

Abstract
Microneedles are a relatively simple, minimally invasive and painless approach to deliver drugs across the skin. However, there remain limitations with this approach because of the materials most commonly utilized for such systems. Silk protein, with tunable and biocompatibility properties, is a useful biomaterial to overcome the current limitations with microneedles. Silk devices preserve drug activity, offer superior mechanical properties and biocompatibility, can be tuned for biodegradability, and can be processed under aqueous, benign conditions. In the present work, we report the fabrication of dense microneedle arrays from silk with different drug release kinetics. The mechanical properties of the microneedle patches are tuned by post-fabrication treatments or by loading the needles with silk microparticles to increase capacity and mechanical strength. Drug release is further enhanced by the encapsulation of the drugs in the silk matrix and coating with a thin dissolvable drug layer. The microneedles are used on human cadaver skin and drugs were delivered successfully. The various attributes demonstrated suggest that silk-based microneedle devices can provide significant benefit as a platform material for transdermal drug delivery.
Keywords: Silk, Micromachining, Transdermal Drug Delivery, Skin, Microneedle
1. Introduction
In the late 1990s, microelectronics technology enabled the idea of microneedles (MN) that were originally proposed in the 1970s for transdermal drug delivery. MN technology has grown with various techniques for fabricating MN arrays with a variety of different materials[1–4]. MN are a safe and promising technology to overcome the skin barrier (stratum corneum) for the delivery of any size drug, including particles[5]. Currently, a number of drugs can be delivered transdermally[6, 7] and are amenable to MN use. A 25% annual growth rate has been experienced for transdermal drug delivery compared to 2% for oral drug delivery, and this figure is expected to rise as novel devices emerge.[8]
The first MN devices were made of silicon prepared using microfabrication techniques.[9] Silicon can be etched isotropically by wet or dry etching techniques or anisotropically either with deep reactive ion etching or in KOH (potassium hydroxide).[9–12] These techniques provide limited control over the geometry of the fabricated needles. This was resolved by combining the two etching techniques (e.g. isotropic and anisotropic) approaches,[13] which resulted in more complex processing steps and a relatively expensive process. Photosensitive polymers have been used to fabricate MN in a clean room environment using photolithography. SU8 (an epoxy-base negative photoresist) has been used to generate templates for MN devices[14] and poly-methyl methacrylate (PMMA) combined with LIGA (lithography, electroplating and molding) processing by vertical X-ray exposure generated high aspect ratio MN.[14] Two photon polymerization was also utilized to fabricate out of plane MN in a sequential layer by layer fabrication process.[15–17] However, most of the above techniques are expensive, tedious, and not suitable for mass production.
Laser ablation, laser cutting and wet etching have been utilized to generate microneedle templates in metals, followed by bending MN templates out of plane (90°) to generate three dimensional arrays.[18, 19] Metallic MN are available as commercial devices for enhancing skin permeability for cosmetics, such as Dermaroller,[6] or drug molecules pre-coated onto the tips of the microneedles. Drug-coated MN can deliver a fixed but limited amount of drug across the skin.[20]
To improve control of the release of drugs, encapsulation of the drug in the MN matrix is needed, as opposed to coating the tips. The most common method used in fabricating polymeric MN is micromolding, in which the MN mold is either filled with concentrated polymer solution and allowed to dry or filled with melted polymer and solidified. Water-based materials such as carboxymethylcellulose (CMC),[21, 22] dextrin,[23, 24] sugar,[25, 26] polyvinyl pyrrolidone (PVP), polyvinyl pyrrolidone-methacrylic acid (PVP-MAA)[27] and chondroitin sulfate,[24, 28] have been used to fabricate MN. Melted maltose or galactose,[25, 29] and poly(lactic-co-glycolic acid (PLGA)[4, 30, 31] have been used in MN molds. Polydimethylsiloxane (PDMS) is often used in prototyping biomedical devices[32–35] and also utilized as a molding material for MN. MN devices fabricated from these materials advanced the field yet limitations remain, such as high temperature processing, quick dissolution of the MN resulting in less control over drug release, instability under humid conditions, UV cross linking requirements, and drug materials/process incompatibility that can impact the potency of the compounds to be delivered.
Silk is a remarkable protein biopolymer that has gained attention for biomedical devices due to its robust mechanical features, biocompatibility, controllable processing in aqueous conditions, as well as its ability to be shaped into various formats.[36] Silk devices for biological implants, drug delivery systems, cell cultivation substrates, gels and photonics materials have been demonstrated.[37–43] The strength and toughness of silk materials can be tailored for particular applications, [44] such as to control drug release kinetics and degradation rate. [45] We have previously shown the fabrication of silk-based MN using micromolding techniques.[46]
In the present study we significantly extend our earlier findings with silk MN by demonstrating the fabrication of dense silk MN arrays in different shapes and sizes using a new fabrication technique. The utility of this technique is demonstrated in the context of silk MN devices, but can be used with other polymers as well. We show different strategies to enhance the mechanical strength of the silk MN along with improved drug release characteristics, including loading and release. The silk based MN patches were evaluated on human cadaver skin for drug delivery. These new silk-based MN systems address the limitations cited above with other current materials used in MNs.
2. Results and Discussion
2.1. Fabrication
Conventional machining can be used to generate MN molds with resolution of tens of microns using fine drilling/milling tool and the depth of feature can be controlled in range of hundreds of microns. To fabricate a MN mold, a 5 mm chamber was drilled into a machinable wax followed by drilling a MN template into the chamber Figure 1A shows the process flow, an SEM of the drilling bits and a final wax mold. A heat cure epoxy was casted onto fabricated wax mold to create MN template that was then use to make PDMS MN negative molds as shown in Figure 1B. Once the PDMS molds were prepared, silk solution was poured into the molds and centrifuged for 20 minutes at 5000 rpm followed by application of vacuum for 30 minutes to fill the MN trenches with the silk solution. After drying, the high aspect micro features of the structures were successfully transferred into the silk material. There were 400 MN tips in a 1.5 × 1.5 cm2 patch that is the dense MN array achieved through this relatively simple process.
Figure 1.

A) (i – iv) Process flow of mold fabrication, showing a machinable wax mold. Left Bottom left is showing SEM images of drilling bits with different tip sizes and bottom right is the fabricated negative MN mold in machinable wax material. B) (i) Schematic of epoxy casting onto the wax mold and optical image of epoxy MN, (ii) Polydimethylsiloxane (PDMS) negative MN mold fabricated from an epoxy-based MN generated from machinable wax, (iii) digital image of pure silk MN prepared in a PDMS mold.
