

Abstract
The nucleus is the defining feature of eukaryotic cells and often represents the largest organelle. Over the past decade, it has become apparent that the nucleus is tightly integrated into the structural network of the cell through so-called LINC (Linker of the nucleoskeleton and cytoskeleton) complexes, which enable transmission of forces between the nucleus and cytoskeleton. This physical connection between the nucleus and the cytoskeleton is essential for a broad range of cellular functions, including intracellular nuclear movement and positioning, cytoskeletal organization, cell polarization, and cell migration. Recent reports further indicate that forces transmitted from the extracellular matrix to the nucleus via they cytoskeleton may also directly contribute to the cell's ability to probe its mechanical environment by triggering force-induced changes in nuclear structures. In addition, it is now emerging that the physical properties of the nucleus play a crucial role during cell migration in three-dimensional (3-D) environments, where cells often have to transit through narrow constrictions smaller than the nuclear diameter, e.g., during development, wound healing, or cancer metastasis. In this review, we provide a brief overview of how LINC complex proteins and lamins facilitate nucleo-cytoskeletal coupling, highlight recent findings regarding the role of the nucleus in cellular mechanotransduction and cell motility in 3-D environments, and discuss how mutations and/or changes in the expression of these nuclear envelope proteins can result in a broad range of human diseases, including muscular dystrophy, dilated cardiomyopathy, and premature aging.
Introduction
Mechanotransduction defines the process by which cells `translate' mechanical stimuli into biochemical signals, enabling cells to sense their physical environment and adjust their structure and function accordingly. While mechanotransduction was first studied in specialized sensory cells such as the inner hair cells involved in hearing, we now know that virtually all cells respond to mechanical stimulation. A growing body of work over the past two decades suggest that rather than relying on a single central `mechanosensor', cells utilize a variety of mechanosensitive elements, ranging from stretch-activated ion channels in the plasma membrane, conformational changes in proteins at focal adhesions and inside the cytoskeleton to force-induced unfolding of extracellular matrix proteins, to sense applied forces and substrate stiffness [1–3]. Recent findings have further fueled the speculation that the nucleus itself may act as a cellular mechanosensor, bypassing diffusion based mechano-signaling through the cytoplasm to directly modulate expression of mechanosensitive genes [3].
A central role in this process has been attributed to lamins, type V nuclear intermediate filaments that constitute the major components of the nuclear lamina, a dense protein network underlying the inner nuclear membrane, and that also form stable structures within the nucleoplasm [4]. Lamins can be separated into A-type and B-type lamins, with lamins A and C as the major A-type isoforms, and lamins B1, and B2 the major B-type isoforms in somatic cells [4]. Lamins interact with a variety of nuclear envelope proteins, including emerin, lamin B receptor (LBR), and the nesprin and SUN protein families [5], as well as numerous transcriptional regulators [4, 5]. Lamins can also directly interact with chromatin [6] and help tether specific chromatin regions known as lamina-associated domains (LADs) to the nuclear periphery [7]; loss of lamins results in changes in chromatin organization, including loss of peripheral heterochromatin [8]. Lamins, in particular lamins A and C, provide structural support to the nucleus [9, 10] and play an important role in physically connecting the nucleus to the cytoskeleton, thereby enabling forces to be transmitted from the cytoskeleton and extracellular matrix to the nuclear interior [11–14].
Lamins are an extended part of the LINC (Linker of Nucleoskeleton and Cytoskeleton) complex [15], which enables force transmission across the nuclear envelope. The LINC complex itself is composed of two protein families, SUN proteins at the inner nuclear membrane and KASH-domain containing proteins at the outer nuclear membrane, which engage across the luminal space via their conserved SUN and KASH domains (Fig. 1). SUN proteins interact with the nuclear lamina, nuclear pore proteins, and other nuclear proteins at the nuclear interior; in the cytoplasm, KASH-domain containing proteins can bind to all major cytoskeletal filament networks, including actin filaments (through the actin-binding domain of the giant isoforms of nesprins -1 and-2), intermediate filaments (via interaction of nesprin-3 with the cytoskeletal linker plectin), and microtubules (via kinesin and dynein motor proteins binding to nesprins-1, -2, -4 and KASH5)[16]. We refer the reader to excellent recent reviews regarding the detailed molecular organization of the LINC complex [16], its evolutionary conserved history [17], and the diverse role of lamins and other nuclear envelope proteins in other cellular functions [18].
Figure 1. Schematic overview of LINC complex proteins and their connections to the cytoskeleton and nuclear interior.
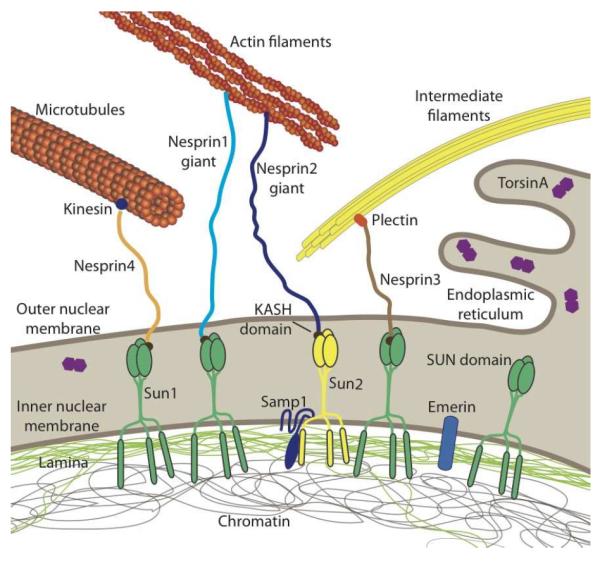
SUN proteins at the inner nuclear membrane bind to the nuclear lamina and other nucleoplasmic proteins while interacting with KASH-domain containing proteins at the outer nuclear membrane. KASH-domain containing proteins directly or indirectly interact with cytoskeletal filaments, thereby forming a physical connection between the nuclear interior and cytoskeleton. Please note that SUN- and KASH domain proteins can exist in multiple isoforms encoded by several genes. In human somatic cells, the most predominant KASH-domain proteins are nesprin-1, -2, and -3 and their various isoforms, and Sun1 and Sun2 as the predominant SUN proteins [16]. Illustrated are only the largest isoforms for nesprins1–4; cells express many additional shorter nesprin isoforms, including some lacking the KASH domain. Smaller nesprin isoform may also be located on the inner nuclear membrane. Note that nesprin-1, -2, -4 and KASH5 can also interact with kinesin and/or dynein. Samp1 and torsinA are involved in the regulation of the LINC complex. Not depicted are KASH5 and the SUN protein isoforms Sun3–5, as their expression is restricted to germ cells. The nuclear lamina comprises A-type and B-type lamins. Note that torsinA can be localized in the endoplasmic reticulum and the perinuclear space, with the distribution varying depending on expression levels.
The importance of nuclear mechanics and nucleo-cytoskeletal coupling in cellular function has become strikingly evident over the past decade by the identification of a growing number of diseases resulting from mutations in lamins and LINC complex components. In particular, mutations in the LMNA gene, encoding the nuclear envelope proteins lamin A and C, cause a variety of human diseases (laminopathies) that include Emery-Dreifuss muscular dystrophy, dilated cardiomyopathy, limb-girdle muscular dystrophy, and Hutchinson-Gilford progeria syndrome [18]. For many of these diseases, the molecular disease mechanism remains incompletely understood, but recent reports demonstrate that mutations in lamins A/C can disrupt LINC complex function and cause defects in skeletal and cardiac muscle [16, 19, 20]. In addition to its role in muscle cells and tissue, proper nucleo-cytoskeletal coupling is also essential in cell migration, for example, during wound healing, inflammation, cancer metastasis, and development [13, 16, 21]. Cytoskeletal forces are required to dynamically position the nucleus during migration on 2-D substrates [21]. In 3-D environments, the cell and nucleus face additional challenges, as the dense fibrous extracellular matrix network and tight interstitial spaces often create constrictions smaller than the size of the nucleus, so that the deformation of the typically large and relatively stiff nucleus can become a rate-limiting step [22].
