

ABSTRACT
Measurements of the presence of prions in biological tissues or fluids rely more and more on cell-free assays. Although protein misfolding cyclic amplification (PMCA) has emerged as a valuable, sensitive tool, it is currently hampered by its lack of robustness and rapidity for high-throughput purposes. Here, we made a number of improvements making it possible to amplify the maximum levels of scrapie prions in a single 48-h round and in a microplate format. The amplification rates and the infectious titer of the PMCA-formed prions appeared similar to those derived from the in vivo laboratory bioassays. This enhanced technique also amplified efficiently prions from different species, including those responsible for human variant Creutzfeldt-Jakob disease. This new format should help in developing ultrasensitive, high-throughput prion assays for cognitive, diagnostic, and therapeutic applications.
IMPORTANCE
The method developed here allows large-scale, fast, and reliable cell-free amplification of subinfectious levels of prions from different species. The sensitivity and rapidity achieved approach or equal those of other recently developed prion-seeded conversion assays. Our simplified assay may be amenable to high-throughput, automated purposes and serve in a complementary manner with other recently developed assays for urgently needed antemortem diagnostic tests, by using bodily fluids containing small amounts of prion infectivity. Such a combination of assays is of paramount importance to reduce the transfusion risk in the human population and to identify asymptomatic carriers of variant Creutzfeldt-Jakob disease.
INTRODUCTION
Prion diseases are infectious neurodegenerative disorders affecting a broad range of mammalian species, including Creutzfeldt-Jakob disease (CJD) in humans, scrapie in sheep and goats, bovine spongiform encephalopathy (BSE) in cattle, and chronic wasting disease in cervids (1). The causal proteinaceous agent, termed a prion, propagates by converting the α-helix-rich host-encoded prion protein PrPC into a misfolded, β-sheet-enriched conformer designated PrPSc. The abnormal form PrPSc is believed to replicate by recruiting and converting PrPC into higher-order aggregates, through a so-called seeded polymerization process (2, 3). Fragmentation of PrPSc assemblies is thought to generate new PrPSc seeds to sustain the conversion (4). Distinct strains of prions are recognized phenotypically, based on different incubation times, neuropathological features, and PrPSc biochemical properties in experimentally infected rodents. Strain biological properties are believed to be encoded in different, specific PrPSc conformers (5–8). Several lines of evidence support PrPSc as the main molecular determinant of prion replication and infectivity (2, 3). Among them is the possibility to generate prion infectivity by PrPSc-templated conversion of PrPC under cell-free conditions using protein misfolding cyclic amplification (PMCA) assays. This technique has emerged in the last decade as a very efficient procedure to amplify prions in a test tube (9). PMCA exploits the ability of PrPSc to template the conversion of PrPC by repetitive cycles of incubation and sonication, leading to the amplification of minute amounts of PrPSc. The sensitivity achieved allows detection of PrPSc present at low levels in biological tissue or fluid samples, including blood, urine, feces, or cerebrospinal fluid (10–14). The mechanisms by which such efficient in vitro amplification is achieved are essentially unknown. By analogy with the nucleation/polymerization process, incubation of PrPSc seeds with PrPC-containing substrate is thought to favor PrPC conversion and growth of PrPSc aggregates. Sonication is thought to fragment the polymers, thus providing new seeds for conversion. The PMCA-generated products are infectious and share (in general) similar biochemical and structural properties and biological strain properties with the prion strain seed that serves for amplification (15–19). There are, however, some clear discrepancies in the amounts of prion infectivity generated among the studies (16, 18–20). Significant variations in the laboratory-specific methodologies employed to amplify PrPSc could explain the discrepancies observed. Importantly, the PMCA reaction mixtures employed were not always supplemented with beads, which were shown to significantly improve the level and the reproducibility of the amplification (21, 22), by putatively favoring the fragmentation of the generated polymers. The infectious titer of the amplicon was also frequently measured after several rounds of amplification. Repetitive, long-term incubations and sonications may alter infectivity. Besides, the higher number of rounds increases the probability of generating false-positive amplifications (23).
In this study, we first report significant simplification and improvements of the PMCA technique, leading to high throughput and highly efficient amplification of PrPSc from several prion strains from different species in a single 48-h round. We then demonstrate by endpoint dilution quantitation in reporter mice that this method restores an infectivity titer comparable to that of in vivo brain-derived prions, whatever the input dilution seeding the PMCA reaction.
RESULTS
Endpoint titration of 127S prions using mb-PMCA.
The experimental conditions leading to miniaturized bead-PMCA (mb-PMCA) were established with brain material from transgenic tg338 mice overexpressing ovine PrPC. These mice were infected or not with the 127S scrapie strain, a prototypal “fast” strain killing the mice within 2 months (24, 25). The procedure was performed by seeding tg338 mouse brain lysate containing ovine PrPC substrate with serial 10-fold dilutions of 127S-infected brain homogenate and running 96 sonication/incubation cycles for a round of 48 h. The amplicons were treated with proteinase K (PK) to eliminate PrPC before detection of PK-resistant PrPSc (PrPres) signals by dot blotting and Western blotting. Without amplification, PrPres is detected until the brain homogenate is diluted 103-fold (Fig. 1a). A series of technical changes were employed, improving by 107-fold the sensitivity of the technique. “Standard” PMCA protocols (26) were initially followed using individual PCR tubes, brain from perfused tg338 mice to prepare the substrate, phosphate-buffered saline (PBS)–Triton as PMCA buffer, no beads added, and a final reaction volume of 100 µl. In our hands, this often resulted in variations in the amplification efficiency and difficulties in completely digesting PrPC with PK, even in the unseeded control samples (data not shown). When PBS was replaced with Tris in the PMCA buffer, better PrPSc amplification and reduced PrPC undigested background were obtained. Under these conditions, PrPres was detected in PMCA reaction mixtures seeded with 10−4-diluted 127S brain homogenate (Fig. 1b). Addition of 3 ceramic beads during the reaction markedly increased the sensitivity and the reproducibility of the amplification; PrPres was detected routinely from reaction mixtures seeded with 10−6-diluted inoculum (Fig. 1b). Addition of one Teflon bead was also beneficial, as previously described (21). Use of strips of 8 PCR tubes instead of individual tubes placed in rigorously defined positions in the cup horn led to systematic and robust amplification of PrPSc in 10−7-diluted inoculum (Fig. 1b). Encouraged by these observations, we shifted to 96-well PCR microplates. The final PMCA volume was reduced to 36 µl. Using this new experimental design (referred to as mb-PMCA), highly efficient and reproducible detection of PrPres was achieved in reaction mixtures seeded with 10−11-diluted brain material (Fig. 1c). PrPres was occasionally detected from amplification of 10−12 dilutions (see, for example, Fig. 2). Reactions seeded with higher dilutions (up to 10−15) did not show any positive PrPres signal (Fig. 1c). Specificity was further assessed by submitting the mb-PMCA samples seeded with 10−13-diluted material to a second round with fresh substrate. No PrPres could be detected (data not shown). Two to six unseeded control samples per plate were systematically included in each mb-PMCA. No PrPres was detected in these control samples in a total of more than 220 mb-PMCA reactions (i.e., the equivalent of 21,000 individual tubes) (for example, Fig. 1c), thus highlighting the high specificity of the reaction. Taken together, these findings suggest that the input seed limiting dilution of 127S prions should be established at 10−11 to 10−12 and can be amplified by mb-PMCA in one 48-h round of 96 cycles of sonication/incubation.
FIG 1.

