

Abstract
Chronic beryllium disease (CBD) is a granulomatous lung disorder caused by a hypersensitivity to beryllium and characterized by the accumulation of beryllium-specific CD4+ T cells in the lung. Genetic susceptibility to beryllium-induced disease is strongly associated with HLA-DP alleles possessing a glutamic acid at the 69th position of the β-chain (βGlu69). The structure of HLA-DP2, the most prevalent βGlu69-containing molecule, revealed a unique solvent-exposed acidic pocket that includes βGlu69 and represents the putative beryllium binding site. The delineation of mimotopes and endogenous self-peptides that complete the αβTCR ligand for beryllium-specific CD4+ T cells suggests a unique role of these peptides in metal ion coordination and the generation of altered self-peptides, blurring the distinction between hypersensitivity and autoimmunity.
Introduction
The characterization of ligands recognized by αβ T cell receptors (TCRs) has predominantly focused on peptide antigens. However, in addition to peptides, αβ TCRs recognize a variety of other ligands, including lipids, superantigens, small organic haptens and metal ions [1]. In this regard, repeated exposure to metals such as beryllium (Be), nickel (Ni) and gold (Au) may result in hypersensitivity. Be-induced hypersensitivity is one of the best characterized hypersensitivity syndromes. Approximately 2–16% of Be-exposed workers develop Be sensitization [2–6], and a subset of Be-sensitized subjects progress to chronic Be disease (CBD) [7]. The diagnosis of CBD hinges on the detection of a Be-specific adaptive immune response in blood and/or lung and the presence of noncaseating granulomatous inflammation on a lung biopsy specimen [8]. The histopathology of CBD is identical to that seen in sarcoidosis, a more common granulomatous lung disease of unknown etiology [9,10]. With the persistence of Be in the lung years after exposure cessation [11,12], the natural history of disease is characterized by a gradual decline in lung function, with one-third of untreated patients progressing to end-stage respiratory insufficiency [13]. Current therapeutic approaches include the use of immunosuppressive medications, such as prednisone, but no evidence exists that any treatment regimen altered the natural history of the disease.
Despite the presence of a small subset of Be-responsive T cells in blood, the majority of Be-specific CD4+ T cells are compartmentalized to the lung [14] and express a polarized Th1 phenotype [14,15]. These cells also express an oligoclonal TCR repertoire [16], are predominantly composed of effector memory T cells [17] and recognize Be in a CD28 independent manner [18]. Conversely, CD8+ T cells do not respond to Be stimulation in culture [17,19], suggesting that CD4+ T cells are the critical T cell subset involved in the immunopathogenesis of CBD.
This review focuses on recent advances in our understanding of the immunopathogenesis of Be-induced disease, in particular those involving the recognition of the MHCII-peptide/Be complex by pathogenic CD4+ T cells derived from the lungs of CBD patients.
Genetic Susceptibility to Beryllium-Induced Disease
In addition to workplace exposure to Be, genetic susceptibility plays a key role in the immunopathogenesis of Be-induced disease. Susceptibility has been most strongly associated with MHCII molecules, in particular HLA-DP [20–24]. Richeldi et al. [20] showed that HLA-DPB1 alleles with a glutamic acid (E) at position 69 of the β-chain (βGlu69) were strongly linked to disease susceptibility, with the most prevalent βGlu69-containing allele being HLA-DPB1*02:01 [20]. Multiple studies have corroborated these findings, documenting the presence of βGlu69-containing DPB1 alleles in 73–95% of CBD patients compared to 30–48% of exposed controls [21–26]. In addition, differential risk of disease development has been associated with certain βGlu69-containing DPB1 alleles [22–24,26].