In the following, we provide an overview of the current understanding of the role of the nucleus and the nuclear envelope in cellular mechanosensing and mechanotransduction signaling and discuss how changes in nuclear structure and disturbed nucleo-cytoskeletal coupling can contribute to human disease. We conclude with a brief outlook at new directions in this exciting research field and how improved insights into nucleo-cytoskeletal coupling and nuclear mechanosensing may eventually point to novel therapeutic approaches for the various nuclear envelopathies.
The role of the nucleus in mechanotransduction
In its literal definition, mechanotransduction refers only to the immediate cellular processes in which mechanical stimuli are transduced into biochemical signals; however, the term mechanotransduction is often applied more broadly to describe the overall cellular response to changes in its mechanical environment, for example, activation of specific genes or changes in cellular structure and organization. In the following, we use the term `mechanosensing' to describe the initiating mechanotransduction events, while denoting the downstream signaling and changes in gene expression as `mechanotransduction signaling'.
Given the central role of the nucleus in transcriptional regulation, it has long been speculated that the nucleus could act as a cellular mechanosensor that can directly modulate gene expression in response to mechanical disturbances. It is well established that external forces applied to a cell are transmitted from the plasma membrane via the cytoskeleton to the nucleus, resulting in (intra-) nuclear deformations [23–25]. These deformations could alter chromatin structure or induce conformational changes in nuclear proteins, e.g., release of transcriptional regulators or translocation of chromatin segments away from transcriptionally repressive regions, thereby activating (or repressing) mechanosensitive genes (Fig. 2). Support for this idea comes from three recent studies. Dahl and colleagues [26] found that fluid shear stress and compressive stress application increase intranuclear movement of fluorescent fusion proteins binding to ribosomal DNA and RNA in a number of cell lines, indicating that externally applied forces can indeed alter chromatin organization and accessibility. Going a step further, Wang and co-workers [27] reported that application of approximately nanoNewton forces to the surface of HeLa cells via magnetic microspheres results in rapid (less than 1 s) dissociation of two major structural Cajal body proteins, coilin and SMN (survival motor neuron protein), and that disruption of the actin cytoskeleton or depletion of lamins A/C abolishes this response. Most recently, Discher and colleagues [28] revealed an additional mechanism by which force-induced nuclear deformation could initiate biochemical responses, focusing on the role of nuclear lamins. Application of fluid shear stress to isolated nuclei caused the Ig-domain of lamin A to unfold, exposing a previously buried cysteine residue [28]. While these findings indicate that the nuclear lamina could function as a nuclear force sensor, in their current study, the authors did not observe any exposed cysteines in intact cells, which may suggest that forces acting on the nucleus under physiological conditions are insufficient to cause (partial) protein unfolding. Furthermore, it remains to be seen whether any partial unfolding of lamins could alter the interaction with their diverse binding partners to initiate further changes in transcriptional regulation.
Figure 2. Potential mechanisms of nuclear mechanosensing.
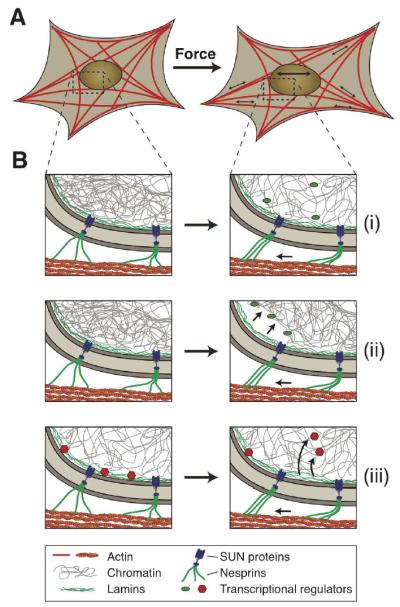
Schematic illustration of how force-induced nuclear deformation could modulate expression of mechano-responsive genes. (A) This example shows a cell exposed to a uniaxial stretch, resulting in nuclear deformation by forces transmitted from focal adhesions through the (actin) cytoskeleton to the nucleus. (B) Potential molecular mechanisms for nuclear mechanosensing: (i) Opening of chromatin structures under force, enabling access of transcriptional regulators to the chromatin. (ii) Chromatin detachment from the lamina, freeing genes from the often transcriptionally repressive nuclear periphery. This process could also result in further changes in chromatin structure, promoting access to transcriptional regulators. (iii) Stretching the lamina could result in conformational changes or partial unfolding of lamins, altering their interaction with transcriptional regulators. Shown here is the release of transcription factors, which can then interact with their target genes. Phosphorylation and other post-translational modifications of nuclear envelope proteins could further contribute to nuclear mechanosensing.
Interestingly, the same study also investigated the expression levels and phosphorylation state of lamins in response to changes in the cellular mechanical environment, revealing that the expression of lamins A and C (relative to B-type lamins) scales with the substrate stiffness in vitro and in vivo [28]. In addition, softer substrates, which correspond to reduced cytoskeletal tension, were associated with higher levels of lamin A/C phosphorylation [28], indicative of a more soluble and mechanically weaker lamin network. As lamins A and C are the main contributors to nuclear stiffness and stability, it is easily conceivable that cells adapt the expression and organization of lamins to their mechanical environment, for example, resulting in high levels of lamins A/C in mechanically stressed tissues such as skeletal and cardiac muscle and low levels of lamins A/C in brain or adipose tissue, thereby normalizing the mechanical stress acting on the lamin network. However, at the current time, it remains to be seen whether this intriguing correlation is caused by a direct role of lamins in mechanosensing and a corresponding feedback loop to control lamin levels, or whether transcriptional regulation of lamins in response to substrate stiffness is downstream of other mechanotransduction signaling pathways.
Arguing (at least in part) against the idea that induced nuclear deformations are essential for cellular mechanosensing and mechanotransduction signaling is a recent study that found that disruption of LINC complex proteins by dominant negative nesprin and SUN constructs almost completely abolishes nuclear deformation when cells are subjected to substrate strain, yet the mechanoresponsive genes tested by the authors were activated normally [24]. While these experiments do not exclude the possibility that some mechanosensitive genes exist that directly respond to nuclear deformation, they suggests that mechanosensors in the plasma membrane and/or the cytoskeleton may be sufficient to initiate mechanotransduction pathways that are then transmitted via biochemical signals to the nucleus.
On the other hand, nuclear envelope proteins undoubtedly play an important role in cellular mechanotransduction signaling. LINC complex disruption impairs intracellular force transmission from the cytoskeleton to the nucleus, and at least in C2C12 myoblasts, LINC complex disruption can interfere with stretch-induced proliferation [29]. In endothelial cells, nesprins play an important role in the response to fluid shear stress, with depletion of nesprin-3 causing altered cell morphology and impaired cell polarization and migration in the direction of the fluid flow [30]. Even more dramatic changes are observed in cells lacking lamins A/C or emerin, which have impaired activation of mechano-responsive genes in vitro and in vivo [10, 31–33]. The molecular details underlying impaired activation of the mechanosensitive transcription factor MRTF-A (myocardin-related transcription factor-A, also known as MKL1 or MAL), were recently elucidated [32]. MRTF-A, which plays a critical role in cardiac development and function, is normally sequestered in the cytoplasm by interaction with monomeric actin; stimulation by mechanical stress or serum induces the assembly of actin filaments, resulting in the release of MRTF-A and its translocation to the nucleus, where it serves as co-activator for the transcription factor SRF (serum response factor) to initiate expression of genes with a serum response element (SRE) that include vinculin, actin, and SRF itself [34]. Nuclear activity and export of MRTF-A are further modulated by polymerization of nuclear actin [34, 35]. Since emerin, which can directly bind actin and promote its polymerization [36], requires lamin A/C for its localization to the inner nuclear membrane, functional loss of lamins A/C or emerin reduces nuclear and cytoskeletal actin dynamics and results in impaired translocation and activation of MRTF-A [32], demonstrating how structural changes mediated by lamin A/C and emerin can affect gene regulation.
