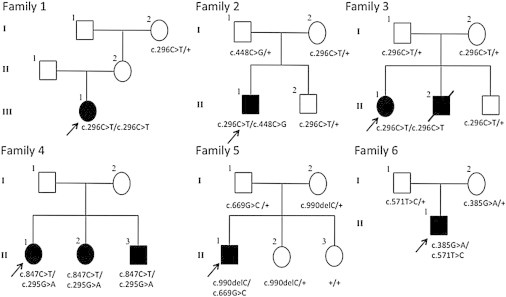
Pedigrees of Families 1–6 Showing the Segregation of PCYT1A Mutant Alleles
Alleles with the wild-type genotype indicated by plus sign. Samples were not available for individuals lacking a genotype designation.
Table 1.
Demographic, Clinical, and Molecular Findings
|
Family 1 |
Family 2 |
Family 3 |
Family 4 |
Family 5 |
Family 6 |
|||
|---|---|---|---|---|---|---|---|---|
| Subject 1 (BH2265_1) | Subject 2 (BH2283_1) | Subject 3 (BH2233_1) | Subject 4 | Subject 5 | Subject 6 | Subject 7 | Subject 8 | |
| Previously reported | Walters et al.2(case 1) | Walters et al.2(case 2) | no | Walters et al.2(case 3) | Walters et al.2(case 4) | Walters et al.2(case 5) | Kitoh et al.5 | no |
| Family origin | North Europe | North Europe | Greece | North Europe | North Europe | North Europe | Japan | Korea |
| Age of initial recognition | 7 months | 13 months | 51 years | 36 months | 27 months | 24 months | 6 months | 23 months |
| Best corrected visual acuity | 20/100 | 20/180 | 8/80 | 5/250 | 20/250 | 10/250 | NA | NA |
| Pigmentary maculopathy | + | + | + | + | + | + | + | + |
| Cone-rod dystrophy (age of diagnosis) | + (13.5 years) | + (17 months) | + (51 years) | + (36 months) | + (27 months) | ND | + (11 years) | + |
| Height SD (most recent measurement available) | −10.7 SD | −5.3 SD | −8 0.4 SD | NA | −6.1 SD | NA | −7.9 SD | −7.4 SD |
| Bowing of long bones | + | + | + | + | + | + | + | + |
| Platyspondyly | + | + | + | + | + | + | + | + |
| Metaphyseal irregularity and cupping | + | + | + | + | + | + | + | + |
| PCTY1A genotype | c.296C>T, c.296C>T | c.296C>T, c.448C>G | c.296C>T, c.296C>T | c.847C>T, c.295G>A | c.847C>T, c.295G>A | c.847C>T, c.295G>A | c.990delC, c.669G>C | c.385G>A, c.571T>C |
| PCTY1A protein change | p.Ala99Val, p.Ala99Val | p.Ala99Val, p.Pro150Ala | p.Ala99Val, p.Ala99Val | p.Arg283∗, p.Ala99Thr | p.Arg283∗, p.Ala99Thr | p.Arg283∗, p.Ala99Thr | p.Ser331Profs∗?, p.Arg223Ser | p.Glu129Lys, p.Phe191Leu |
| SIFT | 0.04 | 0.04, 0 | 0.04 | 0, 0.04 | 0, 0.04 | 0, 0.04 | NA, 0 | 0.02, 0 |
| PolyPhen | 1 | 1, 1 | 1 | 1, 1 | 1, 1 | 1, 1 | NA, 0.791 | 1, 1 |
Subject 1 (BH2265_1; family 1; Figure 2A) was reported when she was 20 years old.2 Now age 29, she has done well with continued linear growth to an adult height of 93.9 cm (−10.7 SD) and modest progression of limitation of range of motion. Visual impairment has not progressed since around age 10. Subject 2 (BH2283_1; family 2, Figure 2E), originally described at age 11 years, is now 20 years old with a current adult height of 139 cm (−5.3 SD) and some progression of joint stiffening. His visual function declined during his second decade and he now requires low-vision aids. Subject 3 (BH2233_1; family 3) is a previously unreported 61-year-old female who was first seen at age 51. Although her skeletal phenotype is similar to that of the others described here, she had a late-onset retinal phenotype (Figure 2B). Radiographs from childhood were reported to show platyspondyly and metaphyseal changes; adult radiographs show hypoplasia of the posterior vertebral bodies but no anterior vertebral protrusions. Adult height (measured at age 54) is 108.3 cm (−8.4 SD). Visual symptoms were not apparent until middle age. An ERG at age 43 (performed because she had a brother with visual impairment) was said to be normal. By age 51, however, her ERG showed evidence of cone-rod dysfunction and her current vision is limited (Table 1). At age 54, her examination showed short stature, rhizomelic limb