

Abstract
Mammalian cells can generate ATP via glycolysis or mitochondrial respiration. Oncogene activation and hypoxia promote glycolysis and lactate secretion. The significance of these metabolic changes to ATP production remains however ill defined. Here, we integrate LC-MS-based isotope tracer studies with oxygen uptake measurements in a quantitative redox-balanced metabolic flux model of mammalian cellular metabolism. We then apply this approach to assess the impact of Ras and Akt activation and hypoxia on energy metabolism. Both oncogene activation and hypoxia induce roughly a twofold increase in glycolytic flux. Ras activation and hypoxia also strongly decrease glucose oxidation. Oxidative phosphorylation, powered substantially by glutamine-driven TCA turning, however, persists and accounts for the majority of ATP production. Consistent with this, in all cases, pharmacological inhibition of oxidative phosphorylation markedly reduces energy charge, and glutamine but not glucose removal markedly lowers oxygen uptake. Thus, glutamine-driven oxidative phosphorylation is a major means of ATP production even in hypoxic cancer cells.
Keywords: cancer bioenergetics, isotope tracing, metabolic flux analysis
Introduction
The seminal work of Otto Warburg revealed that cancer cells avidly ferment glucose even in the presence of oxygen, a phenomenon called aerobic glycolysis or the Warburg effect (Warburg, 1956). This fundamental effect has been repeatedly confirmed in vitro and also in vivo where it accounts for the effectiveness of flurodeoxyglucose PET imaging. Warburg originally attributed aerobic glycolysis to impaired mitochondrial function (Warburg, 1956); however, it subsequently became clear that most cancers display the Warburg effect despite intact mitochondrial respiration capacity. In fact, the contribution of oxidative phosphorylation to total ATP production was recently claimed by Zu and Guppy (2004) to exceed that of aerobic glycolysis in many cancer cells . In light of this, it has been suggested that the shift to aerobic glycolysis serves to maximize ATP production per unit of enzyme synthesized (at the expense of ATP per glucose) or to increase total ATP production without requiring increased mitochondrial capacity (Pfeiffer et al, 2001; Vazquez et al, 2010; Shlomi et al, 2011). Alternatively, instead of functioning to increase ATP production, aerobic glycolysis may promote tumor growth by increasing the concentration of central carbon metabolites available to drive biosynthesis (Vander Heiden et al, 2009).
Whatever its function, the occurrence of the Warburg effect reflects the activation of oncogenic signaling pathways whose physiological function is to promote glucose uptake and anabolic metabolism. These include the PI3K-Akt pathway, the natural effector pathway of insulin signaling, which induces glucose uptake and lipogenesis (Elstrom et al, 2004; Robey and Hay, 2009). The PI3K-Akt pathway is frequently mutated in cancer. In addition, it can be activated by Ras, whose mutation underlies most pancreatic cancer and many other lethal cancers (Wallace, 2005). In addition to the PI3K-Akt pathway, Ras triggers several other pro-growth signaling cascades such as the MAPK pathway. Moreover, it has multifarious metabolic effects including induction of autophagy and macropinocytosis and inhibition of oxidative phosphorylation (Bar-Sagi and Feramisco, 1986; Chun et al, 2010; Yang et al, 2010; Gaglio et al, 2011; Lock et al, 2011).
Like oncogenes, hypoxia promotes glycolytic flux, in part due to the activation of hypoxia-induced factor (HIF) and its downstream target genes, which include many glycolytic enzymes (Tennant et al, 2009; Semenza, 2010). Both Ras and hypoxia decrease flux of glucose through pyruvate dehydrogenase (PDH) into the TCA cycle, in part through activation of pyruvate dehydrogenase kinase (PDK). In such cases, the TCA cycle can be fed by alternative substrates including glutamine, whose importance for cell growth and survival is increased by both Ras activation and hypoxia. This may reflect enhanced reliance on glutamine as a bioenergetic substrate (Le et al, 2012) or as an anabolic precursor to amino acids or acetyl-CoA/ lipids (Gaglio et al, 2011; Metallo et al, 2012).
Here, we study how oncogene activation and hypoxia affect energy metabolism, specifically (i) the contribution of aerobic glycolysis versus oxidative phosphorylation to total ATP production, and (ii) the relative contribution of glucose, glutamine versus other nutrients to producing the reducing power that drives oxidative phosphorylation. Toward this end, we combine LC-MS-based isotope tracer data with oxygen consumption measurements in a quantitative redox-balanced metabolic flux model. Notably, although oxygen consumption rate measurements were previously shown to be valuable for metabolic flux analysis in microbes (Varma and Palsson, 1994), oxygen uptake has not been used together with isotope tracer data to facilitate flux inference in mammalian cells. We apply this approach to study the effect of Ras and Akt activation and hypoxia on fluxes, providing a comprehensive and quantitative view of the impact of these factors on ATP production routes. Through this approach, we find that glutamine-driven oxidative phosphorylation is a major ATP source even in oncogene-expressing or hypoxic cells.
Results and discussion
Quantifying ATP production routes via a redox-balanced metabolic flux model
To study ATP production routes, we used Bax−/−, Bak−/− murine renal epithelial cells immortalized by expression of adenovirus E1A and dominant-negative p53 (Degenhardt et al, 2002) (iBMK cells). Isogenic cell lines were generated by transfecting cells with vector expressing either oncogenic H-RasV12G or myr-Akt (Degenhardt et al, 2006; Degenhardt and White, 2006). Note that activation of these specific genes may not result in identical metabolic consequences to that of other related family members that are frequently mutated in cancer (e.g., K-Ras, PI3KCA). Introduction of either oncogene did not substantially impact cellular growth in vitro (Supplementary Table 1), but greatly enhances tumorigenicity in vivo, as evident by faster allograft growth, with the effect of Ras yet stronger than that of Akt (Degenhardt and White, 2006).
To quantify fluxes in central metabolism, we combined three types of measurements (Figure 1A, see also Methods): (i) uptake and excretion rates of major nutrients (glucose, glutamine, and oxygen, including the fraction of oxygen consumed by oxidative phosphorylation as measured by respiratory chain inhibition) and waste products (lactate, glutamate, pyruvate, and alanine); (ii) cellular DNA, RNA, protein, and fatty acid content (Supplementary Table 2) together with cellular growth rate to determine the flux of metabolic building blocks into biomass; and (iii) steady-state labeling of intracellular metabolites determined by LC-MS when cells are fed media with [U-13C]-glucose or [U-13C]-glutamine (Supplementary Figures 1–4).
Figure 1.