Endpoint titration of 127S prions by a single round of mb-PMCA. Brain homogenate from tg338 mice infected with 127S prions was serially diluted in tg338 healthy brain lysate as indicated. Each dilution was directly analyzed by Western blotting (a) or served as seed for a single 48-h round of PMCA using different experimental conditions (b and c). Samples were amplified in the absence or presence of 3 ceramic beads (as indicated) in individual PCR tubes or an 8-PCR-tube strip containing 100 µl of PMCA mixtures (b) or in a PCR microplate containing 36 µl of PMCA mixture (c). Unseeded samples (U) were run on the same microplate. All samples were digested with PK before Western blot analysis using Sha31 anti-PrP antibody. Undigested normal brain homogenate (PrPC) and PK-digested, infected brain homogenate (PrPres) are provided as electrophoretic references. Molecular mass markers (kDa) are indicated to the left of each panel.
FIG 2.
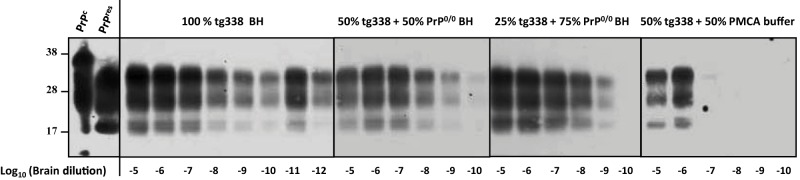
mb-PMCA of 127S prions by using different concentrations of PrPC substrate. Brain homogenate from tg338 mice infected with 127S prions was serially diluted in substrate containing either pure tg338 mouse brain homogenate (BH) or tg338 BH mixed with PrP0/0 BH or PMCA buffer, as indicated. The mixture was then subjected to a single round of mb-PMCA. All samples were digested with PK before Western blot analysis. Undigested normal brain homogenate (PrPC) and PK-digested, infected brain homogenate (PrPres) are provided as electrophoretic references. Molecular mass markers (kDa) are indicated on the left.
PrPC and other brain factors are limiting mb-PMCA efficacy.
We next examined whether the high sensitivity of the mb-PMCA procedure was linked to the amount of PrPC present in the substrate lysate, as tg338 mouse brain overexpresses approximately 8-fold PrPC compared to sheep brain (27). Performing mb-PMCA with serial 10-fold dilutions of 127S input seeds mixed with substrate containing 100%, 50%, or 25% tg338 brain lysate (the latter dilutions were performed in 10% PrP-knockout brain lysates made in PMCA buffer) led to a 2-log10 reduction of the efficacy of the amplification (Fig. 2). In marked contrast, diluting tg338 brain lysate by 1:2 in PMCA buffer reduced by 6 log10 the PMCA efficiency (Fig. 2), suggesting that PrPC was not the sole limiting factor in PMCA conversion efficacy.
We estimated the PrPC conversion yield during the 48-h mb-PMCA procedure. 127S PrPSc, but not PrPC, resists thermolysin digestion (28). Measuring the ratio of thermolysin-resistant PrPSc to total PrP would thus provide information on the percentage of mb-PMCA-converted PrPC. Samples from two independent mb-PMCAs, seeded with serial dilutions of 127S prions, were treated with 200 µg/ml thermolysin, a concentration degrading PrPC in unseeded PMCA samples. We observed substantial variations in the amounts of PrPC converted among the mb-PMCA products, regardless of the dilution of the input seed. Approximately 20% of PrPC was converted in one round as shown in the representative Fig. 3. Taken together, these data suggest that all the PrPC substrate is not converted and that non-PrP brain components may be involved in the PrP conversion process.
FIG 3.
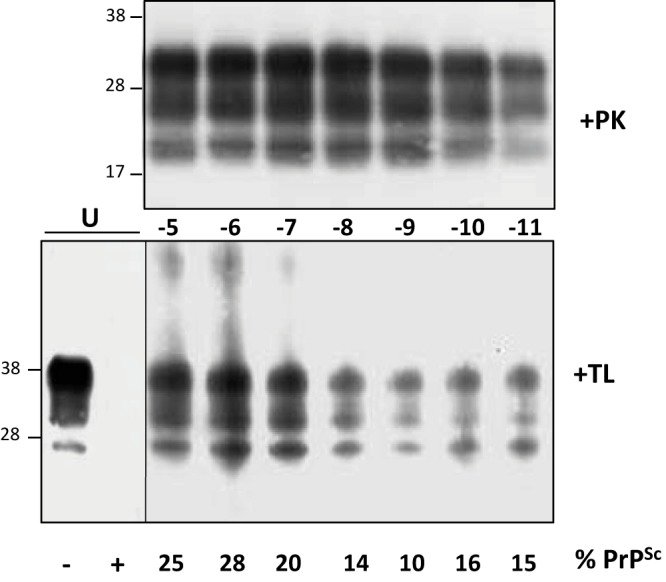
Western blot analysis of thermolysin-resistant PrPSc generated by mb-PMCA. Brain homogenate from tg338 mice infected with 127S prions was serially diluted in uninfected tg338 brain lysate, as indicated, and submitted to a single round of mb-PMCA. The resulting products were treated with either PK (+PK) or thermolysin (+TL) before Western blot analysis. The thermolysin-resistant signals were quantified so as to measure the ratio of thermolysin-resistant PrPSc to total PrP present in the unseeded samples (U). The ratios are indicated (% PrPSc). Unseeded sample has been 10-fold diluted before loading on the Western blot. The figure is representative of the ratios observed in two independent experiments. Molecular masses (kDa) are indicated at left.
Efficient amplification of mouse, hamster, and human prions by mb-PMCA.
To examine whether mb-PMCA sensitivity was specific to 127S prions, we applied this single-round protocol to other prions and PrP species combinations. Previously, all the prions tested have been serially passaged and typed phenotypically on the ad hoc transgenic mice. As summarized in Table 1, mouse prions 139A, 22L, RML, and Chandler were efficiently amplified using transgenic tga20 mouse brain (29) as the substrate for mouse PrPC. PrPres was routinely detected from input seeds diluted up to 109-fold (n ≥ 6 experiments). ME7 prion amplification was 1,000 times less efficient. The PrPres glycoprofile of the amplified products resembled that of the inoculum, with a prominent monoglycosylated PrP form typical of these mouse prions (Fig. 4 and data not shown). PrPres from 263K, Sc237, and HY hamster prions was amplified from 10−7-diluted input seeds in one round using hamster PrP transgenic mouse brain (tg7 line [25]) as the substrate (n ≥ 10 experiments). The DY hamster strain was less efficiently amplified (10−5). The PrPres electrophoretic and glycoform profiles of the nonamplified and mb-PMCA-amplified prions were similar (Fig. 4 and data not shown). Finally, we assessed the efficacy of mb-PMCA for human variant CJD (vCJD) prions using human PrP transgenic mice (Met129 allele; tg650 line [30]). In one round, the limiting dilution of the input seed was reproducibly established at 10−8 (n ≥ 6 experiments) (Fig. 4). The glycoform ratio of the mb-PMCA PrPvCJD products closely resembled that of the input seed (Fig. 4). However, the mb-PMCA PrPvCJD of vCJD exhibited a slightly lower electrophoretic mobility than did brain-derived PrPres (Fig. 4a), as previously reported for hamster prion strains (31). Taken together, these findings highlight mb-PMCA as a versatile protocol to amplify minute amounts of PrPSc from different prion strains in a single round.
TABLE 1.