Approximately 15% of CBD patients do not possess a βGlu69-containing HLA-DP allele, suggesting that other MHCII molecules may be important in genetic susceptibility to Be-induced disease [23,24]. In this population of CBD patients, an increased frequency of HLA-DR13 alleles, which possess an analogous glutamic acid residue at position 71 of the β-chain (βGlu71), was seen [24]. In CBD subjects without a βGlu69-containing HLA-DP allele, Rosenman et al. [27] showed that all of these subjects possessed at least one βGlu71-expressing HLA-DR allele. Collectively, the genetic studies suggest an important role of this negatively-charged glutamic acid in conferring risk of disease development in Be-exposed workers.
Structural Basis of CBD
To characterize the structural features of βGlu69-containing HLA-DP molecules that explain disease association, HLA-DP2 (DPA1*01:03, DPB1*02:01) with a bound self-peptide derived from the HLA-DR α-chain (pDRA) was crystallized and its structure solved to a resolution of 3.25Å [28]. Although the overall structure of the HLA-DP2-pDRA complex was similar to that of other MHCII/peptide complexes, several unique features were evident that might contribute to the role of this molecule in the development of CBD. For example, HLA-DP2 had one of the widest binding grooves (16.18Å) compared to 28 other published human and mouse MHCII structures in the Protein Data Base (PDB) (e.g., width averaged 15.01Å and varied from 13.59Å to 16.73Å) [28]. Importantly, this widening occurred between the peptide and β-chain α-helix (Figure 1). To confirm that the widened peptide binding groove was not a result of the crystallographic conditions, the structures of two additional HLA-DP2 molecules with different peptides in the binding groove (e.g., HLA-DP2-pRas (resolved to 2.7Å) and HLA-DP2-pA28 (1.9Å)) were solved, and nearly identical widths were documented (unpublished data). These findings suggest that the HLA-DP2 β-chain α-helix has rolled away from the peptide and the floor of the binding groove, and recent studies have shown that this region of the β-chain may be quite flexible [29], contributing to the variation in the width of this part of the binding groove.
Figure 1. HLA-DP2 possesses a widened peptide binding groove.

Three MHCII structures were overlaid on the basis of their α1 domains: DP2-pDRA (PDB ID code 3LQZ, magenta), HLA-DR3 bound to the invariant chain CLIP peptide (PDB ID code 1A6A, green) and mouse IAb bound to 3K peptide (PDB ID code 1LNU, blue). Human or mouse MHCII with a bound peptide were analyzed for the distance (Å) between the Cα of the p5 amino acid position of the peptide and the Cα of the MHCII β71 amino acid (equivalent to β69 of DP2): 7.62 Å (IAb-p3K), 9.00 Å (DR3-pCLIP), and 10.94 Å (DP2-pDRA). The distance between the peptide and DP2 β-chain α-helix is among the widest of all MHCII molecules.
Another unique structural feature of the HLA-DP2-pDRA complex was the elevated position of the peptide in the binding groove. This spacing resulted in an inability of the p4Leu of pDRA to occupy the p4 pocket of HLA-DP2. Compared to two other MHC-peptide structures also having a leucine at the p4 position of the peptide [30,31], the backbone of pDRA had risen up in the binding groove at the p4 position (Figure 2A) and the side chain of the p4Leu of pDRA was surface-exposed (Figure 2B). This structure suggests that the p4Leu contributes minimally to the binding affinity of pDRA for HLA-DP2.
Figure 2. Unusual configuration of the HLA-DP2 peptide binding groove.
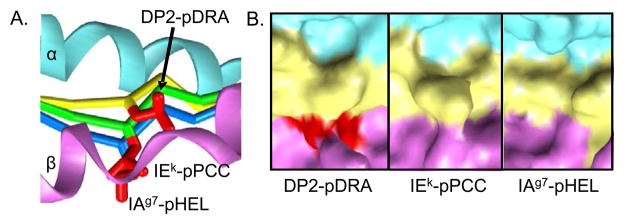
A, A side view of the DP2 β1 helix (magenta) toward the α1 helix (cyan) is shown. The pDRA backbone is shown as a Cα trace (yellow). Two other MHCII/peptide structures, which also have leucines at p4, were overlaid on the DP2-pDRA structure: IEk-pPCC (PDB ID code 1KTD) and IAg7-pHEL (PDB ID code 1F3J). Their peptide Cα traces are shown in green and blue, respectively. For all three peptides, the side chain of the p4Leu is shown as a red wireframe. B, For the same three structures shown in A, the solvent exposed surface of the MHC/peptide complex is shown in the region of p4Leu, α1 - cyan, β1 - magenta and peptide - yellow, expect for the side chain of p4Leu -red.
