

Abstract
Prion diseases, also known as transmissible spongiform encephalopathies (TSEs), are a group of fatal neurodegenerative disorders affecting humans and other mammalian species. The central event in TSE pathogenesis is the conformational conversion of the cellular prion protein, PrPC, into the aggregate, β-sheet rich, amyloidogenic form, PrPSc. Increasing evidence indicates that distinct PrPSc conformers, forming distinct ordered aggregates, can encipher the phenotypic TSE variants related to prion strains. Prion strains are TSE isolates that, after inoculation into syngenic hosts, cause disease with distinct characteristics, such as incubation period, pattern of PrPSc distribution, and regional severity of histopathological changes in the brain. In analogy with other amyloid forming proteins, PrPSc toxicity is thought to derive from the existence of various intermediate structures prior to the amyloid fiber formation and/or their specific interaction with membranes. The latter appears particularly relevant for the pathogenesis of TSEs associated with GPI-anchored PrPSc, which involves major cellular membrane distortions in neurons. In this review, we update the current knowledge on the molecular mechanisms underlying three fundamental aspects of the basic biology of prions such as the putative mechanism of prion protein conversion to the pathogenic form PrPSc and its propagation, the molecular basis of prion strains, and the mechanism of induced neurotoxicity by PrPSc aggregates.
1. Introduction
Prion diseases, also known as transmissible spongiform encephalopathies (TSEs), are rapidly progressive neurodegenerative disorders that affect many species of mammals. In humans, they comprise Creutzfeldt-Jakob disease (CJD), fatal familial insomnia (FFI), kuru, Gerstmann-Sträussler-Scheinker disease (GSS), and the recently described variably protease-sensitive prionopathy (VPSPr), whereas natural TSEs in animals include scrapie of sheep and goats, bovine spongiform encephalopathy (BSE), and chronic wasting disease (CWD) in deer and elk.
Prion diseases belong to the growing group of disorders that are attributed to misfolding and ordered aggregation of proteins, which include Alzheimer's disease, Parkinson's disease, systemic amyloidosis, and many others. In prion disease, in particular, the cellular prion protein, PrPC, after partial misfolding, converts into a partially protease-resistant disease-associated isoform, PrPSc, which aggregates in the brain and forms deposits that are associated with the neurodegenerative changes.
Distinguishing features of prion diseases among these disorders, however, are their wide phenotypic spectrum, the multiple apparent ethiologies (e.g., sporadic, genetic, and acquired), and the transmissibility between individuals, a characteristic which has allowed the early development of experimental models. This has led to the important discovery that mammalian prions occur, like conventional infectious agents, in a variety of different strains: these are defined as natural isolates of infectious prions characterized by distinctive clinical and neuropathological features, which are faithfully recapitulated upon serial passage within the same host genotype. The different strains of the TSE agent or prion are believed to be the main cause of TSE phenotypic diversity. In addition, the host variability in the gene encoding PrPC (PRNP), as determined by polymorphisms or mutations, also modulates the disease phenotype. In this review, we focus on three fundamental aspects of the basic biology of prions, which, despite the significant recent advances, remain unsolved. They include the molecular mechanisms of PrPC to PrPSc conversion, the role of PrPSc in strain determination, and the mechanism of PrPSc aggregate-induced neurotoxicity. Due to the space constraint and the main expertise of the authors, emphasis is given to evidence obtained from the study of naturally occurring diseases, particularly in humans, and from animal models.
2. PrPC-PrPSc Conversion
2.1. Structural Changes Associated with PrPC to PrPSc Conversion
Understanding the structural features of PrPSc remains a key issue to gain the ultimate insight into the molecular basis of prion formation and propagation. Unfortunately, the insoluble nature of PrPSc has hampered most efforts to determine its structure by preventing the use of high-resolution techniques such as NMR or X-ray crystallography. Therefore, only partial structural information is available from low-resolution approaches such as Fourier transform infrared spectroscopy (FTIR), electron microscopy (EM), immunoassays, fiber X-ray diffraction, and limited proteolysis [1–9]. Full-length PrPC encompasses a poorly definite domain at the N-terminal end of the protein (which spans ~100 residues), a globular domain in the central portion (residues 125–228), and a short flexible C-terminal domain, ending with the GPI anchor (residues 229-230/231) [10]. The globular domain is composed of three α-helices and two antiparallel β-sheets, separated by short loops and kept together in their final tertiary structure by interactions between the exposed amino acidic lateral chains that are in close contact with each other when the protein is correctly folded [10]. The conversion of PrPC into the pathological conformer PrPSc is characterized by a significant increase of β-sheet secondary structure. Indeed, FTIR and circular dichroism (CD) spectroscopy experiments indicate a dramatic difference in the secondary structure between the two isoforms. While PrPC contains 47% α-helix and 3% β-structure, PrPSc holds 17–30% α-helix and 43–54% extended β-structure, the range being partially due to the multiple forms and lengths of PrPSc [2, 11].
Taking advantage of the available low-resolution structural information and constraints about PrPSc and of computational techniques, different theoretical models have been proposed to describe the putative PrPSc structure. The β-helical model is based on fiber X-ray diffraction and computer modeling techniques and proposes that the segment ~90–175 forms a four-stranded β-sheet core organized in a β-helical configuration, whereas helices α2 and α3 would retain their native conformation [3]. An alternative “spiral” model is based on molecular dynamics simulations and indicates that during PrPC conversion a longer single β-strand is generated from the elongation of the two native β-sheets. The newly formed β-strand would interact with other PrP molecules and, in turn, lead to polymerization [12]. In both models the basic subunit of the oligomers is considered a trimer. According to the authors who have proposed the spiral model, however, the β-helical model is in disagreement with several critical constraints: notably, it would not fit within the unit cell packing dimensions of the EM data for which it was modeled and would be inconsistent with antibody mapping studies, enzyme cleavage sites, and fibril disaggregation profiles [12]. Furthermore, the results of recent deuterium exchange experiments on brain-derived PrPSc showed that the region from residue ~90 to the entire C-terminus displays slow exchange rates that are typical for a structure consisting of a continuum of β-strands [13]. These findings from Surewicz's group appear inconsistent with both the “β-helical” and the “spiral” models, which are assuming an incomplete conversion of the α-helical structures into β-sheet and add further controversy to the issue. Of course, current models do not rule out the possibility that there are other structures that would satisfy the experimental constraints. Indeed, given that in mammals more than a dozen of different prion strains are documented, a higher structural heterogeneity is expected and should be explained.
2.2. Effects of PRNP Mutations
Several mutations in the PrP gene (PRNP) account for the genetic or familial form of human prion disease, in which the conversion of PrPC into PrPSc is thought to occur spontaneously, triggered by the mutation. About forty mutations linked to familial CJD, GSS, FFI, or other atypical phenotypes have been identified to date [15]; they have been linked to a plethora of effects at both structural and clinicopathological levels. Based on their position in the gene, their effect, and the type of residue replaced, PRNP mutations can be classified in several groups: N-terminal or C-terminal mutations, missense, insert, or STOP-codon mutations, salt bridge-affecting, polar mutations, and hydrophobic or GPI-signal-peptide mutations [16].
Based on in vitro studies it has been proposed that disease-linked mutations increase the likelihood of PrPC misfolding by thermodynamically destabilizing the protein [17–20]. However, this cannot be taken as a general mechanism because individual mutations differently (or barely) affect PrPC stability. Besides influencing the stability of PrPC, mutations may also alter its surface properties, thus triggering an abnormal interaction with other not yet identified cofactors, or causing an aberrant trafficking and accumulation inside the cell [16].