This fabrication approach was versatile in allowing changes in needle dimensions and spacing as a way to assess prototype MN with various designs and shapes. Previously, we had developed a metal (Aluminum, (Al)) based a MN master template using a milling tool, followed by an isotropic (wet) etching of the Al template and casting of PDMS onto the Al template to generate the MN molds.[46] For comparative studies we further modified the design, shape and geometry of the MN from the previous data. Figure 2A shows an SEM of the modified silk MN fabricated with the previous technique with the needles in a pyramid shape, while the base of the pyramid is a square, 850 micron on each side. Figure 2J shows a magnified image of the pyramid shape needles loaded with microparticles. The spacing and the height between these needles was restricted by the milling tool diameter (i.e. >1 mm). In the new fabrication technique from the present study, the density (needles/unit area) of the needles was increased almost 2.5 times; in Figure 2 (B) – (I) all the needles were fabricated by this new method. Images (B) through (F) in Figure 2 were collected from MN prepared from the aqueous silk solution, while (Figure 3G – I) were MN fabricated using the hexafluoroisopropanol (HFIP) silk solution. The tips of the fabricate needle range from 40 μm to 15μm depend on the size of the drilling tool. No significant difference was observed in the needles fabricated from an aqueous or solvent solubilized silk, showing that the technique can be applied to both types of solution.
Figure 2.

A) SEM digital image of aqueous silk MN (pyramid shape) fabricated by our previous method [43]. B) – F) SEM images of cone shaped MN prepared from an aqueous silk solution with different shapes and geometries. G) – I) SEM images of cone shaped MN made from solvent-based (hexafluoroisopropanol) silk solution with various shapes and sizes. J) and K) Microparticle-loaded aqueous silk MN from pyramid and cone shaped needles, respectively.
Figure 3.
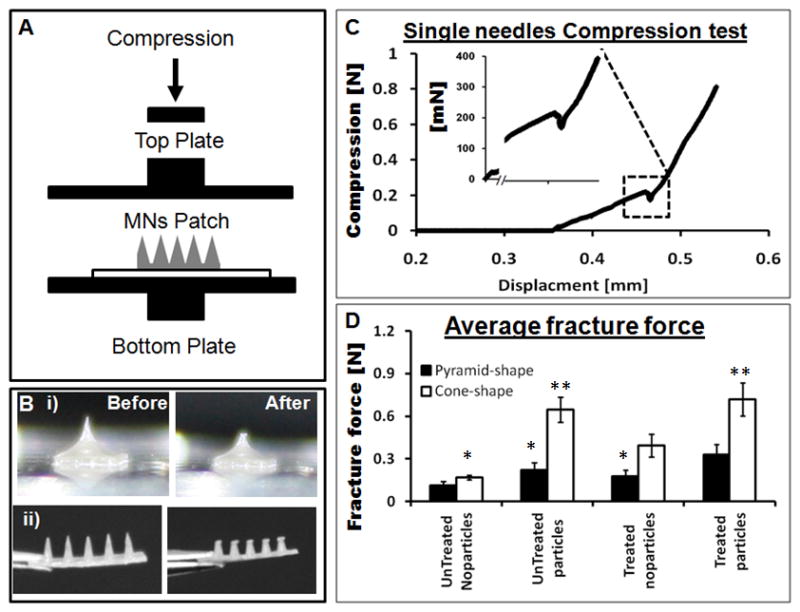
A) Schematic of an axial mechanical test setup to assess MN device performance with two horizontal parallel plates. B) (i) and (ii) Digital images of pyramid and a cone shaped needles, respectively, before and after mechanical testing. C) A single needle compression test that breaks around 225 mN during compression. D) Fracture force of all sub-groups of the pyramid and the cone shaped MN fabricated by micromolding. (N = 10, error bars represent standard deviations) Significant effects are indicated by one (p < 0.05) or two stars (p < 0.01), the one start belong to same group and two stars belong to second group on the bases of 95% confidence interval using ANOVA.
The dimensions of the MN, the diameter of the tip, base and the angle of the cone for individual needles was controlled by the selection of the appropriate milling tool, while the length of the needles was determined by the depth of drilling. Highly magnified MN tips are inserted in each image in Figure 2 to show the dimensions. With the current technique the height of each needle can be controlled in the range from couple of hundreds microns to more than millimeters in size. The microparticles-loaded cone shaped MN is shown in Figure 2K. The MN cone shapes shown in Figure 2B were fabricated from an aqueous silk solution and compared with the pyramid shape (Figure 2A) prepared from same silk solution and fabricated with the same height (700 μm). The size of the tip was approximately 15 microns and the angle of the cone for the MN in Figure 2B was approximately 30 degrees with the base diameter around 360 microns. This size was less than half of the side (base) of the pyramid shaped needles.
2.2. Mechanical properties
A critical characteristic of MN is mechanical stability to overcome the skin barrier and deliver therapeutics, while also avoiding tip fracture during the process. MN with fracture force above 100 mN/needle can successfully pierce the skin barrier.[31] Previously reported, MN fabricated from PLGA failed at approximately 160 mN,[4] while MN from polyvinyl pyrrolidone (PVP) and polyvinyl pyrrolidone-co-methacrylic acid (PVP-MAA) blends experienced failure in a range from 100 mN to 400 mN.[47] The MN fabricated here from aqueous silk solutions had mechanical strength in range from 100 mN to 700 mN based on the specific design, treatment and fabrication method utilized. For mechanical testing the silk MN patches were compressed between two metallic parallel (horizontal) plates shown in Figure 3A, while Figure 3B (i) and (ii) are representative images of the pyramid and cone shaped MN, respectively, before and after compression. The profile of a single microneedle compression test is shown in Figure 3C where the starting position of the top plate was at 0 mm and compression zero (in air) until the plate touched the needle tip at around 0.37 mm. As the top plate engaged with the MN and resistance was observed, the compression value rose (with a uniform slope) until the needle broke at around 0.47 mm. A sharp drop in compression was observed at 225 mN which was the mechanical strength (or fracture force) for that particular needle.