Glutamine-driven oxidative phosphorylation supports ATP production in iBMK-parental cells. (A) Redox-balanced metabolic flux analysis scheme. Experimental measurements including uptake/excretion rates and steady-state intracellular labeling patterns were input into a redox-balanced metabolic flux analysis model. Fluxes with confidence intervals were obtained by optimizing the simulation to fit the steady-state experimental observations. The resulting fluxes also fit kinetic labeling data well. (B) Metabolic fluxes in the parental iBMK cell line. Numbers indicate metabolic flux in nmole/h/μl cells. Colors indicate the contribution of glucose (yellow) or glutamine (blue) in driving each redox reaction. Reactions that do not produce high energy electrons are shown in black. Font size reflects absolute metabolite concentrations. Pyr, pyruvate; AKG, α-ketoglutarate; OAA, oxaloacetate. (C) Effect of oxidative phosphorylation inhibitors on the ATP/ADP ratio and NADH/NAD+ ratio. Absolute concentrations of ATP, ADP, NADH, and NAD+ were measured by isotope ratio-based MS 5 min after addition of vehicle (DMSO), the complex III inhibitor antimycin A (4 μg/ml) or the ATP synthase inhibitor oligomycin (8 μg/ml) (mean ± s.d. of N=3). (D) Oxygen consumption rates and NADH/NAD+ ratio measured after switching cells to complete media or media lacking glucose or glutamine for 8 h (mean±s.d. of N=3).
To infer intracellular metabolic fluxes, we constructed a metabolic network model of glycolysis and TCA cycle (Supplementary Table 3) and applied metabolic flux analysis (MFA) to identify a flux distribution that would optimally fit the experimental data sets. The computational flux model is redox balanced, i.e., the high energy electron (NADH or FADH2) production rate matches the consumption rate by oxidative phosphorylation. Flux confidence intervals were derived by directly computing the range of possible fluxes for each reaction that enable close-to-optimal fit with the experimental data, by iteratively running the MFA while constraining the flux through each reaction to increasing (and then decreasing) values (Methods; note that reactions not included in the metabolic network model may introduce additional error beyond that reflected in the computed confidence intervals). In all cases, we obtained fluxes that resulted in good agreement with the steady-state labeling data (Supplementary Figures 1–4). As a further validation of the inferred fluxes, we measured the kinetic labeling of intracellular metabolites (for 72 h) as well as their absolute concentrations (Supplementary Table 4). The fluxes obtained solely by fitting the steady-state data resulted also in good agreement with the experimentally observed kinetic labeling patterns (Methods; Supplementary Figures 1–4).
Predicted fluxes were used to calculate the contribution of glycolysis and oxidative phorphorylation to ATP production, considering that glycolysis makes 2 ATPs per glucose molecule and the oxidative phosphorylation makes on average 2.5 ATPs per NADH oxidation and 1.5 ATP per FADH2 oxidation. To assess the contribution of glucose, glutamine, or acetyl-CoA (produced by the catabolism of fatty acids or amino acids) to generating the reducing power that drives oxidative phosphorylation, we calculated the rate of high-energy electron donation from each of these nutrients. To this end, we computationally inferred for each NADH/FADH2-producing reaction the abundance of substrate carbons being oxidized that originate from glucose, glutamine, and other sources of acetyl-CoA (see Methods).
Our flux-based approach for inferring the contribution of specific nutrient oxidation to generating reducing power has much in common with a classical approach involving feeding cells with radioactive-labeled nutrients and tracking the release of radioactive CO2(Guppy et al, 1997, 2002). However, these previous methods only tracked reactions that make CO2, while many NADH and FADH2-producing reactions were not included. Moreover, these previous methods were blind to oxidation of alternative non-radioactive substrates, which are visible in our analysis as we track both labeled and non-labeled carbon atoms. Thus, our approach enables both the quantitation of the total generated reducing power and the contribution of individual reactions.
Glutamine-supported oxidative phosphorylation is a major source of ATP in the parental iBMK cell line
The total ATP production rate in the parental iBMK cell line was found to be 861 nmole/μl cells/h (where μl refers to the packed cell volume, with 1 μl equal to ∼106 cells). The relative contribution of oxidative phosphorylation and aerobic glycolysis was ∼80 and 20%, respectively (Supplementary Table 3), consistent with previous results(Zu and Guppy, 2004). The major contribution of oxidative phosphorylation to total ATP production in this cell line is evident when treating the cells with either the complex III inhibitor antimycin A or the ATP synthase inhibitor oligomycin, both of which lead to ∼5-fold increase in the NADH/NAD+ ratio and ∼10-fold decrease in the ATP/ADP ratio (Figure 1C).
Tracking the source of reducing power, we found that oxidation of glutamine, glucose, and acetyl-CoA derived from other sources (such as unlabeled fatty acids or amino acids) contributes 60, 30, and 10% of the total NADH/FADH2 production, respectively. Glutamine’s uptake rate is ∼30% that of glucose, and it directly feeds TCA cycle through α-ketoglutarate. Glucose-derived two-carbon flux into TCA cycle (via pyruvate dehydrogenase) is 60% lower than that of glutamine flux into TCA cycle. Glucose-derived four-carbon flux into TCA cycle (via pyruvate carboxylase) amounts to ∼2% of glutamine flux into TCA cycle.
In addition to glucose-derived carbon atoms being a contributor to TCA turning, glucose-driven glycolysis and serine synthesis in the cytosol can both produce high-energy electrons in the form of cytosolic NADH. To evaluate the potential contribution of the serine pathway to high-energy electron generation, we conducted studies with a U-13C-serine tracer, which revealed that NADH production via this pathway in the tested cell lines is ∼3% of glycolytic flux (see Methods). Moreover, we observed that, of total NADH generated in the cytosol, 84% is consumed to reduce pyruvate to lactate. For the remaining 16% of cytosolic high-energy electrons to contribute to ATP production, they must be imported into the mitochondrion via the malate-aspartate shuttle, which produces mitochondrial NADH, or the glycerol-phosphate shuttle, which converts cytosolic NADH into mitochondrial FADH2. For simplicity, we assume exclusive use of the malate-aspartate shuttle. If cells were instead to exclusively use the glycerol-phosphate shuttle, oxidative ATP production would decrease by 3% (Supplementary Figure 5).
To validate the inferred greater contribution of glutamine than glucose to oxidative metabolism, we measured O2 uptake in cells deprived of either glucose or glutamine (Figure 1D). We find that deprivation of either glucose or glutamine decreases the whole cell NADH/NAD+ ratio, with the effect of glutamine removal being greater (P=0.02, Methods). Note that the observed change in the NADH/NAD+ ratio reflects the combination of cytosolic and mitochondrial pools. The effect on different compartments may vary by nutrient, a topic that merits further investigation using compartment-specific measurement methods (Hung et al, 2011; Zhao et al, 2011). Similar to the greater effect of glutamine on the NADH/NAD+ ratio, glutamine removal was also found to more strongly decrease oxygen uptake (P<0.001). These results are in agreement with those of Le et al (2012) showing a significant drop in ATP level when cells are treated with a glutaminase inhibitor.