Atomic structural details, obtained using solution-state NMR spectroscopy, are available only for a few pathological human (Hu) PrP mutants. Based on the structural comparison of the folded domain (residues 125 to 228) of HuPrP carrying the CJD-linked E200K or V210I [21] mutations and the GSS-linked Q212P [22] mutation, it has been proposed that pathological mutants affects the aromatic and hydrophobic interactions between residues clustered at the interface of the β2-α2 loop and the C-terminal half of the α3 helix. The disruption of these interactions and the consequent exposure to the solvent of the hydrophobic core may represent a common effect of the three mutants, which has led to the proposal that the early stage of prion conversion possibly involves the critical epitope formed by the β2-α2 loop and the α3 helix. Similar findings have been obtained with the X-ray crystal structure of both F198S and D178N mutants [23] and molecular dynamics experiments [24, 25].
HuPrP pathological mutants were also explored in several murine models. In particular, various transgenic (Tg) mouse models overexpressing mutated PrP constructs (or wild-type PrP) were developed in order to determine whether PrP is per se sufficient to give rise to disease and generate infectivity. In an early controversial study Hsiao and colleagues reported that Tg mice overexpressing the mutated PrP P101L, an homologous of the P102L substitution associated with the GSS syndrome in humans, spontaneously develop a clinical-pathological phenotype which propagated disease in inoculated Tg 196 mice expressing lower levels of mutant protein, suggesting that pathogenic PrP gene mutations resulted in the spontaneous formation of PrPSc and de novo production of prions [26]. Subsequent studies, however, have shown that the Tg 196 mice also spontaneously develop the disease in late life as a consequence of PrP overexpression, making the apparent prion propagation observed in this model more accurately characterized as disease acceleration rather than transmission [27]. Remarkably, disease transmission of brain extracts from Tg animals overexpressing the P101L mutation neither occurred to wild-type nor to Tg mice expressing MoPrP-P101L from two transgene copies that do not develop disease spontaneously in their natural lifespan [27], which is in full agreement with a previous study from Manson's group showing that PRNP gene-targeted 101LL mice expressing MoPrPP101L failed to develop the neurodegenerative disease spontaneously [28].
In line with the concept expressed above, several subsequent studies reported that Tg mice overexpressing PrP mutants often develop neuropathological features reminiscent of human TSEs, although in most cases the inoculation of their brain extracts in wild-type animals neither reproduced the main feature of the disease nor generated infectivity [29–34].
Results contradicting this general observation, however, have also been reported. Transgenic mice moderately overexpressing a mutant mouse PrP carrying two point mutations (170N and 174T) that are found as normal variants in the rigid loop of elk PrP spontaneously develop spongiform encephalopathy and PrP plaque deposition in the brain [35]. Repeated subpassages in Tg20 mice showed transmission of disease to wild-type mice and propagation of protease-resistant PrPSc. Similarly, Lindquist and collaborators were able to generate knock-in mice expressing the mouse equivalent of the PrP mutation (i.e., D178N-M129) associated with FFI. These mice developed de novo prion diseases with neuropathological traits similar to FFI that was transmissible to wild-type mice carrying the same 3F4 epitope [36]. A very similar result has been recently obtained by the same group using knock-in mice carrying the mouse equivalent of the most common human mutation (i.e., E200K) associated with genetic CJD. These mice developed the hallmark features of CJD, namely, spongiosis and proteinase K (PK)-resistant PrP aggregates. Furthermore, brain extracts from these mice caused a transmissible neurodegenerative disease after intracerebral inoculation in WT mice [37]. Finally, infectious prions were also reported to form spontaneously, even before the onset of the clinical symptoms, in chimeric mouse/human transgenic mice (called TgMHu2M), also expressing the CJD-linked E200K mutation [38]. Thus, according to these four studies, the introduction of a single (or two) amino acidic change(s) in PRNP in a critical position can cause remarkably different neurodegenerative diseases and may be sufficient to create distinct protein-based infectious prions.
Tg mice lines expressing human PRNP mutations were also used to study the effect of the mutation on disease susceptibility. Transgenic mice carrying the P101L mutation in PrP had remarkable differences in incubation time compared with wild-type littermates, following inoculation with several prion strains from human, hamster, sheep, and murine sources, suggesting a critical role for the structurally “flexible” region of PrP in agent replication [30]. In another study, Asante and collaborators [29] studied mouse lines homozygous for the human PrP102L, 129 M or for human PrP200 K, 129 M transgenes both expressed on PRNP null background. Although both lines did not develop spontaneous neurodegeneration, they showed a different susceptibility to inherited prion diseases. While PrP102L, 129 M were permissive to homotypic P102L prions and not to sCJD prions, PrP200 K, 129 M showed a similar susceptibility to both the E200K inoculum and classical sCJD prions [29]. Tg mice lines carrying PRNP mutations have also been used to unveil molecular pathways that are activated by the expression of mutant PrP, which may lead to neuronal dysfunction. In a recent study Senatore et al. shed light on the effects of insertional mutants on synaptic transmission [39]. Using Tg mice expressing a PrP insertional mutation linked to familial prion disease [31], they pinpointed the existing relationship between the early motor behavioral abnormalities and the impaired glutamatergic neurotransmission in cerebellar granule neurons. In particular, they showed that the misfolded mutant PrP undergoes an aberrant intracellular trafficking causing the intracellular accumulation of the voltage gated calcium channel α2δ-1 subunit, which results in the disruption of the cerebellar glutamatergic neurotransmission [39].
2.3. Cellular Cofactors Featuring in PrPC Conversion and Prion Propagation
Several lines of evidence suggest that different classes of cofactors, possibly acting as chaperones, can influence PrPC conversion and prion propagation [40, 41]. To date, two types of cofactors, lipids and polyanions, have been implicated (Table 1), although their precise mechanism of action remains unclear. Among linear polyanions, glycosaminoglycans (GAGs) and sulfated polysaccharides such as pentosan polysulfate or heparan sulfate were shown to influence prion conversion in vitro [42–45] (Table 1) possibly by facilitating the formation of PrPC-PrPSc complexes through multiple simultaneous interactions with several PrP molecules [42].
Table 1.
Cofactors enhancing PrPC conversion in vitro.