shortening, brachydactyly, stiffness of large joints, and internal tibial torsion. Her family history includes an affected brother, who died at age 45 and was known to have short stature and a confirmed CRD. Her parents may be distantly related. Subjects 4–6 were reported by Walters et al.2 as their cases 3–5 and have not been formally assessed since that time but provided updated information regarding visual function (Table 1, Figures 2C and 2D). Subject 7 (family 5) was previously described.5 Subject 8 (family 6) is a 23-month-old previously unreported Korean male referred for evaluation of growth failure and disproportionate shortening of the limbs. He has an increased antero-posterior thoracic diameter, rhizomelic shortening of his extremities, bilateral bowing of the legs, and mildly limited elbow extension, knee extension, and hip abduction. No visual or hearing impairment was noted at his first examination. Linear growth was impaired (by North American standards): at 23 months he was 68.4 cm (−5.2 SD) and at 48 months, 71.2 cm (−7.4 SD). From age 2 years he had frequent pneumonias with episodes of desaturation and O2 dependency thought to be due to chest wall deformity. He had a waddling gait because of coxa vara deformity. Radiographs at 23 months showed short, bowed long bones with flared, cupped, and spurred metaphyses, and the adjacent epiphyses were large and rounded. In the hands and feet, the metaphyses of the short tubular bones had mild cupping, widening, and flaring and the diaphyses were short. The vertebral bodies were ovoid, mildly flattened, with anterior projections. These radiographic abnormalities were more severe at age 45 months. Mild scoliosis developed. Although no visual impairment was noted at age 23 months, by age 45 months fundus examination showed hypopigmented macular atrophy in both eyes with markedly decreased photopic and moderately decreased scotopic ERGs.
Figure 2.
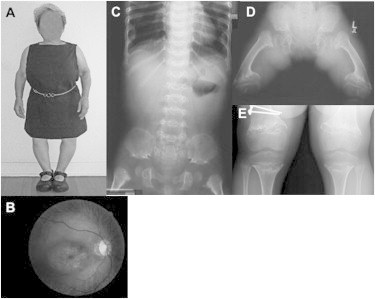
Clinical and Radiographic Features of SMD-CRD
(A) General physical phenotype (subject 1 at age 16 years 5 months).
(B) Fundus photograph showing pigmentary maculopathy (subject 3 at age 61 years).
(C) Spine and pelvis radiograph demonstrating platyspondyly, characteristic pelvic configuration, and proximal femoral metaphyseal changes (subject 6 at 3 years 8 months).
(D) Pelvis and femurs showing the marked metaphyseal changes that are typical (subject 4 at age 11 years 1 month).
(E) Knee radiographs, similarly showing the marked metaphyseal changes that are typical in this diagnosis (subject 2 at 13 years).
For our molecular studies, we isolated genomic DNA from fresh whole blood via the Gentra Puregene Kit (QIAGEN Sciences). Subjects 1, 2, and 3 were genotyped on Illumina’s ExomeChip1.1 GWAS array. This allows us to estimate inbreeding coefficient based on the observed and expected homozygous genotypes at genome-wide level among 199 samples from unrelated white individuals (defined by PCA) after LD pruning (PLINK). The inbreeding coefficient was 0.1535, −0.007, and 0.035 for subjects 1, 2, and 3, respectively, suggesting that subject 1 (BH2265_1) is the product of an unrecognized consanguineous union. Although subject 3 is also homozygous for p.Ala99Val mutation, B allele frequency plot showed multiple loss-of-heterozygosity segments across genome for subject 1 but not subject 3. This result suggests that homozygosity for p.Ala99Val in subject 1 is a result of consanguinity whereas in subject 3 it is the result of recurrent mutation (see below).