Endpoint titration of mouse, hamster, and human prions by a single round of mb-PMCA
| Species | Prion strain | PrPC substrate (mouse line) | Limiting dilution of brain material |
|---|---|---|---|
| Mouse | 139A | Mouse (tga20) | 10−8 |
| RML | 10−9 | ||
| Chandler | 10−7 | ||
| 22L | 10−9 | ||
| BSE | 10−10 | ||
| ME7 | 10−6 | ||
| Hamster | Sc237 | Hamster (tg7) | 10−7 |
| 263K | 10−7 | ||
| HY | 10−7 | ||
| DY | 10−5 | ||
| Human | vCJD | Human M129 (tg650) | 10−8 |
FIG 4.

Highly efficient amplification of murine, hamster, and human prions by a single round of mb-PMCA. Western blot analysis (a) and ratio (b) of high- and low-molecular-mass PrPres glycoforms of mb-PMCA products. Brain homogenates from tga20 mice infected with Chandler prions, tg7 mice infected with 263K prions, and tg650 mice infected with vCJD prions were serially diluted in homotypic brain lysates as indicated. Each dilution was directly analyzed by Western blotting or was submitted to a single round of mb-PMCA, as indicated (a). All samples were treated with PK before SDS-PAGE and Western blotting. Data in panel b are plotted as means ± standard errors of the means. Nonamplified, brain prions are represented by red triangles; mb-PMCA amplicons generated from the indicated input seed are indicated by blue symbols. The ratios were established from 3 independent mb-PMCA experiments.
mb-PMCA generates highly infectious prions.
We finally sought to determine whether highly efficient mb-PMCA of PrPSc was associated with similarly efficient amplification of infectivity. This was done with 127S prions using the tg338 mouse bioassay. According to endpoint titration by the intracerebral (IC) route, the infectious titer of 127S prions in tg338 mouse brain is 109.2 lethal dose 50 (LD50 IC) per gram (25). These experiments allowed us to determine that the lowest dose resulting in positive transmission (as based on the appearance of clinical signs and detection of PrPSc in brain) was observed at the 10−7 dilution of 127S brain inoculum (Table 2; Fig. 5). mb-PMCA products were generated from reaction mixtures seeded with this limiting dilution or with 100-fold-more-diluted or -more-concentrated seeds. The amplicon generated with the 10−9 seed was 10-fold diluted up to a 10−7 dilution for complete endpoint titration while the amplicons obtained with the 10−5 and 10−7 seeds were diluted 101-, 103-, 105-, and 107-fold. Two unseeded samples run in parallel were 10-fold diluted. All the dilutions were prepared separately and immediately inoculated into recipient tg338 mice by the intracerebral route. The results are summarized in Table 2. At the time of writing, more than 400 days after the experimental infection, mice inoculated with unseeded controls were still alive and healthy. Whatever the initial dilution of the 127S seeds that served for mb-PMCA, an attack rate of 100% was observed in tg338 mice inoculated with all the amplicons diluted up to 105- to 106-fold. At the 10−7 dilution, 1/5 (10−7 and 10−9 seed) or 2/5 (10−5 seed) tg338 mice were still infected, an attack rate reminiscent of that observed for nonamplified 127S-infected brain. In other words, one round of PMCA was sufficient to regenerate infectivity to levels identical to those reached in the brains of intracerebrally inoculated tg338 mice at the terminal stage of disease. For the fully titrated amplicon generated with the 10−9 input seed, the relationship between prion concentration and mouse incubation period appeared superimposable on that observed with prions derived from terminally sick mouse brains (Fig. 5), suggesting similar multiplication rates between cell-free and brain-derived prions upon injection in the tg338 mouse brain.
TABLE 2 .
Incubation times of tg338 mice inoculated with serial 10-fold dilutions of brain-derived or mb-PMCA-generated 127S prionsa
| Dilution | Incubation time in days ± SEM (n/n0) |
|||
|---|---|---|---|---|
| PMCA generated |
Brain derived | |||
| 10−5 seed | 10−7 seed | 10−9 seed | ||
| 10−1 | 63 ± 2 (5/5) | 60 ± 1 (4/4) | 59 ± 1 (5/5) | 64 ± 1 (5/5) |
| 10−3 | 74 ± 1 (5/5) | 72 ± 2 (5/5) | 74 ± 3 (5/5) | 79 ± 2 (5/5) |
| 10−4 | ND | ND | 82 ± 2 (5/5) | 85 ± 1 (5/5) |
| 10−5 | 91 ± 2 (5/5) | 98 ± 3 (5/5) | 111 ± 11 (5/5) | 99 ± 2 (5/5) |
| 10−6 | ND | ND | 117 ± 11 (5/5) | 119 ± 7 (4/5) |
| 10−7 | 134; 147 (2/5) | 124 (1/5) | 139 (1/5) | 157 (1/5) |
| Unseeded no. 1 | >400 | |||
| Unseeded no. 2 | >400 | |||
n/n0, number of affected/number of inoculated tg338 mice; ND, not done.
FIG 5.
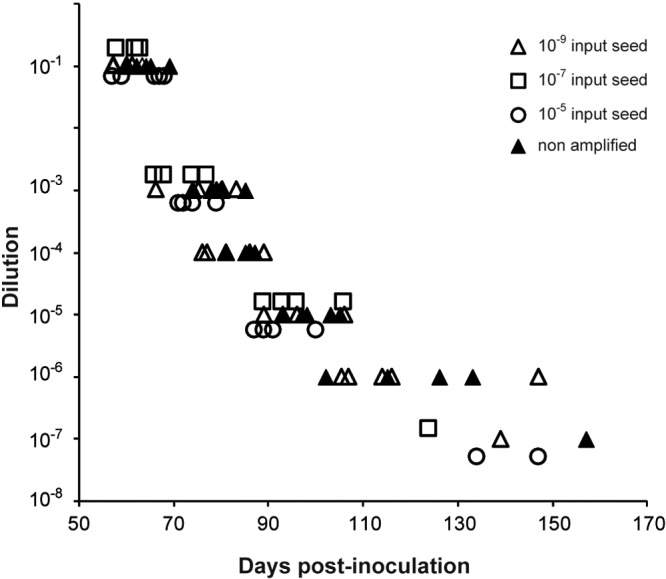
Endpoint titration of nonamplified and mb-PMCA-generated 127S prion infectivity in tg338 mice. Brain homogenates from infected tg338 mice and mb-PMCA-generated amplicons were 10-fold diluted (y axis) and inoculated intracerebrally into groups of tg338 mice. Individual survival times are shown on the x axis as days postinoculation. tg338 mice were inoculated with brain-derived 127S prions (filled triangles) or mb-PMCA amplicons generated from 10−5 (open circles), 10−7 (open squares), and 10−9 (open triangles) input seed.
The clinical signs of tg338 mice inoculated with serially diluted amplicons were identical to those of mice infected intracerebrally with 127S prions, including notably hyperexcitability, waddling, and rolling gait. Brain and spleen samples from these animals accumulated PrPres with a glycosylation and tissue-specific (28) mobility profile similar to that found in 127S-infected brain and spleen, respectively (Fig. 6a and b and data not shown). The regional distribution of PrPres in the brains of tg338 mice infected with the amplicons resembled that observed with 127S inoculated intracerebrally at equivalent dilutions (24) (Fig. 6c and data not shown). The PrPres staining was pronounced in the septum, corpus callosum, habenula, hypothalamus, lateral and posterior hypothalamic area, and brain stem.
FIG 6.