Ras induces aerobic glycolysis without increasing the total ATP production rate
Activation of either Ras or Akt induces aerobic glycolysis, as evident by a roughly two-fold increase in glucose uptake and lactate secretion (Figure 2A). However, following the activation of either oncogene, most cellular ATP is still produced oxidatively, with Ras but not Akt activation causing a decrease in oxidative ATP production (Figure 2B). Oxygen consumption was significantly decreased by Ras but not Akt activation (P=0.01 and P=0.97, respectively) with the Ras versus Akt difference also significant (P=0.02). Consistent with this, treating the oncogene-activated cells with mitochondrial inhibitors leads to a rise in the NADH/NAD+ ratio of 6.0 and a drop in ATP/ADP ratio of ∼88% following Akt activation and to a lesser rise in NADH/NAD+ of 3.9 and smaller drop of ATP/ADP ∼76% for Ras (P=0.05 for greater fractional drop in ATP/ADP ratio in Akt-driven than Ras-driven cells for antimycin, P=0.03 for oligomycin) (Supplementary Figure 6A, Figure 2C). Our finding of Ras activation decreasing oxidative ATP production is in agreement with previous reports of Ras inhibiting both oxygen consumption and glycolytic two-carbon flux entering TCA cycle (Chun et al, 2010; Yang et al, 2010; Gaglio et al, 2011).
Figure 2.

Energy metabolism in oncogene-driven cells. (A) Metabolic fluxes in iBMK cells expressing H-RasV12G or myr-Akt. Colors indicate flux changes in the oncogene-driven cell lines compared with parental cells, showing only reactions whose flux confidence interval in the oncogene-activated cells does not overlap the corresponding confidence interval in parental cell line. (B) ATP production rates from oxidative phosphorylation and glycolysis in parental, Ras, and Akt iBMK cells. (C) Effect of oxidative phosphorylation inhibitors on the ATP/ADP ratio. ATP and ADP were measured 5 min after addition of vehicle (DMSO), the complex III inhibitor antimycin A (4 μg/ml) or the ATP synthase inhibitor oligomycin (8 μg/ml) (mean ± s.d. of N=3). (D) Contribution of various nutrients to driving cofactor reduction (sum of NAD+ and FAD). (E) Oxygen consumption rates measured after switching cells to complete media or media lacking glucose or glutamine for 8 h (mean±s.d. of N=3).
Glutamine oxidation remains the main source of reducing power following the activation of both Ras and Akt (Figure 2D). Similar to the parental cell line, removal of glutamine decreases the consumption of oxygen (Figure 2E) and the NADH/NAD+ ratio (Supplementary Figure 6B) to a greater extent than glucose removal (P=0.01 for oxygen uptake measurements in Ras and P=0.003 in Akt). Moreover, for the Ras cell line, the importance of glutamine as a TCA substrate is increased, with the relative contribution of glutamine oxidation to reducing power rising from 62% to 75%. The increased reliance on glutamine results from unchanged glutamine flux into the TCA cycle via α-ketoglutarate, paired with decreased influx of other substrates, most importantly decreased glycolytic two-carbon flux (Figure 2D).
Interestingly, while Ras activation induces aerobic glycolysis, it proportionally reduces oxidative phosphorylation, such that the total ATP production rate remains the same. This suggests that the induction of aerobic glycolysis by Ras, at least in normoxia, is not for the purpose of increasing ATP production, arguing against claims that Warburg effect promotes tumorigenesis by accelerating the production of usable energy (Pfeiffer et al, 2001; Shlomi et al, 2011).
Glutamine-supported oxidative phosphorylation is a major source of ATP also in hypoxia
To quantify metabolic flux in hypoxia, we repeated the analysis described above, examining the parental iBMK cells grown in a hypoxic chamber with 1% oxygen. Similarly to oncogene activation, hypoxia induces aerobic glycolysis (Figure 3A). More interestingly, in hypoxia, the oxygen consumption rate is reduced by only 30% compared with normoxia. Our observation of persistent oxygen uptake in hypoxia is consistent with Frezza et al (2011), who reported an oxygen consumption rate of the HCT116 cell line in hypoxia that is 50% of the normoxic rate . Hence, we find that 60% of cellular ATP is still made oxidatively in hypoxia (Figure 3B), which is confirmed by profound ATP/ADP ratio drop and NADH/NAD+ ratio increase upon treatment with mitochondrial inhibitors (Figure 3C). As expected, the drop in ATP/ADP due to the oxidative phosphorylation inhibitors is less severe in hypoxia (P=0.002 for antimycin and P=0.002 for oligomycin).
Figure 3.

Energy metabolism in iBMK parental cells in hypoxia (1% O2). (A) Metabolic fluxes in the parental iBMK cell line in hypoxia. Colors indicate flux changes in hypoxia compared with normoxia, showing only reactions whose flux confidence interval in hypoxia does not overlap the corresponding confidence interval in normoxia. (B) ATP production rates from oxidative phosphorylation and glycolysis in iBMK parental cells in normoxia versus hypoxia. (C) Effect of oxidative phosphorylation inhibitors on the ATP/ADP ratio and the NADH/NAD+ ratio in parental cells in hypoxia. ATP, ADP, NADH, and NAD+ levels were measured 5 min after addition of vehicle (DMSO), the complex III inhibitor antimycin A (4 μg/ml) or the ATP synthase inhibitor oligomycin (8 μg/ml). (D) Contribution of various nutrients to driving cofactor reduction (sum of NAD+ and FAD). (E) Oxygen consumption rates and NADH/NAD+ ratio in parental cells in hypoxia measured after switching cells to complete media or media lacking glucose or glutamine for 8 h (mean ± s.d. of N=3). (F) Effect of oxidative phosphorylation inhibitors on the ATP/ADP ratio and NADH/NAD+ ratio in Ras and Akt cells in hypoxia. (G) Oxygen consumption rates and NADH/NAD+ ratio in Ras and Akt cells in hypoxia measured after switching cells to complete media or media lacking glucose or glutamine for 8 h (mean±s.d. of N=3).
