

Abstract
Joubert syndrome and related disorders (JSRDs) are genetically heterogeneous and characterized by a distinctive mid-hindbrain malformation. Causative mutations lead to primary cilia dysfunction, which often results in variable involvement of other organs such as the liver, retina, and kidney. We identified predicted null mutations in CSPP1 in six individuals affected by classical JSRDs. CSPP1 encodes a protein localized to centrosomes and spindle poles, as well as to the primary cilium. Despite the known interaction between CSPP1 and nephronophthisis-associated proteins, none of the affected individuals in our cohort presented with kidney disease, and further, screening of a large cohort of individuals with nephronophthisis demonstrated no mutations. CSPP1 is broadly expressed in neural tissue, and its encoded protein localizes to the primary cilium in an in vitro model of human neurogenesis. Here, we show abrogated protein levels and ciliogenesis in affected fibroblasts. Our data thus suggest that CSPP1 is involved in neural-specific functions of primary cilia.
Main Text
Joubert syndrome (JBTS [MIM 213300]) and related disorders (JSRDs) are a group of congenital conditions characterized by the presence of a unique brainstem and cerebellar malformation, including cerebellar vermis hypoplasia and/or dysplasia, elongated superior cerebellar peduncles, and deepened interpeduncular fossa, which together are recognized as the “molar tooth sign” on brain imaging (MTI).1 The most common clinical features comprise cerebellar ataxia, hypotonia, oculomotor apraxia, abnormal respiratory patterns in the neonatal periods, and delayed psychomotor development. Neurological findings are frequently accompanied by additional major-organ abnormalities, such as retinal degeneration, cystic kidney, liver fibrosis, and polydactyly.2,3
To date, biallelic mutations in 20 genes, all involved in primary cilium biology, are known to compose the genetic spectrum of JSRDs. The primary cilium is a microtubule-based organelle that protrudes from most epithelial-derived cells in animals to act as a sensory hub and coordinate intracellular responses to morphogens and signaling molecules.4 All cilia arise from the mother centriole, the older centriole inherited during mitosis, which after docking to the plasma membrane protrudes a microtubule extension apically to form the ciliary axoneme.5 Protein modules located at the base of the cilium form a specialized region (termed the transition zone) that functions as a ciliary gate and tightly regulates the molecular composition of the ciliary compartment. This region is prone to JSRD-related mutations that often associate with tissue-specific defects in a protein-module-dependent manner.6 However, genetic causes of nearly 50% of JSRD-affected individuals are still unknown.
In order to uncover further genetic causes of JSRDs, we recruited a worldwide cohort of 526 unique probands on the basis of neurological findings from regions with high consanguinity rates and focused on those individuals whose brain MRI documented MTI as the major anomaly. Of those, we evaluated a total of 287 individuals for mutations in protein-coding genomic regions by using whole-exome sequencing of at least one, but often two (when available), affected members in a family (or in some instances, parents or unaffected siblings) for a total of 426 sequenced individuals. Recruitment of affected individuals was focused in parts of the world with the highest consanguinity, and overall, 72% of the families had documented parental consanguinity. The study was approved by the institutional review board at the University of California, San Diego, and the families provided age-dependent and cognition-appropriate informed consent or assent.
Coding genomic regions (exons) were captured from blood DNA with the use of the NimbleGen Exome 44 Mb Library 2.0 or Agilent SureSelect Human All Exome 50 Mb Kit and were subsequently sequenced on an Illumina HiSeq2000 instrument, resulting in ∼94% recovery at >10× coverage. Raw paired-end sequencing data were processed according to the Broad Institute’s Genome Sequencing and Analysis Program recommendations. Reads were aligned to NCBI Genome build 37 of the human reference genome with the Burrows-Wheeler Aligner (BWA) with standard parameters. PCR duplicates were removed, scored for quality, and recalibrated with Picard. The Genome Analysis Toolkit (GATK) software (version 2.12) was used for realigning around indels for repairing any misalignment by the BWA. Single-nucleotide variants, genotypes, and quality scores were identified, called, and recalibrated, respectively, with GATK. Variants were annotated with SeattleSeq, OMIM, Phastcons, and PolyPhen-2 and profiled in a custom mySQL database for integrating data across multiple sequencing batches.7 Standard variant filters, including SNP quality (≥20), proximity to indels (≥5 bp away), and read depth (>4×), were applied.