The net effect of these structural changes is a solvent-exposed acidic pocket that is flanked by leucines at the p4 and p7 positions of the pDRA (Figure 3A). The acidity of this pocket is due to the presence of three HLA-DP2 β-chain amino acids: βGlu68 and βGlu69 from the β-chain α-helix and βGlu26 from the floor of the peptide binding groove (Figure 3B). This cluster of glutamic acids was also solvent-exposed in the HLA-DP2-pRas and HLA-DP2-pA28 structures (unpublished data). Since βGlu69 is the most important polymorphism associated with the genetic susceptibility to CBD and solved structures of other proteins associated with Be generally show Be coordination by acidic amino acids [32], these findings strongly suggest that this acidic pocket is the Be binding site within the TCR footprint of HLA-DP2.
Figure 3. βGlu69 lies in a solvent-exposed acidic pocket.

A, The electrostatic surface charge of the HLA-DP2 molecule (with bound pDRA) is shown colored by the relative charge of the surface atoms (red - negative and blue - positive). A wireframe representation of the peptide is also shown with CPK coloring. B, View from above the surface-exposed acidic pocket of the HLA-DP2-pDRA structure. The β strand floors of DP2 α- and β-chain domains are colored light cyan and light magenta, respectively. Wireframe representations of the side chains of β26Glu, β68Glu, and β69Glu are shown with magenta carbon and red oxygen. The wireframe representations of p4 to p7 of the peptides are shown with white carbon, red oxygen and blue nitrogen.
Beryllium Presentation to CD4+ T Cells
Using antigen-specific T cell lines and hybridomas expressing Be-specific TCRs, investigators have shown that most Be presentation occurs through HLA-DP, with HLA-DR playing a minor role [33,34]. Conversely, only certain HLA-DP molecules are capable of presenting Be to pathogenic CD4+ T cells [33], and the HLA-DPB1 alleles that mediate Be presentation match those implicated in disease susceptibility, confirming that the contribution of βGlu69-containing HLA-DP molecules is based on the ability of those proteins to bind and present Be to pathogenic CD4+ T cells [33,34].
In addition to βGlu69, the HLA-DP2 crystal structure suggests that βGlu26 and βGlu68 may also be involved in Be coordination and presentation (Figure 3B). Because these two amino acids are invariant among HLA-DP alleles [35], they were not identified in genetic analyses of the linkage between HLA-DPB1 alleles and CBD, and their presence is not sufficient for Be presentation in the absence of βGlu69. Using fibroblasts expressing HLA-DP2 molecules mutated at each of these three positions, Dai et al. [28] showed that the mutant HLA-DP2 molecules were no longer able to induce Be-specific T cell activation, suggesting that these three glutamic acid residues are critical for Be coordination and T cell activation. Similarly, HLA-DR-restricted T cell recognition of Be recognition was also dependent on βGlu71 [36].
The unique structural features of HLA-DP2 raised the possibility that Be-responsive T cells may not recognize the HLA-DP2-peptide/Be complex in a conventional manner with a diagonal TCR orientation centered over the pMHCII complex. In this regard, unconventional binding interactions have been reported for the majority of human autoimmune complexes solved to date [37–39]. Using site-directed mutagenesis of the CDRs of Be-specific TCRs, Bowerman et al. [40] showed that Be-specific T cells recognize antigen using an unconventional binding topology, with the majority of interactions contributed by TCR Vβ CDR3 and the HLA-DP2 β1-chain. Thus, unusual docking topologies are not exclusively used by autoreactive T cells, but also for the recognition of metal antigens.