For WES, we captured the CCDS exonic regions and flanking intronic regions totaling ∼51 Mb by using the Agilent SureSelect Human All Exon V4 51Mb Kit and performed paired end 100 bp reads on subjects 1–3 with the Illumina HiSeq2000 platform. We aligned each read to the reference genome (NCBI human genome assembly build 37; Ensembl core database release 50_36110) with the Burrows-Wheeler Alignment (BWA) tool11 and identified single-nucleotide variants (SNVs) and small insertion-deletions (indels) with SAMtools.12 We also performed local realignment and base call quality recalibration by using GATK.13,14 We identified potentially causal variants by standard filtering criteria: SNV and indel minimal depth of 8×, root mean square mapping quality of 25, strand bias p value below 10−4, end distance bias below 10−4, and filtering out SNVs within 3 bp of an indel and indels within 10 bp of each other; followed by the use of the Analysis Tool of PhenoDB9 to design the prioritization strategy (N.S., unpublished data). We prioritized rare functional variants (missense, nonsense, splice site variants, and indels) that were homozygous or compound heterozygous in each of the three subjects and excluded variants with a MAF > 0.01 in dbSNP 126, 129, and 131 or in the Exome Variant Server (release ESP6500SI-V2) or 1000 Genomes Project.15 We also excluded all variants found in our in-house controls (CIDRVar 51Mb). We generated a homozygous and a compound heterozygous variant list for each subject and merged them to identify genes that were mutated in both alleles in all three subjects. We designed PCR primers to amplify exons and flanking intronic splice sites followed by direct Sanger sequencing to validate candidate causative variants, determine their segregation within families, and sequence PCYT1A in subjects 4–8. All the variants described here were based on the RefSeq transcript NM_005017.2 and NCBI human genome assembly build 37 (Table 1).
Analysis of WES data in subjects 1–3 identified two genes containing candidate causal mutations in all three subjects: TTN (MIM 188840) and PCYT1A. TTN is a large gene (313 exons) expressed primarily in skeletal and cardiac muscle.16 TTN variants are found frequently in controls17 and have been implicated as causative in various cardiac and skeletal myopathies,17–24 but not in individuals with retinal or skeletal dysplasia phenotypes. For these reasons, we removed TTN from consideration, leaving PCYT1A as the only gene with rare variants in both alleles in all three subjects. Subjects 1 and 3 are homozygous for the PCYT1A missense variant p.Ala99Val (c.296C>T) in exon 5 and subject 2 is a compound heterozygote for p.Ala99Val and p.Pro150Ala (c.448C>G) in exon 6 of PCYT1A. These variants are not present in the >6,000 individuals in the Exome Variant Server nor in the 1,092 individuals whose sequence is currently available from the 1000 Genomes. Direct Sanger sequencing of PCR-amplified products confirmed appropriate Mendelian segregation of these variants in available family members (Figure 1). Based on these results, we used PCR and bidirectional Sanger sequencing to interrogate all PCYT1A exons and flanking intronic sequence in subjects 4–8 (three probands and two affected sibs). All were compound heterozygotes for rare PCYT1A variants.
Thus, in total, we identified eight rare PCYT1A variants (one nonsense, one frame-shifting indel, and six missense variants) present either in the homozygous or compound heterozygous state in eight individuals with SMD-CRD in six families from around the world (Table 1). The missense mutations all change highly conserved residues, including one, Ala99, that is altered by two different variants (p.Ala99Val and p.Ala99Thr) and all are predicted to be damaging by SIFT25 and either probably damaging or possibly damaging by PolyPhen-2 (Table 1).26 The c.296C>T variant producing the p.Ala99Val change occurs at a CpG dinucleotide on the reverse strand, whereas the mutation in the same codon producing p.Ala99Thr (c.295G>A) in family 4 does not involve a CpG dinucleotide. To determine whether the three unrelated subjects with the p.Ala99Val change have a shared or recurrent mutation, we utilized the SNP genotyping data to analyze IBD sharing and runs of homozygosity to show that, as predicted by the different geographical origins of the three families, the p.Ala99Val variants in families 1–3 are on different haplotypes, indicating that recurrent CpG mutations are responsible.
Located at 3q29, PCYT1A (Figure 3) contains 10 exons, is ubiquitously expressed, and encodes CCTα, an amphitropic enzyme that catalyzes the synthesis of CDP-choline from phosphocholine and CTP. The CCTα reaction is the rate-limiting step in the major pathway for phosphatidylcholine synthesis (Figure 4).7,8 In mammals, phosphatidylcholine can also be synthesized from phosphatidylethanolamine in a reaction catalyzed by phosphatidylethanolamine N-methyltransferase (PEMT) but the expression of the enzyme is limited to liver (Figure 4).7,8,27 Phosphatidylcholine is the predominant phospholipid in eukaryotic membranes.7,8 Consistent with the importance of CCTα in phosphatidylcholine synthesis, a mouse knockout of Pcyt1a is early embryonic lethal in the homozygous state.28 An X-linked PCYT1A paralog, PCYT1B (MIM 604926), encodes three isoforms (CCTβ1, CCTβ2, and CCTβ3) with activities similar to CCTα but whose expression is limited to the central nervous system.8,29
Figure 3.