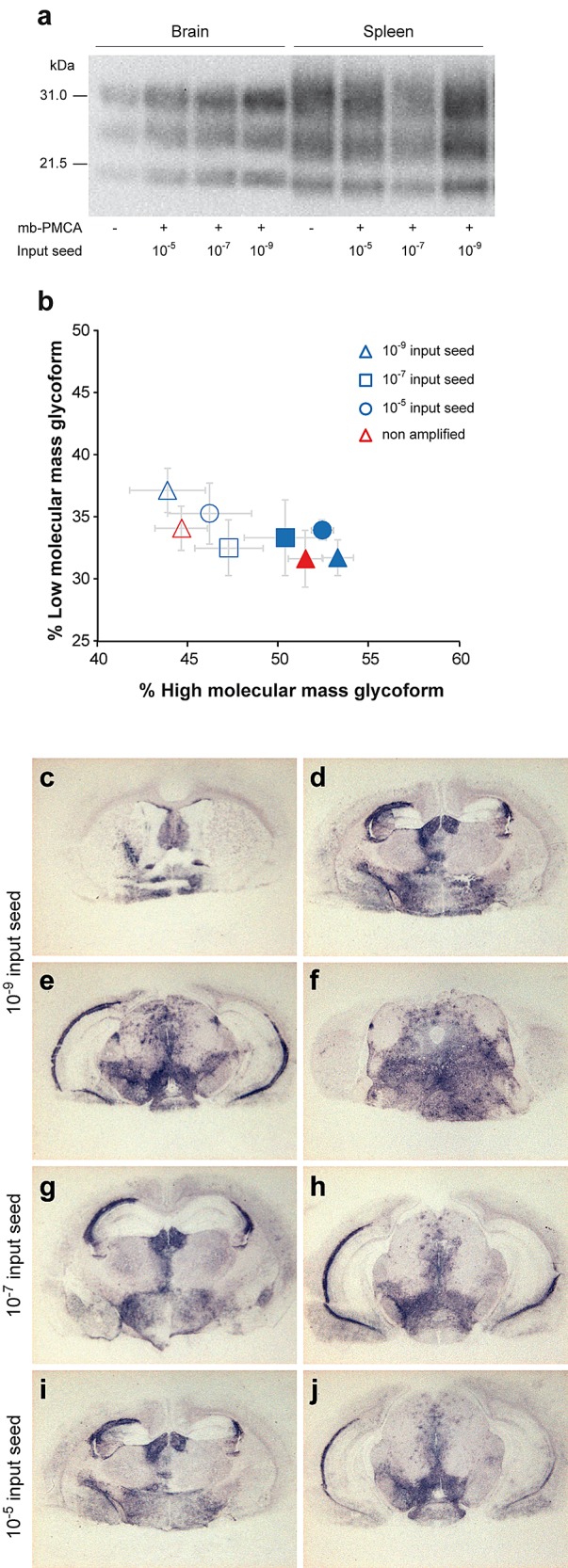
PrPres glycopattern and PrPres regional deposition in the brains of tg338 mice inoculated with mb-PMCA-generated 127S prions. Western blot analysis (a) and ratios (b) of high- and low-molecular-mass PrPres glycoforms in the brains and spleens of tg338 mice following challenge with brain-derived or mb-PMCA-generated 127S prions. Samples in panel a are from mice inoculated with 10-fold-diluted amplicons. Data in panel b are plotted as means ± standard errors of the means. Brains and spleen are represented by filled and open symbols, respectively. Brain-derived 127S prions are represented by red triangles; mb-PMCA amplicons generated from 10−5, 10−7, and 10−9 input seed are indicated by blue circles, squares, and triangles, respectively. The ratios were established from tissue samples of mice inoculated with all the amplicon dilutions. As no differences were observed among the amplicon dilutions, the ratios were combined for the sake of clarity. Histoblotting of representative coronal sections of tg338 mice inoculated with mb-PMCA amplicons at the level of the septum (c), hippocampus (d), midbrain (e), and brain stem (f) (10−9 input seed) and hippocampus (g and i) and midbrain (h and j) (10−7 and 10−5 input seed).
Together, our data led to the conclusion that the mb-PMCA 127S amplicons are strongly related, if not identical, to 127S prions phenotypically and that one round of mb-PMCA restores infectivity levels similar to that of 127S original brain material.
DISCUSSION
The sensitivity of PMCA in amplifying prions has long been recognized. By using 96-well microplates, beads, and a reduced reaction volume, we now show for the first time specific and highly sensitive prion amplification in a single round, thus greatly limiting the probability of false-positive samples. We also provide evidence that the PMCA-amplified products can be highly infectious, exhibiting a titer similar to that found in the parental brain material.
The PMCA method is constantly under continuous adaptation and improvements, due to inconsistent robustness and reproducibility, thus compromising its use for large-scale purposes. Two important technical limitations were circumvented in our study: (i) the repetition of incubation/sonication cycles and substrate refreshment over several rounds, which appeared necessary to attain high amplification levels (17, 32, 33) but was shown to be one of the main factors responsible for inadvertent cross-contamination events or so-called spontaneous de novo generation of prions (23, 34), and (ii) the position of the tubes within the sonicator and their distance from the center of the horn (21, 26, 35), which greatly limited the number of amplified samples per round. These improvements were achieved by using ceramic beads during the incubation/sonication cycles and by decreasing the volume necessary to the reaction with the PCR microplate format. This led to the use of the maximum surface offered by the sonicator, allowing more than 110 samples to be run at the same time (see Fig. S1 in the supplemental material). The observation that ceramic beads greatly improved the efficiency of PMCA is congruent with previous observations made with Teflon beads (21, 22). Beads are believed to promote efficient fragmentation of PrPSc polymers, thus increasing the number of seeds available for conversion (21). The observation that the number and the size of beads added to the conversion appeared less important than their chemical composition (our unpublished data and reference 21) might suggest a more complex physical involvement of the beads in the PMCA reaction. This effect appears optimal in a reduced reaction volume, suggesting a link with the distribution of sonication energy.
Using the so-called mb-PMCA protocol, a single 48-h round was sufficient to achieve amplification of the highest dilution of 127S prions to levels detectable by conventional Western blotting. The sensitivity reached by the mb-PMCA assay exceeded that of the tg338 bioassay by 104- to 105-fold. This difference is consistent with other studies (17, 33). A significant part of the inoculated or generated prions might be degraded in animal bioassays (36, 37), a phenomenon that would be limited in cell-free assays. Remarkably, the technical improvements employed were beneficial to other prion strains, such as mouse, hamster, and human vCJD prions, without, at variance with previous studies (35, 38, 39), an apparent need to adapt the conditions to each prion strain. Variation in the power amplitude of the sonicator (10 to 80%) was without notable influence on the sensitivity achieved with 127S, 263K, Chandler, and vCJD prions (data not shown), suggesting that these samples received optimal sonication energy in the mb-PMCA format. However, some prion sources, notably those responsible for certain CJD subtypes, fairly resist mb-PMCA (unpublished observations), suggesting that certain cell-free prion polymerizations would necessitate cofactors or different experimental conditions. The maximum dilution of prion brain inoculum detected by mb-PMCA varied with certain strain types (Table 1). This may reflect differences in infectious titers among strains. Comparatively, ME7 and DY are at least 100-fold less infectious than RML/Chandler and HY prions in tga20 mice (40) and hamsters (41), respectively. The sensitivity of detection achieved with vCJD prions would be compatible with that necessary for reliable detection of this agent in blood (42), an important public health concern given the current uncertainties about the number of individuals incubating the disease (43–45).