Variants were analyzed by an automated prioritization workflow taking into account familial inheritance patterns, variant severity (nonsense, splice, and indel > missense), homozygous haplotypes, and allele frequency in ethnically matched control data sets for identifying possible homozygous and compound-heterozygous deleterious candidates, which were then analyzed for the most likely deleterious. For families affected by obvious deleterious mutations in known JSRD-associated genes, the single variant was tested for segregation in the family. For other families, a list of all possible deleterious variants passing the threshold of GERP score > 4 or Phastcon score > 0.8 were tested for segregation (average of four alleles per family), and only those in which a single deleterious variant segregated were marked as potentially causative. Each potentially causative mutation was tested by Sanger sequencing in the whole family for excluding exome sequencing error or variants not passing segregation analysis (i.e., according to a recessive inheritance model).
Approximately 40% of the 287 evaluated families were found to be affected by mutations in genes already associated with JSRDs. With 22, 20, and 18 identified biallelic mutations, CEP290 (MIM 610142), C5orf42 (MIM 614571), and AHI1 (MIM 608894), respectively, were the most recurrent (Figure 1A). Most of these were predicted to alter splice sites and/or generate premature stop codons. With a few exceptions, where renal and retinal findings were documented, individuals with CEP290, C5orf42, and AHI1 mutations did not show overt extraneural findings other than the typical JSRD mid-hindbrain malformation, supporting the finding that mutations in these genes associate with cerebello-oculo or cerebello-oculo-renal syndrome. Two individuals with AHI1 mutations had corpus callosum hypoplasia on brain MRI, and one showed an abnormally thin brainstem. Mutations in CC2D2A (MIM 612013) (11 individuals) and TMEM67 (MIM 609884) (ten individuals) were also quite prevalent in our cohort, and despite lower representation, most of the already known JSRD-associated genes—including TCTN2 (MIM 613846) (five individuals), INPP5E (MIM 613037), MKS1 (MIM 609883), TMEM237 (MIM 614423), and NPHP8 (MIM 610937) (four individuals each), TMEM138 (MIM 614459) (three individuals), KIF7 (MIM 611254), OFD1 (MIM 300170), TCTN1 (MIM 609863), and TCTN3 (MIM 613847) (two individuals each), and NPHP1 (MIM 607100) (one individual)—were also present in our cohort. These results compose a reliable representation of the distribution of disease-associated genes in the general JSRD population and validate our recruitment strategy.
Figure 1.
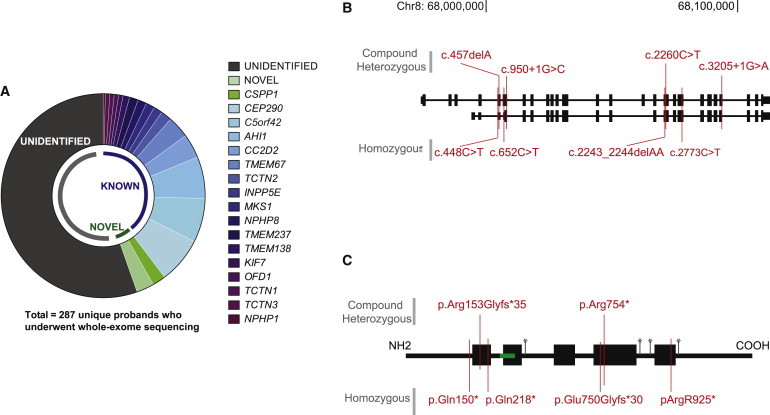
CSPP1 Is Recurrently Mutated in JBTS
(A) Whole-exome sequencing results summarized for 287 unique probands indicate genes demonstrating evidence of mutations. The most commonly mutated genes were CEP290, C5orf42, and AHI1. Six unique probands displayed mutations in CSPP1 (light green). In more than half of affected individuals, a causative mutation could not be identified.