Beryllium-Dependent Peptides in Metal Ion Coordination
In addition to Be, specific peptides are required to complete the Be-specific αβTCR ligand [28]. However, a set of known HLA-DP2-binding peptides [41], including those derived from HLA-DR α-chain, Ras and HLA-A28, did not induce IL-2 secretion by T cell hybridomas expressing Be-specific TCRs [40], demonstrating that HLA-DP2-restricted T cell recognition of Be depends on a limited number of specific peptides. Falta et al. [42] identified Be-dependent mimotopes that bind to HLA-DP2 and Be, forming a complex recognized by pathogenic CD4+ T cells in CBD. Important characteristics of Be-dependent mimotopes included a preference for bulky hydrophobic or nonpolar amino acids at the p1 and p6 anchor positions that match the known HLA-DP2-binding motif [43], independence of TCR recognition on the C-terminus of the peptide, and negatively-charged aspartic and glutamic acid residues at p4 and p7 of the peptide that represent potential Be coordination sites [42]. The location of these two negatively-charged amino acids in addition to the three glutamic acids contributed by the HLA-DP2 β-chain strongly suggests their role in capturing and coordinating Be for T cell recognition.
In a search for endogenously-derived peptides with homology to the mimotope sequences and an ability to bind to HLA-DP2/Be and stimulate pathogenic CD4+ T cells from CBD patients, plexin A peptides were identified. Plexins are transmembrane proteins encoded by nine genes (PLXNA1-4, B1-3, C1 and D1) that are involved in cell movement and response [44]. Only the plexin A family contains the stimulatory epitope for the Be-responsive TCRs that includes the acidic amino acids at both the p4 and p7 positions [44]. Several plexin A proteins are expressed in BAL fluid and lung tissue; thus, they are readily available as a source of antigen in the target organ of CBD patients [42]. Using HLA-DP2-plexin A4 tetramers soaked in Be, Falta et al. [42] showed tetramer-binding CD4+ T cells in the BAL of all HLA-DP2-expressing CBD patients who possessed a Be-specific immune response in lung, strongly supporting a role of plexin A as an endogenous antigen for a set of Be-specific TCRs.
Models for Beryllium Presentation to CD4+ T Cells
Currently, the mechanism(s) by which Be binds to the MHCII/peptide remains an important unanswered question. The various possibilities by which Be may interact with this complex are shown in Figure 4. Be may bind to either the MHCII/peptide complex (Figure 4A) or peptide alone (Figure 4B), and both of these models would require direct interaction between Be and the TCR. An indirect interaction between Be and TCR is shown in Figure 4C and D. The presence of Be in the binding groove may neutralize the acidic environment of the p4 pocket and alter the repertoire of peptides capable of binding to the MHCII (Figure 4C). The ability of endogenous peptides, such as those derived from plexin A, to participate in metal ion capture may result in the conversion of a self-peptide into a neoantigen with an altered conformation (Figure 4D). The creation of neoantigens that are absent in the thymus and arise in a target organ plays a key role in the genesis of autoimmunity [45] and hypersensitivity [46]. In CBD, self-peptides bound in the HLA-DP2 binding groove may be altered by the presence of Be, resulting in the conversion of these endogenous peptides into neoantigens and culminating in Be-specific adaptive immunity. Although Figure 4D depicts the preferred model, ultimate confirmation will await structural analysis of the TCR-pMHCII/Be complex.
Figure 4.

Models of Be presentation to CD4+ T cells in the context of MHCII molecules. A, A schematic model for the direct binding of Be to the MHCII/peptide complex is shown. B, A direct model of interaction is depicted where the metal binds to the antigenic peptide alone. C, The potential ability of Be to alter the MHCII peptide repertoire that is capable of bindings due to the presence of Be in the peptide-biding groove is shown. D, The preferred model depicting the ability of Be to alter the conformation of self-peptides that are subsequently recognized as neoantigens by the TCR is shown. It remains unknown whether the Be-specific TCR directly contacts Be or recognizes an altered self-peptide with no direct contacts between the metal and the TCR (as shown in Figure 4D).