Importantly, lamins and other proteins involved in nucleo-cytoskeletal coupling also directly interact with chromatin and numerous transcriptional regulators, including retinoblastoma protein (pRb), c-Fos, and ERK1/2 for lamins A/C, α-catenin and ERK1/2 for nesprin2, and β-catenin, BAF, GCL and the splicing-associated factor YT521-B for emerin [4, 5, 37]. Consequently, defects in mechanotransduction signaling in lamin A/C- or emerin-deficient cells may also be attributed to the interaction of lamins or emerin with these transcriptional modulators, rather than their role in nucleo-cytoskeletal coupling and nuclear deformability, although more experimental evidence is needed to distinguish between these (non-mutually exclusive) hypotheses.
As these findings demonstrate, nuclear structure and deformability, as well as force transmission between the cytoskeleton and nucleus play crucial roles in activating or modulating cellular mechanotransduction signaling. At the same time, nuclear mechanics and nucleo-cytoskeletal coupling can also directly affect other cellular functions that require the physical movement and positioning/anchoring of the nucleus within the cell. Examples include the rearward nuclear position in (most) migrating cells, the peripheral nuclear placement in striated muscle cells, or the basal nuclear position in stem cells asymmetrically dividing in their niche [16].
Nuclear positioning in 2-D cell migration
Many cells cultured on flat substrates show a characteristic cellular reorientation (polarization) before initiating migrating [38]. Scratch wound assays reveal that during the polarization process, the nucleus moves rearwards, away from the wound edge, resulting in the centrally located centrosome to be positioned ahead of the nucleus, towards the wound edge (Fig. 3). This process requires intact nucleo-cytoskeletal coupling, as LINC complex disruption or depletion of lamins prevents rearward nuclear movement [12, 21, 24].
Figure 3. Nuclear positioning during cell polarization via TAN lines.
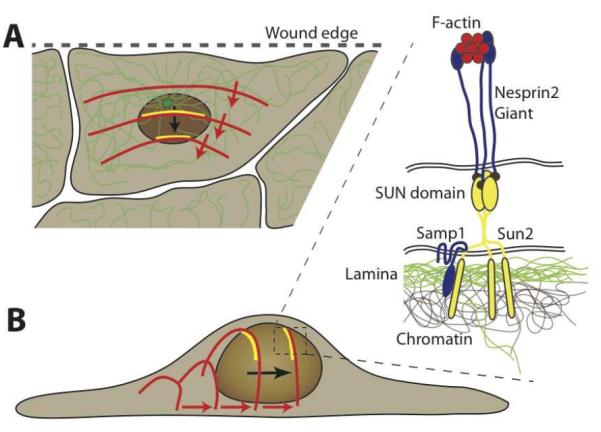
Schematic depiction of retrograde nuclear movement during early polarization in a scratch wound assay. (A) The nucleus moves to the rear end of the cell, resulting in the centrosome (green, with microtubule network) to become located towards the leading edge (i.e., the wound edge) of the cell. Nuclear translocation is mediated by rearward moving dorsal actin cables (red), which form stable connections to complexes of nesprin2, Sun2 and Samp1 (yellow), referred to as TAN lines. (B) Schematic side view of the process by which rearward moving actin cables move the nucleus towards the rear of the cell. The inset shows a close-up of the molecular structure of the TAN lines: F-actin cables interact with the actin-binding domain of nesprin-2 molecules, which bind to Sun2 homotrimers across the perinuclear space. Sun2 also interacts with Samp1 and the underlying nuclear lamina and chromatin.
A seminal study by Luxton and colleagues [21] uncovered that the nuclear repositioning during cell polarization is mediated by coupling the nucleus to dorsal actin cables that—driven by Cdc42 and actin-myosin II interactions—originate near the leading edge of the cell and move rearward, thereby dragging the nucleus backwards (Fig. 3)[21]. These so called TAN (Transmembrane Actin-associated Nuclear) lines are comprised of actin filaments, nesprin-2 giant at the outer nuclear membrane, and Sun2 at the inner nuclear membrane [21], and, as recently discovered, Samp1 [39]. The mobility of nesprins that are part of the TAN lines is significantly lower than in other parts of the nucleus, indicating that they are part of a stable complex [21]. This complex formation may be mediated by Samp1, as depletion of Samp1 results in failure to reposition the nucleus [39]. Similarly, when the LINC complex is disrupted by RNAi mediated depletion of lamin A or Sun2, the TAN lines drift across the nuclear envelope without becoming sufficiently anchored, resulting in lack of nuclear movement and defects in cell polarization and migration [21]. In single cell migration assays, LINC complex disruption causes reduced migration speed and decreased directional persistence [24], further demonstrating the importance of intact nucleo-cytoskeletal coupling. We refer the reader to a recent review [16] for a more detailed discussion of nucleocytoskeletal coupling in 2-D cell migration.
Cell migration in 3-D environments
Most in vitro migration assays are conducted on 2-D surfaces; in contrast, cell motility in vivo—for example, cell migration during early development, infiltration of immune cells into sites of infection, or invasion of cancer cells into adjacent tissues—typically takes place in 3-D environments. An emerging field of research suggests that cell migration in 3-D environments differs substantially from 2-D migration (discussed in [40]).
Nuclear deformability as rate limiting step in 3-D cell migration
While much of the research in cell migration—both in 2-D and 3-D environments—has been focused on processes at the leading edge, particularly the dynamics of the actin cytoskeleton, it is now becoming evident that the mechanical properties of the cell nucleus and its connection to the cytoskeleton play an essential role in 3-D migration [22, 41]. When cells encounter constrictions in the interstitial space that are smaller than their nuclear diameter, cells can either proteolytically degrade the constricting extracellular matrix or attempt to squeeze through the narrow opening, requiring substantial cellular deformation. During non-proteolytic migration, the highly adaptable and dynamic cytoskeleton and plasma membrane can penetrate spaces less than 1 μm in diameter [22], but the large and stiff nucleus is much more resistant to large deformations and imposes a rate-limiting step during migration through narrow constrictions [22, 42]. Findings from recent studies with cells migrating through 3-D collagen matrices, polycarbonate filters, or microfabricated channels with well-defined pore sizes demonstrate that decreasing pore sizes beyond 20 μm gradually reduces migration speed [22, 43]. Movement of the cell body and nucleus stalls completely when encountering constrictions smaller than ~10% of the initial nuclear diameter [22], suggesting a finite limit of the compressibility of the nucleus [22, 44].
Given the prominent role of nuclear envelope proteins, particularly lamins A and C, in determining nuclear deformability, it is intriguing to speculate to what extent nuclear envelope composition can affect cell migration in 3-D environments. Cells expressing a lamin A mutation that increases nuclear stiffness [45, 46] have difficulties navigating through 6 μm wide constrictions, even though they have similar migration speeds in unconfined spaces as control cells [47]. Conversely, neutrophils have evolved highly lobulated nuclei almost completely lacking lamins A/C, making them well suited to pass through narrow capillaries and narrow constrictions during extravasation and interstitial migration [48]. Ectopic expression of lamin A in neutrophil-differentiated cells causes rounder nuclei and an impaired ability to pass through narrow constrictions during perfusion and migration [42], further illustrating the importance of nuclear deformability in 3-D cell motility.
Pulled or pushed? How does the cytoskeleton move the nucleus during 3-D migration?