We confirm here that PMCA sensitivity is dependent on the concentration of PrPC in the PMCA substrate (46–48). However, despite its remarkable sensitivity, the PrPC conversion yield achieved by the mb-PMCA assay was approximately 20%. This low conversion yield might suggest that all brain PrPC species are not convertible, a hypothesis consistent with the deposition of PrPSc in (strain-dependent) specific brain areas in infected animals (49–51). Alternatively but not exclusively, other brain factors might be a limiting factor. Supporting this hypothesis, a 2-fold dilution of PMCA substrate in PMCA buffer instead of PrP0/0 brain lysate dramatically decreased the mb-PMCA efficiency. RNAs or poly(A) and lipids have been shown to be instrumental in efficient PMCA conversion (52–54). The high-throughput screening capacities of mb-PMCA will be helpful to investigate this further and to search for potent cellular factors involved in the PrPC conversion on a large scale.
Our 20% conversion yield sharply contrasted with the 100% conversion yield previously reported by using beads in the PMCA reaction (21). These differences might be due to different prion sources/PrP substrate combinations and/or to the quantification methods used. Here, we quantified the densitometric ratio of thermolysin-resistant PrPSc generated by mb-PMCA to the total amount of PrP. In a mixture such as a PMCA product containing both PrPSc and PrPC, thermolysin is known to preserve protease-sensitive and protease-resistant PrPSc species while destroying PrPC (55, 56) and thus may allow reliable assessment of the conversion yield.
Remarkably, the rates of multiplication and terminal prion titers appeared similar between mb-PMCA-generated and brain-derived 127S prions when injected in reporter tg338 mice. The use of mb-PMCA has immediate potential application as a surrogate for 127S infectivity quantitation. The infectivity of the mb-PMCA amplicons was determined by endpoint titration in tg338 mice, which is regarded as the most accurate method for measuring prion infectious titer in infected samples (57, 58). Some titrated amplicons were generated with input seeds diluted 100-fold more than the 127S prion limiting dilution in the tg338 bioassay, thus excluding any contribution of the parental seed to the infectivity measured. This tight relationship between prion replication dynamics in cell-free and animal assays contrasts with previously published results. Shikiya and Bartz (18) found, as we did, that their PMCA protocol generated high-titer prions; however, the rate of prion amplification was significantly altered. Notably, they titrated PMCA-generated HY prions after the 10th round of amplification, which may have altered the amplicon infectious properties. Other contradictory studies, which were also based on multiround PMCA, divided the infectivity titer by the amount of PrPres generated by the PMCA reaction, so as to calculate PrPres specific infectivity (16, 19). A number of pieces of experimental evidence suggest a quantitative disconnection between infectivity and PrPres in the brains of prion-infected animals (59). Specifically for 127S and the extensively used Sc237 hamster prions, a subpopulation of PrPSc assemblies would support most of the infectivity, making a significant proportion of PrPres assemblies relatively innocuous (25, 60). We may have regenerated high-titer 127S prions by a single round of mb-PMCA because this “most infectious” PrPSc subpopulation was preferentially amplified. Accordingly, we demonstrated with another “fast” ovine prion strain, exhibiting a behavior similar to that of 127S in tg338 mice (25), that the subset of PrPSc assemblies that carried the major part of prion infectivity also exhibited by far the highest templating activity by mb-PMCA (61).
In summary, the mb-PMCA assay allows large-scale, fast, and reliable cell-free amplification of subinfectious levels of prions from different species. The sensitivity and rapidity achieved approach or equal those of other prion-seeded conversion assays (62), such as the quaking-induced conversion (QuIC) assay (63). At variance with the latter (62, 64), mb-PMCA can regenerate large amounts of prion infectivity. Such a simplified assay may be amenable to high-throughput, automated purposes and serve, in a complementary manner with QuIC-like (62) or solid-phase binding (42) assays, for urgently needed preclinical diagnostic tests, by using bodily fluids containing small amounts of prion infectivity.
MATERIALS AND METHODS
Ethics statement.
All animal experiments have been performed in strict accordance with EU directive 2010/2063 and were approved by the local ethics committees of the authors’ institutions (Comethea; permit number 12/034).
Transgenic mice and prion strains.
The ovine (tg338 line; Val136-Arg154-Gln171 allele), human (tg650 line; Met129 allele), hamster (tg7 line), and mouse (tga20) PrP transgenic lines have been described previously (24, 25, 29, 30, 43, 65). These lines are homozygous and overexpress about 8-, 6-, 4-, and 10-fold the heterologous PrPC level on a mouse PrP-null background, respectively.
The 127S scrapie prion strain has been obtained through serial transmission and subsequent biological cloning by limiting dilutions of PG127 field scrapie isolate to tg338 mice (24, 27). The 127S infectious titer is 109.2 50% lethal doses (LD50)/g of tg338 brain (25). Pools of 127S-infected tg338 mouse brains were prepared as 20% (wt/vol) homogenate in 5% glucose by use of a tissue homogenizer (Precellys 24 Ribolyzer; Ozyme; Bertin Technologies, France). The homogenate was diluted half to 10% in PMCA buffer (see below) to obtain the 10−1 dilution of the inoculum and stored at −80°C. All subsequent dilutions refer to this 10% homogenate starting material.
Mouse prion strains 139A, 22L, RML, Chandler, ME7, and mouse-adapted bovine spongiform encephalopathy (BSE); hamster prion strains 263K, Sc237, HY, and DY; and human vCJD prions have been serially passaged on tga20, tg7 (25), and tg650 mice (30), respectively.
PMCA.
Mouse brain lysate from uninfected tg338 mice was used as the substrate for 127S scrapie prions. Mouse brain lysates from tga20 mice, tg7 mice, and tg650 mice were used as the substrates for mouse, hamster, and human prions, respectively. Two- to 12-month-old mice were euthanized by CO2 exposure. Brains were rapidly removed and washed twice in Ca2+/Mg2+-free PBS containing 5 mM EDTA. They were either used immediately to prepare the PrPC substrate lysate or stored at −80°C until use. PrPC substrate (10% brain lysate) was prepared using cold PMCA buffer (50 mM Tris-HCl, pH 7.4, 5 mM EDTA, 300 mM NaCl, 1% Triton X-100) and Dounce cooled in ice with 20 to 30 strokes to completely homogenize the brain tissue. The lysate was left at 4°C for 30 min and briefly clarified by centrifugation at 1,000 × g for 2 min at 4°C. The resulting supernatant, corresponding to the PrPC substrate lysate, was collected, aliquoted, and stored at −80°C. Protein misfolding cyclic amplification (PMCA) performed with either young (6- to 10-week) or old (1-year) mouse brain homogenates yielded the same results. Moreover, brain perfusion prior to collection and homogenization appeared not to be of any influence on the performance of the mb-PMCA (data not shown).
PMCA was performed in a final volume of either 100 µl or 36 µl of lysate per well, in either single PCR tubes (conventional method); in 2-, 4-, or 8-PCR-tube strips; or with a 96-well PCR microplate (Axygen, Union City, CA, USA). Each tube or well was first filled with ceramic beads (3 beads of 1.23 mm in diameter; Mineralex, France). Two ceramic beads of 2.4 mm or one Teflon bead of 2.381 mm in diameter (Marteau et Lemarié, Pantin, France) was also efficient. A 4-µl aliquot of the analyte inoculum (10−2 dilution) was suspended in 36 µl of healthy tg338 brain lysate to obtain the 10−3 dilution. Then, a series of 10-fold dilutions was made by adding 4 µl from the previous inoculum dilution to the next 36-µl-containing tube or well. Individual tubes, tube strips, or microplates were placed on a Plexiglas rack designated for the cup horn of the S3000 or Q700 sonicator (Misonix, Farmingdale, NY, USA, or Delta Labo, Colombelles, France) and subjected to 96 cycles of 30 s of sonication at 220- to 240-W power (level 6 to 7 for the S3000 or 30% amplitude of the Q700 sonicator) followed by 29 min 30 s of incubation at 37°C. The cup horn was filled with 300 ml of water (or 4 M guanidium hydrochloride solution) circulating with rubber tubing in a water bath maintained at a temperature of 35 to 36°C. When needed, subsequent rounds of PMCA were realized using a 1/10 dilution of the products of the previous PMCA round as the template. At the end of the PMCA, the tubes or microplates were removed and aliquots from each sample were taken to be analyzed for their PrPres content.