Conclusions
The unique structural features of HLA-DP2 and the ability of particular peptides to participate in Be ion capture and coordination provides an explanation for the generation of CBD in a genetically-susceptible host exposed to an environmental antigen. Similar to T cell recognition of self-peptide in autoimmunity, our findings suggest that Be-specific CD4+ T cells utilize an altered binding topology to recognize the HLA-DP2-peptide landscape created by the addition of Be. Thus, we believe that recent findings in Be-induced hypersensitivity are relevant to autoimmune diseases and the role of neoantigens in driving both hypersensitivity and autoimmunity.
Highlights.
CBD is a classic example of a disorder resulting from a gene-environment interaction.
HLA-DP2 is linked for the generation of beryllium-induced hypersensitivity.
HLA-DP2 possesses unique structural features, including a beryllium-binding site.
Beryllium’s binding to MHCII-peptide generates an abnormal landscape for TCR interaction.
Acknowledgments
This work was supported by the following NIH grants: HL062410, ES011810, HL102245 (to A.P.F) and TR000156 (to SD). SD is also supported by the Boettcher Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1•.Yin L, Crawford F, Marrack P, Kappler JW, Dai S. T-cell receptor (TCR) interaction with peptides that mimic nickel offers insight into nickel contact allergy. Proc Natl Acad Sci U S A. 2012;109:18517–18522. doi: 10.1073/pnas.1215928109. This study provided insight into T cell recognition of nickel in the context of HLA-DR52c and suggested a common site for cation binding in metal allergies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kreiss K, Newman LS, Mroz M, Campbell PA. Screening blood test identifies subclinical beryllium disease. J Occup Med. 1989;31:603–608. doi: 10.1097/00043764-198907000-00011. [DOI] [PubMed] [Google Scholar]
- 3.Kreiss K, Wasserman S, Mroz MM, Newman LS. Beryllium disease screening in the ceramics industry: blood test performance and exposure-disease relations. J Occup Med. 1993;35:267–274. [PubMed] [Google Scholar]
- 4.Kreiss K, Mroz MM, Zhen B, Martyny JW, Newman LS. Epidemiology of beryllium sensitization and disease in nuclear workers. Am Rev Respir Dis. 1993;148:985–991. doi: 10.1164/ajrccm/148.4_Pt_1.985. [DOI] [PubMed] [Google Scholar]
- 5.Kreiss K, Mroz MM, Newman LS, Martyny J, Zhen B. Machining risk of beryllium disease and sensitization with median exposures below 2 μg/m3. Am J Ind Med. 1996;30:16–25. doi: 10.1002/(SICI)1097-0274(199607)30:1<16::AID-AJIM3>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 6.Henneberger PK, Cumro D, Deubner DD, Kent MS, McCawley M, Kreiss K. Beryllium sensitization and disease among long-term and short-term workers in a beryllium ceramics plant. Int Arch Occup Environ Health. 2001;74:167–176. doi: 10.1007/s004200100237. [DOI] [PubMed] [Google Scholar]
- 7.Newman LS, Mroz MM, Balkissoon R, Maier LA. Beryllium sensitization progresses to chronic beryllium disease: a longitudinal study of disease risk. Am J Respir Crit Care Med. 2004;171:54–60. doi: 10.1164/rccm.200402-190OC. [DOI] [PubMed] [Google Scholar]
- 8.Newman LS, Kreiss K, King TE, Jr, Seay S, Campbell PA. Pathologic and immunologic alterations in early stages of beryllium disease: re-examination of disease definition and natural history. Am Rev Respir Dis. 1989;139:1479–1486. doi: 10.1164/ajrccm/139.6.1479. [DOI] [PubMed] [Google Scholar]