The nuclear deformation during cell passage through narrow constrictions requires substantial cytoskeletal forces acting on the nucleus. One can imagine several non-mutual exclusive possibilities explaining how forces could be applied to the nucleus to move it through tight constrictions. The cytoskeleton could exert forces from the cell front, pulling on the nucleus, or it could apply contractile forces from the rear, pushing and squeezing the nucleus through the constriction (Fig. 4). Pulling forces could result from molecular motors such as dynein attached to the nuclear surface via LINC complex proteins, moving the nucleus along the microtubules network towards the centrosome on the other side of the constriction. Actin-myosin interactions could exert contractile forces between forward-based focal adhesions and the anterior nuclear side. Contribution of pulling forces is supported by the finding that integrin and actomyosin-dependent force generation is required for non-proteolytic cell migration through dense collagen matrices [22] and observations of herniations of the nuclear membrane at the anterior edge of the nucleus in lamin B1-mutant neurons during migration and detachment of the chromatin from the nuclear envelope [49].
Figure 4. Nuclear deformation during cell migration through tight constrictions.
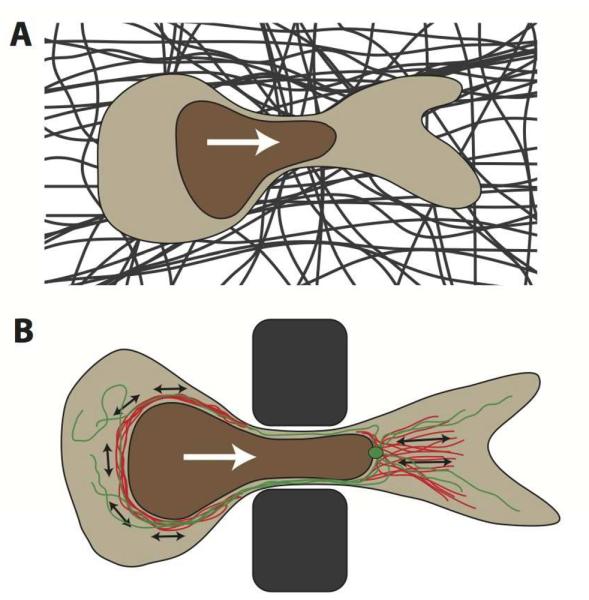
(A) Schematic depiction of a cross-section of a cell migrating through a constriction in the dense extracellular matrix (dark fibers) that is smaller than the nuclear diameter. The white arrow denotes the direction of cell migration. The nucleus is depicted in brown. (B) Sideview of a cell migrating through a polycarbonate filter or microfabricated device used to study nuclear deformation during cell migration through precisely defined pores. Illustrated in red are actinmyosin networks, applying contractile forces (black arrows) to the nucleus, either posterior to the nucleus, resulting in a pushing force, or anterior, pulling on the nucleus. Molecular motors on the microtubule network (green, with centrosome) may apply additional forces to the nucleus, particularly during neuronal migration. White arrow indicates migration direction.
At the same time, actomyosin-generated contraction can also serve as the pushing force for the nucleus, as seen in the interkinetic nuclear migration of neurons in the retina of zebrafish [50]. Unlike in mammalian cells, where interkinetic nuclear movement is mainly driven by microtubule-associated motors [51], Zebrafish neurons rely on myosin II activity at the rear of the nucleus to push the nucleus forward [50], possibly reflecting species- or cell shape-dependent differences [51]. While non-muscle myosin-IIa is located near the leading edge of cells [52–54], non-muscle myosin-IIb is present in the actin network surrounding the nucleus [55]. The idea of a contractile network consisting of F-actin and myosin-II at the side and rear of the cell responsible for pushing the nucleus through the constriction is consistent with the data observed by Wolf et al. [22] and further supported by the finding that in breast cancer cells invading Matrigel scaffolds, actomyosin-based cytoskeletal contraction is limited to the rear of the cells, and inhibiting actomyosin-contraction abolishes invasion [56].
As squeezing the fluid-filled nucleus from the rear may produce similar nuclear protrusions into the constriction as expected in a pulling model (Fig. 4B), it is challenging to distinguish between the two major modes, i.e., pulling or pushing the nucleus through the constriction, by observation of nuclear deformations alone. Further research is necessary to elucidate the molecular details involved in overcoming the nuclear resistance during cell migration in 3-D environments. Importantly, it remains to be seen to what extent these processes require nucleo-cytoskeletal coupling through the LINC complex. While at least one study reported that LINC complex disruption impairs cell migration in 3-D environments [57], a contractile actomyosin network at the rear of the nucleus may not necessarily require LINC complex function to transmit forces to the nucleus. Furthermore, a LINC complex independent nuclear positioning mechanism has been observed in the migration of nuclei in drosophila oocytes, where polymerizing microtubules at the rear of the nucleus propel the nucleus forward [58].
In light of the emerging importance of nuclear mechanics during cell migration in 3-D environments, it is intriguing to speculate whether cells are capable of dynamically adjusting the mechanical properties of the nucleus. An example of long-term adjustment can be seen during granulopoiesis, when cells downregulate expression of lamins while increasing expression of LBR, resulting in highly lobulated and deformable nuclei in granulocytes that promote passage through tight spaces [42, 59]. Given the recent finding of changes in lamin expression and phosphorylation in response to substrate stiffness [28], it is not too far-fetched to envision that cells may dynamically reduce or partially depolymerize the nuclear lamin network to transiently increase nuclear deformability, similar to the process of nuclear envelope breakdown during mitosis. Alternatively, cells could enhance migration through narrow constrictions by increasing the cytoskeletal tension, thereby exerting more forces on the nucleus. This could be particularly relevant in the spreading of cancer cells, as cells with increased metastatic potential were recently shown to generate higher cytoskeletal forces [60].
LINC complex related diseases
Given the broad role of cellular functions that require intact nucleo-cytoskeletal coupling, it comes as no surprise that mutations in LINC complex-associated proteins can result in a large number of human diseases (Table 1). The majority of diseases are caused by mutations in the LMNA gene, encoding lamins A/C. These laminopathies range from highly tissue-specific diseases affecting striated muscle, adipose tissue, or peripheral nerves to systemic disorders and include Emery-Dreifuss muscular dystrophy, limb-girdle muscular dystrophy, dilated cardiomyopathy, familial partial lipodystrophy, Charcot-Marie-Tooth and the accelerated aging disorder Hutchinson-Gilford progeria syndrome (reviewed in [18]).
Table 1.
List of proteins/genes involved in nucleo-cytoskeletal coupling and the diseases associated with specific mutations. Not included here are mutations in cytoskeletal and motor proteins, which can result in muscular dystrophies, cardiomyopathies, and lissencephaly due to impaired neuronal migration [16].