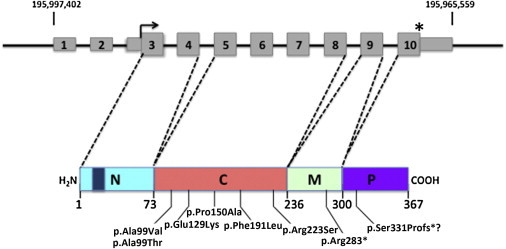
PCYT1A Structure and Domain Organization of CCTα
PCYT1A is located at 3q29 and includes ten exons (gray rectangles) as shown on the top line of the diagram. The thin rectangles encode the 5′ and 3′ UTR sequences. The right angle arrow denotes the start of the open reading frame and the asterisk indicates the location of the stop codon. The genomic coordinates on chromosome 3 are shown above. Below in color is a diagram of CCTα showing the domain structure with the diagonal dashed lines indicating the exons that contribute to each domain. The N-terminal domain (N) is in light blue with a dark blue rectangle indicating the position of the nuclear localization signal. The catalytic domain (C) is in pink. The amphitrophic membrane-binding domain (M) is in green and the C-terminal phosphorylated domain (P) is in purple. The numbers below indicate the residues at the boundaries of the domains. The mutations and their locations are shown below.
Figure 4.
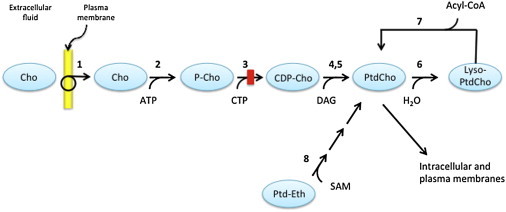
Pathways of Phosphatidylcholine Biosynthesis
Enzymes are indicated by numbers as defined below. Choline (Cho) is transported from the extracellular fluid across the plasma membrane by Na+-dependent, ATP-requiring choline transporter-like proteins (1). Free intracellular choline is phosphorylated by choline kinase (2) to produce phosphocholine (P-Cho). The latter is converted to cytidine-diphosphate choline (CDP-Cho) in a reaction catalyzed by phosphocholine cytidylyltransferase (CCTα, 3). CDP-choline is esterified with diacylglycerol (DAG) to phosphatidylcholine (PtdCho) in reactions catalyzed either by cholinephosphotransferase (4) or choline/ethanolaminephosphotransferase (5). PtdCho is converted to lyso-phosphatidylcholine (Lyso-PtdCho) by phospholipase A (6) which, in turn, is converted back to PtdCho, in a reaction catalyzed by acylglycerophosphate acyltransferase (7). In mammals, a second pathway of PtdCho synthesis is limited to liver, where PtdCho is synthesized from phosphatidylethanolamine (Ptd-Eth) in a series of methylation reactions catalyzed by phosphatidylethanolamine N-methyltransferase (8) with S-adenosylmethionine (SAM) as the methyl donor. The red rectangle indicates the position of the block in PtdCho synthesis caused by deficiency of CCT.
The 367 residue CCTα has 4 domains: a 73 residue N-terminal domain containing the nuclear localizing signal, a 163 residue catalytic domain, a 64 residue membrane-binding M domain, and a C-terminal 67 residue unordered domain with multiple residues that undergo phosphorylation (Figure 3).30,31 CCTα is found as an inactive homodimer in the nucleoplasm where, in response to lipid signaling, it binds to the nuclear membrane through interactions mediated by the amphipathic M domain and becomes catalytically active.8,30 The sequence and domain structure of CCTα is highly conserved across phylogeny and the crystal structure of the catalytic domain of rat CCTα, which has 100% sequence identity with human CCTα, has been solved.30
The PCYT1A missense alleles identified in the subjects with SMD-CRD all involve residues conserved across vertebrate and most of invertebrate and prokaryotic phylogeny. All mutations change residues in the catalytic domain of CCTα: Ala99 (altered in subjects 1–6) is adjacent to Gln98, which is directly involved in binding CDP; Pro150 (altered in proband 2) is adjacent to Trp151, which is also directly involved in binding choline; and Phe191 (altered in proband 8) is located three residues N-terminal to Thr194, which plays a role in coordinating CDP. The p.Glu129Lys and p.Arg223Ser substitutions alter residues that are linearly more distant from those that interact directly with substrates.30 The nonsense mutation, c.847C>T, occurs in the penultimate exon 9, 50 bp from the 3′ end of the exon, and possibly results in nonsense-mediated RNA decay of the mutant transcript. The 1 bp indel allele in subject 7 (c.990delC) changes the frame in sequence encoded by the last exon in a way that predicts continued translation all the way 3′ to the site of poly(A) addition. Such mutations have been termed “nonstop” alleles and are associated with dramatic reduction in mRNA abundance.32 We conclude that these variants, on the basis of the their extremely low frequency, the high conservation of the involved residues and predicted deleterious consequences for enzymatic activity, their occurrence in homozygosity or compound heterozygosity in multiple unrelated affected individuals in six families, and their segregation fitting an autosomal-recessive model, are PCYT1A loss-of-function alleles responsible for the SMD-CRD phenotype.