The average value for the fracture force measured for the water vapor treated pyramid shaped MN without microparticles was approximately 175 mN/needles (Figure 4D). When tested before water vapor treatment, the same needles showed reduced mechanical strength of around 110 mN/needle. Under the same conditions the cone shaped silk MN had an average value of 400 mN/needle measured for the water vapor-treated MN containing no microparticles. The microparticles improved mechanical stability of the MN even without water vapor treatment, while in combination with water vapor treatment the fracture force improved further to 330 mN/needle in the case of the pyramid MN. By mixing silk solution and beta sheet induced microparticles created a silk composite material, with the interfacial compatibility between silk liquid (continues phase) and particle phase. This silk composite formulation reinforced the the bulk silk material and improved mechanical stability of.[44, 48, 49] The cone shaped MN had superior mechanical properties than the pyramid shaped needles, with an average fracture force of 720 mN/needle in the case of the microparticle-loaded MN with water vapor treatment (Figure 3D). The water vapor treatments increase beta sheet (crystalline) content in the silk material resulting in the improved mechanical properties as well as insolubility in water.[50] The mechanical stability of the needles can be compromised by increasing the amount of drug that can be overcome by adding microparticles into the silk matrix (see below).
Figure 4.
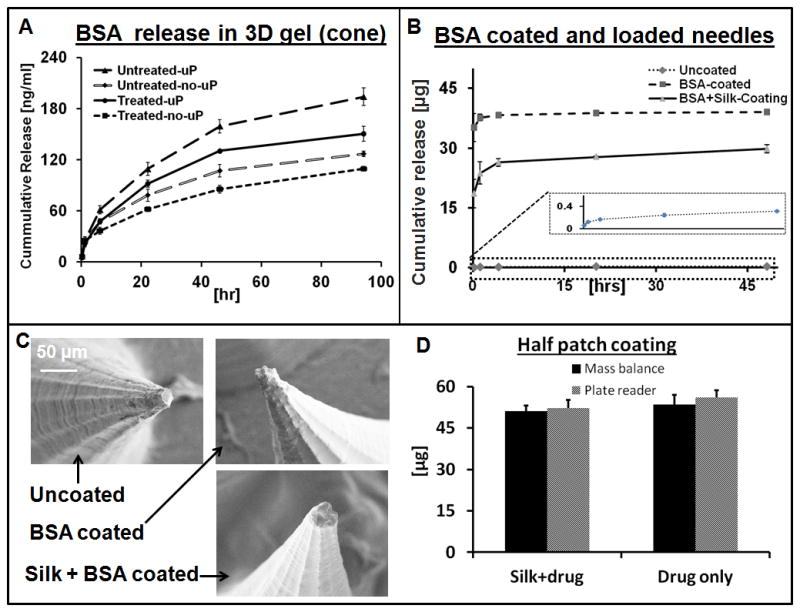
BSA release from the silk MN in 3D collagen hydrogels, determined by digestion of collagen in collagenase solution followed by optical spectroscopy of the released BSA. A) Plots showing the release profile from cone shaped microneedles loaded with drug only and also microparticle containing same drug. B) Release kinetics of Drug loaded and coated MN patches. C) SEM digital images of the MN before and after coating with different drug solutions. D) Quantification of coated drug onto MN patch and verified by two separate techniques. (N = 3, error bars represent standard deviations).
2.3. Drug release in 3D gel
Various studies have shown the utilization of silk biomaterials for release of different therapeutics in a controlled manner from the silk matrix.[51] Post fabrication treatments improve the mechanical stability of the silk, but also provide control of the degradation rate and drug release kinetics.[45] In the current study we released bovine serum albumin labeled with Texas-red (BSA) and compared drug release from the same material prepared in MN format with the various techniques. The release of BSA form the MN was performed into a 3D collagen gel (contain >85% water) matrix at room temperature. The cone-shaped MN loaded with BSA released three times more drug than the pyramid shaped MN patches (Figure 1S), while more overall drug was released from the untreated (not water vapor annealed) MN patches (Figure 4A). The increase drug flux was related to an increase in number of needles per patch and the treatment conditions. Compared to MN containing BSA the treated (water vapor annealed) MN with no microparticles released 6.5 time less drug than the untreated versions (Figure 4A). The microparticles loaded MN patch contained 1.8 times more drug without compromising its mechanical integrity. The controlled release experiment was performed over 2 days and a maximum of ~160 ng drug was released from a 7 × 7 mm2 patch (high density needles/patch, cone shaped needles, no treatment, microparticles loaded). The actual patch size was a 15 × 15 mm2, which approximates the release of 0.8 μg of drug in the same time period under these conditions.
Though in a cone shaped design the number of needles per patch increased by 2.5 times, which increased the influx of the drug into the collagen gel. Still most of the drug was entrapped in the silk patch after two days period. In order to enhance the delivery of drug in a shorter period of time, coating of additional drug was performed (as discussed below)
Galactose MN fabricated by micromolding was able to release 5 μg of drug (aminolevulinic acid) when initially loaded with 25 μg of drug. The issue with sugar-based MN is the instability and high temperature processing that leads to loss of active drug.[25] Vinyl pyrrolidone and its copolymer have been processed under UV light to fabricate MN, and the mechanical strength of these needles can be adjusted.[47] However, as with the sugar MN, these needles dissolve quickly in water and release 80 to 99% of the loaded drug within 5 to 20 minutes.[52] A single vaccine dose delivered through PVP MN induced a protective immune response, which showed the advantage of MN over intramuscular delivery. [27] A maximum of 1 mg of a low molecular weight model drug (i.e. sulforhodamine) was delivered through carboxymethylcellulose MN over 2 days.[21] Coated MN systems released almost the entire coated drug within 5 to 15 minutes. Titanium MN delivered 22 μg of desmopression in 15 min when coated with 23 μg of the drug.[53] The same system delivered 9.9 μg of ovalbumin (per cm2 area of patch) when coated with 238 μg of ovalbumin in the same surface area.[7] Stainless steel MN coated with 1.6 μg of plasmid (hepatitis C) delivered 1.2 μg in one minute and produce an immune response comparable to the plasmid when delivered using a gene gun.[54] In the present study we combined both coating and encapsulation in a single MN system that is challenging or not possible to achieve with existing materials used for dissolvable MN technology. In the release studies an initial burst of the coated drug from the MN device was followed by slow release from the MN matrix. Figure 4B shows that almost 70% of the coated drug was released in first 10 minutes, but in case of silk and drug (formulation) coated MN (2nd type coating) only 40% of the coated drug was released due to the slow dissolution of thin silk film. The release of drug was slower in the case when silk and drug formulation was coated on the MN patch while direct drug coating gave an abrupt release in first 1 hour. The drug release profile of the 2nd type (silk and drug formulation) of coating can be adjusted and tailored for specific applications by controlling film thinness, beta sheet content in the coated film and also by using different processing conditions for the silk used in the coating. Drug release was performed from a quarter of the whole patch, which released approximately 38 μg in the case of the direct coating and 30 μg from the 2nd formulation in 2 days. In SEM images there was no significant observable difference between the tips of coated and uncoated needles (Figure 4C). Figure 4D shows the quantification of drug coated on silk MN and was characterized by two independent techniques (micro plate reader and mass balance), which gave us similar results. Various sizes of the patches were coated with the same drug solution and as the patch size increased the amount of coated drug increased (Figure 2S)
2.4. In Vitro cadaver skin test
Human cadaver skin was tested with the silk MN (with and without drug) patches to determine skin permeation and drug delivery. Pyramid and cone shaped MN successfully breached the stratum corneum. Histological assays (Figure 3S and Figure 5A) showed that both designs successfully penetrated into the epidermis. In the case of the sulforhodamine sodium salt (sRh) loaded MN, all the patches from the different sub-groups (see Experimental Section) released the model drug into the human cadaver skin. Under the same scenario (Figure 3S and 5C) the cone shaped needles released more sRh into the skin compared to the pyramid needles. The maximum amount of drug released from the patches was with those loaded with microparticles containing the sRh and no further post fabrication treatment (Figure 5B). As the needles were more closely packed in the cone shaped design, the sRh was more uniformly distributed throughout the epidermis. Silk patches (films) without MN released sRh only at the top surface of the skin, which demonstrated the need of MN to deliver drug across the stratum corneum (Figure 5D).