| Cofactor | Experimental setting | Results | Refs. | |
|---|---|---|---|---|
| Pentosan polysulfate (PPS) |
Cell-free conversion assay | Hamster and mouse [35S] GPI(-) PrPC seeded with brain derived PrPres from infected hamsters (263 K) and mice (87 V) | (i) PPS increases the rate of formation and the yield of [35S] PrPres
(ii) PPS facilitates conversion of both Mo and SHa [35S] GPI(-) PrPC at different temperatures |
[44] |
|
| ||||
| Heparin | Cell-PMCA | Cell lysates plus exogenously expressed HuPrP seeded with sCJD, vCJD, and hamster-adapted scrapie 263 K | (i) Both low and high molecular weight heparin enhance PMCA efficiency (ii) Seed-dependent effect of heparin on amplification efficiency |
[45] |
|
| ||||
| Sulfated dextran compounds | PMCA | PrPSc derived from BSE-infected cattle brain diluted in PrPC substrate | (i) Enhanced BSE PrPSc amplification (ii) Amplified PrPSc induce lesions typical of prion disease in TgBoPrP |
[135] |
|
| ||||
| Synthetic poly (A) RNA | PMCA | Normal and diluted scrapie brain homogenate | (i) Stochastic de novo formation of PrPSc molecules from unseeded purified substrates (ii) Both amplified Sc237 or 139H PrPSc and de novo PrPSc molecules cause scrapie in inoculated Syrian hamsters |
[136] |
|
| ||||
| Phosphatidylethanolamine (PE) | PMCA | recPrP substrate with a recPrPSc seed | (i) Generation of infectious prions (ii) PE supports prion propagation using PrP molecules from multiple animal species |
[55] |
|
| ||||
| RNA from normal mouse liver plus POPG | PMCA | Normal mouse brain homogenate seeded with recPrP | (i) In vitro generated recPrPres
(ii) recPrPres propagates its PK-resistant conformation to endogenous PrPC (iii) recPrPres causes bona fide prion disease in wild-type mice |
[50] |
Most significantly, host-encoded RNA was shown to facilitate the prion-seeded conversion of PrPC to PrPSc in vitro [46–50]. However, whether RNA acts as a mere catalyst of the PrP misfolding process or, alternatively, is associated with the infectious particle and contribute to determine the prion strain specificity is still unsolved. A recent study showed that the requirement of RNA for in vitro amplification of PrPSc is species dependent, with only hamster-derived PrPSc being largely dependent on the presence of RNA, whereas mouse-derived PrPSc is not [51]. Another study showed similar RNA-dependent amplifications of six hamster prion strains [52]. DNA and phospholipids have also been implicated as cofactors modulating prion replication in vitro. The polymerization of the mouse recombinant PrP (rPrP) was enhanced in presence of nucleic acids and sequence-specific DNA binding to rPrP converted it from a α-helical conformation to a soluble, β-sheet enriched isoform similar to that found in the fibrillar PrPSc state [53, 54]. Unlike RNA, the essential membrane phospholipid phosphatidylethanolamine (PE) was described as a highly promiscuous cofactor that can promote prion propagation using rPrP molecules from different mammalian species [55]. Critical questions, which are still far from being fully answered, concerns the role, if any, of cofactors in modulating prion infectivity and the specific properties of prion strains. Preliminary data on in vitro reconstitute prions seem to indicate that the presence of cofactors enhances in vivo prion infectivity, whereas the data collected to date on the issue of strains appear to be inconsistent. For example, while the use of PE as unique cofactor in the propagation process allowed the adaption of two different native prion strains into the same unique output strain, suggesting that a single cofactor is able to force the conversion of different strains into a single strain having its own phenotypic features [56], in another study it was found that replication under RNA-depleted conditions does not modify RML prion strain properties [57].
2.4. Cellular Sites of PrPSc Formation
Being PrPC a GPI-anchored protein, it mainly localizes in lipid rafts of cellular membranes where it can interact in trans with a variety of signaling molecules, including caveolin-1, Fyn, and Src tyrosine kinases [58], or with other cell-surface proteins as NCAM [59], stress-inducible protein 1 [60–62], vitronectin, lipoprotein receptor-related protein 1 [63, 64], or reelin [65].
Several lines of evidence suggest that lipid rafts are critically involved in the conversion of PrPC into the pathological form PrPSc. Using immortalized neuroblastoma cells ScN2a, chronically infected by the Rocky Mountain Laboratory (RML) prion strain, Naslavsky et al. showed that PrPSc is attached to lipid rafts [66] and that the amount of the abnormal protein inversely correlates with sphingomyelin levels [67]. Furthermore, using thin-layer chromatography and mass spectrometry, it has been found that the insoluble aggregates of N-terminally truncated PrPSc (i.e., PrP 27–30) contain small amounts of two host sphingolipids, galactosylceramide and sphingomyelin [68], which also supports the localization of PrPSc in rafts. Other data pointing to a raft-mediated conversion include the observations that depletion of cellular cholesterol or the replacement of PrPC GPI-anchor with the transmembrane and cytosolic domain from nonrafts proteins diminished or prevented the formation of PrPSc [58]. More recent studies, however, highlighted the possibility that lipid rafts favour the conversion by bringing together PrPSc and PrPC, rather than by triggering PrPC refolding [69]. Indeed, given their role in PrPC folding and stabilization of its conformation, lipid rafts may even prevent PrPC transconformation. According to this view the conversion would occur only after PrPC exits in these domains. Finally, other studies also suggested that lipid rafts do not provide the environment in which PrPC-PrPSc refolding occurs, but rather promote PrPSc aggregation and fibrillization once the pathogenic misfolded protein has been produced elsewhere (reviewed in [70]).
Concerning the precise cellular site of conversion of PrPC to PrPSc, early studies pointed to the cell surface [71], which appears a plausible location particularly for the case of transmitted prion diseases, or to the endocytic pathway [72–75]. Subsequent studies further underlined the potential role in the conversion process of intracellular compartments such as the endosomal or lysosomal pathways, or even the ER [76–79]. Evidence for the conversion of PrPC to PrPSc occurring shortly after internalization, during an endocytic process, is indeed numerous. After treatment of both scrapie-infected Syrian hamster brain and ScN2a cell lines with guanidine-hydrochloride, which allows epitope unmasking in native PrPSc, the abnormal protein was primarily described intracellularly [75], where it was found to accumulate in lysosomes. In another study, using cryo-immunogold electron microscopy, PrPSc was found to be concentrated in early/recycling endosomes of neuritis of prion infected hippocampal neurons [77, 79]. Similarly, in three different neuronal cell lines infected with different prion strains more than 25% of PrPSc has been observed to colocalize with a marker for the early recycling compartment. Classic studies have also shown that PrPSc accumulates intracellularly as an N-terminal truncated form, which is generated after proteolytic cleavage in both endosomes and lysosomes [73, 74]. Supporting evidence for the role of endosomes or lysosomes in PrPC conversion is also provided by the observations that an acidic pH triggers the conformational change of PrPC to a PrPSc-like form and that the lowering of the temperature to 18°C, supposedly by slowing the rate of PrPC endocytosis, reduces PrPSc formation (reviewed in [80]).
Finally, the main cellular site of PrPC and PrPSc location was also found to differ depending on the investigated cell line. In ScN2a cells, for example, PrPC and PrPSc colocalize in the late-endosomial compartments, whereas in scrapie-infected hypothalamic (GT1—7) cells PrPSc is present in an additional vesicular compartment which is flotillin-1-positive [81].
As a whole, the data collected indicate that in most infected cell lines the conversion event occurs either on the cell surface or along the endocytic pathway, with PrPSc ultimately mainly accumulating in lysosomes. Nevertheless, other cellular sites might be also involved depending on the cell type, the prion strain, or the disease etiology.
3. PrPSc and the Strain Phenomenon
The first demonstration of prion strains was obtained after transmission of distinct scrapie isolates [82]. When these sheep brain extracts were passaged to goats, a drowsy syndrome developed in some animals, while others had a scratching syndrome. A variety of scrapie strains were subsequently identified after passage through inbred mouse lines [83]. Properties that differentiate the strains are the length of incubation time following inoculation, the type and distribution of lesions (neuropathologic profile), and the pattern of intracerebral deposition of PrPSc [83–85]. The wide variety of scrapie strains has been traditionally seen as the major challenge to the protein only hypothesis [86, 87]. While in classical infectious diseases different strains of the agent are associated with variations in their nucleic acid genomes, the prion hypothesis implicates that PrPSc itself would encode the phenotypic properties of the strains.