Like the deficiency of choline kinase B caused by mutations in CHKB (MIM 612395) responsible for muscular dystrophy, congenital megaconial type (MIM 602541),33 SMD-CRD is the second inborn error in the phosphatidylcholine synthetic pathway described. The central importance of this lipid in membrane biology suggests that complete loss of function of this pathway would result in embryonic lethality, as observed in mice homozygous for Pcyt1a-null alleles.28 Thus, it is puzzling that individuals with SMD-CRD are healthy aside from their skeletal and retinal involvement. One possible explanation is that despite the predicted severe functional consequences of the mutations we describe, there remains some residual CCTα function. This may be augmented by activity of the CCTβ isozymes and by synthesis of phosphatidylcholine from phosphatidylethanolamine by the PEMT catalyzed reaction in the liver.8 The activities of the CCTβ isozymes and PEMT were increased in cells derived from conditional Pcyt1a knockout mice.34,35 These possibilities should be explored because they may suggest therapeutic strategies that could be effective, especially in younger SMD-CRD-affected individuals.
Our results suggest a connection between bone and retinal lipid metabolism that would explain the sensitivity of these two tissues to deficiency of CCTα and impaired phosphatidylcholine synthesis. Photoreceptors have an especially high demand on membrane biosynthesis because of the daily shedding of outer segment discs.36 Therefore, it is not surprising that these cells might be especially susceptible to defects in the biosynthetic pathway for the most abundant membrane phospholipid. Indeed, photoreceptor degeneration in the rd11 mouse has been shown to be due to loss-of-function mutations in the gene encoding lysophosphatidylcholine acyltransferase 1 (Lpcat1), a phospholipid remodeling enzyme.37,38 Interestingly, bone formation is abnormal in certain Mendelian disorders caused by mutations in genes encoding proteins involved in fatty acid metabolism, including rhizomelic chondrodysplasia punctata (MIM 215100)39 and the Conradi-Hunermann form of chondrodysplasia punctata (MIM 302960).40 These observations suggest key functions for membrane lipids in bone formation and homeostasis.
In our study, there was no obvious genotype-phenotype correlation apparent from evaluation of the six affected individuals in four families with mutations altering codon Ala99. Subjects 1 and 3, both homozygous for c.296C>T (p.Ala99Val), have strikingly dissimilar age at onset of visual phenotype, of clinical characteristics, and of radiologic features. This broad range of phenotypic severity in individuals with the same mutations at the disease gene locus suggests that variation in other components of the phosphatidylcholine pathways and/or environmental variables may have a strong influence on phenotypic manifestations and severity. Understanding these variables may lead to improved management of individuals with SMD-CRD. Interestingly, one of the individuals described here, subject 8, has had frequent pneumonias and has required supplemental oxygen from age 2. We speculate that this might relate to the high demand for phosphatidylcholine in surfactant biosynthesis by the alveolar type II cells.41,42 These cells are known to express PCYT1A at high levels and phosphatidylcholine is the major phospholipid component of surfactant.41–43 A conditional, epithelial cell-specific Pcyt1a knockout mouse had severe respiratory failure at birth and reduced levels of surfactant.44 Although only one of the individuals described here has had this problem, the potential biological connections suggest that additional studies of pulmonary function and surfactant status in proband 8 and possibly other cases of SMD-CRD is warranted.
To our knowledge, SMD-CRD is the only described disorder in which CRD is associated with a skeletal dysplasia. By contrast, there are several examples of RP associated with bony abnormalities including some for which the responsible gene is yet to be identified.45,46 CRD and RP may be part of the same phenotypic spectrum of retinal degeneration. Mutations in certain genes have been reported to cause either phenotype.47,48 For this reason, we suggest that PCYT1A should be assessed in any individual with CRD or RP associated with any form of spondylometaphyseal dysplasia or spondyloepiphyseal dysplasia, including in particular individuals with axial SMD with retinal degeneration phenotype (MIM 602271).