- 9.Newman LS, Kreiss K. Nonoccupational beryllium disease masquerading as sarcoidosis: identification by blood lymphocyte proliferation response to beryllium. Am Rev Respir Dis. 1992;145:1212–1214. doi: 10.1164/ajrccm/145.5.1212. [DOI] [PubMed] [Google Scholar]
- 10.Newman LS, Rose CS, Maier LA. Sarcoidosis. N Engl J Med. 1997;336:1224–1234. doi: 10.1056/NEJM199704243361706. [DOI] [PubMed] [Google Scholar]
- 11.Jones-Williams W, Kelland D. New aid for diagnosing chronic beryllium disease (CBD): laser ion mass analysis (LIMA) J Clin Pathol. 1986;39:900–901. doi: 10.1136/jcp.39.8.900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones-Williams W, Wallach ER. Laser microprobe mass spectrometry (LAMMS) analysis of beryllium, sarcoidosis, and other granulomatous diseases. Sarcoidosis. 1989;6:111–117. [PubMed] [Google Scholar]
- 13.Newman LS, Lloyd J, Daniloff E. The natural history of beryllium sensitization and chronic beryllium disease. Environ Health Perspect. 1996;104:937S–943S. doi: 10.1289/ehp.96104s5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fontenot AP, Canavera SJ, Gharavi L, Newman LS, Kotzin BL. Target organ localization of memory CD4+ T cells in patients with chronic beryllium disease. J Clin Invest. 2002;110:1473–1482. doi: 10.1172/JCI15846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tinkle SS, Kittle LA, Schumacher BA, Newman LS. Beryllium induces IL-2 and IFN-γ in berylliosis. J Immunol. 1997;158:518–526. [PubMed] [Google Scholar]
- 16.Fontenot AP, Falta MT, Freed BM, Newman LS, Kotzin BL. Identification of pathogenic T cells in patients with beryllium-induced lung disease. J Immunol. 1999;163:1019–1026. [PubMed] [Google Scholar]
- 17.Fontenot AP, Palmer BE, Sullivan AK, Joslin FG, Wilson CC, Maier LA, Newman LS, Kotzin BL. Frequency of beryllium-specific, central memory CD4+ T cells in blood determines proliferative response. J Clin Invest. 2005;115:2886–2893. doi: 10.1172/JCI24908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fontenot AP, Gharavi L, Bennett SR, Canavera SJ, Newman LS, Kotzin BL. CD28 costimulation independence of target organ versus circulating memory antigen-specific CD4+ T cells. J Clin Invest. 2003;112:776–784. doi: 10.1172/JCI18317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saltini C, Winestock K, Kirby M, Pinkston P, Crystal RG. Maintenance of alveolitis in patients with chronic beryllium disease by beryllium-specific helper T cells. N Engl J Med. 1989;320:1103–1109. doi: 10.1056/NEJM198904273201702. [DOI] [PubMed] [Google Scholar]
- 20.Richeldi L, Sorrentino R, Saltini C. HLA-DPB1 glutamate 69: a genetic marker of beryllium disease. Science. 1993;262:242–244. doi: 10.1126/science.8105536. [DOI] [PubMed] [Google Scholar]
- 21.Richeldi L, Kreiss K, Mroz MM, Zhen B, Tartoni P, Saltini C. Interaction of genetic and exposure factors in the prevalence of berylliosis. Am J Ind Med. 1997;32:337–340. doi: 10.1002/(sici)1097-0274(199710)32:4<337::aid-ajim3>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 22.Wang Z, White PS, Petrovic M, Tatum OL, Newman LS, Maier LA, Marrone BL. Differential susceptibilities to chronic beryllium disease contributed by different Glu69 HLA-DPB1 and -DPA1 alleles. J Immunol. 1999;163:1647–1653. [PubMed] [Google Scholar]
- 23.Rossman MD, Stubbs J, Lee CW, Argyris E, Magira E, Monos D. Human leukocyte antigen Class II amino acid epitopes: susceptibility and progression markers for beryllium hypersensitivity. Am J Respir Crit Care Med. 2002;165:788–794. doi: 10.1164/ajrccm.165.6.2104002. [DOI] [PubMed] [Google Scholar]