Kascsak et al. [88] originally documented that the relative proportion of PrPSc glycoforms, the so called “glycoform ratio,” was associated with strain variability and could be used to differentiate strains of the scrapie agent when isolated in inbred mice. At about the same time, mouse strains ME7 and 139A scrapie associated fibrils (SAF) were shown to differ from hamster strain 263K SAF in terms of morphology, sedimentation rate, and sensitivity to PK digestion [89]. Noteworthy, these distinctive PrPSc physicochemical properties were initially considered an effect of the scrapie agent on PrP rather than an evidence for a role of PrPSc itself in strain determination. Indeed, the idea that the molecular basis of strain variation may lie in the structure of PrPSc, as predicted by the prion hypothesis, was fully embraced only after Bessen and Marsh found that two strains of transmissible mink encephalopathy (TME), transmitted to inbred Syrian hamsters, give rise to PrPSc molecules with distinct electrophoretic mobility and degree of resistance to protease digestion [90]. The two TME strain-specific PrPSc have been subsequently propagated in vitro through nongenetic mechanisms [91], which has further strengthened the view that the self-propagation of distinct PrPSc conformers may represent the basis of the prion strain phenomenon.
Experiments of FFI transmission to Tg mice gave additional support to the idea that the diversity of prion strains is enciphered in the PrPSc structure [92]. Brain homogenates from subjects affected by FFI, which contained a PrPSc fragment after PK digestion (PrPres) of 19 kDa, and from subjects with sporadic CJD (sCJD) or a genetic CJD (gCJD) subtype linked to the E200K-129M haplotype (CJDE200K-129M), which contained a PrPres fragment with a relative molecular mass of 21 kDa, were inoculated to syngenic mice. The endogenous PrPres recovered in the affected animals consistently and precisely replicated the size of the corresponding human PrPres.
In 1998 Safar et al. [93] introduced the conformation-dependent immunoassay (CDI), which measures the extent of epitope exposure after GndHCl denaturation and is therefore assumed to measure indirectly the relative percent of PrPSc β-sheet and α-helical content. Eight mouse-passaged scrapie strains were analyzed for strain-specific differences in secondary structure [93]. By plotting the ratio of antibody binding to the denatured/native proteins as a function of the concentration of PrPSc, the authors observed that each strain occupies a unique position, suggesting a distinct conformation.
FTIR spectroscopy has also been used to measure the secondary structure of both PK-treated and full-length PrPSc. Caughey and colleagues have originally compared the conformations of PrPSc in the HY, DY, and 263K hamster TSE strains and found striking differences in their secondary structures [1]. Similarly, another team [94, 95] has subsequently found strain-specific differences in secondary structure, temperature stability, and hydrogen-deuterium exchange characteristics between purified PrPSc preparations obtained from three scrapie strains and the classical BSE strain after passage in hamster.
More recently, the issue of the relationship between PrPSc conformational stability and strain-specific properties, such as incubation time and in vitro replication efficiency, has been addressed. In 2006 Legname et al. [96] reported that a reduced resistance to GndHCl denaturation, indicative of a reduced conformational stability, correlates with a shorter incubation time in mouse adapted prion strains. Similarly, the stability of PrPSc aggregates both in terms of resistance to GndHCL induced denaturation and thermostability was inversely correlated with the capacity to induce a rapidly lethal disease [97]. The provided explanation for these observations is that a decrease of PrPSc stability increases PrPSc aggregate fragmentation resulting in an increase in agent replication that produces a correspondingly shorter incubation period and a more aggressive disease. The relationship between the stability of PrPSc aggregates and PrPSc replication investigated in vitro using the protein misfolding cyclic amplification (PMCA) paradigm [52] also supports a link between PrPSc conformational stability and fragmentation rate of PrPSc aggregates. Other data, however, suggest a more complex picture, especially in vivo, where additional factors, related to cellular processing, may also play a significant role. In apparent contrast with what was observed in mice, Ayers et al. [98] found that hamster-adapted scrapie strains with a short incubation period were more efficiently replicated, had a more stable conformation, and were more resistant to clearance from the soma of neurons than those with a longer incubation time which, in contrast, predominantly accumulated in glial cells. These results suggest that the progression of prion disease is also influenced by the balance between replication and clearance of PrPSc in neurons.
A potential new perspective to the study of PrPSc properties and their relationship to prion strains was opened by the characterization of the so-called “sensitive PrPSc” (sPrPSc), an isoform of abnormal PrP which is fully degraded at a PK activity comparable to that necessary to digest PrPC, despite maintaining other properties that are specific for PrPSc [99–101]. Evidence for sPrPSc being a biologically relevant species originally came from the study of PrPSc properties in naturally occurring prion diseases. Indeed, a fully PK-sensitive PrPSc has been detected in various phenotypically atypical variants of both human and animal prion diseases [102–107]. Furthermore, according to some studies [100, 108], sPrPSc represents an invariable and quantitatively significant component of prions, contributing up to 90% of the whole PrPSc signal even in classic TSEs such as sCJD and classical scrapie. Recent studies have also found a correlation between the relative amount of sPrPSc with strain-specific properties such as the incubation period after inoculation or the clinical duration of the disease [109, 110]. We also recently looked for sPrPSc in purified detergent-insoluble PrPSc sCJD preparations [111]. At variance with the findings above, however, our results showed that, irrespectively of the human prion strain, this slowly sedimenting sPrPSc represents a relatively minor component of abnormal PrP not exceeding 10% of total detergent-insoluble PrPSc. Thus, this significant discrepancy, which may depend at least partially on methodological aspects or data interpretation [111], needs to be further explored and explained.
Although not essential for prion propagation [112], PrP glycosylation of asparagine residues at positions 181 and 197 represents another factor likely contributing to the diversity of mammalian prions. Indeed, differences in ratios of di-, mono-, and unglycosylated PrPSc have been detected among phenotypic subtypes of both human and animal TSEs and are commonly used to differentiate specific strains [113–116]. This is consistent with the notion that glycosylation is critical in determining and maintaining conformation and interaction of glycoproteins [117, 118]. However, it is at present unclear whether glycans affect the backbone conformation of PrPSc molecules or rather modulate the interaction of these molecules by introducing specific steric constraints or by forming crucial intermolecular contact sites between PrPSc monomers [119]. In a recent elegant study Cancellotti et al. [120] have demonstrated that the passage in Tg mice expressing a PrP partially or completely lacking the N-glycan moieties affected the phenotypic characteristics of at least one TSE agent strain. Given that these changes could be successfully retained on passage in wild-type mice, it has been concluded that infectious properties of a TSE strain can be altered by posttranslational changes to host PrP, possibly as the result of the selection of mutant TSE strain.
Taken together all these pieces of evidence provide strong support to the argument that different PrPSc conformers encipher the prion “strains.” Nevertheless the direct proof for this contention is not yet available. Until a higher resolution picture of PrPSc provides the precise molecular-level details surrounding the puzzling phenomenon of prion strains and the conformational adaptability of PrP observed upon cross-species transmission, questions and alternative interpretations of the data will remain. For example, we cannot yet be sure of whether the distinctive properties of PrPSc directly reflect the tertiary conformation of monomers or are determined by interactions between PrPSc and other molecules acting as cofactors. PrPSc is extracted from the brain in a highly aggregated state and the heterogeneity in size of the PK digested protein core may well reflect the quaternary rather than the tertiary structure of the molecule. Similarly, the extent of conversion of each glycoform of PrPSc, which ultimately determines the glycoform ratio of PrPres, may also represent a signature imparted by another molecule that interacts with PrP. Finally, the central question that still remains to be answered is how an identical primary sequence can drive different tertiary conformations in the prion protein, if no other informational molecule exist. Even more difficult to explain in terms of PrPSc structural plasticity are other two fundamental aspects of the biology of prions, the so-called “species barrier,” that is, the phenomenon for which a strain must adapt to a new species host with a typical delay in incubation time, or even the loss of infection ability in that species, and, above all, the fact that prion strains, like conventional infectious agent strains, incur in spontaneous “mutations.” The latter phenomenon is often explained with the quasispecies hypothesis [121], which predicts that PrPSc with different conformations may be present at low levels in an infectious inoculum and that the variant most suitable for replication in a particular host is selected to become the dominant component of the population [122, 123]. However, evidence for large numbers of conformations is still lacking nor is it clear whether the required multiple conformations would be plausible in terms of thermodynamic stability.