(B) CSPP1 mutation locations relative to the genomic exon locations in hg19. Compound-heterozygous mutations are shown on the top, and homozygous mutations are shown on the bottom.
(C) CSPP1 alteration locations relative to the predicted protein. Blocks represent coiled-coil domains, green indicates the nuclear localization signal, and asterisks indicate predicted phosphorylation sites.
Of the remaining screened cases, we were able to identify causative mutations in genes previously not associated with JSRDs in 5%. For the unidentified 55%, we anticipate that either mutations fall in poorly covered exonic regions (with null or low numbers of reads) or noncoding genomic regions (thus not accessible to exome sequencing) or these individuals display multiple potential candidates or more subtle exomic mutations that are not distinguishable with the prediction tools we applied. From the 5% in which we identified mutations, 3% (eight probands) were in families affected by a single rare most likely deleterious variant, were in genes predicted to be involved in ciliary function, were validated by Sanger sequencing, and segregated with the disease in the families. By far, the most highly mutated gene was CSPP1 (RefSeq accession number NM_024790.6) (six probands), which had eight unique mutations. Four were homozygous mutations occurring within blocks of homozygosity8 in each of three consanguineous families from India, Libya, and Egypt and one nonconsanguineous Mexican family (Figure S1, available online). The additional four mutations were present in compound heterozygosity in two nonconsanguineous families from the United States and China (Figures 1B and 1C).
The clinical phenotype of the affected children included hypotonia, developmental delay, intellectual disability, hyperpnea, and apnea episodes in the neonatal period and the MTI (Figure 2). Half of the affected children also had ataxia (3/6) and variable ophthalmologic findings such as retinopathy (1/6), oculomotor apraxia, nystagmus, and bilateral ptosis (4/6), as well as hypoplasia of the corpus callosum and the brain stem (3/6) (Table1). Hepatic fibrosis, nephronophthisis, and polydactyly were not present in any individual with CSPP1 mutations. Coexistent obesity, which is linked to ciliopathies, was only observed in MTI-136. The fact that there was little extra-CNS involvement suggests that CSPP1 mutations primarily associate with the classical form of JBTS.9
Figure 2.
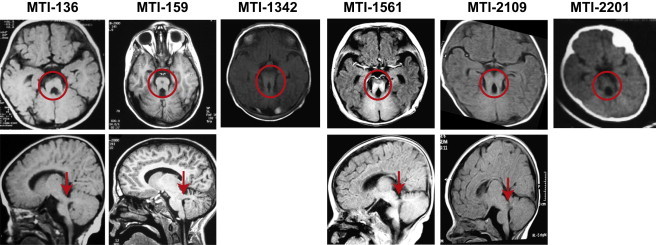
Brain Imaging Demonstrates Molar Tooth Sign in Individuals with CSPP1 Muations
Axial MRI (upper panels) and parasagittal MRI (lower panels, where available). Images show molar tooth sign (red circle) with elongated superior cerebellar peduncles, deepened interhemispheric fossa, and cerebellar vermis hypoplasia. Lower panels show thick and horizontally oriented superior cerebellar peduncles in all affected children (red arrows) and thin corpus callosum in MTI-136, MTI-1561, and MTI-2109.
Table 1.