Acknowledgments
We are grateful to the families for participating in this project. The Kathryn and Alan Greenberg Center for Skeletal Dysplasias and a grant from the National Human Genome Research Institute (1U54HG006493) provided support for this work. The authors acknowledge intellectual contributions from all members of the Baylor-Hopkins Center for Mendelian Genomics (BHCMG). We also thank Michael Bamshad for making the connection of BHCMG with W.-Y.P. and his colleagues in Korea and Japan, Gerald A. Fishman for providing fundus photographs of subject 3, and Larry Nogee and Dan Raben for helpful discussions regarding phospholipid and surfactant metabolism.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
Baylor-Hopkins Center for Mendelian Genomics, https://mendeliangenomics.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Picard, http://picard.sourceforge.net/
PLINK IBS/IBD estimation, http://pngu.mgh.harvard.edu/∼purcell/plink/ibdibs.shtml
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
References
- 1.Warman M.L., Cormier-Daire V., Hall C., Krakow D., Lachman R., LeMerrer M., Mortier G., Mundlos S., Nishimura G., Rimoin D.L. Nosology and classification of genetic skeletal disorders: 2010 revision. Am. J. Med. Genet. A. 2011;155A:943–968. doi: 10.1002/ajmg.a.33909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walters B.A., Raff M.L., Hoeve J.V., Tesser R., Langer L.O., France T.D., Glass I.A., Pauli R.M. Spondylometaphyseal dysplasia with cone-rod dystrophy. Am. J. Med. Genet. A. 2004;129A:265–276. doi: 10.1002/ajmg.a.30145. [DOI] [PubMed] [Google Scholar]
- 3.Sousa S.B., Russell-Eggitt I., Hall C., Hall B.D., Hennekam R.C. Further delineation of spondylometaphyseal dysplasia with cone-rod dystrophy. Am. J. Med. Genet. A. 2008;146A:3186–3194. doi: 10.1002/ajmg.a.32576. [DOI] [PubMed] [Google Scholar]
- 4.Turell M., Morrison S., Traboulsi E.I. Spondylometaphyseal dysplasia with cone-rod dystrophy. Ophthalmic Genet. 2010;31:12–17. doi: 10.3109/13816810903397812. [DOI] [PubMed] [Google Scholar]
- 5.Kitoh H., Kaneko H., Kondo M., Yamamoto T., Ishiguro N., Nishimura G. Spondylometaphyseal dysplasia with cone-rod dystrophy. Am. J. Med. Genet. A. 2011;155A:845–849. doi: 10.1002/ajmg.a.33898. [DOI] [PubMed] [Google Scholar]
- 6.Hamel C.P. Cone rod dystrophies. Orphanet J. Rare Dis. 2007;2:7. doi: 10.1186/1750-1172-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kent C. Regulatory enzymes of phosphatidylcholine biosynthesis: a personal perspective. Biochim. Biophys. Acta. 2005;1733:53–66. doi: 10.1016/j.bbalip.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 8.Fagone P., Jackowski S. Phosphatidylcholine and the CDP-choline cycle. Biochim. Biophys. Acta. 2013;1831:523–532. doi: 10.1016/j.bbalip.2012.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamosh A., Sobreira N., Hoover-Fong J., Sutton V.R., Boehm C., Schiettecatte F., Valle D. PhenoDB: a new web-based tool for the collection, storage, and analysis of phenotypic features. Hum. Mutat. 2013;34:566–571. doi: 10.1002/humu.22283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hubbard T.J., Aken B.L., Ayling S., Ballester B., Beal K., Bragin E., Brent S., Chen Y., Clapham P., Clarke L. Ensembl 2009. Nucleic Acids Res. 2009;37(Database issue):D690–D697. doi: 10.1093/nar/gkn828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.The 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y., Luoh S.M., Hon L.S., Baertsch R., Wood W.I., Zhang Z. GeneHub-GEPIS: digital expression profiling for normal and cancer tissues based on an integrated gene database. Nucleic Acids Res. 2007;35(Web Server issue):W152–W158. doi: 10.1093/nar/gkm381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopes L.R., Zekavati A., Syrris P., Hubank M., Giambartolomei C., Dalageorgou C., Jenkins S., McKenna W., Plagnol V., Elliott P.M., Uk10k Consortium Genetic complexity in hypertrophic cardiomyopathy revealed by high-throughput sequencing. J. Med. Genet. 2013;50:228–239. doi: 10.1136/jmedgenet-2012-101270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerull B., Gramlich M., Atherton J., McNabb M., Trombitás K., Sasse-Klaassen S., Seidman J.G., Seidman C., Granzier H., Labeit S. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat. Genet. 2002;30:201–204. doi: 10.1038/ng815. [DOI] [PubMed] [Google Scholar]