In terms of reducing power, we find that, also in hypoxia, more than 60% of NADH/FADH2 is produced by glutamine oxidation. The contribution of glucose oxidation decreases from 32% in normoxia to 17% in hypoxia (Figure 3D). The decreased contribution of glucose oxidation to producing reductive power results from a 4-fold decrease in pyruvate dehydrogenase (PDH) flux in hypoxia, which is consistent with the known PDH inhibition by hypoxia (Papandreou et al, 2006). In contrast, the contribution of other sources increases in hypoxia. The role of glutamine-supported oxidative phosphorylation in hypoxia is further demonstrated via a significant decrease in oxygen consumption and NADH/NAD+ ratio upon glutamine removal in hypoxia (P=0.03 for oxygen consumption and P=0.01 for NADH/NAD+ ratio; Figure 3E).
The changes in ATP production routes induced by hypoxia resemble those induced by Ras activation: that is, the increased reliance on aerobic glycolysis for ATP production, as well as the decreased reliance on glucose oxidation for generating mitochondrial reducing power. These changes may provide Ras-driven tumor cells an advantage in hypoxia.
To examine the generality of our observation of persistent oxidative ATP production in hypoxia, we analyzed the impact of antimycin and oligomycin treatment on hypoxic Ras- and Akt-driven cells (Figure 3F), with both drugs decreasing the ATP/ADP ratio by ∼ 50% in both cell lines, while also substantially increasing the NADH/NAD+ ratio. Consistent with these observations, we observed persistent oxygen uptake also in these cell lines in hypoxia. Similar to that in the parental cells, in hypoxia, glutamine deprivation was found to significantly decrease oxygen consumption and the NADH/NAD+ ratio in these cell lines (Figure 3G), emphasizing the general dependence of both normoxic- and hypoxic-cultured murine renal epithelial cells on glutamine to power oxidative phosphorylation irrespective of oncogene activation.
ATP production routes in cancer cell lines in both normoxia and hypoxia
To study the importance of glutamine-supported oxidative phosphorylation in other cell lines, we extended our analysis to two common cancer cell lines: 4T1, Akt-driven mouse mammary tumor cell line and ASPC1, a K-Ras-driven human pancreatic cancer cell line, in both normoxia and hypoxia. Similar to the iBMK cell line, in both 4T1 and ASPC, the majority of ATP is made oxidatively. Hypoxia decreases the oxidative phosphorylation rate by 60% in 4T1 and 40% in ASPC1, while increasing the glycolytic rate in both cell lines, and increases the fraction of glucose excreted as lactate instead of entering TCA cycle. However, in both cases, oxidative phosphorylation continues to account for more than 50% of total energy production (Figure 4A). To assess the importance of glucose and glutamine oxidation, we measured the oxygen consumption rate after glucose or glutamine removal (Figure 4B). In both cell lines, in either hypoxia or normoxia, glutamine removal more strongly reduced oxygen consumption than glucose removal, confirming an essential role for glutamine in supporting oxidative phosphorylation. Interestingly, in ASPC1 cells, especially in hypoxia, when glucose is removed from media, oxygen consumption increases. This suggests that, in glucose starvation, cells can upregulate oxidative phosphorylation, driven by glutamine and other sources, to compensate for the decreased glycolytic ATP production.
Figure 4.

Glutamine-driven oxidative ATP production in normoxic and hypoxic cancer cells. (A) ATP production rates from oxidative phosphorylation and glycolysis in 4T1 and ASPC1 cancer cells. (B) Oxygen consumption rates in hypoxia measured after switching cells to complete media or media lacking glucose or glutamine for 8 h (mean±s.d. of N=3, significant differences (P<0.05) by T-test marked with *). (C) Contribution of oxidative phosphorylation to ATP production in NCI-60 cell lines as predicted by nutrient uptake rate-constrained flux balance analysis.
To analyze the role of oxidative phosphorylation in a broader set of cell lines, we applied flux balance analysis (FBA) to predict metabolic flux rates through glycolysis and oxidative phosphorylation in each of the NCI-60 cell lines based on available metabolite uptake and secretion rates from Jain et al (2012) (see Methods), which did not include the oxygen uptake rate (preventing redox balancing in this analysis). Utilizing the same metabolic network model of glycolysis and TCA cycle as for the 13C-based flux analysis (Supplementary Table 3), FBA was applied to search for flux rates that maintain stoichiometric mass balance, optimally match the measured uptake and secretion rates (of glucose, lactate, glutamine, and glutamate), and have no thermodynamically infeasible cycles (Methods). Our FBA analysis shows that oxidative phosphorylation contributes 88% of the total ATP production on average across the 60 cell lines (Figure 4C). Notably, the predicted mitochondrial ATP production rates for all 60 cell lines analyzed here are consistent with cellular solvent capacity constraint, which gives rise to an upper threshold of 8.4 μmole/μl cells/h (Supplementary Figure 7) (Vazquez and Oltvai, 2011). Reassuringly, applying the same FBA approach to predict ATP production routes in the iBMK cell lines (using just corresponding uptake and secretion measurements) resulted in qualitatively similar results to those obtained with the isotope tracer data (with an average error of 5% in the prediction of glycolytic versus oxidative phosphorylation ATP production).
To further validate the predicted contribution of oxidative phosphorylation to ATP production, we repeated the flux analysis in the NCI60 cell lines using a genome-scale human metabolic network model (Duarte et al, 2007). We constrained metabolite uptake and secretion rates using data from Jain et al (as in the above analysis with the reduced network model of Supplementary Table 3) and further included a biomass reaction constraint (Methods) (Folger et al, 2011). Applying FBA to predict fluxes that maximize the ATP production rate resulted in an average contribution of oxidative phosphorylation to total ATP production of 84% across cell lines. Alternatively, constraining oxygen consumption rate to a range of likely rates (around the measured oxygen consumption in the iBMK cell lines) without maximizing ATP production predicted an average contribution of 70% (Supplementary Figure 8). In both cases, the predictions obtained with the genome-scale metabolic network model support the major contribution of oxidative phosphorylation to ATP production.
In summary, we have developed an approach to analyzing cellular metabolic fluxes including ATP production routes, which combines quantitatively 13C-tracer and oxygen uptake measurements. The results contribute to growing literature on the importance of glutamine in driving TCA cycle flux, especially in cells with activated Ras or grown in a hypoxic environment (Frezza et al, 2011; Gaglio et al, 2011; Le et al, 2012; Metallo et al, 2012). Importantly, even in such circumstances, we show that oxidative phosphorylation remains the largest quantitative contributor to ATP production. Moreover, we find that Ras has no net effect on ATP production, as the increase in glycolysis is offset by decreased oxidative phosphorylation. Taken in totality, these observations argue for a primary role of oxidative metabolism in most cancers.