| Protein (Gene) | Diseases [Reference] |
|---|---|
|
| |
| Lamin A/C (LMNA) | Emery-Dreifuss muscular dystrophy [78] |
| Limb-girdle muscular dystrophy [79] | |
| Dilated cardiomyopathy [80] | |
| Congenital muscular dystrophy (dropped head) [81] | |
| Heart-hand syndrome [82] | |
| Dunnigan-type familial partial lipodystrophy [83] | |
| Generalized lipoatrophy [84] | |
| Mandibuloacral dysplasia [85] | |
| Charcot-Marie-Tooth syndrome [86] | |
| Atypical Werner Syndrome [87] | |
| Hutchinson-Gilford progeria syndrome [86, 88] | |
| Restrictive dermopathy [89] | |
|
| |
| Lamin B1 (LMNB1) | Adult onset leukodystrophy (caused by duplication) [66] |
|
| |
| Lamin B2 (LMNB2) | Partial lipodystrophy [67, 68] |
|
| |
| Emerin (STA/EMD) | Emery-Dreifuss muscular dystrophy [90] |
|
| |
| Nesprin1 (SYNE1) | Emery-Dreifuss muscular dystrophy [91] |
| Dilated Cardiomyopathy [92] | |
| Cerebellar ataxia [61] | |
| Arthrogryposis [62] | |
|
| |
| Nesprin2 (SYNE2) | Emery-Dreifuss muscular dystrophy [91] |
| Dilated Cardiomyopathy [91] | |
|
| |
| Nesprin3 (SYNE3) | None reported to date |
|
| |
| Nesprin4 (NESP4) | Hearing loss [64] |
|
| |
| SUN1 (SUN1) | None reported to date |
|
| |
| SUN2 (SUN2) | Emery-Dreifuss muscular dystrophy (patient also carried other mutations) [65] |
|
| |
| TorsinA (TOR1A) | Early-onset generalized torsion dystonia [93] |
Interestingly, diseases affecting striated muscle, i.e., Emery-Dreifuss muscular dystrophy and dilated cardiomyopathy, can also be caused by mutations in emerin (STA or EMD gene), nesprin-1 (SYNE1), and nesprin-2 (SYNE2), suggesting a LINC complex-associated disease mechanism [16]. In addition to these muscular phenotypes, nesprin-1 mutations are also responsible for autosomal recessive cerebellar ataxia [61] and arthrogryposis [62], which is characterized by congenital joint contractures resulting from reduced fetal movements. Mutations in nesprin-4, for which expression is limited to secretory epithelial cells and hair cells of the inner ear [63], result in progressive high-frequency hearing loss, a phenotype that can be recapitulated in mice lacking either nesprin-4 or Sun1 [64]. In contrast, no disease-causing mutations have been reported for either of the SUN proteins, although a novel mutation in Sun2 was recently described in a patient with Emery-Dreifuss muscular dystrophy who was also carrying a mutation in nesprin-1α1, which by itself is considered non-pathogenic [65]. Interestingly, the same study also identified a patient with Duchenne muscular dystrophy caused by a mutation in the dystrophin gene (DMD) carrying a nesprin-1α2 mutation, suggesting that mutations in LINC complex proteins can act as modifier genes in other muscular dystrophies. Mutations and gene duplications have also been described for B-type lamins [18]. Duplication of LMNB1 results in adult onset leukodystrophy [66], characterized by demyelination in the central nervous system. Mutations in LMNB2 causes acquired partial lipodystrophy, which involves a progressive loss of subcutaneous fat tissue [67, 68].
The disease etiology for the broad spectrum of nuclear envelopathies remains incompletely understood. Patient cells are often characterized by abnormal nuclear morphology and altered distribution of nuclear envelope proteins, including mislocalization of lamins, nesprins, and SUN proteins [16], and lamin mutations linked to striated muscle diseases result in impaired nucleocytoskeletal force transmission and reduced nuclear stability [11, 20]. These findings suggest that at least for the diseases affecting cardiac and skeletal muscle, which are exposed to particularly high levels of mechanical stress, defects in nucleo-cytoskeletal coupling and nuclear mechanics could directly contribute to the disease phenotype. Nonetheless, it is likely that additional mechanisms, such as impaired mechanotransduction signaling, disturbed transcriptional regulation, or impaired stem cell function, further contribute to the disease development and are responsible for the broad spectrum of human diseases [18].
One interesting and unexpected disease mechanism emerged from the recent crossing of lamin A/C-deficient and Sun1-deficient mouse models. Mice that lack lamins A and C develop severe muscular dystrophy and dilated cardiomyopathy and die at 4–8 weeks of age [69]. Surprisingly, when crossed with Sun1-deficient mice, which lack an overt phenotype, the resulting double deletion of lamin A/C and Sun1 expands the lifetime of the animals, possibly by preventing toxic accumulation of Sun1 in the Golgi apparatus [70]. Similarly increased survival was observed in mice lacking exon 9 of the Lmna gene, which causes a progeria like phenotype when crossed with Sun1-deficient mice [70]. These findings suggest that in addition to disrupting their normal role in nucleo-cytoskeletal coupling, displacement of nuclear envelope proteins may cause further cellular defects by inducing Golgi stress and compromising Golgi functionality.
Recently, altered expression of nuclear envelope proteins, particularly lamins, has been reported in a number of cancers. For example, lamins A/C are downregulated in breast cancer, leukemias, lymphomas, colon cancer, and gastric carcinoma, whereas expression of A-type lamins is upregulated in prostate, skin and ovarian cancers [4, 71, 72]. Furthermore, a recent genome-wide analysis of 100 cancer patients identified mutations in lamins A/C, nesprin-1, and nesprin-2, which, albeit unlikely to be driver mutations, could represent modulators of cancer progression [73]. In cancer cells, altered lamin function could directly affect nuclear deformability required for interstitial migration or act through diverse signaling pathways that promote cell motility [72, 74]. These changes in nuclear envelope composition, which may provide an explanation for the often severe abnormal nuclear shape in cancer cells, could directly contribute to the disease progression, either by altering the mechanical properties of the cell nucleus [41] or by modulating signaling pathways and cytoskeletal organization associated with changes in lamin expression [75].
Outlook
Over the past decade, numerous novel nuclear envelope proteins involved in nucleo-cytoskeletal coupling and force transmission to the nucleus have been identified, including nesprins and SUN proteins, the core components of the LINC complex. Nonetheless, many questions remain unanswered. What is the role of nuclear envelope proteins in cellular mechanotransduction? Can these proteins act as nuclear mechanosensors, or do they primarily serve as processing hubs in the cellular mechanotransduction signaling network? In the context of intracellular force transmission, given the broad distribution of nesprins and SUN proteins along the nuclear surface, how is the interaction of LINC complex proteins regulated to promote (dynamic) anchoring to specific cytoskeletal structures while avoiding `locking up' the nucleus by unwanted interaction with other cytoskeletal elements? Which proteins are involved in this regulation? Where does the regulation take place – at the cytoplasm, the nucleoplasm, or the luminal interaction between the SUN and KASH domains? Are there other, yet to be characterized proteins involved in linking the nucleus to the cytoskeleton independent of LINC complex proteins?
Answering these questions will not only advance our understanding of normal cellular processes but also aid in the development of therapeutic approaches, targeting the many diseases resulting from mutations in LINC complex-associated proteins. As of now, it remains unclear to what extent direct mechanical defects such as impaired nuclear anchoring as opposed to impaired transcriptional regulation or stem cell dysfunction contribute to the disease mechanisms, and whether these defects are interrelated [11, 18]. Treating impaired signaling provides a more rapidly attainable goal and has already produced some promising in cardiac laminopathies [76], but may be insufficient to overcome structural defects.
Twenty years from now, we will probably look back with a smile at the limitations of our current knowledge of nucleo-cytoskeletal coupling and nuclear mechanotransduction. The concept of transmembrane connections between the actin cytoskeleton and the extracellular matrix, leading to the discovery of integrins, is almost 40 years old [77]. That work has evolved into a tremendously successful research field spanning cell migration, stem cell differentiation and anti-cancer therapies. Is nucleo-cytoskeletal coupling headed the same way? We will not find out for a while, but it is certainly an exciting ride, wherever it may lead us.