Protease digestion of PMCA products.
To analyze the production of proteinase K (PK)-resistant PrPSc species during PMCA, 10 to 18 µl of each sample was supplemented with SDS (0.3 to 0.6% final concentration) and treated with PK (115-µg/ml final concentration) at 37°C for 1 h. The PK digestion was stopped by adding an equal volume of 2× Laemmli denaturation sample buffer and heating at 100°C for 5 min. The samples were then stored at −20°C until dot blotting and Western blotting.
To analyze the levels of thermolysin-resistant species, PMCA products were first diluted 1/5 in TNT buffer (50 mM Tris-HCl, pH 7.4, 5 mM EDTA, 150 mM NaCl, 1% Triton X-100) to decrease the EDTA concentration which prevents thermolysin digestion. The samples were then treated with 200 µg/ml of thermolysin (Sigma, St. Louis, MO, USA) at 37°C for 1 h before denaturation as described above.
Dot blotting analysis of PrPSc.
Given the significant number of samples generated with the PMCA procedure, a quick dot blot analysis was set up and turned out to be practical and efficient in getting an overview of the PMCA results. Ten to 20 µl of each PK-digested PMCA sample was removed and transferred into a 96-well microplate containing 50 µl of 1% SDS and 20% glycerol. The samples were then transferred onto a nitrocellulose membrane placed on the dot blot apparatus (Whatman, France) connected to a vacuum system. The membrane was removed and rinsed twice with Tris-buffered saline (TBS)–0.1% Tween 20 before incubation with the primary antibody (biotinylated Sha31 [66] anti-PrP monoclonal antibody) for 15 to 30 min at room temperature. After 3 washes of 5 min each, the membrane was incubated with streptavidin-coupled horseradish peroxidase for 15 to 30 min at room temperature and processed for detection with the enhanced chemiluminescence (ECL) reagent (GE Healthcare, Saclay, France).
SDS-PAGE and Western blotting.
PMCA samples were run on either 4 to 12% or 12% Bis-Tris NuPAGE precast gels (Invitrogen, Cergy-Pontoise, France) or on Criterion XT 12% Bis-Tris precast gels (Bio-Rad, Hercules, CA, USA), electrotransferred onto nitrocellulose membranes with the semidry electrotransfer system (Bio-Rad), and probed with biotinylated Sha31 anti-PrP monoclonal antibody. Secondary antibody incubation and ECL detection were performed as described above. When necessary, the PrPres content of PMCA samples was determined with GeneTools software after acquisition of the signals with a GeneGnome digital imager (Syngene, Frederick, MD, USA).
Endpoint titration of PMCA products in tg338 mice.
A strict protocol based on the use of disposable equipment and preparation of all inocula in a class II microbiological cabinet was followed to avoid any cross-contamination. Serial 10-fold dilutions of PMCA products were prepared in sterile 5% glucose containing 5% bovine serum albumin, and 20 μl of each dilution was immediately inoculated into individually identified 6- to 10-week-old tg338 recipient mice (n = 5 mice per dilution) by the intracerebral route. The inoculated animals were observed daily for the appearance of prion disease. Animals at the terminal stage of disease were euthanized. The survival time was defined as the number of days from inoculation to euthanasia. Their brains and spleens were removed for PrPres analysis by Western blotting and histoblotting as previously described (24, 25). For the histoblotting procedure, brains were rapidly removed from euthanized mice and frozen on dry ice. Cryosections were cut at 8 to 10 mm, transferred onto Superfrost slides, and kept at −20° C until use. Histoblot analyses were performed on 3 brains per dilution per amplicon, using the 12F10 anti-PrP antibody (67).
SUPPLEMENTAL MATERIAL
mb-PMCA efficiency does not rely on the position of the samples in the PCR microplate wells. Serial 10-fold dilutions of 127S, 263K, 139A, and vCJD prions were prepared in duplicate and subjected to a single round of mb-PMCA. Four unseeded mouse brain lysates (tg338, tga20, tg7, and tg650) were run in the same plate. At the end of mb-PMCA, samples were treated with PK and analyzed by dot blotting as described in Materials and Methods. Download
ACKNOWLEDGMENTS
We thank J. Castilla and C. Soto (University of Texas Medical Branch, Galveston, TX, USA) for introducing us to PMCA and the staff of Animalerie Rongeurs (INRA, Jouy-en-Josas, France) for excellent animal care.
This work was supported by fellowship (F.L.) and grants from the Ile-de-France region (DIM MALINF, France).
Footnotes
Citation Moudjou M, Sibille P, Fichet G, Reine F, Chapuis J, Herzog L, Jaumain E, Laferrière F, Richard C-A, Laude H, Andréoletti O, Rezaei H, Béringue V. 2013. Highly infectious prions generated by a single round of microplate-based protein misfolding cyclic amplification. mBio 5(1):e00829-13. doi:10.1128/mBio.00829-13.