Materials and methods
Cell lines and culture conditions
Immortalized baby mouse kidney (iBMK) epithelial cells were generated as described previously (Degenhardt et al, 2002). Briefly, primary kidney epithelial cells from mice double deficient for Bax and Bak (Bax−/−/Bak−/−) were immortalized by E1A and dominant-negative p53 expression (Degenhardt et al, 2002; Mathew et al, 2008). iBMK cells expressing human oncogenic H-RasV12G or myr-Akt were derived by electroporation with pcDNA1.H-RasV12G (Lin et al, 1995) or pcDNA3.myr-Akt (Plas et al, 2001), respectively, followed by zeocin selection. The resulting cell lines were grown in Dulbecco’s modified eagle media (DMEM) without pyruvate (Cellgro), supplemented with 10% dialyzed fetal bovine serum (HyClone) in all metabolomics experiments. For normoxia experiments, cells are grown in an incubator containing 5% CO2 and ambient oxygen at 37°C. For hypoxia experiments, cells are grown and all experiments are completed inside a hypoxia glove box (Coy Lab) containing 1% oxygen and 5% CO2 at 37°C. For labeling experiments, medium was prepared from DMEM without glucose or glutamine (Cellgro), with the desired isotopic form of glucose and/or glutamine added to a final concentration of 4.5 g/l glucose and 0.584 g/l glutamine. Short-term experiments (e.g., nutrient uptake and kinetic flux profiling) were conducted at 70–80% confluency; for longer-term labeling experiments, confluency varied as the cells multiplied.
Exchange rate measurements
Media samples were collected at various time points. Glucose, glutamine, and lactate were measured by enzymatic assay with electrochemical detection on a YSI7200 instrument (YSI, Yellow Springs, OH). Alanine and pyruvate were measured by LC-MS. Oxygen consumption was measured using a Seahorse XF24 flux analyzer (Seahorse Bioscience, North Billerica, MA). To measure oxygen uptake in hypoxia, the Seahorse instrument was placed in the hypoxia chamber with 1% oxygen. To probe the fraction of oxygen consumption, which is effectively coupled with the electron transport chain, we measured the oxygen consumption rate when cells were treated with an electron transport chain inhibitor, antimycin A. We observed that ∼80% of oxygen consumption across the studied cell lines and growth conditions was used for oxidative phosphorylation.
Metabolomic experiments and LC-MS analysis
For all metabolomic and isotope-tracer experiments, metabolism was quenched, and metabolites were extracted by quickly aspirating the media and immediately adding a 80:20 methanol:water extraction solution at −80°C.
Samples were analyzed using multiple LC-MS systems (each from Thermo Scientific and fed by electrospray ionization), as described previously (Munger et al, 2008; Lemons et al, 2010; Lu et al, 2010). In brief, a stand-alone orbitrap mass spectrometer (Exactive) operating in negative-ion mode was coupled to reversed-phase ion-pairing chromatography and used to scan from m/z 85–1000 at 1 Hz and 100 000 resolution; a TSQ Quantum Ultra triple-quadrupole mass spectrometer operating in positive-ion mode was coupled to hydrophilic interaction chromatography on an aminopropyl column and used to analyze selected compounds by multiple reaction monitoring; and a TSQ Quantum Discovery triple-quadrupole mass spectrometer operating in negative-ion mode was coupled to reverse-phase ion-pairing chromatography and used to analyze selected compounds by multiple reaction monitoring. Data were analyzed using the MAVEN software suite (Melamud et al, 2010). The results are adjusted for natural 13C abundance and enrichment impurity of labeled substrate supplied to cells.
Absolute metabolite levels were quantified as previously described (Bennett et al, 2008) and normalized by packed cell volume.
De novo serine synthesis rate
To quantify the rate of serine synthesis, cells were cultured in DMEM media containing U-13C-serine. The steady-state labeling pattern of intracellular 3-phosphoglycerate, serine, and glycine was measured by extracting metabolites after washing three times with ice-cold PBS. Labeled 3-phosphoglycerate was never observed, confirming that reverse serine synthesis pathway flux is negligible. Glycine was observed in two forms: unlabeled and M+2 (whose relative fractions are referred to as G0 and G2 below). Serine was observed in four forms: unlabeled, M+1, M+2, and M+3, with fractions S0, S1, S2, S3, respectively. These forms arise via de novo synthesis from 3-phosphoglycerate (making unlabeled via F1, see Supplementary Figure 9), uptake from media (making M+3 via F2), and reverse SHMT flux from glycine (making all possible forms, depending on the labeling of glycine and the methylene-THF methyl group, via F3). The fraction of methylene-THF with the reactive one-carbon unit unlabeled is denoted T0 and that labeled on the one-carbon unit is denoted T1. We also measured net serine uptake flux F2 by monitoring the change of serine concentration in the media.
Under isotopic steady state, the balance equations for the four labeling forms of serine can be formulated as following:
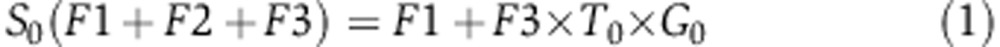 |
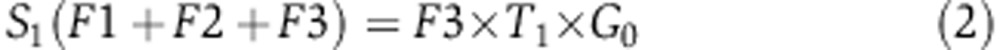 |
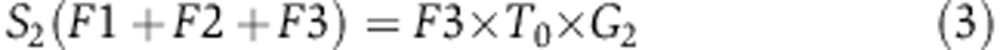 |
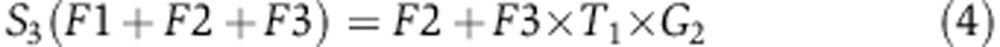 |
The de novo serine biosynthesis flux is derived by solving the above equations for F1:
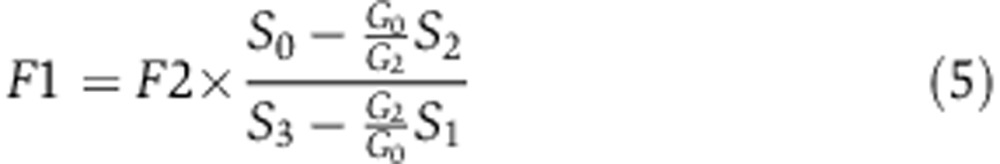 |
Redox-balanced metabolic flux analysis
To infer intracellular metabolic fluxes, we constructed a metabolic network model of glycolysis and TCA cycle (Supplementary Table 3) and applied metabolic flux analysis (MFA) to identify a flux distribution that optimally fits the following experimental data sets (Supplementary Figures 1–4 and Supplementary Table 5): (i) measured uptake and secretion rates of glucose, glutamine, lactate, pyruvate, and alanine; (ii) measured oxygen consumption rate used by oxidative phosphorylation; (iii) steady-state isotopic labeling pattern of pyruvate, α-ketoglutarate, citrate, malate, and cytosolic acetyl-CoA, where the labeling pattern of cytosolic acetyl-CoA was inferred based on the steady-state labeling pattern of fatty acids via isotopomer spectral analysis (ISA)(Kharroubi et al, 1992); (iv) consumption of metabolic intermediates for biomass production based on measured growth rate and biomass component contents. Specifically, we measured the DNA, RNA, and protein contents to be equal to 11, 14, and 88 μg/μl cells, respectively, in iBMK cells. Cellular demands for acetyl-CoA based on steady-state fatty acid concentrations, cellular growth rate, and fatty acid uptake rates are shown in Supplementary Table 5; and (v) measured flux in the serine biosynthesis pathway.