Acknowledgements
We apologize to all authors whose work could not be cited due to space constraints. This work was supported by National Institutes of Health awards [R01 NS059348, R01 HL082792], a National Science Foundation CAREER award [CBET-1254846], the Department of Defense Breast Cancer Idea Award [BC102152], an award from the Progeria Research Foundation [PRF2011-0035], and a Pilot Project Award by the Cornell Center on the Microenvironment & Metastasis through Award Number U54CA143876 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jaalouk DE, Lammerding J. Mechanotransduction gone awry. Nat Rev Mol Cell Biol. 2009;10:63–73. doi: 10.1038/nrm2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eyckmans J, Boudou T, Yu X, Chen CS. A hitchhiker's guide to mechanobiology. Developmental cell. 2011;21:35–47. doi: 10.1016/j.devcel.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang N, Tytell JD, Ingber DE. Mechanotransduction at a distance: mechanically coupling the extracellular matrix with the nucleus. Nat Rev Mol Cell Biol. 2009;10:75–82. doi: 10.1038/nrm2594. [DOI] [PubMed] [Google Scholar]
- 4.Ho CY, Lammerding J. Lamins at a glance. J Cell Sci. 2012;125:2087–2093. doi: 10.1242/jcs.087288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson KL, Berk JM. The nuclear envelope at a glance. J Cell Sci. 2010;123:1973–1978. doi: 10.1242/jcs.019042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, Goldman RD. Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes & development. 2008;22:832–853. doi: 10.1101/gad.1652708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bickmore WA, van Steensel B. Genome architecture: domain organization of interphase chromosomes. Cell. 2013;152:1270–1284. doi: 10.1016/j.cell.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 8.Solovei I, Wang AS, Thanisch K, Schmidt CS, Krebs S, Zwerger M, Cohen TV, Devys D, Foisner R, Peichl L, et al. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell. 2013;152:584–598. doi: 10.1016/j.cell.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 9.Lammerding J, Fong LG, Ji JY, Reue K, Stewart CL, Young SG, Lee RT. Lamins A and C but not lamin B1 regulate nuclear mechanics. J Biol Chem. 2006;281:25768–25780. doi: 10.1074/jbc.M513511200. [DOI] [PubMed] [Google Scholar]
- 10.Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. The Journal of clinical investigation. 2004;113:370–378. doi: 10.1172/JCI19670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zwerger M, Jaalouk DE, Lombardi ML, Isermann P, Mauermann M, Dialynas G, Herrmann H, Wallrath LL, Lammerding J. Myopathic lamin mutations impair nuclear stability in cells and tissue and disrupt nucleo-cytoskeletal coupling. Hum Mol Genet. 2013;22:2335–2349. doi: 10.1093/hmg/ddt079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Folker ES, Ostlund C, Luxton GW, Worman HJ, Gundersen GG. Lamin A variants that cause striated muscle disease are defective in anchoring transmembrane actin-associated nuclear lines for nuclear movement. Proc Natl Acad Sci U S A. 2011;108:131–136. doi: 10.1073/pnas.1000824108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luxton GG, Gomes ER, Folker ES, Worman HJ, Gundersen GG. TAN lines: a novel nuclear envelope structure involved in nuclear positioning. Nucleus. 2011;2:173–181. doi: 10.4161/nucl.2.3.16243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ostlund C, Folker ES, Choi JC, Gomes ER, Gundersen GG, Worman HJ. Dynamics and molecular interactions of linker of nucleoskeleton and cytoskeleton (LINC) complex proteins. J Cell Sci. 2009;122:4099–4108. doi: 10.1242/jcs.057075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crisp M, Liu Q, Roux K, Rattner JB, Shanahan C, Burke B, Stahl PD, Hodzic D. Coupling of the nucleus and cytoplasm: role of the LINC complex. J Cell Biol. 2006;172:41–53. doi: 10.1083/jcb.200509124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gundersen GG, Worman HJ. Nuclear positioning. Cell. 2013;152:1376–1389. doi: 10.1016/j.cell.2013.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rothballer A, Kutay U. The diverse functional LINCs of the nuclear envelope to the cytoskeleton and chromatin. Chromosoma. 2013;122:415–429. doi: 10.1007/s00412-013-0417-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schreiber KH, Kennedy BK. When lamins go bad: nuclear structure and disease. Cell. 2013;152:1365–1375. doi: 10.1016/j.cell.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mejat A, Misteli T. LINC complexes in health and disease. Nucleus. 2010;1:40–52. doi: 10.4161/nucl.1.1.10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Folker ES, Ostlund C, Luxton GW, Worman HJ, Gundersen GG. Lamin A variants that cause striated muscle disease are defective in anchoring transmembrane actin-associated nuclear lines for nuclear movement. Proc Natl Acad Sci U S A. 2010;108:131–136. doi: 10.1073/pnas.1000824108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luxton GW, Gomes ER, Folker ES, Vintinner E, Gundersen GG. Linear arrays of nuclear envelope proteins harness retrograde actin flow for nuclear movement. Science. 2010;329:956–959. doi: 10.1126/science.1189072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolf K, Te Lindert M, Krause M, Alexander S, Te Riet J, Willis AL, Hoffman RM, Figdor CG, Weiss SJ, Friedl P. Physical limits of cell migration: Control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J Cell Biol. 2013;201:1069–1084. doi: 10.1083/jcb.201210152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morimoto A, Shibuya H, Zhu X, Kim J, Ishiguro K, Han M, Watanabe Y. A conserved KASH domain protein associates with telomeres, SUN1, and dynactin during mammalian meiosis. J Cell Biol. 2012;198:165–172. doi: 10.1083/jcb.201204085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lombardi ML, Jaalouk DE, Shanahan CM, Burke B, Roux KJ, Lammerding J. The interaction between nesprins and sun proteins at the nuclear envelope is critical for force transmission between the nucleus and cytoskeleton. J Biol Chem. 2011;286:26743–26753. doi: 10.1074/jbc.M111.233700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caille N, Tardy Y, Meister JJ. Assessment of strain field in endothelial cells subjected to uniaxial deformation of their substrate. Ann Biomed Eng. 1998;26:409–416. doi: 10.1114/1.132. [DOI] [PubMed] [Google Scholar]
- 26.Booth-Gauthier EA, Alcoser TA, Yang G, Dahl KN. Force-induced changes in subnuclear movement and rheology. Biophysical journal. 2012;103:2423–2431. doi: 10.1016/j.bpj.2012.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poh YC, Shevtsov SP, Chowdhury F, Wu DC, Na S, Dundr M, Wang N. Dynamic force-induced direct dissociation of protein complexes in a nuclear body in living cells. Nature communications. 2012;3:866. doi: 10.1038/ncomms1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swift J, Ivanovska IL, Buxboim A, Harada T, Dingal PC, Pinter J, Pajerowski JD, Spinler KR, Shin JW, Tewari M, et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. 2013;341:1240104. doi: 10.1126/science.1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brosig M, Ferralli J, Gelman L, Chiquet M, Chiquet-Ehrismann R. Interfering with the connection between the nucleus and the cytoskeleton affects nuclear rotation, mechanotransduction and myogenesis. The international journal of biochemistry & cell biology. 2010;42:1717–1728. doi: 10.1016/j.biocel.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 30.Morgan JT, Pfeiffer ER, Thirkill TL, Kumar P, Peng G, Fridolfsson HN, Douglas GC, Starr DA, Barakat AI. Nesprin-3 regulates endothelial cell morphology, perinuclear cytoskeletal architecture, and flow-induced polarization. Molecular biology of the cell. 2011;22:4324–4334. doi: 10.1091/mbc.E11-04-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cupesi M, Yoshioka J, Gannon J, Kudinova A, Stewart CL, Lammerding J. Attenuated hypertrophic response to pressure overload in a lamin A/C haploinsufficiency mouse. Journal of molecular and cellular cardiology. 2010;48:1290–1297. doi: 10.1016/j.yjmcc.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ho CY, Jaalouk DE, Vartiainen MK, Lammerding J. Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature. 2013;497:507–511. doi: 10.1038/nature12105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lammerding J, Hsiao J, Schulze PC, Kozlov S, Stewart CL, Lee RT. Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. J Cell Biol. 2005;170:781–791. doi: 10.1083/jcb.200502148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol. 2010;11:353–365. doi: 10.1038/nrm2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baarlink C, Wang H, Grosse R. Nuclear actin network assembly by formins regulates the SRF coactivator MAL. Science. 2013;340:864–867. doi: 10.1126/science.1235038. [DOI] [PubMed] [Google Scholar]