REFERENCES
- 1. Collinge J. 2001. Prion diseases of humans and animals: their causes and molecular basis. Annu. Rev. Neurosci. 24:519–550 [DOI] [PubMed] [Google Scholar]
- 2. Diaz-Espinoza R, Soto C. 2012. High-resolution structure of infectious prion protein: the final frontier. Nat. Struct. Mol. Biol. 19:370–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Colby DW, Prusiner SB. 2011. Prions. Cold Spring Harb. Perspect. Biol 3:a006833. 10.1101/cshperspect.a006833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Knowles TP, Waudby CA, Devlin GL, Cohen SI, Aguzzi A, Vendruscolo M, Terentjev EM, Welland ME, Dobson CM. 2009. An analytical solution to the kinetics of breakable filament assembly. Science 326:1533–1537 [DOI] [PubMed] [Google Scholar]
- 5. Bessen RA, Marsh RF. 1994. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J. Virol. 68:7859–7868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sim VL, Caughey B. 2009. Ultrastructures and strain comparison of under-glycosylated scrapie prion fibrils. Neurobiol. Aging 30:2031–2042 [DOI] [PubMed] [Google Scholar]
- 7. Spassov S, Beekes M, Naumann D. 2006. Structural differences between TSEs strains investigated by FT-IR spectroscopy. Biochim. Biophys. Acta 1760:1138–1149 [DOI] [PubMed] [Google Scholar]
- 8. Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Mastrianni J, Lugaresi E, Gambetti P, Prusiner SB. 1996. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 274:2079–2082 [DOI] [PubMed] [Google Scholar]
- 9. Saborio GP, Permanne B, Soto C. 2001. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411:810–813 [DOI] [PubMed] [Google Scholar]
- 10. Atarashi R, Moore RA, Sim VL, Hughson AG, Dorward DW, Onwubiko HA, Priola SA, Caughey B. 2007. Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat. Methods 4:645–650 [DOI] [PubMed] [Google Scholar]
- 11. Bannach O, Birkmann E, Reinartz E, Jaeger KE, Langeveld JP, Rohwer RG, Gregori L, Terry LA, Willbold D, Riesner D. 2012. Detection of prion protein particles in blood plasma of scrapie infected sheep. PLoS One 7:e36620. 10.1371/journal.pone.0036620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Castilla J, Saa P, Soto C. 2005. Detection of prions in blood. Nat. Med. 11:982–985 [DOI] [PubMed] [Google Scholar]
- 13. Lacroux C, Vilette D, Fernandez-Borges N, Litaise C, Lugan S, Morel N, Corbiere F, Simon S, Simmons H, Costes P, Weisbecker JL, Lantier I, Lantier F, Schelcher F, Grassi J, Castilla J, Andreoletti O. 2012. Prionemia and leukocyte-platelet-associated infectivity in sheep transmissible spongiform encephalopathy models. J. Virol. 86:2056–2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Safar JG, Lessard P, Tamguney G, Freyman Y, Deering C, Letessier F, Dearmond SJ, Prusiner SB. 2008. Transmission and detection of prions in feces. J. Infect. Dis. 198:81–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Castilla J, Gonzalez-Romero D, Saa P, Morales R, De Castro J, Soto C. 2008. Crossing the species barrier by PrP(Sc) replication in vitro generates unique infectious prions. Cell 134:757–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Klingeborn M, Race B, Meade-White KD, Chesebro B. 2011. Lower specific infectivity of protease-resistant prion protein generated in cell-free reactions. Proc. Natl. Acad. Sci. U. S. A. 108:E1244–E1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Saa P, Castilla J, Soto C. 2006. Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J. Biol. Chem. 281:35245–35252 [DOI] [PubMed] [Google Scholar]
- 18. Shikiya RA, Bartz JC. 2011. In vitro generation of high-titer prions. J. Virol. 85:13439–13442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Weber P, Giese A, Piening N, Mitteregger G, Thomzig A, Beekes M, Kretzschmar HA. 2006. Cell-free formation of misfolded prion protein with authentic prion infectivity. Proc. Natl. Acad. Sci. U. S. A. 103:15818–15823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Daus ML, Wagenfuhr K, Thomzig A, Boerner S, Hermann P, Hermelink A, Beekes M, Lasch P. 25 October 2013. Infrared microspectroscopy detects protein misfolding cyclic amplification (PMCA)-induced conformational alterations in hamster scrapie progeny seeds. J. Biol. Chem. 10.1074/jbc.M113.497131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gonzalez-Montalban N, Makarava N, Ostapchenko VG, Savtchenk R, Alexeeva I, Rohwer RG, Baskakov IV. 2011. Highly efficient protein misfolding cyclic amplification. PLoS Pathog. 7:e1001277. 10.1371/journal.ppat.1001277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johnson CJ, Aiken JM, McKenzie D, Samuel MD, Pedersen JA. 2012. Highly efficient amplification of chronic wasting disease agent by protein misfolding cyclic amplification with beads (PMCAb). PLoS One 7:e35383. 10.1371/journal.pone.0035383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cosseddu GM, Nonno R, Vaccari G, Bucalossi C, Fernandez-Borges N, Di Bari MA, Castilla J, Agrimi U. 2011. Ultra-efficient PrP(Sc) amplification highlights potentialities and pitfalls of PMCA technology. PLoS Pathog. 7:e1002370. 10.1371/journal.ppat.1002370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Langevin C, Andreoletti O, Le Dur A, Laude H, Beringue V. 2011. Marked influence of the route of infection on prion strain apparent phenotype in a scrapie transgenic mouse model. Neurobiol. Dis. 41:219–225 [DOI] [PubMed] [Google Scholar]
- 25. Tixador P, Herzog L, Reine F, Jaumain E, Chapuis J, Le Dur A, Laude H, Beringue V. 2010. The physical relationship between infectivity and prion protein aggregates is strain-dependent. PLoS Pathog. 6:e1000859. 10.1371/journal.ppat.1000859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Castilla J, Saa P, Morales R, Abid K, Maundrell K, Soto C. 2006. Protein misfolding cyclic amplification for diagnosis and prion propagation studies. Methods Enzymol. 412:3–21 [DOI] [PubMed] [Google Scholar]
- 27. Vilotte JL, Soulier S, Essalmani R, Stinnakre MG, Vaiman D, Lepourry L, Da Silva JC, Besnard N, Dawson M, Buschmann A, Groschup M, Petit S, Madelaine MF, Rakatobe S, Le Dur A, Vilette D, Laude H. 2001. Markedly increased susceptibility to natural sheep scrapie of transgenic mice expressing ovine PrP. J. Virol. 75:5977–5984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dron M, Moudjou M, Chapuis J, Salamat MK, Bernard J, Cronier S, Langevin C, Laude H. 2010. Endogenous proteolytic cleavage of disease-associated prion protein to produce C2 fragments is strongly cell- and tissue-dependent. J. Biol. Chem. 285:10252–10264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fischer M, Rulicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. 1996. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 15:1255–1264 [PMC free article] [PubMed] [Google Scholar]
- 30. Beringue V, Le Dur A, Tixador P, Reine F, Lepourry L, Perret-Liaudet A, Haik S, Vilotte JL, Fontes M, Laude H. 2008. Prominent and persistent extraneural infection in human PrP transgenic mice infected with variant CJD. PLoS One 3:e1419. 10.1371/journal.pone.0001419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gonzalez-Montalban N, Baskakov IV. 2012. Assessment of strain-specific PrP(Sc) elongation rates revealed a transformation of PrP(Sc) properties during protein misfolding cyclic amplification. PLoS One 7:e41210. 10.1371/journal.pone.0041210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim C, Haldiman T, Surewicz K, Cohen Y, Chen W, Blevins J, Sy MS, Cohen M, Kong Q, Telling GC, Surewicz WK, Safar JG. 2012. Small protease sensitive oligomers of PrPSc in distinct human prions determine conversion rate of PrP(C). PLoS Pathog. 8:e1002835. 10.1371/journal.ppat.1002835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Makarava N, Savtchenko R, Alexeeva I, Rohwer RG, Baskakov IV. 2012. Fast and ultrasensitive method for quantitating prion infectivity titre. Nat. Commun. 3:741. 10.1038/ncomms1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barria MA, Mukherjee A, Gonzalez-Romero D, Morales R, Soto C. 2009. De novo generation of infectious prions in vitro produces a new disease phenotype. PLoS Pathog. 5:e1000421. 10.1371/journal.ppat.1000421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morales R, Duran-Aniotz C, Diaz-Espinoza R, Camacho MV, Soto C. 2012. Protein misfolding cyclic amplification of infectious prions. Nat. Protoc. 7:1397–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Safar JG, DeArmond SJ, Kociuba K, Deering C, Didorenko S, Bouzamondo-Bernstein E, Prusiner SB, Tremblay P. 2005. Prion clearance in bigenic mice. J. Gen. Virol. 86:2913–2923 [DOI] [PubMed] [Google Scholar]