Clinical Features of Individuals with CSPP1 Mutations in the Indicated Families
|
Family ID |
||||||
|---|---|---|---|---|---|---|
| MTI-136 | MTI-159 | MTI-1342 | MTI-1561 | MTI-2109 | MTI-2201 | |
| Country of origin | Mexico | United States | India | China | Libya | Egypt |
| Gender | male | female | male | male | male | female |
| Consanguinity | − | − | + | − | + | + |
| cDNA mutation | c.652C>T | c.[950+1G>C];[3205+1G>A] | c.2243_2244delAA | c.[2260C>T];[457delA] | c.2773C>T | c.448C>T |
| Protein alteration | p.Gln218∗ | splice; splice | p.Glu750Glyfs∗30 | p.[Arg754∗];[Arg153Glyfs∗35] | p.Arg925∗ | p.Gln150∗ |
| Neurological Findings | ||||||
| Hypotonia | + | + | + | + | + | + |
| Ataxia | + | + | + | − | − | − |
| Psychomotor delay | + | + | + | + | + | + |
| Intellectual disability | + | + | + | − | + | + |
| Breathing abnormalities | − | + | + | − | + | + |
| Seizures | − | − | − | − | − | − |
| Macrocephaly | − | − | − | − | − | − |
| Ophthalmologic Findings | ||||||
| Retinopathy | + | − | − | − | − | − |
| Coloboma | − | − | − | − | − | − |
| Oculomotor apraxia | + | + | + | − | − | + |
| Nystagmus | + | + | − | − | + | + |
| Other | bilateral ptosis | − | − | bilateral ptosis | bilateral ptosis and exotropia | bilateral ptosis |
| Miscellaneous Findings | ||||||
| Nephronophthisis | − | − | − | NA | − | − |
| Hepatic fibrosis | NA | − | − | NA | − | − |
| Polydactyly | − | − | − | − | − | − |
| Obesity | + | − | − | − | − | − |
| Other | mild SNHL | − | exitus due to pneumonia | moderate SNHL | vesicoureteral reflux | − |
| Cranial MRI Findings | ||||||
| Molar tooth sign | + | + | + | + | + | + |
| Cerebellar vermis dysgenesis | + | + | + | + | + | + |
| Hypoplasia of the brainstem | + | + | − | + | − | − |
| Occipital meningocele | − | − | − | − | − | − |
| Thin corpus callosum | + | − | NA | + | + | NA |
The following abbreviations are used: NA, not available; and SNHL, sensorineural hearing loss.
A finding that merits attention in our otherwise classical JBTS is the mild to moderate sensorineural hearing loss (SNHL) that was present in two of the affected children (MTI-136 and MTI-1561). The presence of hearing loss as a feature of JBTS was assessed in 22 Dutch individuals,10 and in only three cases (aged 17–26 years) was there mild SNHL with no compelling evidence of significant impairment. Previously, hearing thresholds were reported to be subclinically increased in a group of adolescent individuals with the ciliopathy Bardet-Biedl syndrome.11 Contrary to these two groups, children with CSPP1 mutations were fairly young when hearing loss was detected (5 years old and 6 months old). As more cases of CSPP1-linked JBTS are reported, it should be possible to determine whether this association is greater than expected by chance.
We confirmed CSPP1 mutations in all the affected individuals by Sanger sequencing and validated segregation on the basis of a strict recessive model of inheritance with full penetrance by sequencing the whole family (Figure S2). Two were substitutions of conserved first nucleotides of splice donor sites, two were deletions of one or two nucleotides, and the remaining four were nonsense mutations; all were predicted to result in premature stop codons. With the exception of CSPP1 c.2243_2244delAA, which was found in heterozygosity in the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Server with an allele frequency less than 0.001, none of the other mutations were present in any publically available data set or in our in-house exome sequences of 2,500 individuals. These data suggest that biallelic CSPP1 loss-of-function mutations result in JSRDs.
CSPP1 generates two well-documented splice variants that differ by the transcriptional start site and the alternative splicing of an exon, resulting in both long and short isoforms (Figure 1B). Five mutations occur within the coding exons of the two isoforms, whereas the three most upstream mutations alter coding exons of the longer isoform; these latter coding exons correspond to the 5′ UTR of the shorter isoform. Previous work has suggested that the two isoforms show largely redundant activity; however, with the exception of some variability in the expression of the two isoforms along the cell cycle, the longer isoform is the more abundant in all tested cell lines.12 These data indicate that the long isoform could be the more physiologically relevant. Although we do not know how mutations in the 5′ UTR might affect protein levels of the short isoform, the similar clinical features of children with CSPP1 mutations suggest that all the mutations equally affect protein function.