3.1. PrPSc Characterization and Strain Variation in Natural Hosts: CJD, FI, GSS, and VPSPr
Five major clinicopathological phenotypes of human prion disease are currently recognized. These are CJD, FI, GSS, PrP-cerebral amyloid angiopathy, and VPSPr (phenotypic features of each form are reviewed in [15, 124–128]). The vast majority of human prion cases belong to CJD and occur in a sporadic fashion and worldwide. Only a small proportion of CJD cases are associated with PRNP mutations, in the form of familial or more properly genetic CJD (gCJD). Secondary CJD associated with inadvertent medical transmission is termed iatrogenic CJD (iCJD), and the only known zoonotic form of CJD, which is associated with exposure to BSE, is termed variant CJD (vCJD). VPSPr is a very recently described rare sporadic phenotype resembling GSS, FI can either occur sporadically or in a familiar form (FFI) associated with the D178N-129 M PRNP haplotype, while GSS and PrP-CAA phenotypes are tightly associated with mutations in the PRNP gene. In CJD, the prototype of human prion diseases, the characterization of PrPSc after PK treatment has led to the discovery of two major fragments of protease-resistant PrPSc (PrPres). The largest of these peptides, named type 1, has a relative electrophoretic mobility of 21 kDa and a primary PK cleavage site at residue 82 while the smallest, or type 2, has a relative molecular mass of 19 kDa and a primary cleavage site at residue 97 [114, 115, 129] (Figure 1). Based on the analysis of a large series of 300 sCJD cases it was shown that the two different PrPres types can be associated with each of the three possible PRNP genotypes determined by the polymorphic codon 129 (methionine, M, or valine, V) and that the six different possible combinations between these two molecular variables significantly correlate with the clinico-pathological heterogeneity of sCJD [130]. Intriguingly, the two PrPres types were also detected in the genetic and acquired forms of CJD, including vCJD, thus independently from the apparent etiology of the disease, that is, sporadic, inherited or acquired by infection [114, 129, 131], suggesting that the same prion strains are contributing to all forms of human TSEs. Furthermore, PrPres types 1 and 2 were also found to cooccur in the same brain in about one-third of all sCJD cases [130, 132–134]. The results obtained in large series of cases indicate that the deposition of either type 1 or 2, when concurrent, is not random and is always characterized by the coexistence of phenotypic features previously described for the “pure” subtypes, a finding which strongly suggests that these cases harbour a mixture of prion strains.
Figure 1.
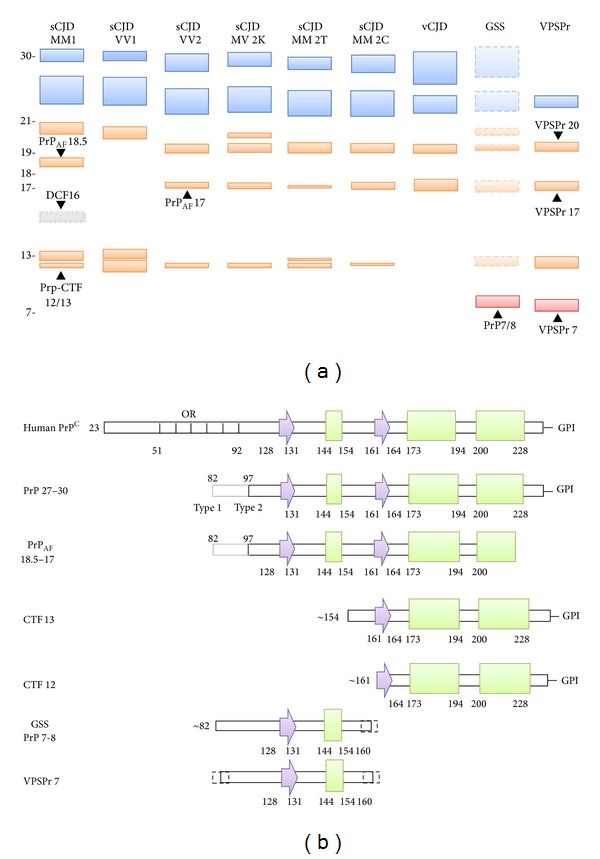
(a) Schematic representation of the spectrum of PrPres fragments observed in human prion diseases and their electrophoretic profile. The unglycosylated forms of all PrPres fragments with the glycosylation sites in their sequence are indicated in orange, while the fragments lacking these sites are shown in red. Among the glycosylated peptides, only the mono- and the diglycosylated forms of PrPres 27–30 (18–21 kDa range) fragments are shown (in blue). The DCF16 fragment, which is generated only in partially denaturing conditions is labeled with a dotted line and a gray color. For GSS, the fragments that have been described only associated with specific PRNP mutations (e.g., P102L or A117V) are shown with dotted lines and in transparency. Molecular weights are indicated on the left in kDa. (b) Diagrams of the secondary structural elements of human PrPC and of the PrPres fragments observed in human prion diseases. Arrows are representative of β-strands and rectangles of α-helices and OR indicates the octapeptide repeats region. The secondary structure numbering has been derived from pdb (Protein Data Bank) id 2LSB (human PrP).
The identification of an excess of pathological phenotypes (i.e., at least six) with respect to PrPres types 1 and 2 dichotomy has prompted further attempts to identify PrPres properties that would correlate with each disease phenotype. Using a standardized high buffer strength for brain homogenization, PK digestion at pH 6.9 with a high enzyme concentration, and long running gels, Notari et al. [137] showed that distinctive PrPres properties can indeed be found in sCJD phenotypes sharing the same PrPres type. For example, (i) PrPres type 2 from MV cases shows a unique doublet band that differs from PrPSc type 2 in MM and VV cases, and (ii) type 1 PrPres from VV cases migrates faster than type 1 PrPres from MM1 and MV1 samples when PK digestion is performed at pH under 7.2 (Figure 1(a)).
A further fine tuning of the PrPSc signature associated with each CJD-associated strain has been obtained with the discovery that PrPSc aggregates include PrPres C-terminal fragments with a relative mass of about 12 and 13 kDa (PrP-CTF12/13), in addition to PrP 27–30 (Figure 1). These fragments originate from the cleavage of PrPSc at residues 162–167 and 154–156 and vary in relative abundance among sCJD subtypes; in particular the peptide CTF-13 is present in significant amount in MM1 cases and is particularly abundant in VV1 subjects, whereas all PrPres type 2-associated sCJD subtypes but the MM 2T, as well as vCJD, show only traces of this fragment [138]. Notari et al. [138] also identified a novel C-terminally truncated PrPres fragment showing an apparent molecular mass of either ~18.5 kDa (when associated with type 1) or ~17 kDa (when associated with type 2). This fragment shares the primary N-terminal sequence with either type 1 or type 2 but lacks the very end of the C-terminus together with the GPI anchor (PrPAF 18.5-17) (Figure 1). Finally, a fragment with an apparent molecular mass of about 16 kDa, which is only generated in partially denaturing conditions (DCF 16), has been detected in sCJD MM1/MV1 (Figure 1). Epitope mapping indicates that the fragment has an intact C-terminal end and is truncated in the region between residue 112 and residue 144. Taken together, these data suggest that each sCJD subtype can be associated with a specific profile of PrPres fragments (PrP 27–30, PrPAF 18.5-17, DCF 16, PrP-CTF12/13), possibly reflecting subtype-specific structural characteristics of the protein aggregate [138] (Figure 1).