Figure 5.
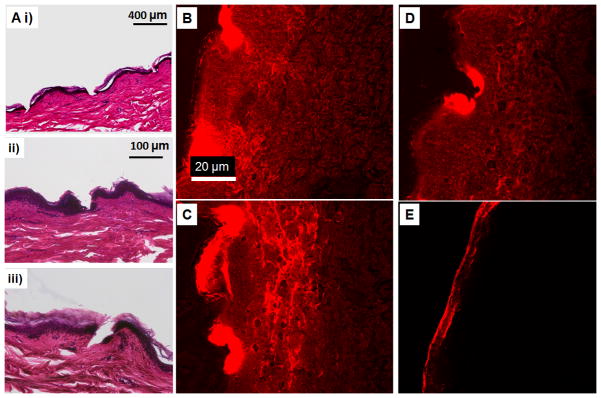
A) H&E micrographs of human cadaver skin treated with cone shaped silk MN, (i) MN loaded with microparticles and no post treatment, (ii) MN with no microparticles and post treatment with water vapor and (iii) MN loaded with microparticles and post treated with water vapor. B) Sulforhodamine released into the skin from the cone shaped silk MN loaded with the microparticles and no further post fabrication treatment. C) and D) Sulforhodamine released into human cadaver skin from MN patch with no post fabrication treatment and water vapor annealing treatment, respectively. The needles contained no microparticles. E) Human cadaver skin treated with the control patch (silk film without microneedles).
2.5. Drug release in cadaver skin
To quantify the drug release in a physiological relevant environment, MN patches were tested on human cadaver skin at room temperature. Figure 6A shows the skin treated with MN patch, under UV light the position of the needles was fluorescent because of the delivery of model drug. In case of MN drug loaded (without coating) minimum amount of drug was delivered (i.e. ~0.4% in 16 hr), while maximum drug was released from the patch loaded plus coated with drug. Figure 6B showed that more than 5 μg of drug was delivered into the skin in 3hr if patch was coated with drug solution or with silk plus drug solution. In 16 hr almost 10 μg of drug was delivered from the drug only coated patched. Figure 6C showed the percent drug retained in the patch after the skin test, while Figure 6D is the total amount of drug retained in a patch at different times of release measured spectroscopically after dissolving the patch.
Figure 6.
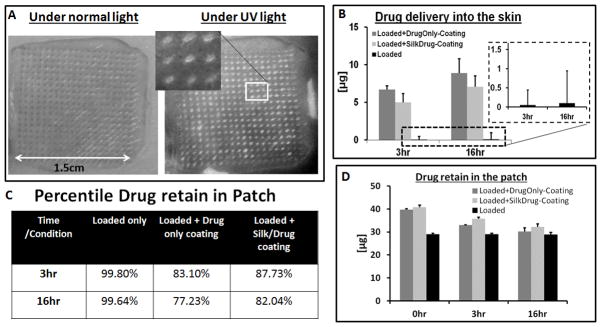
A) Optical image of human cadaver skin treated with MN patch under normal and UV light, inserted image showing the magnified view of the position of needles fluorescent under UV light. B) Amount of drug delivered into the cadaver skin, inserted plot is the drug delivery from drug loaded patches without coating. C) Table of the drug retains in the MN patches after cadaver skin application. D) Total amount of drug per patch at different time point.
The results demonstrated a simple fabrication technique to produce dense and sharp MN arrays with the ability to penetrate the skin. This method is desirable for rapid prototyping for design optimization with control over the density of needles per patch, the size of the patch and the shape and size of each needle. The technique is not limited to computer numerical control (CNC) machining but other tools such as circuit board plotters or other prototyping machines used for engraving, suggesting affordable manufacturing techniques. The technique developed here for MN technology can also be employed to generate other microstructures. Utilization of silk as a biomaterial for transdermal drug delivery, with tunable mechanical properties and drug release, adds control to this drug delivery technology. Aside from water vapor annealing, other techniques such as autoclaving and methanol treatment can be utilized to further modify the secondary structure of the silk for additional control of drug release.[50] The technique used would depend on the sensitivity of the drug molecules.
A new method to fabricate dense MN devices with sharp tips was demonstrated, where mechanical strength was enhanced by the introduction of microparticles into the silk matrix. The post fabrication modifications of secondary structure of silk MN allowed the integration of dissolvable MN with drug coating on the tips. The combination of techniques helped incorporate more drug into a single patch. Further opportunities arise due to the remarkable stabilization of proteins and other compounds in silk, suggesting that this platform is not only useful for transdermal drug delivery, but also concurrently as a stabilization, distribution and delivery matrix for drugs. In addition to the benign processing and drug preservation characteristics of the silk, two different approaches (drug encapsulation and coating) were integrated into one device that offer the possibility to deliver two different drugs from one MN system.