- 19.Itoh-Satoh M., Hayashi T., Nishi H., Koga Y., Arimura T., Koyanagi T., Takahashi M., Hohda S., Ueda K., Nouchi T. Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2002;291:385–393. doi: 10.1006/bbrc.2002.6448. [DOI] [PubMed] [Google Scholar]
- 20.Carmignac V., Salih M.A., Quijano-Roy S., Marchand S., Al Rayess M.M., Mukhtar M.M., Urtizberea J.A., Labeit S., Guicheney P., Leturcq F. C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann. Neurol. 2007;61:340–351. doi: 10.1002/ana.21089. [DOI] [PubMed] [Google Scholar]
- 21.Herman D.S., Lam L., Taylor M.R., Wang L., Teekakirikul P., Christodoulou D., Conner L., DePalma S.R., McDonough B., Sparks E. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012;366:619–628. doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Satoh M., Takahashi M., Sakamoto T., Hiroe M., Marumo F., Kimura A. Structural analysis of the titin gene in hypertrophic cardiomyopathy: identification of a novel disease gene. Biochem. Biophys. Res. Commun. 1999;262:411–417. doi: 10.1006/bbrc.1999.1221. [DOI] [PubMed] [Google Scholar]
- 23.Hackman P., Vihola A., Haravuori H., Marchand S., Sarparanta J., De Seze J., Labeit S., Witt C., Peltonen L., Richard I., Udd B. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am. J. Hum. Genet. 2002;71:492–500. doi: 10.1086/342380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lange S., Xiang F., Yakovenko A., Vihola A., Hackman P., Rostkova E., Kristensen J., Brandmeier B., Franzen G., Hedberg B. The kinase domain of titin controls muscle gene expression and protein turnover. Science. 2005;308:1599–1603. doi: 10.1126/science.1110463. [DOI] [PubMed] [Google Scholar]
- 25.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 26.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glunde K., Bhujwalla Z.M., Ronen S.M. Choline metabolism in malignant transformation. Nat. Rev. Cancer. 2011;11:835–848. doi: 10.1038/nrc3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang L., Magdaleno S., Tabas I., Jackowski S. Early embryonic lethality in mice with targeted deletion of the CTP:phosphocholine cytidylyltransferase alpha gene (Pcyt1a) Mol. Cell. Biol. 2005;25:3357–3363. doi: 10.1128/MCB.25.8.3357-3363.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carter J.M., Demizieux L., Campenot R.B., Vance D.E., Vance J.E. Phosphatidylcholine biosynthesis via CTP:phosphocholine cytidylyltransferase 2 facilitates neurite outgrowth and branching. J. Biol. Chem. 2008;283:202–212. doi: 10.1074/jbc.M706531200. [DOI] [PubMed] [Google Scholar]
- 30.Lee J., Johnson J., Ding Z., Paetzel M., Cornell R.B. Crystal structure of a mammalian CTP: phosphocholine cytidylyltransferase catalytic domain reveals novel active site residues within a highly conserved nucleotidyltransferase fold. J. Biol. Chem. 2009;284:33535–33548. doi: 10.1074/jbc.M109.053363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ding Z., Taneva S.G., Huang H.K., Campbell S.A., Semenec L., Chen N., Cornell R.B. A 22-mer segment in the structurally pliable regulatory domain of metazoan CTP: phosphocholine cytidylyltransferase facilitates both silencing and activating functions. J. Biol. Chem. 2012;287:38980–38991. doi: 10.1074/jbc.M112.402081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frischmeyer P.A., van Hoof A., O’Donnell K., Guerrerio A.L., Parker R., Dietz H.C. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science. 2002;295:2258–2261. doi: 10.1126/science.1067338. [DOI] [PubMed] [Google Scholar]
- 33.Mitsuhashi S., Ohkuma A., Talim B., Karahashi M., Koumura T., Aoyama C., Kurihara M., Quinlivan R., Sewry C., Mitsuhashi H. A congenital muscular dystrophy with mitochondrial structural abnormalities caused by defective de novo phosphatidylcholine biosynthesis. Am. J. Hum. Genet. 2011;88:845–851. doi: 10.1016/j.ajhg.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacobs R.L., Devlin C., Tabas I., Vance D.E. Targeted deletion of hepatic CTP:phosphocholine cytidylyltransferase alpha in mice decreases plasma high density and very low density lipoproteins. J. Biol. Chem. 2004;279:47402–47410. doi: 10.1074/jbc.M404027200. [DOI] [PubMed] [Google Scholar]