- 24.Maier LA, McGrath DS, Sato H, Lympany P, Welsh K, Du Bois R, Silveira L, Fontenot AP, Sawyer RT, Wilcox E, et al. Influence of MHC class II in susceptibility to beryllium sensitization and chronic beryllium disease. J Immunol. 2003;171:6910–6918. doi: 10.4049/jimmunol.171.12.6910. [DOI] [PubMed] [Google Scholar]
- 25.McCanlies EC, Ensey JS, Schuler CR, Kreiss K, Weston A. The association between HLA-DPB1Glu69 and chronic beryllium disease and beryllium sensitization. Am J Ind Med. 2004;46:95–103. doi: 10.1002/ajim.20045. [DOI] [PubMed] [Google Scholar]
- 26.Silveira LJ, McCanlies EC, Fingerlin TE, Van Dyke MV, Mroz MM, Strand M, Fontenot AP, Bowerman N, Dabelea DM, Schuler CR, et al. Chronic beryllium disease, HLA-DPB1, and the DP peptide binding groove. J Immunol. 2012;189:4014–4023. doi: 10.4049/jimmunol.1200798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenman KD, Rossman M, Hertzberg V, Reilly MJ, Rice C, Kanterakis E, Monos D. HLA class II DPB1 and DRB1 polymorphisms associated with genetic susceptibility to beryllium toxicity. Occup Environ Med. 2011;68:487–493. doi: 10.1136/oem.2010.055046. [DOI] [PubMed] [Google Scholar]
- 28••.Dai S, Murphy GA, Crawford F, Mack DG, Falta MT, Marrack P, Kappler JW, Fontenot AP. Crystal structure of HLA-DP2: Implications for chronic beryllium disease. Proc Natl Acad Sci U S A. 2010;107:7425–7430. doi: 10.1073/pnas.1001772107. This study represents the first structural analysis of an HLA-DP molecule and identified the putative beryllium binding site. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Painter CA, Cruz A, Lopez GE, Stern LJ, Zavala-Ruiz Z. Model for the peptide-free conformation of class II MHC proteins. PLoS ONE. 2008;3:e2403. doi: 10.1371/journal.pone.0002403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fremont DH, Dai S, Chiang H, Crawford F, Marrack P, Kappler J. Structural basis of cytochrome c presentation by IE(k) J Exp Med. 2002;195:1043–1052. doi: 10.1084/jem.20011971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Latek RR, Suri A, Petzold SJ, Nelson CA, Kanagawa O, Unanue ER, Fremont DH. Structural basis of peptide binding and presentation by the type I diabetes-associated MHC class II molecule of NOD mice. Immunity. 2000;12:699–710. doi: 10.1016/s1074-7613(00)80220-4. [DOI] [PubMed] [Google Scholar]
- 32.Cho H, Wang W, Kim R, Yokota H, Damo S, Kim SH, Wemmer D, Kustu S, Yan D. BeF3− acts as a phosphate analog in proteins phosphorylated on aspartate: structure of a BeF3− complex with phosphoserine phosphatase. Proc Natl Acad Sci U S A. 2001;98:8525–8530. doi: 10.1073/pnas.131213698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fontenot AP, Torres M, Marshall WH, Newman LS, Kotzin BL. Beryllium presentation to CD4+ T cells underlies disease susceptibility HLA-DP alleles in chronic beryllium disease. Proc Natl Acad Sci U S A. 2000;97:12717–12722. doi: 10.1073/pnas.220430797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lombardi G, Germain C, Uren J, Fiorillo MT, du Bois RM, Jones-Williams W, Saltini C, Sorrentino R, Lechler R. HLA-DP allele-specific T cell responses to beryllium account for DP-associated susceptibility to chronic beryllium disease. J Immunol. 2001;166:3549–3555. doi: 10.4049/jimmunol.166.5.3549. [DOI] [PubMed] [Google Scholar]
- 35.Gilchrist FC, Bunce M, Lympany PA, Welsh KI, du Bois RM. Comprehensive HLA-DP typing using polymerase chain reaction with sequence-specific primers and 95 sequence-specific primer mixes. Tissue Antigens. 1998;51:51–61. doi: 10.1111/j.1399-0039.1998.tb02946.x. [DOI] [PubMed] [Google Scholar]