3. Conclusions
Dense, high aspect ratio, microstructures in silk protein were generated for MN using a simple non clean room fabrication technique. Aqueous and solvent formats of silk protein were successfully used to generate the MN patch. Various strategies such as post fabrication treatments, microparticle loading into the MN structure and drug coating were studied to improve mechanical stability and drug release kinetics. MN devices were successfully tested on human cadaver skin for drug delivery and skin penetration. The results expand both the utility of MN systems as well as the tunability of drug loading and drug release kinetics.
4. Experimental Section
4.1. MN mold design and fabrication
Machinable wax-based negative microneedles (MN) molds were fabricated using computer numerical control (CNC) machining. A carbide end mill tool (McMaster-Carr, Atlanta, GA) of diameter 3.17 mm was used with the vertical milling machine (Bridgeport model-Prototrak MX3, Elmira, NY) at 4,000 rpm (milling rate: 250 mm/min) to generate a 5 mm deep square (22 mm × 22 mm) solution holding chamber. A 500 to 700 micron deep, 20 × 20 MN hole array (in a 2.25 cm2 area) was drilled into the a machinable wax chamber using a carbide engraving tool from the Bits & Bits Company, Silverton, OR. The tip sizes ranged from 10 to 40 μm and total angles of each tip were 20 to 50 degrees, for the final cone of same angle. An array of 400 MN points was drilled into the chamber at 4,000 rpm with the feed rate of approximately 250 mm/min. Finished molds were washed with soapy water followed by thorough rinsing in DI-water.
4.2. PDMS mold for microneedles
The mold patterns were transferred into the PDMS, first by creating a MN template in an epoxy followed by casting the PDMS onto the epoxy MN array. Briefly, the embedding epoxy (from Electron Microscopy Sciences, Hatfield, PA) was mixed according to supplier specifications and degassed under vacuum for 1 hour. The uncured epoxy was poured into the machinable wax MN molds and centrifuged (in a swing-bucket rotor) at 5,000 rpm for 20 minutes. The epoxy was cured at 80°C for 24 hours and then removed from the mold showed in Figure 1B (i). The MN pattern was then transferred into PDMS by casting Sylgard® 184 (silicon elastomeric base and curing agent mixed in 10:1 ratio) and poured onto an epoxy template and degassed under vacuum followed by polymerization for 2 hours at 80°C, final PDMS Mold is shown in Figure 1B (ii).
4.3. Silk solution preparation
An aqueous silk [36] solution was prepared by dissolving 30 minutes degummed silk fibroin in 9.3 moles/liter lithium bromide solution for 3 hours. The dissolved silk fibroin solution was then dialyzed in dialysis cassettes with molecular weight cut off less than 3.5 KDa. The dialysis was performed in DI-water for 3 days with fresh water replaced after every 10 hr. The dialyzed silk was then centrifuged three times at 10,000 rpm in 50 ml conical tubes to remove any debris or particles from the silk processing step. The aqueous silk solution was then stored in the refrigerator until used. To prepare solvent-based silk, the aqueous silk solution was lyophilized (Labconco, company, Kansas city, MO) and then dissolved in hexafluoroisopropanol (HFIP) (Sigma-Aldrich, St. Louis, MO) at 12 % wt/vol. All the processing for the HFIP was performed in a chemical hood.
4.4. Microparticle fabrication and drug loading
Silk microparticles were prepared with silk and polyvinyl alcohol (PVA) (Sigma-Aldrich, St. Louis, MO) using a phase separation method.[55] Briefly, 3% wt/vol silk solution was mixed in 3% w/v PVA solution at a ratio of 1 to 4 and stirred at 3,000 rpm for 2 hours using a magnetic stirrer bar. Then 24 ml of the silk-PVA blend was poured into a 100 mm petri dish and dried overnight in a chemical hood under air flow. Silk/PVA films were then dissolved in DI-water and particles were separated by centrifugation at high speed (10,000 rpm). The particles were rinsed 3 times with DI-water and one time with methanol. Model drugs, either bovine serum albumin labeled with texas-red (BSA) or sulforhodamine sodium salt (sRh) (Sigma-Aldrich, St. Louis, MO), were loaded into the silk particles by a concentration equilibrium technique. First, silk particles were lyophilized into powder form followed by re-suspension into a solution containing the model drug and allowed to incubate at room temperature with gentle stirring for 12 hours. Drug-loaded particles were then centrifuged at 12,000 rpm in an Eppendorf tube (2 ml) so that the particles collected at the bottom and the supernatant (drug solution) were removed from the tube. The drug loaded particles were rinsed twice with DI-water and then lyophilized and stored at −20°C until used. To quantify the drug loading in the silk microparticles, particles were dissolved in protease XIV and the amount of drug was measured spectroscopically. Approximately, 200 μg of drug was loaded in 1 mg of silk particles.
4.5. Drug loading microneedles patch
Two groups of silk MN patches were fabricated: “silk + drug” and “silk + drug + microparticle loaded drug.” Each group was further divided into two sub-groups based on the type of treatment. The aqueous silk solution (7%) was mixed with the model drug (with or without particles) and cast into the MN molds, 5mm deep and area 22 mm × 22 mm To fill the deep MN trenches, lower concentration (0.5% silk + drug) was first poured onto the molds and centrifuged for 20 minutes (at 5000rpm) followed by the application of vacuum to degas. Once the molds were completely degassed the 0.5% silk drug solution was replaced with a high concentration (~7%) silk + drug. The silk solution was allowed to dry in the MN mold at room temperature and peeled off after drying showed in Figure 1 B (iii). In the case of microparticle-loaded MN, 4 mg of drug-loaded microparticles were resuspended in a 10 ml silk solution and cast (~3 ml) onto the mold as above. After peel off from molds the needle areas were cut from whole film to get the final patches. As dried MN patches were considered as a first sub-group. When these patches were treated for 24 hours by water vapor annealing,[47] the treated patches were considered a second sub-group. The final loading of either BSA or sRh in silk each patch was approximately 130 μg. In the case of microparticles loaded MN, each patch 1.5cm × 1.5cm had approximately 0.5 mg of drug loaded particles that contributed almost 100 μg more drug (per patch). Solvent (HFIP)-based silk MN were fabricated using the same degassing technique.