In order to determine relative CSPP1 abundance in human tissues, we performed RT-PCR on a collection of RNA from fetal brain, cortex, and cerebellum and adult brain structures and nonneural major body organs by using primers that overlap coding exons of both CSPP1 isoforms. After 30 cycles of amplification, neural tissue showed the most abundant CSPP1 signal, which was clearly enriched in the adult and fetal cerebellum (Figure 3A). These data support CSPP1 relevance in human brain development, especially in the developing cerebellum, which correlates well with the specific mid-hindbrain malformation found in children with CSPP1 mutations.
Figure 3.
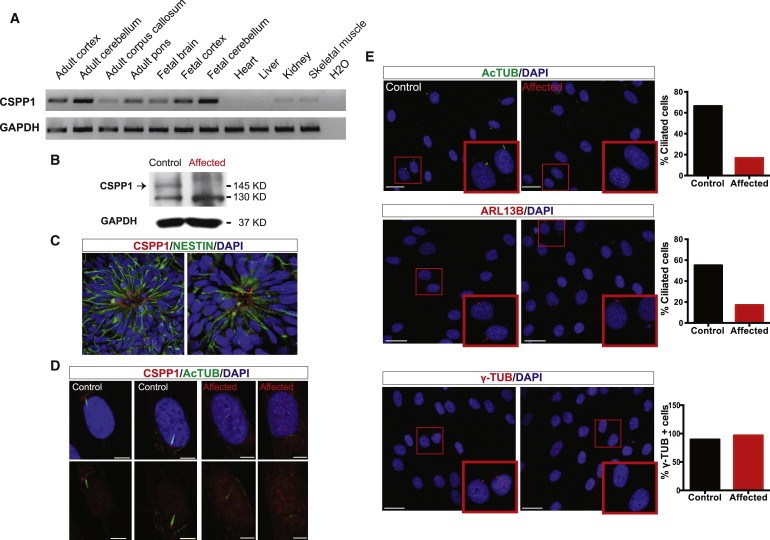
CSPP1 Is Expressed Predominantly in Neural Tissues Localized to Cilia, and the Truncating Mutation Abrogates Ciliogenesis
(A) RNA from human tissue RNA collection (Clontech) was retrotranscribed to cDNA with the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen) with oligodT. PCR with primers specific to CSPP1 demonstrated that human neural tissues, and especially the cerebellum, have the highest expression.
(B) Immunoblot with CSPP1 antibody (Proteintech) (arrow) in MTI-2109 control and affected fibroblasts showed severely reduced protein levels in the presence of the mutation. GAPDH (Millipore) was used as a loading control.
(C) CSPP1 immunostaining (red) in human-embryonic-stem-cell-derived neural rosettes showed localization in the apical region (close to the lumen), suggestive of colocalization with primary cilium.
(D) CSPP1 (red) localized to the base of the primary cilium (green) in control fibroblasts and was absent in MTI-2109 affected fibroblasts. The scale bar represents 5 μm.
(E) Acetylated tubulin (AcTUB) (Sigma) immunostaining (upper panel, green) and ARL13B immunostaining (middle panel, red) in MTI-2109 control and affected fibroblasts indicated a ciliary defect in mutated cells. γ-tubulin (γ-TUB) immunostaining (lower panel, red) indicated that the centrosome was equally present in affected and control cells. Graphs show the percentage of fibroblasts (n = 105–256) with AcTUB-positive (upper panel) and ARL13B-positive (middle panel) cilia and a γ-TUB-positive centrosome (lower panel). Scale bars represent 25 μm. DAPI (blue) was used for labeling DNA.
CSPP1, as the name denotes, is localized to the centrosomes and mitotic spindle. The longer predicted isoform is 1,221 amino acids and has five coiled-coil domains, a nuclear localization signal, and four serine phosphorylation sites, but no other notable protein domains. As mitosis progresses, it migrates from the spindle poles in metaphase to the central spindle in anaphase.12,13 During this process, CSPP1 interacts with Myo-GEF, and the depletion of either protein results in mitotic furrow regression and cell-cycle arrest in a low but significant proportion of cultured HeLa cells.14 In children with homozygous CSPP1 mutations, however, normal body and brain size (Table 1 and Figure 2), as well as unremarkable general organ size, argue against a required role for CSPP1 in general cell proliferation and rather suggest alternative physiological roles. To test this, we cultured skin fibroblasts from the heterozygous father (obligate carrier control) and affected children (homozygous null mutation) from family MTI-2109 and found unremarkable proliferation rates (data not shown). Immunoblot analysis showed the absence of CSPP1 in affected individuals’ cells (Figure 3B). We conclude that CSPP1 depletion does not grossly impair cell proliferation, although we cannot exclude that CSPP1 regulates proliferation in specific cell types, such as, perhaps, in granule cells that compose the cerebellar vermis.