- 36.Bill JR, Mack DG, Falta MT, Maier LA, Sullivan AK, Joslin FG, Martin AK, Freed BM, Kotzin BL, Fontenot AP. Beryllium presentation to CD4+ T cells is dependent on a single amino acid residue of the MHC class II β-chain. J Immunol. 2005;175:7029–7037. doi: 10.4049/jimmunol.175.10.7029. [DOI] [PubMed] [Google Scholar]
- 37.Sethi DK, Schubert DA, Anders AK, Heroux A, Bonsor DA, Thomas CP, Sundberg EJ, Pyrdol J, Wucherpfennig KW. A highly tilted binding mode by a self-reactive T cell receptor results in altered engagement of peptide and MHC. J Exp Med. 2011;208:91–102. doi: 10.1084/jem.20100725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hahn M, Nicholson MJ, Pyrdol J, Wucherpfennig KW. Unconventional topology of self peptide-major histocompatibility complex binding by a human autoimmune T cell receptor. Nat Immunol. 2005;6:490–496. doi: 10.1038/ni1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y, Huang Y, Lue J, Quandt JA, Martin R, Mariuzza RA. Structure of a human autoimmune TCR bound to a myelin basic protein self-peptide and a multiple sclerosis-associated MHC class II molecule. EMBO J. 2005;24:2968–2979. doi: 10.1038/sj.emboj.7600771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40••.Bowerman NA, Falta MT, Mack DG, Kappler JW, Fontenot AP. Mutagenesis of beryllium-specific TCRs suggests an unusual binding topology for antigen recognition. J Immunol. 2011;187:3694–3703. doi: 10.4049/jimmunol.1101872. This study was the first to suggest that beryllium-specific CD4+ T cells utilize an unconventional binding topology in the recognition of HLA-DP2-peptide/Be complexes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Diaz G, Canas B, Vazquez J, Nombela C, Arroyo J. Characterization of natural peptide ligands from HLA-DP2: new insights into HLA-DP peptide-binding motifs. Immunogenetics. 2005;56:754–759. doi: 10.1007/s00251-004-0735-5. [DOI] [PubMed] [Google Scholar]
- 42••.Falta ML, Pinilla C, Mack DG, Tinega A, Crawford F, Guilianotti M, Santos R, Clayton GM, Wang Y, Zhang X, et al. Identification of peptides recognized by CD4 T cells in chronic beryllium disease. J Exp Med. 210:1403–1418. doi: 10.1084/jem.20122426. This study was the first to delineate a complete αβTCR ligand for a metal-specific CD4+ T cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sidney J, Steen A, Moore C, Ngo S, Chung J, Peters B, Sette A. Five HLA-DP molecules frequently expressed in the worldwide human population share a common HLA supertypic binding specificity. J Immunol. 2010;184:2492–2503. doi: 10.4049/jimmunol.0903655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roney K, Holl E, Ting J. Immune plexins and semaphorins: old proteins, new immune functions. Protein Cell. 2013;4:17–26. doi: 10.1007/s13238-012-2108-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marrack P, Kappler JW. Do MHCII-presented neoantigens drive type 1 diabetes and other autoimmune diseases? Cold Spring Harb Perspect Med. 2012;2:2–17. doi: 10.1101/cshperspect.a007765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46•.Illing PT, Vivian JP, Dudek NL, Kostenko L, Chen Z, Bharadwaj M, Miles JJ, Kjer-Nielsen L, Gras S, Williamson NA, et al. Immune self-reactivity triggered by drug-modified HLA-peptide repertoire. Nature. 2012;486:554–558. doi: 10.1038/nature11147. Outstanding study that delineated the structural basis of the abacavir hypersensitivity syndrome. [DOI] [PubMed] [Google Scholar]