The MFA method was formulated as an optimization problem aiming to select a vector of fluxes v that maximizes the log-likelihood of measured mass-isotopomer distributions in both 13C-glucose and 13C-glutamine experiments, denoted 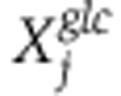 and
and 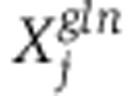 . The mass-isotopomer distributions produced by the set of fluxes v given the metabolic network in Supplementary Table 3 are denoted
. The mass-isotopomer distributions produced by the set of fluxes v given the metabolic network in Supplementary Table 3 are denoted 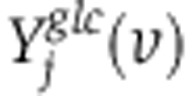 and
and 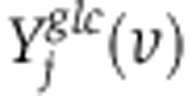 . Assuming a Gaussian error model for measured isotope labeling data, maximum log-likelihood is obtained by minimizing the variance-weighted sum of squared residuals between measured and computed mass-isotopomer distributions(Antoniewicz et al, 2006, 2007), where
. Assuming a Gaussian error model for measured isotope labeling data, maximum log-likelihood is obtained by minimizing the variance-weighted sum of squared residuals between measured and computed mass-isotopomer distributions(Antoniewicz et al, 2006, 2007), where 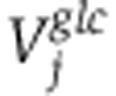 and
and 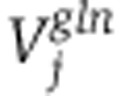 are diagonal matrices with the inverse of experimental variance for the labeling pattern of metabolite j:
are diagonal matrices with the inverse of experimental variance for the labeling pattern of metabolite j:
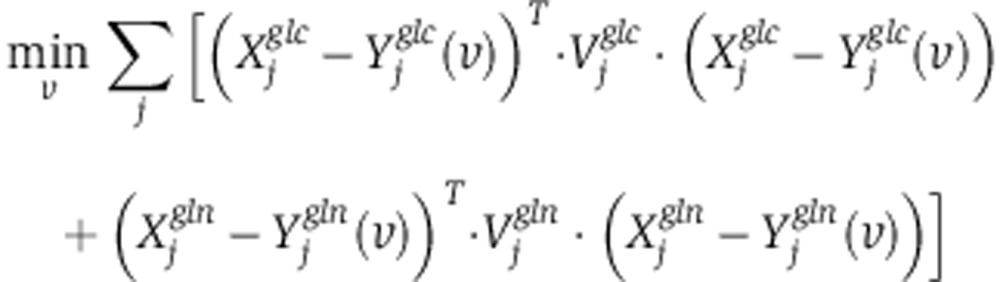 |
s. t.
 |
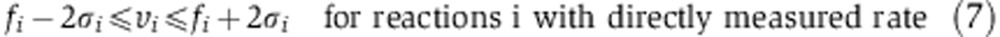 |
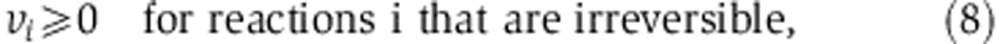 |
where S represents a stoichiometric matrix (with Sji representing the stoichiometric coefficient of the jth metabolite in the ith reaction), and fi and σi represent the measured flux through reaction i and s.d., respectively. Stoichiometric mass and redox-balance constraint is enforced by Equation 6. Metabolite uptake and secretion rates, biomass demand fluxes, serine biosynthesis rate, and oxidative phosphorylation rate (based on oxygen consumption rate) were constrained to experimental measurements, allowing them to vary by no more than two experimental s.d. from the mean measured fluxes (Equation 7; Supplementary Tables 3 and 5). Irreversible reactions are constrained to have positive flux (Equation 8). To efficiently compute the mass-isotopomer distributions for both the glucose- and glutamine labeling experiments (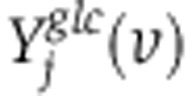 and
and 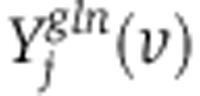 ) per each candidate flux vector (v) as part of the optimization, we employed elementary metabolite units (EMU)(Antoniewicz et al, 2007). The non-convex optimization problem was solved using Matlab’s Sequential Quadratic Optimization (SQP). To overcome potential local minima in SQP algorithm, the optimization problem was solved several times, starting from different sets of random fluxes.
) per each candidate flux vector (v) as part of the optimization, we employed elementary metabolite units (EMU)(Antoniewicz et al, 2007). The non-convex optimization problem was solved using Matlab’s Sequential Quadratic Optimization (SQP). To overcome potential local minima in SQP algorithm, the optimization problem was solved several times, starting from different sets of random fluxes.
To compute flux confidence intervals, we used the likelihood ratio test to compare the maximum log-likelihood estimation, computed by the above SQP optimization, with that obtained when constraining the flux to higher or lower values. Specifically, we iteratively ran the SQP optimization to compute the maximum log-likelihood estimation while constraining the flux to increasing (and then decreasing) values (with a step size equal to 5% of the flux predicted in the initial maximum log-likelihood estimation). The confidence interval bounds were determined based on the 95% quantile of χ2-distribution with one degree of freedom (Antoniewicz et al, 2006). The results of this iterative process (i.e., the probability of a reaction having a range of possible flux rates) were also used to calculate the s.d. of flux estimates. This computationally intensive computation of accurate flux confidence intervals took a total of ∼48 h on a standard laptop computer using Matlab.