CSPP1 was found to localize at the base and transition zone of the primary cilium and to interact with RPGRIP1L (NPHP8),15 a protein often associated with nephronophthisis or JBTS. Although the clinical features of affected individuals clearly suggest a defect in primary cilium function, none displayed clinically impaired renal function. In addition, the screening of an additional 768 individuals with a spectrum of nephronophthisis-related phenotypes demonstrated no CSPP1 mutations, suggesting a tight link specifically with JSRDs and neural-specific functions. Consistent with the brain-specific phenotype of individuals with CSPP1 mutations, the protein was found to localize to the apical zone of neural progenitors in human-embryonic-stem-cell-derived neural rosettes, the site of the neuronal primary cilium16 (Figure 3C).
Finally, we assessed the effect of CSPP1 depletion on primary cilia biogenesis. We cultured fibroblasts from affected children and controls under serum starvation for 24 hr and immunostained them with acetylated tubulin antibody, enriched in primary cilia. As expected, most of the control fibroblasts displayed an evident primary cilium with CSPP1 localized at the base (Figure 3D). In the few mutant fibroblasts where we found structures resembling primary cilia (longer than 1 μm), CSPP1 was absent, which correlates with the immunoblot analysis. Furthermore, quantification demonstrated a defect in ciliogenesis by showing less ciliary acetylated tubulin and ARL13B17 staining but the same number of centrosomes labeled with γ-tubulin (Figure 3E). All together, our data suggest that CSPP1 is required for proper primary cilium formation or stability and that CSPP1 mutations result in abnormal mid-hindbrain development.
In summary, we report that, in our cohort, CSPP1 is the most commonly mutated gene not previously associated with JSRDs. Although mutated in just six probands, this gene ranks among the top handful of most commonly mutated genes in our JSRD cohort, suggesting that it should be considered especially in individuals with the classical form of JBTS and in the setting of hearing loss. Clarifying a required role in mitosis and cell-cycle progression will require future work, but the clinical and cellular phenotypes we uncovered suggest that CSPP1 plays a role in cilia function, probably by interacting with other ciliopathy proteins at the basal body and perhaps also at the transition zone.
Acknowledgments
We thank the affected children and their families for their invaluable contributions to this study, supported by National Institutes of Health grants R01NS041537, R01NS048453, R01NS052455, P01HD070494, and P30NS047101 (J.G.G.) and DK068306 (F.H.), the Howard Hughes Medical Institute (J.G.G. and F.H.), and the California Institute of Regenerative Medicine (N.A.). We thank the Broad Institute (U54HG003067 to E. Lander), the Yale Center for Mendelian Disorders (U54HG006504 to R. Lifton and M. Gunel) for sequencing support, T. Caspary for ARL13B antibody, and J. Santini at the University of California, San Diego Neuroscience Microscopy Core. CSPP1 has been designated as JBTS21 by the HUGO Gene Nomenclature Committee.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
SeattleSeq Annotation 137, http://snp.gs.washington.edu/SeattleSeqAnnotation137/
Accession Numbers
The dbGaP accession number for the exome data reported in this paper is phs000288.