4.6. Drug coating and quantification
Dip coating was utilized to coat drug molecules onto the MN device. Direct drug coating was performed by dipping the (water vapor treated) MN patches into the drug solution (2.5 mg/ml). Silk and drug solution was also prepared as a second drug coating formulation, with the final concentration of 3.5 % silk and 2.5 mg/ml of drug. The patches were removed from the coating solutions after two minutes and the back side of the patch was blotted dry. The patch was dried under air flow in a hood. To quantify the amount of drug coating two independent techniques were utilized that is micro plate reader and mass balance technique. In mass balance technique first the mass of the coating solution (with known drug concentration) was determined by mass balance. A standard curve was generated to co-relate mass of coated solution vs. volume, for both of the coating formulations. The equation from the standard curve was used to determine the mass of coated drug on each patch. In the second technique (i.e. plate reader) the same patches were dissolved in lithium bromide (LiBr) solution.[45] The dissolved samples with standard (made in same silk/LiBr solution) were put in 96 well plates and the intensities were measured using plate reader (Molecular Device, LLC, model: SpectraMax M12, Sunnyvale, CA). The standard curve was then used to measure the unknown samples. In the case of direct BSA coating, approximately 50 (±15) μg of drug was coated onto the single patch, while 50 (±10) μg of drug was coated onto each patch using silk and drug as a coating solution. The amount of drug encapsulated in each patch was approximately 175 μg. While in case of sRh 40 (± 12) μg and 40 (± 8) μg was coating with drug only and drug plus silk formulation, respectively, on the quarter of the patch.
7. Imaging
Scanning electron micrographs (SEMs) were taken using a JEOL SEM (model: LSM-840, Peabody, MA). Samples were prepared by coating with a thin layer (a few nanometers) of gold and mounted onto sample holders. Bright field and fluorescent images of the tissue samples were taken using a fluorescent microscope (Leica Microsystems, model: DMIL, Buffalo Grove, IL). White light was used to take phase contrast images while fluorescent images of the tissue samples were taken using UV light (ex/em: 560nm/645nm). A digital camera (Canon, model: EOS, Rebel XSi, Lake Success, NY) was used to take the digital pictures of the molds and MN.
4.8. Mechanical testing
The axial strength of the MN was characterized using a uniaxial mechanical tester (Instron, Model: 3366, Norwood, MA) by pressing the MN patch between two parallel metal plates. Small patches of MN (1 to 9 MN per patch) were mounted on a flat hard surface (~1 mm thick glass slide) and placed on the horizontal bottom plate of the mechanical tester and pressed with the top horizontal metal plate (Figure 3A). The maximum capacity of the load cell used in the compression tests was 10 N with resolution of 0.1 mN. The rate of compression was 1 mm per minute. Ten different MN patches were tested from each sub-group (total of 4 sub-groups per fabrication method) and 40 to 60 MN were tested per sub-group for each fabrication method. The final data were plotted as fracture force per needle by normalizing values with the total number of MN per patch. Error bars (±) represent the standard deviation
4.9. Drug release in 3d gel
BSA release was performed in a 3 dimensional collagen gel as mentioned previously.[46] Briefly, a 2 mm thick 10% gelatin gel (Knox™ unflavored) was cast into a 100 mm Petri dish from which collagen blocks were cut and similarly size of MN patches (from both pyramid and cone shaped designs) were gently pressed onto the collagen gel. The whole construct was sandwiched between two glass slides, to keep the microneedle tips inserted into the gel, and placed in Petri dish with the lid closed along with a hydrated sponge to avoid dehydration of the gel. At different time points (0.16, 1, 6, 15, 24, 48 hours) the patches were transferred onto the new gel block, and the treated collagen blocks were collected in 2 ml centrifuge tubes and kept refrigerated. The collagen blocks were then degraded in 350 μl of collagenase (0.5 mg/ml) solution (Sigma-Aldrich, St. Louis, MO) at 37°C for 2 hours. The fluorescence of the released BSA was measured in plate reader (Molecular Device, LLC, model: SpectraMax M12, Sunnyvale, CA) along with controls and standards (in the same collagen/collagenase solution). All experiments were repeated three times, and in each experiment 5 different samples were measured per sub-group. MN patches with no BSA were used as a negative control. The release data was plotted as a cumulative release; error bars (±) represent the standard deviation.
4.10. In vitro human cadaver skin
MN patches (pyramid and cone) were prepared as described above with or without microparticles and used as dried or 24 hr water vapor annealed patches. sRh loaded MN were used on full thickness human cadaver skin with adipose layers (Anatomy Gifts Registry, Hanover, MD). Skin tissues were collected within the 8 hours post-mortem from the donor (<50 years) and kept frozen at −80°C until used. The skin tissue was prepared by removing the fat layer (using a surgical blade) and thawed completely (for 30 minutes) to room temperature. The cadaver skin was flattened onto a wooden block with 10 to 12 layers of Kimwipes™ underneath. Silk MN patches were applied on the flat skin surface for 1 minute using 1 kg cylindrical shape weight. In drug release experiments the drug loaded MN patches were left on the cadaver skin for two hours at room temperature. Control patches consisted of silk films with no MN arrays and plain silk films containing the drug.
4.11. Skin tissue processing for imaging
The human cadaver skin treated with silk MN patches (with or without microparticles) was fixed in 10% formaldehyde (Thermo Fisher Scientific Inc. Pittsburg, PA) for 24 hours and embedded in paraffin wax using tissue processing with stepwise replacement of organic solvent, in a Tissue-Tek VIP, Model 300, Torrance, CA. The embedded skin was then cut into 15 to 20 micron thick sections using a rotary microtome (Leica microsystem, Buffalo Grove, IL, RM2255) and samples were collected on glass slides. The sectioned skin tissues were stained with hematoxylin and eosin (H&E) and embedded in a Permount SP15–500 Toluene solution (Thermo Fisher Scientific Inc. Pittsburg, PA) followed by capping with a glass cover slip and imaged under a microscope.
Tissues treated with drug-loaded MN patches (with or without microparticles) were prepared for cryostat sectioning. After MN treatment, the skin was frozen in the embedding medium (Tissue-Tek, OCT (optimal cutting temperature) compound) at −196°C. Briefly, a plastic container filled (three quarters) with embedding medium was placed in a metal beaker (one third) filled with isopropanol (Thermo Fisher Scientific Inc., Pittsburg, PA) surrounded by liquid nitrogen. The tissue samples were vertically aligned in the embedding medium, and the medium was allowed to freeze completely. Embedded frozen samples were kept at −80°C until sectioning in a cryostat machine (Leica microsystem, Buffalo Grove, IL, Ag Protect, CM1950) at −20°C. The tissues were section into 20 micron slices and imaged under microscope.