PrPSc from different prion strains can also be typed through its glycoform ratio, that is the ratio between the three differently glycosylated isoforms of PrPres 27–30 (i.e., diglycosylated, monoglycosylated, and unglycosylated) (Figure 1). In the large majority of CJD cases, PrPres glycosylation is characterized by an overrepresentation of the monoglycosylated form [115, 130]. A rather grossly major distinction with diagnostic relevance has been introduced to distinguish the above-described “pattern A” from “pattern B” characterized by a predominance of the fully glycosylated form, the latter being found in vCJD [114, 116] or in gCJD and FFI linked to the E200K or D178N mutations, respectively [129, 139]. However, finer significant differences in PrPres glycoform ratio have also been described among CJD subtypes with either “pattern A” or “pattern B” using either mono- or two-dimensional gel electrophoresis [130, 140].
Besides the strain typing approaches based on the analysis of the PrPres fragments generated by PK cleavage and glycoform ratio, other approaches have focused on PrPSc detergent solubility and aggregate size, degree of protease-resistance, and conformational stability [141–143]. Kobayashi et al. [142] studied PrPSc aggregation in MM 1 and MM 2T sCJD (sFI) cases and found that the former has a larger aggregation size than that of the latter, a result which they also confirmed in case with the cooccurrence of PrPSc types 1 and 2. More recently, Saverioni et al. [111] have analyzed PrPSc protease resistance and aggregate size across the whole spectrum of human prions (all sCJD subtypes, sporadic FI (sFI), vCJD, and VPSPr) and found that the strain-specific PrPSc sensitivity varies over a 100-fold range of PK concentration and that these differences stem from both PrPSc aggregate stability and size.
Preliminary data on the conformational stability of PrPSc in CJD subtypes have also become available. Conformational stability assay (CSA), which measures the progressive loss of PrPSc PK-resistance after exposure to increasing concentration of GndHCl, showed that sCJDMM1 PrPSc is more stable than sCJDMM 2C PrPSc [143]. The same result was obtained with the conformation stability and solubility assay (CSSA), which measures the increase in solubility of PrPSc after exposure to increasing concentrations of GndHCl. [144]. Finally, both sCJDMM1 and VV2 PrPSc showed a higher stability than vCJD PrPSc in the conformation dependent immunoassay (CDI), which evaluates the increase in epitopes exposure after GndHCl denaturation [145].
According to Kim et al. [109] sPrPSc concentration and stability is in close correlation with the disease progression rate. This, in turn, would reflect the association between the strain-specific amount and stability of sPrPSc conformers and the efficiency in initiating the replication process in vitro [110].
Preliminary data obtained in three sCJD variants seem to suggest that both levels and stability of sPrPSc are good predictors of the progression rate in sCJD and that small oligomers of protease-sensitive conformers of PrPSc may govern conversion potency. In particular, when sPrPSc is less stable than rPrPSc, as in sCJDMM1 and VV2, the difference in stability would correlate with less accumulated sPrPSc and a shorter duration of the disease, whereas when sPrPSc conformers are more stable than rPrPSc, as in sCJD MM2, it would correlate with more accumulated sPrPSc and a longer disease duration [110]. sPrPSc oligomers, smaller in size than rPrPSc polymers, may be the most powerful in triggering in vitro amplification due to an increased surface availability for recruiting PrPC molecules for conversion. So, the strain in which these sPrPSc conformers are most abundant would be the most efficient in amplification assays. In this regard, it is noteworthy that PMCA requires a sonication phase aiming to reduce the aggregation size of the seed. Although stimulating and sound with the current view of the biology of prions, the scenario depicted above must be taken with caution and definitely awaits confirmation by further investigations.
In addition to classical CJD variants and FI, human prion diseases include GSS and the recently described VPSPr. GSS is a familial disease which has been linked to missense, stop-codon, or insertional mutations in PRNP. The clinical phenotype in GSS is most commonly characterized by a progressive cerebellar syndrome, accompanied by extrapyramidal and pyramidal signs and cognitive decline, which may evolve into severe dementia [124]. However, a clinical variability, with either cognitive decline anticipating ataxia and rigidity or spastic paraplegia as a presenting symptom, has been observed. Neuropathological features associated with GSS disease vary substantially but always include PrP-positive multicentric amyloid plaques in the cerebellum and the cerebral cortex with or without associated spongiform change. Pioneering studies in GSS showed that purified amyloid preparations and the PrPres obtained by in vitro proteolysis mainly comprise atypical unglycosylated 7-8 kDa PrP fragments with ragged N and C termini, primarily composed of mutant PrP, which are lacking in classic TSEs such as CJD and FI (Figure 1) [102, 146–151]. In keeping with the significant phenotypic heterogeneity of the disease, however, it was also shown that the western blot profile of PrPres in GSS may comprise additional PrPres fragments of higher molecular weight, including the CJD-associated PrPres type 1 (Figure 1) [102, 146]. More specifically, GSS affected subjects carrying the most common GSS mutation (P102L) may either show a rapidly progressive CJD-like phenotype with both spongiform changes and amyloid plaques correlating with the cooccurrence PrPSc type 1 and the 8 kDa fragments or show a more slowly progressive “pure” GSS phenotype correlating with the presence of amyloid plaques and the 8 kDa PrP fragment [102, 146]. Finally, GSS associated PrPSc has also been reported to be unusually protease sensitive, at least in a subgroup of cases [102, 105, 106, 152]. Interestingly, when compared with CJD PrPSc, this increased proteolytic sensitivity of PrPSc does not correlate with a distinct aggregate sedimentation profile, suggesting that it is not due to a lower size of aggregates but rather to differences in their conformation [105, 106].
VPSPr is a recently described atypical variant of sporadic human prion disease, clinically characterized by language deficits, cognitive impairment, motor signs, especially Parkinsonism and ataxia, and an average longer clinical course than CJD [104, 153–156]. The disease can apparently affect all 3 codon 129 genotypes, although this genetic variability affects both susceptibility and phenotypic expression [104]. Pathologically, VPSPr is characterized by the spongiform change, which is especially seen in neocortical and subcortical regions of the cerebrum, such as the striatum and thalamus, and PrP-positive amyloid microplaques in the cerebellar molecular layers [104, 153].
Despite the clear differences in the clinicopathological phenotype between VPSPr and GSS, the characterization of PrPSc physicochemical properties has highlighted strong similarities which have led to the hypothesis that the former may represent the sporadic variant of the latter [153]. Indeed, PrPres in VPSPr shows a striking, ladder-like, electrophoretic profile comprising at least 4 bands, including a prominent one migrating at about 8 kDa (Figure 1). Furthermore, the abnormal PrP shows a variable degree of PK-resistance according to the codon 129 genotype; it is highly protease-sensitive in subjects with VV, whereas it shows a degree of resistance comparable to some sCJD types in subjects MV or MM at codon 129 [104, 111]. A very recent study also demonstrated that VPSPr shares PrPSc features with a known familial CJD linked to a valine to isoleucine mutation at residue 180 of PrP (fCJDV180I), exhibiting similar patterns of glycosylation and protease cleavage [157].