References
- 1.Maria B.L., Hoang K.B., Tusa R.J., Mancuso A.A., Hamed L.M., Quisling R.G., Hove M.T., Fennell E.B., Booth-Jones M., Ringdahl D.M. “Joubert syndrome” revisited: key ocular motor signs with magnetic resonance imaging correlation. J. Child Neurol. 1997;12:423–430. doi: 10.1177/088307389701200703. [DOI] [PubMed] [Google Scholar]
- 2.Valente E.M., Brancati F., Dallapiccola B. Genotypes and phenotypes of Joubert syndrome and related disorders. Eur. J. Med. Genet. 2008;51:1–23. doi: 10.1016/j.ejmg.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 3.Sattar S., Gleeson J.G. The ciliopathies in neuronal development: a clinical approach to investigation of Joubert syndrome and Joubert syndrome-related disorders. Dev. Med. Child Neurol. 2011;53:793–798. doi: 10.1111/j.1469-8749.2011.04021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goetz S.C., Anderson K.V. The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet. 2010;11:331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sorokin S. Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. J. Cell Biol. 1962;15:363–377. doi: 10.1083/jcb.15.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garcia-Gonzalo F.R., Corbit K.C., Sirerol-Piquer M.S., Ramaswami G., Otto E.A., Noriega T.R., Seol A.D., Robinson J.F., Bennett C.L., Josifova D.J. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Genet. 2011;43:776–784. doi: 10.1038/ng.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seelow D., Schuelke M., Hildebrandt F., Nürnberg P. HomozygosityMapper—an interactive approach to homozygosity mapping. Nucleic Acids Res. 2009;37(Web Server issue):W593–W599. doi: 10.1093/nar/gkp369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brancati F., Dallapiccola B., Valente E.M. Joubert Syndrome and related disorders. Orphanet J. Rare Dis. 2010;5:20. doi: 10.1186/1750-1172-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kroes H.Y., Van Zanten B.G., De Ru S.A., Boon M., Mancini G.M., Van der Knaap M.S., Poll-The B.T., Lindhout D. Is hearing loss a feature of Joubert syndrome, a ciliopathy? Int. J. Pediatr. Otorhinolaryngol. 2010;74:1034–1038. doi: 10.1016/j.ijporl.2010.05.034. [DOI] [PubMed] [Google Scholar]
- 11.Ross A.J., May-Simera H., Eichers E.R., Kai M., Hill J., Jagger D.J., Leitch C.C., Chapple J.P., Munro P.M., Fisher S. Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat. Genet. 2005;37:1135–1140. doi: 10.1038/ng1644. [DOI] [PubMed] [Google Scholar]
- 12.Patzke S., Stokke T., Aasheim H.C. CSPP and CSPP-L associate with centrosomes and microtubules and differently affect microtubule organization. J. Cell. Physiol. 2006;209:199–210. doi: 10.1002/jcp.20725. [DOI] [PubMed] [Google Scholar]
- 13.Patzke S., Hauge H., Sioud M., Finne E.F., Sivertsen E.A., Delabie J., Stokke T., Aasheim H.-C. Identification of a novel centrosome/microtubule-associated coiled-coil protein involved in cell-cycle progression and spindle organization. Oncogene. 2005;24:1159–1173. doi: 10.1038/sj.onc.1208267. [DOI] [PubMed] [Google Scholar]
- 14.Asiedu M., Wu D., Matsumura F., Wei Q. Centrosome/spindle pole-associated protein regulates cytokinesis via promoting the recruitment of MyoGEF to the central spindle. Mol. Biol. Cell. 2009;20:1428–1440. doi: 10.1091/mbc.E08-01-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patzke S., Redick S., Warsame A., Murga-Zamalloa C.A., Khanna H., Doxsey S., Stokke T. CSPP is a ciliary protein interacting with Nephrocystin 8 and required for cilia formation. Mol. Biol. Cell. 2010;21:2555–2567. doi: 10.1091/mbc.E09-06-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mirzadeh Z., Merkle F.T., Soriano-Navarro M., Garcia-Verdugo J.M., Alvarez-Buylla A. Neural stem cells confer unique pinwheel architecture to the ventricular surface in neurogenic regions of the adult brain. Cell Stem Cell. 2008;3:265–278. doi: 10.1016/j.stem.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caspary T., Larkins C.E., Anderson K.V. The graded response to Sonic Hedgehog depends on cilia architecture. Dev. Cell. 2007;12:767–778. doi: 10.1016/j.devcel.2007.03.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.