4.12. Drug release in cadaver skin
Skin was prepared as described above and silk MN patches loaded and also coated with drug (drug solution and silk plus drug formulation) as discuss above were implanted on the cadaver skin. Total amount of drug per patch at different time point of the experiment was measured by mass balance and spectroscopy (plate reader) techniques (Figure 6D). Cadaver skin was treated for 3 hrs and 16 hrs. Three different patches were tested for each condition at each time point. The patches were dissolved in LiBr after removal from the skin at specific time point and quantified using plate reader along with the standards.[45] The difference between the drug at 3hr and 0 hr and at 16 hr and 0 hr was the amount of drug delivered to the skin.
Supplementary Material
Acknowledgments
We thank the NIH (EB002520), the AFOSR and the NSF for support of this work.
Contributor Information
Dr. Waseem K. Raja, Biomedical Engineering, Science and Technology Center, Tufts University, 4 Colby Street, Medford, MA 02155 USA
Scott MacCorkle, Machine Shop, Science and Technology Center, Tufts University, 4 Colby Street, Medford, MA 02155 USA.
Izzuddin M. Diwan, Biomedical Engineering, Science and Technology Center, Tufts University, 4 Colby Street, Medford, MA 02155 USA
Abdurrahman Abdurrob, Biomedical Engineering, Science and Technology Center, Tufts University, 4 Colby Street, Medford, MA 02155 USA.
Jessica Lu, Biomedical Engineering, Science and Technology Center, Tufts University, 4 Colby Street, Medford, MA 02155 USA.
Prof. Fiorenzo G. Omenetto, Biomedical Engineering, Science and Technology Center, Tufts University, 4 Colby Street, Medford, MA 02155 USA
Prof. David L. Kaplan, Biomedical Engineering, Science and Technology Center, Tufts University, 4 Colby Street, Medford, MA 02155 USA
References
- 1.Donnelly RF, Majithiya R, Singh TR, Morrow DI, Garland MJ, Demir YK, Migalska K, Ryan E, Gillen D, Scott CJ, Woolfso1n AD. Pharm Res. 2011;28:41–57. doi: 10.1007/s11095-010-0169-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kochhar JS, Goh WJ, Chan SY, Kang L. Drug Dev Ind Pharm. 2012;39:299–309. doi: 10.3109/03639045.2012.679361. [DOI] [PubMed] [Google Scholar]
- 3.McAllister DV, Wang PM, Davis SP, Park JH, Canatella PJ, Allen MG, Prausnitz MR. Proc Natl Acad Sci U S A. 2003;100:13755–60. doi: 10.1073/pnas.2331316100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park JH, Allen MG, Prausnitz MR. Pharm Res. 2006;23:1008–19. doi: 10.1007/s11095-006-0028-9. [DOI] [PubMed] [Google Scholar]
- 5.Coulman SA, Anstey A, Gateley C, Morrissey A, McLoughlin P, Allender C, Birchall JC. Int J Pharm. 2009;366:190–200. doi: 10.1016/j.ijpharm.2008.08.040. [DOI] [PubMed] [Google Scholar]
- 6.Badran MM, Kuntsche J, Fahr A. Eur J Pharm Sci. 2009;36:511–23. doi: 10.1016/j.ejps.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 7.Matriano JA, Cormier M, Johnson J, Young WA, Buttery M, Nyam K, Daddona PE. Pharm Res. 2002;19:63–70. doi: 10.1023/a:1013607400040. [DOI] [PubMed] [Google Scholar]
- 8.Brown MB, Martin GP, Jones SA, Akomeah FK. Drug Deliv. 2006;13:175–87. doi: 10.1080/10717540500455975. [DOI] [PubMed] [Google Scholar]
- 9.Henry S, McAllister DV, Allen MG, Prausnitz MR. J Pharm Sci. 1999;88:948. doi: 10.1021/js990783q. [DOI] [PubMed] [Google Scholar]
- 10.Ji J, Tay FE, Miao Ja, Iliescu C. Journal of Physics: Conference Series. 2006;34:1127–1131. [Google Scholar]
- 11.Li WZ, Huo MR, Zhou JP, Zhou YQ, Hao BH, Liu T, Zhang Y. Int J Pharm. 2010;389:122–9. doi: 10.1016/j.ijpharm.2010.01.024. [DOI] [PubMed] [Google Scholar]
- 12.Wilke N, Mulcahy A, Ye S-Ra, Morrissey A. Microelectronics Journal. 2005;36:650–656. [Google Scholar]
- 13.Chen Y-T, Hsu C-C, Tsai C-Ha, Kang S-W. Journal of Marine Science and Technology. 2010;18:243–248. [Google Scholar]
- 14.Park JH, Yoon YK, Choi SO, Prausnitz MR, Allen MG. IEEE Trans Biomed Eng. 2007;54:903–13. doi: 10.1109/TBME.2006.889173. [DOI] [PubMed] [Google Scholar]
- 15.Gittard SD, Ovsianikov A, Chichkov BN, Doraiswamy A, Narayan RJ. Expert Opin Drug Deliv. 2010;7:513–33. doi: 10.1517/17425241003628171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han M, Hyun D-H, Park H-H, Lee SS, Kim C-Ha, Kim C. J Micromech Microeng. 2007;17:1184–1191. [Google Scholar]
- 17.Sachdeva V, Banga AK. Recent Pat Drug Deliv Formul. 2011;5:95–132. doi: 10.2174/187221111795471445. [DOI] [PubMed] [Google Scholar]
- 18.Davis SP, Martanto W, Allen MG, Prausnitz MR. IEEE Trans Biomed Eng. 2005;52:909–15. doi: 10.1109/TBME.2005.845240. [DOI] [PubMed] [Google Scholar]
- 19.Gill HS, Prausnitz MR. J Control Release. 2007;117:227–37. doi: 10.1016/j.jconrel.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gill HS, Prausnitz MR. Pharm Res. 2007;24:1369–80. doi: 10.1007/s11095-007-9286-4. [DOI] [PubMed] [Google Scholar]
- 21.Lee JW, Park JH, Prausnitz MR. Biomaterials. 2008;29:2113–24. doi: 10.1016/j.biomaterials.2007.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raphael AP, Prow TW, Crichton ML, Chen X, Fernando GJ, Kendall MA. Small. 2010;6:1785–93. doi: 10.1002/smll.201000326. [DOI] [PubMed] [Google Scholar]