3.2. Transmission Studies with Human Prions
The first characterization of the transmissible, strain-related properties of human sporadic prion isolates was accomplished in transgenic mice. Inocula from a single sFI (i.e., MM 2T) case produced disease characteristics that differed from those induced by sCJD MM1 as well as from genetic CJD cases carrying the E200K-129M or the V210I-129M haplotypes [158]. Preliminary data concerning the transmission properties of other sCJD subtypes became available a few years later [159, 160], but only recently the reevaluation of the National Institutes of Health series of prion disease transmitted to non-human primates [131, 161] and more comprehensive experimental transmissions to transgenic mice [153, 154] have substantially clarified the issue of the extent of strain variation in sporadic human prion disease and provided answers to the crucial question of how the current classification relates to different strains of sCJD. The results of these studies indicate that, besides the MM 2T variant already mentioned above, four out of five of the other neuropathologic and molecular “pure” types of sCJD defined by the classification of Parchi et al. [130, 133] behave indeed as different strains of agent. Most importantly, sCJD MM1 and MV1 isolates have identical transmission properties, which significantly differ from those of sCJD VV2 or MV 2K. Furthermore, both the sCJD MM 2C and sCJD VV1 subtypes behave differently from each other and from the other isolates after transmission [162]. However, at variance with the sCJD MM1/MV1 and VV2/MV 2K strains, only single cases of sCJD MM 2C, MM 2T, and VV1 have been examined, with the assumption that transmission characteristics of a single case will be representative of the particular subgroup. Thus, the results obtained for these rare subtypes, although clear and somehow expected, await confirmation [131, 162, 163]. Familial and acquired forms (except for vCJD; see below) are likely linked to the same pool of strains isolated from sCJD. For example, inocula from carriers of E200K and V210I mutations affected by the MM1 CJD phenotype showed the same transmission properties of sCJD MM1 inocula when propagated in Tg mice, non-human primates, or bank voles [92, 131, 160]; similarly, experimentally transmitted kuru reproduced the same clinico-pathological and biochemical features of VV2 and MV 2K sCJD [131]. Finally, similar properties have been observed by FFI and sFI prions when propagated into Tg mice [34, 158]. In contrast to prions propagated in classical CJD and kuru, the transmission properties of vCJD prions are strikingly distinct and have established vCJD as a distinct human prion strain [164, 165]. The vCJD prions transmit disease to wild-type mice far more efficiently than any other form of human prion disease [164–166] and in transgenic mice faithful propagation of the vCJD phenotype is dependent upon homozygous expression of human PrP 129 methionine [165, 167–170]. Transgenic mice homozygous for human PrP 129 valine show a pronounced transmission barrier to vCJD prions and propagate a distinct clinical-pathological phenotype [165, 167–169, 171]. As a consequence, the possibility that the BSE-vCJD strain may be associated with other human pathological phenotypes besides that observed in subjects carrying MM at codon 129 should not be dismissed.
With the significant exception of the GSS P102L associated with spongiform changes and PrPSc type 1, which shows CJD-like transmission properties, GSS variants have been more difficult to transmit to animals than CJD or FFI [161, 172]. This has led to the suggestion these GSS phenotypes are not true prion diseases (e.g., TSEs) and are better designated as nontransmissible proteinopathies. In more recent studies, however, the use of transgenic mice carrying GSS mutations such as A117V or the mouse equivalent of P102L has led to the finding that brain tissue from GSS patients carrying the corresponding mutation could induce a pathological phenotype into these mice, although with some significant differences between the two models [173, 174]. More specifically, in the first, the inoculation of brain extracts from a GSSP102L patient with no spongiform change caused almost no clinical disease but induced striking PrP-amyloid deposition in brains of several recipient mice; extracts of those brains failed to transmit neurological disease on further passage but again induced PrP-amyloid plaques in recipient mice [173]. In the second study, instead, the transmission of a more typical TSE phenotype, including the deposition of classic protease-resistant PrPSc 27–30, has been obtained in 117VV HuPrP transgenic mice challenged with A117V prion isolates [174]. Thus, especially according to this latter result, GSS may also be considered a true prion disease, although much less prone than CJD to transmit, possibly because it is characterized by the formation of less stable PrPSc aggregates.
3.3. PrPSc Properties and Strain Variation in Natural Hosts: Scrapie, BSE, and CWD
3.3.1. Scrapie
Biochemical typing of natural scrapie isolates has been largely based on the assessment of PrPres electrophoretic mobility, glycoform ratio, and epitope mapping of PK-cleavage sites using different monoclonal antibodies. The use of other approaches such the analyses of PrPres protease-resistance and conformational stability of PrPres has been, so far, limited.
Despite the known diversity of classical scrapie strains that have been isolated in wild-type mice [83] or hamster [175, 176], the identification of strain-specifc PrPres signatures in sheep with natural scrapie has proved to be challenging [177–182]. Indeed, the molecular signature of most isolates of classical scrapie comprises an unglycosylated PrPres with a “high” (h-type) molecular mass (i.e., in the range of human PrPres type 1 and including the epitope recognized by the N-terminal P4 antibody), whereas only a few cases show a PrPres profile with a “low” (l-type) electrophoretic mobility (i.e., in the range of human PrPres type 2 and not labeled by P4), similar to that seen in BSE or experimental scrapie strain CH1641 (Figure 2). Similarly, PrPres glycoform ratios did not clearly differ from those found in cattle-BSE and did not reveal distinct subgroups of classical scrapie [180–185] (Figure 2). Some evidence for a strain-related heterogeneity of PrPSc associated with classical natural scrapie isolates derives from CDI analysis. It has been shown that PrPSc extracted from sheep with the VRQ/VRQ PRNP genotype has higher levels of PK-sensitive PrPSc than the PrPSc associated with ARQ/ARQ [108]. Furthermore, the two isolates propagated in mice are associated with two PrPSc with distinct conformational stability, with the PrPSc-VRQ inocula being more sensitive to denaturation than the other [186].
Figure 2.
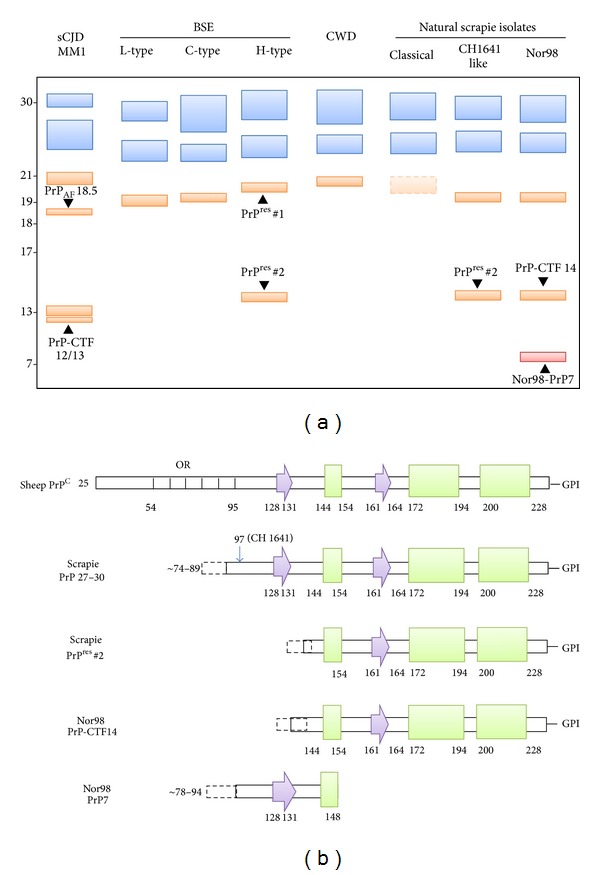
(a) Schematic representation of the spectrum of PrPres fragments observed in animal prion diseases and their electrophoretic profile. The unglycosylated forms of all PrPres fragments with the glycosylation sites in their sequence are indicated in orange, while the fragments lacking these sites are shown in red. Among the glycosylated peptides, only the mono- and the diglycosylated forms of PrPres 27–30 (18–21 kDa range) fragments are shown (in blue). To facilitate the comparison with human forms, the profile of MM1 sCJD associated PrPres is shown; note that the unglycosylated band of sCJDMM1 PrPres has the same electrophoretic mobility of that of CWD as reported by Xie et al. [14]. (b) Diagrams of the secondary structural elements of sheep PrPC and of the PK-resistant PrP fragments observed in classical and atypical Nor98 scrapie. Arrows are representative of β-strands and rectangles of α-helices and OR indicates the octapeptide repeats region. The secondary structure numbering has been derived from pdb (Protein Data Bank) id 1XYU (sheep PrP).