To compute metabolite labeling kinetics given a flux vector (v) derived by the above optimization problem, we considered a system of ordinary differential equations that describe the abundance of isotopomer k of metabolite j at time t (denoted Uj,k(t)) as following(Noack et al, 2011):
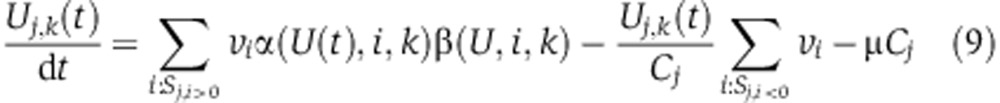 |
where the α(U(t),i,j,k) and β(U(t),i,j,k) are defined as follows, assuming that reaction i that makes metabolite j has two substrates s1 and s2: α(U(t),i,j,k) is equal to the sum of abundances of isotopomers of s1 that produce isotopomer k of metabolite j via reaction i, normalized by the steady-state concentration of metabolite j (denoted Cj). β(U(t),i,j,k) is defined analogously based on the abundance of isotopomers of s2. For reaction i that has one substrate, α(U(t),i,j,k) is defined in the same way, while β(U(t),i,j,k) is equal to 1. Growth rate is denoted μ. The three terms on the right hand side of the equation (from left to right) represent the sum of production rate of isotopomer k of metabolite j by metabolic reactions, the sum of consumption rate of the isotopomer by metabolic reactions, and the consumption of the isotopomer due to cellular growth. Metabolite labeling kinetics in terms of relative abundance of mass-isotopomers was calculated by summing the computed abundance of all isotopomer with a given mass in each time point. Calculated metabolite labeling kinetics for each cell line in normoxia for both glucose and glutamine labeling were compared with experimental measurements (Supplementary Figures 1–4).
Flux balance analysis (FBA) in NCI-60 cell lines
To predict the most likely fluxes for NCI-60 cell lines, we employed the same metabolic network model used in the MFA analysis described above (Supplementary Table 3) and applied the following FBA analysis to search for a steady-state flux distribution (denoted v) that matches metabolite uptake and secretion rates measured by Jain et al (2012):
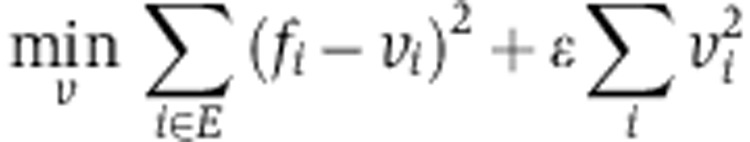 |
s. t.
 |
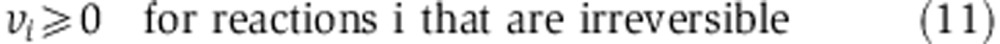 |
where fi denotes the measured uptake or secretion rate through reaction i (for every reaction i in reactions’ set E). Specifically, we used measured reactions for glucose uptake (F1) and lactate secretion (F12) taken from Jain et al (2012). Glutamine flux into TCA cycle (F11) was defined as glutamine uptake minus glutamate secretion on the basis of measurements from Jain et al. An upper bound on acetyl-CoA demand for fatty acid biosynthesis (F14) was calculated based on the cell line-specific growth rate (NCI’s DTP database http://dtp.nci.nih.gov/docs/misc/common_files/cell_list.html), assuming that fatty acids comprise less than 20% of cellular dry weight of 200 μg per 106 cells. S is the same stoichiometric matrix as in the MFA analysis (see Supplementary Table 3). The first term of the optimization function aims to minimize the difference between measured and predicted metabolite uptake and secretion rates, whereas the second term aims to minimize the total sum of square of flux (Schuetz et al, 2007). ε was set to 0.001. Similar results were obtained also for lower values of ε.
We used a similar approach to predict likely fluxes for NCI60 cell lines using a genome-scale metabolic network model (Duarte et al, 2007). Here, we explicitly constrained the flux through nutrient uptake and byproduct secretion reactions (for F1, F11, and F12), based on the experimental measurements. We further defined a growth reaction based on cellular biomass being 60% protein, 10% DNA and RNA, and 10% lipids, and constrained cellular growth rate based on experimental data (from NCI’s DTP database). We then applied FBA in two ways: (i) optimizing for maximal ATP production rate (and then minimizing the total sum of fluxes squared as in the above analysis); (ii) constraining oxygen consumption to be between 75–225 nmol/μl cells/h (where the lower bound is 50% lower and the upper bound is 50% higher than the average oxygen consumption by oxidative phosphorylation measured in the iBMK cell lines) and optimizing for minimal sum of fluxes squared. Varying protein mass between 50 and 90% and DNA/RNA and lipid mass between 3 and 20% changed the predicted contribution of oxidative phosphorylation by less than 1%.
Supplementary Material
Supplementary Figures S1–9, Supplementary Tables S1–5
Acknowledgments
We thank Hilary Coller, Bryson D Bennett, and Johanna MS Lemons for thoughtful discussions. This work was supported by NIH Center grant P50 GM071508, Challenge grant 1RC1 CA147961-02, and RO1 grant CA163591-01A1, and Stand Up To Cancer. JF is supported by an HHMI international student research fellowship. JJK is a Hope Funds for Cancer Research Fellow supported by the Hope Funds for Cancer Research (HFCR-11-03-01). TS was supported by grant from the Israel Science Foundation.
Author Contributions: JDR, JF, TS, and EW designed the research. JF, JJK, RM, and MKC conducted the experiments. TS and JF performed the computational analysis. JDR, TS, and JF wrote the manuscript.
Footnotes
The authors declare that they have no conflict of interest.