- 36.Holaska JM, Kowalski AK, Wilson KL. Emerin caps the pointed end of actin filaments: evidence for an actin cortical network at the nuclear inner membrane. PLoS biology. 2004;2:E231. doi: 10.1371/journal.pbio.0020231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neumann S, Schneider M, Daugherty RL, Gottardi CJ, Eming SA, Beijer A, Noegel AA, Karakesisoglou I. Nesprin-2 interacts with {alpha}-catenin and regulates Wnt signaling at the nuclear envelope. J Biol Chem. 2010;285:34932–34938. doi: 10.1074/jbc.M110.119651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gomes ER, Jani S, Gundersen GG. Nuclear movement regulated by Cdc42, MRCK, myosin, and actin flow establishes MTOC polarization in migrating cells. Cell. 2005;121:451–463. doi: 10.1016/j.cell.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 39.Borrego-Pinto J, Jegou T, Osorio DS, Aurade F, Gorjanacz M, Koch B, Mattaj IW, Gomes ER. Samp1 is a component of TAN lines and is required for nuclear movement. J Cell Sci. 2012;125:1099–1105. doi: 10.1242/jcs.087049. [DOI] [PubMed] [Google Scholar]
- 40.Friedl P, Sahai E, Weiss S, Yamada KM. New dimensions in cell migration. Nat Rev Mol Cell Biol. 2012;13:743–747. doi: 10.1038/nrm3459. [DOI] [PubMed] [Google Scholar]
- 41.Friedl P, Wolf K, Lammerding J. Nuclear mechanics during cell migration. Current opinion in cell biology. 2011;23:55–64. doi: 10.1016/j.ceb.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rowat AC, Jaalouk DE, Zwerger M, Ung WL, Eydelnant IA, Olins DE, Olins AL, Herrmann H, Weitz DA, Lammerding J. Nuclear envelope composition determines the ability of neutrophil-type cells to passage through micron-scale constrictions. J Biol Chem. 2013;288:8610–8618. doi: 10.1074/jbc.M112.441535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tong Z, Balzer EM, Dallas MR, Hung WC, Stebe KJ, Konstantopoulos K. Chemotaxis of cell populations through confined spaces at single-cell resolution. PloS one. 2012;7:e29211. doi: 10.1371/journal.pone.0029211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rowat AC, Lammerding J, Herrmann H, Aebi U. Towards an integrated understanding of the structure and mechanics of the cell nucleus. BioEssays : news and reviews in molecular, cellular and developmental biology. 2008;30:226–236. doi: 10.1002/bies.20720. [DOI] [PubMed] [Google Scholar]
- 45.Dahl KN, Scaffidi P, Islam MF, Yodh AG, Wilson KL, Misteli T. Distinct structural and mechanical properties of the nuclear lamina in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A. 2006;103:10271–10276. doi: 10.1073/pnas.0601058103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verstraeten VL, Ji JY, Cummings KS, Lee RT, Lammerding J. Increased mechanosensitivity and nuclear stiffness in Hutchinson-Gilford progeria cells: effects of farnesyltransferase inhibitors. Aging cell. 2008;7:383–393. doi: 10.1111/j.1474-9726.2008.00382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Booth-Gauthier EA, Du V, Ghibaudo M, Rape AD, Dahl KN, Ladoux B. Hutchinson-Gilford progeria syndrome alters nuclear shape and reduces cell motility in three dimensional model substrates. Integrative biology : quantitative biosciences from nano to macro. 2013;5:569–577. doi: 10.1039/c3ib20231c. [DOI] [PubMed] [Google Scholar]
- 48.Olins AL, Hoang TV, Zwerger M, Herrmann H, Zentgraf H, Noegel AA, Karakesisoglou I, Hodzic D, Olins DE. The LINC-less granulocyte nucleus. European journal of cell biology. 2009;88:203–214. doi: 10.1016/j.ejcb.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jung HJ, Nobumori C, Goulbourne CN, Tu Y, Lee JM, Tatar A, Wu D, Yoshinaga Y, de Jong PJ, Coffinier C, et al. Farnesylation of lamin B1 is important for retention of nuclear chromatin during neuronal migration. Proc Natl Acad Sci U S A. 2013;110:E1923–1932. doi: 10.1073/pnas.1303916110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Norden C, Young S, Link BA, Harris WA. Actomyosin is the main driver of interkinetic nuclear migration in the retina. Cell. 2009;138:1195–1208. doi: 10.1016/j.cell.2009.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cooper JA. Cell biology in neuroscience: mechanisms of cell migration in the nervous system. J Cell Biol. 2013;202:725–734. doi: 10.1083/jcb.201305021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vicente-Manzanares M, Zareno J, Whitmore L, Choi CK, Horwitz AF. Regulation of protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. J Cell Biol. 2007;176:573–580. doi: 10.1083/jcb.200612043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cai Y, Biais N, Giannone G, Tanase M, Jiang G, Hofman JM, Wiggins CH, Silberzan P, Buguin A, Ladoux B, et al. Nonmuscle myosin IIA-dependent force inhibits cell spreading and drives F-actin flow. Biophysical journal. 2006;91:3907–3920. doi: 10.1529/biophysj.106.084806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vicente-Manzanares M, Koach MA, Whitmore L, Lamers ML, Horwitz AF. Segregation and activation of myosin IIB creates a rear in migrating cells. J Cell Biol. 2008;183:543–554. doi: 10.1083/jcb.200806030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lo CM, Buxton DB, Chua GC, Dembo M, Adelstein RS, Wang YL. Nonmuscle myosin IIb is involved in the guidance of fibroblast migration. Molecular biology of the cell. 2004;15:982–989. doi: 10.1091/mbc.E03-06-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Poincloux R, Collin O, Lizarraga F, Romao M, Debray M, Piel M, Chavrier P. Contractility of the cell rear drives invasion of breast tumor cells in 3D Matrigel. Proc Natl Acad Sci U S A. 2011;108:1943–1948. doi: 10.1073/pnas.1010396108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Khatau SB, Bloom RJ, Bajpai S, Razafsky D, Zang S, Giri A, Wu PH, Marchand J, Celedon A, Hale CM, et al. The distinct roles of the nucleus and nucleus-cytoskeleton connections in three-dimensional cell migration. Scientific reports. 2012;2:488. doi: 10.1038/srep00488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao T, Graham OS, Raposo A, St Johnston D. Growing microtubules push the oocyte nucleus to polarize the Drosophila dorsal-ventral axis. Science. 2012;336:999–1003. doi: 10.1126/science.1219147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Olins AL, Zwerger M, Herrmann H, Zentgraf H, Simon AJ, Monestier M, Olins DE. The human granulocyte nucleus: Unusual nuclear envelope and heterochromatin composition. European journal of cell biology. 2008;87:279–290. doi: 10.1016/j.ejcb.2008.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kraning-Rush CM, Califano JP, Reinhart-King CA. Cellular traction stresses increase with increasing metastatic potential. PloS one. 2012;7:e32572. doi: 10.1371/journal.pone.0032572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gros-Louis F, Dupre N, Dion P, Fox MA, Laurent S, Verreault S, Sanes JR, Bouchard JP, Rouleau GA. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat Genet. 2007;39:80–85. doi: 10.1038/ng1927. [DOI] [PubMed] [Google Scholar]
- 62.Attali R, Warwar N, Israel A, Gurt I, McNally E, Puckelwartz M, Glick B, Nevo Y, Ben-Neriah Z, Melki J. Mutation of SYNE-1, encoding an essential component of the nuclear lamina, is responsible for autosomal recessive arthrogryposis. Hum Mol Genet. 2009;18:3462–3469. doi: 10.1093/hmg/ddp290. [DOI] [PubMed] [Google Scholar]