- 23.Ito Y, Hagiwara E, Saeki A, Sugioka N, Takada K. Eur J Pharm Sci. 2006;29:82–8. doi: 10.1016/j.ejps.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 24.Ito Y, Hagiwara E, Saeki A, Sugioka N, Takada K. J Drug Target. 2007;15:323–6. doi: 10.1080/10611860701349794. [DOI] [PubMed] [Google Scholar]
- 25.Donnelly RF, Morrow DI, Singh TR, Migalska K, McCarron PA, O’Mahony C, Woolfson AD. Drug Dev Ind Pharm. 2009;35:1242–54. doi: 10.1080/03639040902882280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin CJ, Allender CJ, Brain KR, Morrissey A, Birchall JC. J Control Release. 2012;158:93–101. doi: 10.1016/j.jconrel.2011.10.024. [DOI] [PubMed] [Google Scholar]
- 27.Sullivan SP, Koutsonanos DG, Del Pilar Martin M, Lee JW, Zarnitsyn V, Choi SO, Murthy N, Compans RW, Skountzou I, Prausnitz MR. Nat Med. 2010;16:915–20. doi: 10.1038/nm.2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fukushima K, Ise A, Morita H, Hasegawa R, Ito Y, Sugioka N, Takada K. Pharm Res. 2011;28:7–21. doi: 10.1007/s11095-010-0097-7. [DOI] [PubMed] [Google Scholar]
- 29.Miyano T, Tobinaga Y, Kanno T, Matsuzaki Y, Takeda H, Wakui M, Hanada K. Biomed Microdevices. 2005;7:185–8. doi: 10.1007/s10544-005-3024-7. [DOI] [PubMed] [Google Scholar]
- 30.Kim M, Jung B, Park JH. Biomaterials. 2012;33:668–78. doi: 10.1016/j.biomaterials.2011.09.074. [DOI] [PubMed] [Google Scholar]
- 31.Park JH, Allen MG, Prausnitz MR. J Control Release. 2005;104:51–66. doi: 10.1016/j.jconrel.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 32.Fan HC, Wang J, Potanina A, Quake SR. Nat Biotechnol. 2011;29:51–7. doi: 10.1038/nbt.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McDonald JC, Whitesides GM. Acc Chem Res. 2002;35:491–9. doi: 10.1021/ar010110q. [DOI] [PubMed] [Google Scholar]
- 34.Raja WK, Gligorijevic B, Wyckoff J, Condeelis JS, Castracane J. Integr Biol (Camb) 2010;2:696–706. doi: 10.1039/c0ib00044b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Regehr KJ, Domenech M, Koepsel JT, Carver KC, Ellison-Zelski SJ, Murphy WL, Schuler LA, Alarid ET, Beebe DJ. Lab Chip. 2009;9:2132–9. doi: 10.1039/b903043c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rockwood DN, Preda RC, Yucel T, Wang X, Lovett ML, Kaplan DL. Nat Protoc. 2011;6:1612–31. doi: 10.1038/nprot.2011.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Altman GH, Diaz F, Jakuba C, Calabro T, Horan RL, Chen J, Lu H, Richmond J, Kaplan DL. Biomaterials. 2003;24:401–16. doi: 10.1016/s0142-9612(02)00353-8. [DOI] [PubMed] [Google Scholar]
- 38.Cronin-Golomb M, Murphy AR, Mondia JP, Kaplan DL, Omenetto FG. Journal of Polymer Science Part B: Polymer Physics. 2012;50:257–262. [Google Scholar]
- 39.Hronik-Tupaj M, Raja WK, Tang-Schomer M, Omenetto FG, Kaplan DL. J Biomed Mater Res A. 2013 doi: 10.1002/jbm.a.34565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kojic N, Panzer MJ, Leisk GG, Raja WK, Kojic M, Kaplan DL. Soft Matter. 2012;8:2897–2905. doi: 10.1039/C2SM25783A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meinel L, Hofmann S, Karageorgiou V, Zichner L, Langer R, Kaplan D, Vunjak-Novakovic G. Biotechnol Bioeng. 2004;88:379–91. doi: 10.1002/bit.20252. [DOI] [PubMed] [Google Scholar]
- 42.Pritchard EM, Szybala C, Boison D, Kaplan DL. J Control Release. 2010;144:159–67. doi: 10.1016/j.jconrel.2010.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uebersax L, Hagenmuller H, Hofmann S, Gruenblatt E, Muller R, Vunjaknovakovic G, Kaplan DL, Merkle HP, Meinel L. Tissue Eng. 2006;12:3417–29. doi: 10.1089/ten.2006.12.3417. [DOI] [PubMed] [Google Scholar]
- 44.Mandal BB, Grinberg A, Seok Gil E, Panilaitis B, Kaplan DL. Proc Natl Acad Sci U S A. 2012;109:7699–704. doi: 10.1073/pnas.1119474109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hines DJ, Kaplan DL. Biomacromolecules. 2011;12:804–12. doi: 10.1021/bm101421r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsioris K, Raja WK, Pritchard EM, Panilaitis B, Kaplan DL, Omenetto FG. Adv Funct Mater. 2012;22:330–335. [Google Scholar]
- 47.Sullivan SP, Murthy Na, Prausnitz MR. Advanced Materials. 2008;20:933–938. doi: 10.1002/adma.200701205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gil ES, Kluge JA, Rockwood DN, Rajkhowa R, Wang L, Wang X, Kaplan DL. J Biomed Mater Res A. 2011;99:16–28. doi: 10.1002/jbm.a.33158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rajkhowa R, Gil ES, Kluge J, Numata K, Wang L, Wang X, Kaplan DL. Macromol Biosci. 2010;10:599–611. doi: 10.1002/mabi.200900358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu X, Kaplan DLa, Cebe P. Macromolecules. 2006;39:10. [Google Scholar]
- 51.Lu Q, Wang X, Hu X, Cebe P, Omenetto F, Kaplan DL. Macromol Biosci. 2010;10:359–68. doi: 10.1002/mabi.200900388. [DOI] [PubMed] [Google Scholar]
- 52.Chu LY, Choi SO, Prausnitz MR. J Pharm Sci. 2010;99:4228–38. doi: 10.1002/jps.22140. [DOI] [PubMed] [Google Scholar]
- 53.Cormier M, Johnson B, Ameri M, Nyam K, Libiran L, Zhang DD, Daddona P. J Control Release. 2004;97:503–11. doi: 10.1016/j.jconrel.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 54.Gill HS, Soderholm J, Prausnitz MR, Sallberg M. Gene Ther. 2010;17:811–4. doi: 10.1038/gt.2010.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang X, Yucel T, Lu Q, Hu X, Kaplan DL. Biomaterials. 2010;31:1025–35. doi: 10.1016/j.biomaterials.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.