- 37. Safar JG, Kellings K, Serban A, Groth D, Cleaver JE, Prusiner SB, Riesner D. 2005. Search for a prion-specific nucleic acid. J. Virol. 79:10796–10806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Deleault NR, Kascsak R, Geoghegan JC, Supattapone S. 2010. Species-dependent differences in cofactor utilization for formation of the protease-resistant prion protein in vitro. Biochemistry 49:3928–3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Murayama Y, Yoshioka M, Masujin K, Okada H, Iwamaru Y, Imamura M, Matsuura Y, Fukuda S, Onoe S, Yokoyama T, Mohri S. 2010. Sulfated dextrans enhance in vitro amplification of bovine spongiform encephalopathy PrP(Sc) and enable ultrasensitive detection of bovine PrP(Sc). PLoS One 5:e13152. 10.1371/journal.pone.0013152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thackray AM, Klein MA, Aguzzi A, Bujdoso R. 2002. Chronic subclinical prion disease induced by low-dose inoculum. J. Virol. 76:2510–2517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bessen RA, Marsh RF. 1992. Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J. Gen. Virol. 73:329–334 [DOI] [PubMed] [Google Scholar]
- 42. Edgeworth JA, Farmer M, Sicilia A, Tavares P, Beck J, Campbell T, Lowe J, Mead S, Rudge P, Collinge J, Jackson GS. 2011. Detection of prion infection in variant Creutzfeldt-Jakob disease: a blood-based assay. Lancet 377:487–493 [DOI] [PubMed] [Google Scholar]
- 43. Beringue V, Herzog L, Jaumain E, Reine F, Sibille P, Le Dur A, Vilotte JL, Laude H. 2012. Facilitated cross-species transmission of prions in extraneural tissue. Science 335:472–475 [DOI] [PubMed] [Google Scholar]
- 44. Collinge J. 2012. Cell biology. The risk of prion zoonoses. Science 335:411–413 [DOI] [PubMed] [Google Scholar]
- 45. Gill ON, Spencer Y, Richard-Loendt A, Kelly C, Dabaghian R, Boyes L, Linehan J, Simmons M, Webb P, Bellerby P, Andrews N, Hilton DA, Ironside JW, Beck J, Poulter M, Mead S, Brandner S. 2013. Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: large scale survey. BMJ 347:f5675. 10.1136/bmj.f5675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kurt TD, Perrott MR, Wilusz CJ, Wilusz J, Supattapone S, Telling GC, Zabel MD, Hoover EA. 2007. Efficient in vitro amplification of chronic wasting disease PrPRES. J. Virol. 81:9605–9608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mays CE, Titlow W, Seward T, Telling GC, Ryou C. 2009. Enhancement of protein misfolding cyclic amplification by using concentrated cellular prion protein source. Biochem. Biophys. Res. Commun. 388:306–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Segarra C, Bougard D, Moudjou M, Laude H, Beringue V, Coste J. 2013. Plasminogen-based capture combined with amplification technology for the detection of PrP(TSE) in the pre-clinical phase of infection. PLoS One 8:e69632. 10.1371/journal.pone.0069632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bruce ME, McBride PA, Jeffrey M, Scott JR. 1994. PrP in pathology and pathogenesis in scrapie-infected mice. Mol. Neurobiol. 8:105–112 [DOI] [PubMed] [Google Scholar]
- 50. DeArmond SJ, Sanchez H, Yehiely F, Qiu Y, Ninchak-Casey A, Daggett V, Camerino AP, Cayetano J, Rogers M, Groth D, Torchia M, Tremblay P, Scott MR, Cohen FE, Prusiner SB. 1997. Selective neuronal targeting in prion disease. Neuron 19:1337–1348 [DOI] [PubMed] [Google Scholar]
- 51. Hecker R, Taraboulos A, Scott M, Pan KM, Yang SL, Torchia M, Jendroska K, DeArmond SJ, Prusiner SB. 1992. Replication of distinct scrapie prion isolates is region specific in brains of transgenic mice and hamsters. Genes Dev. 6:1213–1228 [DOI] [PubMed] [Google Scholar]
- 52. Deleault NR, Walsh DJ, Piro JR, Wang F, Wang X, Ma J, Rees JR, Supattapone S. 2012. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc. Natl. Acad. Sci. U. S. A. 109:E1938–E1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Deleault NR, Harris BT, Rees JR, Supattapone S. 2007. Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. U. S. A. 104:9741–9746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang F, Wang X, Yuan CG, Ma J. 2010. Generating a prion with bacterially expressed recombinant prion protein. Science 327:1132–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cronier S, Gros N, Tattum MH, Jackson GS, Clarke AR, Collinge J, Wadsworth JD. 2008. Detection and characterization of proteinase K-sensitive disease-related prion protein with thermolysin. Biochem. J. 416:297–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Owen JP, Maddison BC, Whitelam GC, Gough KC. 2007. Use of thermolysin in the diagnosis of prion diseases. Mol. Biotechnol. 35:161–170 [DOI] [PubMed] [Google Scholar]
- 57. Dickinson AG, Fraser H. 1969. Genetical control of the concentration of ME7 scrapie agent in mouse spleen. J. Comp. Pathol. 79:363–366 [DOI] [PubMed] [Google Scholar]
- 58. Dickinson AG, Meikle VM, Fraser H. 1969. Genetical control of the concentration of ME7 scrapie agent in the brain of mice. J. Comp. Pathol. 79:15–22 [DOI] [PubMed] [Google Scholar]
- 59. Barron RM, Campbell SL, King D, Bellon A, Chapman KE, Williamson RA, Manson JC. 2007. High titers of transmissible spongiform encephalopathy infectivity associated with extremely low levels of PrPSc in vivo. J. Biol. Chem. 282:35878–35886 [DOI] [PubMed] [Google Scholar]
- 60. Silveira JR, Raymond GJ, Hughson AG, Race RE, Sim VL, Hayes SF, Caughey B. 2005. The most infectious prion protein particles. Nature 437:257–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Laferriere F, Tixador P, Moudjou M, Chapuis J, Sibille P, Herzog L, Reine F, Jaumain E, Laude H, Rezaei H, Beringue V. 2013. Quaternary structure of pathological prion protein as a determining factor of strain-specific prion replication dynamics. PLoS Pathog. 9:e1003702. 10.1371/journal.ppat.1003702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Orru CD, Wilham JM, Vascellari S, Hughson AG, Caughey B. 2012. New generation QuIC assays for prion seeding activity. Prion 6:147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wilham JM, Orru CD, Bessen RA, Atarashi R, Sano K, Race B, Meade-White KD, Taubner LM, Timmes A, Caughey B. 2010. Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog. 6:e1001217. 10.1371/journal.ppat.1001217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. McGuire LI, Peden AH, Orru CD, Wilham JM, Appleford NE, Mallinson G, Andrews M, Head MW, Caughey B, Will RG, Knight RS, Green AJ. 2012. Real time quaking-induced conversion analysis of cerebrospinal fluid in sporadic Creutzfeldt-Jakob disease. Ann. Neurol. 72:278–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cronier S, Beringue V, Bellon A, Peyrin JM, Laude H. 2007. Prion strain- and species-dependent effects of antiprion molecules in primary neuronal cultures. J. Virol. 81:13794–13800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Feraudet C, Morel N, Simon S, Volland H, Frobert Y, Creminon C, Vilette D, Lehmann S, Grassi J. 2005. Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. J. Biol. Chem. 280:11247–11258 [DOI] [PubMed] [Google Scholar]
- 67. Krasemann S, Groschup MH, Harmeyer S, Hunsmann G, Bodemer W. 1996. Generation of monoclonal antibodies against human prion proteins in PrP0/0 mice. Mol. Med. 2:725–734 [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
mb-PMCA efficiency does not rely on the position of the samples in the PCR microplate wells. Serial 10-fold dilutions of 127S, 263K, 139A, and vCJD prions were prepared in duplicate and subjected to a single round of mb-PMCA. Four unseeded mouse brain lysates (tg338, tga20, tg7, and tg650) were run in the same plate. At the end of mb-PMCA, samples were treated with PK and analyzed by dot blotting as described in Materials and Methods. Download