- 63.Roux KJ, Crisp ML, Liu Q, Kim D, Kozlov S, Stewart CL, Burke B. Nesprin 4 is an outer nuclear membrane protein that can induce kinesin-mediated cell polarization. Proc Natl Acad Sci U S A. 2009;106:2194–2199. doi: 10.1073/pnas.0808602106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Horn HF, Brownstein Z, Lenz DR, Shivatzki S, Dror AA, Dagan-Rosenfeld O, Friedman LM, Roux KJ, Kozlov S, Jeang KT, et al. The LINC complex is essential for hearing. The Journal of clinical investigation. 2013;123:740–750. doi: 10.1172/JCI66911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Taranum S, Vaylann E, Meinke P, Abraham S, Yang L, Neumann S, Karakesisoglou I, Wehnert M, Noegel AA. LINC complex alterations in DMD and EDMD/CMT fibroblasts. European journal of cell biology. 2012;91:614–628. doi: 10.1016/j.ejcb.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Padiath QS, Saigoh K, Schiffmann R, Asahara H, Yamada T, Koeppen A, Hogan K, Ptacek LJ, Fu YH. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet. 2006;38:1114–1123. doi: 10.1038/ng1872. [DOI] [PubMed] [Google Scholar]
- 67.Hegele RA, Cao H, Liu DM, Costain GA, Charlton-Menys V, Rodger NW, Durrington PN. Sequencing of the reannotated LMNB2 gene reveals novel mutations in patients with acquired partial lipodystrophy. Am J Hum Genet. 2006;79:383–389. doi: 10.1086/505885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gao J, Li Y, Fu X, Luo X. A Chinese patient with acquired partial lipodystrophy caused by a novel mutation with LMNB2 gene. Journal of pediatric endocrinology & metabolism : JPEM. 2012;25:375–377. doi: 10.1515/jpem-2012-0007. [DOI] [PubMed] [Google Scholar]
- 69.Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, Stewart CL, Burke B. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol. 1999;147:913–920. doi: 10.1083/jcb.147.5.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen CY, Chi YH, Mutalif RA, Starost MF, Myers TG, Anderson SA, Stewart CL, Jeang KT. Accumulation of the inner nuclear envelope protein Sun1 is pathogenic in progeric and dystrophic laminopathies. Cell. 2012;149:565–577. doi: 10.1016/j.cell.2012.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Foster CR, Przyborski SA, Wilson RG, Hutchison CJ. Lamins as cancer biomarkers. Biochemical Society transactions. 2010;38:297–300. doi: 10.1042/BST0380297. [DOI] [PubMed] [Google Scholar]
- 72.de Las Heras JI, Batrakou DG, Schirmer EC. Cancer biology and the nuclear envelope: a convoluted relationship. Seminars in cancer biology. 2013;23:125–137. doi: 10.1016/j.semcancer.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 73.Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kong L, Schafer G, Bu H, Zhang Y, Zhang Y, Klocker H. Lamin A/C protein is overexpressed in tissue-invading prostate cancer and promotes prostate cancer cell growth, migration and invasion through the PI3K/AKT/PTEN pathway. Carcinogenesis. 2012;33:751–759. doi: 10.1093/carcin/bgs022. [DOI] [PubMed] [Google Scholar]
- 75.Foster CR, Robson JL, Simon WJ, Twigg J, Cruikshank D, Wilson RG, Hutchison CJ. The role of Lamin A in cytoskeleton organization in colorectal cancer cells: a proteomic investigation. Nucleus. 2011;2:434–443. doi: 10.4161/nucl.2.5.17775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cattin ME, Muchir A, Bonne G. `State-of-the-heart' of cardiac laminopathies. Current opinion in cardiology. 2013;28:297–304. doi: 10.1097/HCO.0b013e32835f0c79. [DOI] [PubMed] [Google Scholar]
- 77.Hynes RO. The emergence of integrins: a personal and historical perspective. Matrix biology : journal of the International Society for Matrix Biology. 2004;23:333–340. doi: 10.1016/j.matbio.2004.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bonne G, Di Barletta MR, Varnous S, Becane HM, Hammouda EH, Merlini L, Muntoni F, Greenberg CR, Gary F, Urtizberea JA, et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet. 1999;21:285–288. doi: 10.1038/6799. [DOI] [PubMed] [Google Scholar]
- 79.Muchir A, Bonne G, van der Kooi AJ, van Meegen M, Baas F, Bolhuis PA, de Visser M, Schwartz K. Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B) Hum Mol Genet. 2000;9:1453–1459. doi: 10.1093/hmg/9.9.1453. [DOI] [PubMed] [Google Scholar]
- 80.Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ, Jr., Spudich S, De Girolami U, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. The New England journal of medicine. 1999;341:1715–1724. doi: 10.1056/NEJM199912023412302. [DOI] [PubMed] [Google Scholar]
- 81.Quijano-Roy S, Mbieleu B, Bonnemann CG, Jeannet PY, Colomer J, Clarke NF, Cuisset JM, Roper H, De Meirleir L, D'Amico A, et al. De novo LMNA mutations cause a new form of congenital muscular dystrophy. Annals of neurology. 2008;64:177–186. doi: 10.1002/ana.21417. [DOI] [PubMed] [Google Scholar]
- 82.Renou L, Stora S, Yaou RB, Volk M, Sinkovec M, Demay L, Richard P, Peterlin B, Bonne G. Heart-hand syndrome of Slovenian type: a new kind of laminopathy. Journal of medical genetics. 2008;45:666–671. doi: 10.1136/jmg.2008.060020. [DOI] [PubMed] [Google Scholar]
- 83.Shackleton S, Lloyd DJ, Jackson SN, Evans R, Niermeijer MF, Singh BM, Schmidt H, Brabant G, Kumar S, Durrington PN, et al. LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet. 2000;24:153–156. doi: 10.1038/72807. [DOI] [PubMed] [Google Scholar]
- 84.Caux F, Dubosclard E, Lascols O, Buendia B, Chazouilleres O, Cohen A, Courvalin JC, Laroche L, Capeau J, Vigouroux C, et al. A new clinical condition linked to a novel mutation in lamins A and C with generalized lipoatrophy, insulin-resistant diabetes, disseminated leukomelanodermic papules, liver steatosis, and cardiomyopathy. The Journal of clinical endocrinology and metabolism. 2003;88:1006–1013. doi: 10.1210/jc.2002-021506. [DOI] [PubMed] [Google Scholar]
- 85.Novelli G, D'Apice MR. The strange case of the “lumper” lamin A/C gene and human premature ageing. Trends in molecular medicine. 2003;9:370–375. doi: 10.1016/s1471-4914(03)00162-x. [DOI] [PubMed] [Google Scholar]
- 86.De Sandre-Giovannoli A, Chaouch M, Kozlov S, Vallat JM, Tazir M, Kassouri N, Szepetowski P, Hammadouche T, Vandenberghe A, Stewart CL, et al. Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot-Marie-Tooth disorder type 2) and mouse. Am J Hum Genet. 2002;70:726–736. doi: 10.1086/339274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen L, Lee L, Kudlow BA, Dos Santos HG, Sletvold O, Shafeghati Y, Botha EG, Garg A, Hanson NB, Martin GM, et al. LMNA mutations in atypical Werner's syndrome. Lancet. 2003;362:440–445. doi: 10.1016/S0140-6736(03)14069-X. [DOI] [PubMed] [Google Scholar]
- 88.Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Navarro CL, De Sandre-Giovannoli A, Bernard R, Boccaccio I, Boyer A, Genevieve D, Hadj-Rabia S, Gaudy-Marqueste C, Smitt HS, Vabres P, et al. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum Mol Genet. 2004;13:2493–2503. doi: 10.1093/hmg/ddh265. [DOI] [PubMed] [Google Scholar]
- 90.Bione S, Maestrini E, Rivella S, Mancini M, Regis S, Romeo G, Toniolo D. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet. 1994;8:323–327. doi: 10.1038/ng1294-323. [DOI] [PubMed] [Google Scholar]
- 91.Zhang Q, Bethmann C, Worth NF, Davies JD, Wasner C, Feuer A, Ragnauth CD, Yi Q, Mellad JA, Warren DT, et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery-Dreifuss Muscular Dystrophy and are critical for nuclear envelope integrity. Hum Mol Genet. 2007 doi: 10.1093/hmg/ddm238. [DOI] [PubMed] [Google Scholar]
- 92.Puckelwartz MJ, Kessler EJ, Kim G, Dewitt MM, Zhang Y, Earley JU, Depreux FF, Holaska J, Mewborn SK, Pytel P, et al. Nesprin-1 mutations in human and murine cardiomyopathy. Journal of molecular and cellular cardiology. 2010;48:600–608. doi: 10.1016/j.yjmcc.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Breakefield XO, Blood AJ, Li Y, Hallett M, Hanson PI, Standaert DG. The pathophysiological basis of dystonias. Nature reviews. Neuroscience. 2008;9:222–234. doi: 10.1038/nrn2337. [DOI] [PubMed] [Google Scholar]