References
- Antoniewicz MR, Kelleher JK, Stephanopoulos G (2006) Determination of confidence intervals of metabolic fluxes estimated from stable isotope measurements. Metab Eng 8: 324–337 [DOI] [PubMed] [Google Scholar]
- Antoniewicz MR, Kelleher JK, Stephanopoulos G (2007) Elementary metabolite units (EMU): a novel framework for modeling isotopic distributions. Metab Eng 9: 68–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Sagi D, Feramisco JR (1986) Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science 233: 1061–1068 [DOI] [PubMed] [Google Scholar]
- Bennett BD, Yuan J, Kimball EH, Rabinowitz JD (2008) Absolute quantitation of intracellular metabolite concentrations by an isotope ratio-based approach. Nat Protoc 3: 1299–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun SY, Johnson C, Washburn JG, Cruz-Correa MR, Dang DT, Dang LH (2010) Oncogenic KRAS modulates mitochondrial metabolism in human colon cancer cells by inducing HIF-1alpha and HIF-2alpha target genes. Mol Cancer 9: 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degenhardt K, Chen G, Lindsten T, White E (2002) BAX and BAK mediate p53-independent suppression of tumorigenesis. Cancer Cell 2: 193–203 [DOI] [PubMed] [Google Scholar]
- Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, Nelson DA, Jin S, White E (2006) Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 10: 51–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degenhardt K, White E (2006) A mouse model system to genetically dissect the molecular mechanisms regulating tumorigenesis. Clin Cancer Res 12: 5298–5304 [DOI] [PubMed] [Google Scholar]
- Duarte NC, Becker SA, Jamshidi N, Thiele I, Mo ML, Vo TD, Srivas R, Palsson BO (2007) Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc Natl Acad Sci USA 104: 1777–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB (2004) Akt stimulates aerobic glycolysis in cancer cells. Cancer Res 64: 3892–3899 [DOI] [PubMed] [Google Scholar]
- Folger O, Jerby L, Frezza C, Gottlieb E, Ruppin E, Shlomi T (2011) Predicting selective drug targets in cancer through metabolic networks. Mol Syst Biol 7: 501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Zheng L, Tennant DA, Papkovsky DB, Hedley BA, Kalna G, Watson DG, Gottlieb E (2011) Metabolic profiling of hypoxic cells revealed a catabolic signature required for cell survival. PLoS ONE 6: e24411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, Alberghina L, Stephanopoulos G, Chiaradonna F (2011) Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol 7: 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guppy M, Kong SE, Niu X, Busfield S, Klinken SP (1997) Method for measuring a comprehensive energy budget in a proliferating cell system over multiple cell cycles. J Cell Physiol 170: 1–7 [DOI] [PubMed] [Google Scholar]
- Guppy M, Leedman P, Zu X, Russell V (2002) Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem J 364: 309–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung YP, Albeck JG, Tantama M, Yellen G (2011) Imaging cytosolic NADH-NAD(+) redox state with a genetically encoded fluorescent biosensor. Cell Metab 14: 545–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB, Mootha VK (2012) Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 336: 1040–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharroubi AT, Masterson TM, Aldaghlas TA, Kennedy KA, Kelleher JK (1992) Isotopomer spectral analysis of triglyceride fatty acid synthesis in 3T3-L1 cells. Am J Physiol 263: E667–E675 [DOI] [PubMed] [Google Scholar]
- Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, Zimmerman LJ, Liebler DC, Slebos RJ, Lorkiewicz PK, Higashi RM, Fan TW, Dang CV (2012) Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab 15: 110–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemons JM, Feng XJ, Bennett BD, Legesse-Miller A, Johnson EL, Raitman I, Pollina EA, Rabitz HA, Rabinowitz JD, Coller HA (2010) Quiescent fibroblasts exhibit high metabolic activity. PLoS Biol 8: e1000514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HJ, Eviner V, Prendergast GC, White E (1995) Activated H-ras rescues E1A-induced apoptosis and cooperates with E1A to overcome p53-dependent growth arrest. Mol Cell Biol 15: 4536–4544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, Debnath J (2011) Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell 22: 165–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Clasquin MF, Melamud E, Amador-Noguez D, Caudy AA, Rabinowitz JD (2010) Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone orbitrap mass spectrometer. Anal Chem 82: 3212–3221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Degenhardt K, Haramaty L, Karp CM, White E (2008) Immortalized mouse epithelial cell models to study the role of apoptosis in cancer. Methods Enzymol 446: 77–106 [DOI] [PubMed] [Google Scholar]
- Melamud E, Vastag L, Rabinowitz JD (2010) Metabolomic analysis and visualization engine for LC-MS data. Anal Chem 82: 9818–9826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, Kelleher JK, Vander Heiden MG, Iliopoulos O, Stephanopoulos G (2012) Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481: 380–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger J, Bennett BD, Parikh A, Feng XJ, McArdle J, Rabitz HA, Shenk T, Rabinowitz JD (2008) Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat Biotechnol 26: 1179–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noack S, Noh K, Moch M, Oldiges M, Wiechert W (2011) Stationary versus non-stationary (13)C-MFA: a comparison using a consistent dataset. J Biotechnol 154: 179–190 [DOI] [PubMed] [Google Scholar]
- Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC (2006) HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 3: 187–197 [DOI] [PubMed] [Google Scholar]
- Pfeiffer T, Schuster S, Bonhoeffer S (2001) Cooperation and competition in the evolution of ATP-producing pathways. Science 292: 504–507 [DOI] [PubMed] [Google Scholar]
- Plas DR, Talapatra S, Edinger AL, Rathmell JC, Thompson CB (2001) Akt and Bcl-xL promote growth factor-independent survival through distinct effects on mitochondrial physiology. J Biol Chem 276: 12041–12048 [DOI] [PubMed] [Google Scholar]
- Robey RB, Hay N (2009) Is Akt the "Warburg kinase"?-Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol 19: 25–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz R, Kuepfer L, Sauer U (2007) Systematic evaluation of objective functions for predicting intracellular fluxes in Escherichia coli. Mol Syst Biol 3: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL (2010) HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev 20: 51–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlomi T, Benyamini T, Gottlieb E, Sharan R, Ruppin E (2011) Genome-scale metabolic modeling elucidates the role of proliferative adaptation in causing the Warburg effect. PLoS Comput Biol 7: e1002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennant DA, Frezza C, MacKenzie ED, Nguyen QD, Zheng L, Selak MA, Roberts DL, Dive C, Watson DG, Aboagye EO, Gottlieb E (2009) Reactivating HIF prolyl hydroxylases under hypoxia results in metabolic catastrophe and cell death. Oncogene 28: 4009–4021 [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma A, Palsson BO (1994) Stoichiometric flux balance models quantitatively predict growth and metabolic by-product secretion in wild-type Escherichia coli W3110. Appl Environ Microbiol 60: 3724–3731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez A, Liu J, Zhou Y, Oltvai ZN (2010) Catabolic efficiency of aerobic glycolysis: the Warburg effect revisited. BMC Syst Biol 4: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez A, Oltvai ZN (2011) Molecular crowding defines a common origin for the Warburg effect in proliferating cells and the lactate threshold in muscle physiology. PLoS One 6: e19538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC (2005) Mitochondria and cancer: Warburg addressed. Cold Spring Harb Symp Quant Biol 70: 363–374 [DOI] [PubMed] [Google Scholar]
- Warburg O (1956) On the origin of cancer cells. Science 123: 309–314 [DOI] [PubMed] [Google Scholar]
- Yang D, Wang MT, Tang Y, Chen Y, Jiang H, Jones TT, Rao K, Brewer GJ, Singh KK, Nie D (2010) Impairment of mitochondrial respiration in mouse fibroblasts by oncogenic H-RAS(Q61L). Cancer Biol Ther 9: 122–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Jin J, Hu Q, Zhou HM, Yi J, Yu Z, Xu L, Wang X, Yang Y, Loscalzo J (2011) Genetically encoded fluorescent sensors for intracellular NADH detection. Cell Metab 14: 555–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu XL, Guppy M (2004) Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys Res Commun 313: 459–465 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures S1–9, Supplementary Tables S1–5