- 35.Zhang D., Tang W., Yao P.M., Yang C., Xie B., Jackowski S., Tabas I. Macrophages deficient in CTP:Phosphocholine cytidylyltransferase-alpha are viable under normal culture conditions but are highly susceptible to free cholesterol-induced death. Molecular genetic evidence that the induction of phosphatidylcholine biosynthesis in free cholesterol-loaded macrophages is an adaptive response. J. Biol. Chem. 2000;275:35368–35376. doi: 10.1074/jbc.M007099200. [DOI] [PubMed] [Google Scholar]
- 36.LaVail M.M. Rod outer segment disk shedding in rat retina: relationship to cyclic lighting. Science. 1976;194:1071–1074. doi: 10.1126/science.982063. [DOI] [PubMed] [Google Scholar]
- 37.Friedman J.S., Chang B., Krauth D.S., Lopez I., Waseem N.H., Hurd R.E., Feathers K.L., Branham K.E., Shaw M., Thomas G.E. Loss of lysophosphatidylcholine acyltransferase 1 leads to photoreceptor degeneration in rd11 mice. Proc. Natl. Acad. Sci. USA. 2010;107:15523–15528. doi: 10.1073/pnas.1002897107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bridges J.P., Ikegami M., Brilli L.L., Chen X., Mason R.J., Shannon J.M. LPCAT1 regulates surfactant phospholipid synthesis and is required for transitioning to air breathing in mice. J. Clin. Invest. 2010;120:1736–1748. doi: 10.1172/JCI38061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Braverman N., Chen L., Lin P., Obie C., Steel G., Douglas P., Chakraborty P.K., Clarke J.T., Boneh A., Moser A. Mutation analysis of PEX7 in 60 probands with rhizomelic chondrodysplasia punctata and functional correlations of genotype with phenotype. Hum. Mutat. 2002;20:284–297. doi: 10.1002/humu.10124. [DOI] [PubMed] [Google Scholar]
- 40.Herman G.E., Kelley R.I., Pureza V., Smith D., Kopacz K., Pitt J., Sutphen R., Sheffield L.J., Metzenberg A.B. Characterization of mutations in 22 females with X-linked dominant chondrodysplasia punctata (Happle syndrome) Genet. Med. 2002;4:434–438. doi: 10.1097/00125817-200211000-00006. [DOI] [PubMed] [Google Scholar]
- 41.Goss V., Hunt A.N., Postle A.D. Regulation of lung surfactant phospholipid synthesis and metabolism. Biochim. Biophys. Acta. 2013;1831:448–458. doi: 10.1016/j.bbalip.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 42.Agassandian M., Mallampalli R.K. Surfactant phospholipid metabolism. Biochim. Biophys. Acta. 2013;1831:612–625. doi: 10.1016/j.bbalip.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ridsdale R., Tseu I., Wang J., Post M. CTP:phosphocholine cytidylyltransferase alpha is a cytosolic protein in pulmonary epithelial cells and tissues. J. Biol. Chem. 2001;276:49148–49155. doi: 10.1074/jbc.M103566200. [DOI] [PubMed] [Google Scholar]
- 44.Tian Y., Zhou R., Rehg J.E., Jackowski S. Role of phosphocholine cytidylyltransferase alpha in lung development. Mol. Cell. Biol. 2007;27:975–982. doi: 10.1128/MCB.01512-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ehara S., Kim O.H., Maisawa S., Takasago Y., Nishimura G. Axial spondylometaphyseal dysplasia. Eur. J. Pediatr. 1997;156:627–630. doi: 10.1007/s004310050679. [DOI] [PubMed] [Google Scholar]
- 46.Isidor B., Le Merrer M., Ramos E., Baron S., David A. Cone-rod dystrophy, growth hormone deficiency and spondyloepiphyseal dysplasia: report of a new case without nephronophtisis. Am. J. Med. Genet. A. 2009;149A:788–792. doi: 10.1002/ajmg.a.32343. [DOI] [PubMed] [Google Scholar]
- 47.Yang Z., Chen Y., Lillo C., Chien J., Yu Z., Michaelides M., Klein M., Howes K.A., Li Y., Kaminoh Y. Mutant prominin 1 found in patients with macular degeneration disrupts photoreceptor disk morphogenesis in mice. J. Clin. Invest. 2008;118:2908–2916. doi: 10.1172/JCI35891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cremers F.P.M., van de Pol D.J.R., van Driel M., den Hollander A.I., van Haren F.J.J., Knoers N.V.A.M., Tijmes N., Bergen A.A.B., Rohrschneider K., Blankenagel A. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum. Mol. Genet. 1998;7:355–362. doi: 10.1093/hmg/7.3.355. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.