The unusual scrapie isolates with a l-type PrPres profile, designated as CH1641-like, have for some time posed a diagnostic challenge because of the similarities with the PrP molecular properties of experimentally transmitted BSE to sheep. Immunoblot assays have shown that they share a migration pattern similar to the unglycosylated PrPres fragment but have different levels of diglycosylated PrPres [178] (Figure 2). More recently, however, it has been found that the PrPres associated with the CH1641-like isolate clearly differs from BSE-PrPres by the presence of an additional band at approximately 14 kDa, which is specifically recognized by the C-terminal antibody SAF84 (Figure 2) [187]. This additional PrPres fragment was also observed after transmission in a transgenic mouse model (TgOvPrP4) of both the natural CH1641-like isolate [188, 189] and the CH1641 experimental scrapie isolate that was originally isolated from a British scrapie case and maintained by serial transmissions in sheep [190]. Unlike CH1641 this PrPres fragment was not detected in the scrapie strains with h-type PrPres (C506M3, Chandler, and 79A), arguing that PrPres 14 kDa preferentially associates with l-type PrPres [189]. Intriguingly, both l-type and h-type PrPres were detected in the brain of TgOvPrP4 infected with some scrapie isolates [187], which strongly suggests that the two phenotypes found in mice could be the result of the cooccurrence of two strains in these sheep. Indeed the possible existence of a mixture of strains from a single scrapie case, which can only be separated by biological cloning, has been documented following bioassay in mice or hamsters [191, 192].
In 2003 an atypical scrapie strain (Nor98) was described in five sheep from Norway [193]. Scrapie cases similar to Nor98 were later detected in other European countries [194–196] and in the United States [197]. Western blotting analysis of Nor98-affected brain extracts has allowed the identification of a peculiar PrPres electrophoretic profile consisting of multiple protein bands including a prominent band of relatively low molecular mass that was initially reported to migrate around 12 kDa [186].
In particular, the use of different mAbs raised against epitopes located in the middle and in the C-terminal regions of PrP has allowed the identification of two previously unrecognized fragments, respectively, designated as Nor98-PrP7 and PrP-CTF14 (Figure 2). Nor98-PrP7 is a PK resistant N- and C-terminally truncated fragment with a molecular weight of 7 kDa which is not affected by PNGase F treatment, while PrP-CTF14 is a C-terminal fragment migrating at 14 kDa after deglycosylation. Interestingly, both fragments showed an increased protease sensitivity when compared to PrPSc in classical scrapie, suggesting that the PrPSc associated with the two diseases have a different conformation [198].
The intracerebral inoculation of a panel of atypical/Nor98 scrapie isolates into mice overexpressing the ovine prion protein (Tg338) suggests that a single prion strain is responsible for atypical scrapie [199]. Using a set of PrP-specific monoclonal antibodies two distinct C- and N-terminally ragged PK-resistant PrPres fragments of approximately 8 kDa and 5 kDa which are differently truncated at their C-termini were detected, thus confirming the complexity and the specificity of the molecular PrPres phenotype of these atypical scrapie isolates [199] and its similarities with some human TSE variants such as GSS-P102L and VPSPr (Figures 1 and 2) [200].
3.3.2. Bovine Spongiform Encephalopathy (BSE)
On the basis of the electrophoretic profiles of the unglycosylated band of PrPres, three different BSE phenotypes are currently recognized: the classical BSE (C-type) and two atypical BSE variants showing, respectively, a lower (L-type) and a higher (H-Type) relative molecular mass of PrPres in comparison to the C-type [113, 181, 201, 202] (Figure 2(a)).
Early evidence suggested that BSE was caused by a prion strain characterized by an efficient ability to overcome the species barrier and with a PrPres signature featuring a lower relative molecular mass compared to the PrPres associated with classic scrapie (and CWD) and a marked predominance of the high molecular weight glycoform [164, 203].
In 2004, however, a distinct phenotype of bovine amyloidotic spongiform encephalopathy (BASE or L-type) [113], correlating with a PrPres showing a slightly lower electrophoretic mobility than the PrPres of the C-type and a predominant monoglycosylated isoform, was found [201]. The evidence that BSE and BASE are caused by two distinct prion strains is supported by transmission experiments showing that the inoculation of BSE or BASE brain homogenates in transgenic mice (Tgbov XV) causes two distinct phenotypes [204]. Noteworthy, BASE was also shown to convert into the classical BSE strain upon serial transmission to inbred mouse lines, which has raised the hypothesis that BSE originated from BASE [205].
Intriguingly also the H-type BSE, first described by Biacabe et al. in 2004 [201], can recapitulate most of the phenotypic features of classical BSE after cross-species transmission experiments in wild-type mice [206]. Compared with the C-type, the H-type strain is characterized by an extended N-terminus of PrPres and by the presence of two distinct PrPres cleavage products, PrPres#1 (19–30 kDa), showing a slightly higher electrophoretic mobility than the PrPres of the C-type, and PrPres#2 (14–24 kDa), characterized by a more C-terminal cleavage [207] (Figure 2(a)). This typical H-type PrPres banding pattern was also described in a BSE case associated with a PRNP mutation (E211K) [208].
3.3.3. Chronic Wasting Disease (CWD)
CWD, like scrapie, is a prion disease mainly transmitted via an environmental route [209]. Although the horizontal transmission of CWD among cervids by direct or indirect contacts is remarkably efficient, its transmission to different species has yet to be fully clarified [210–212].
The PrPres electrophoretic profiles of CWD-affected animals and of sCJDMM1 have led to the observations that they share some similarities as shown by the conformational stability assay and by the observation that in both samples the unglycosylated PK-resistant isoform migrates at 21 kDa, thus indicating a similar conformation of the PK resistant cores. However, the two PrPres do not display a similar glycoform profile with a prevalence of the diglycosylated isoform in the CWD PrPres, as observed in BSE and in vCJD [14] (Figure 2(a)). The same electrophoretic and glycoform profiles were also observed in two different CWD strains (CWD1 and CWD2) which were identified after the inoculation of different CWD isolates in Tg mice expressing cervid PrP (Tg (CerPrP)1536+/−) [213].
Interestingly the PrPres immunoblot analysis of white-tailed deers orally inoculated with the CWD agent revealed that in Q95H/G96S animals the unglycosylated fragment migrates at lower molecular weight and the level of PK-resistance seems to be reduced, suggesting the generation of a PrPres with different properties which the PrPres generated in the other infected cervids [214].
4. Role of PrPSc in Prion Toxicity and Neurodegeneration
The understanding of the mechanisms of toxicity resulting from misfolding and ordered aggregation of proteins involved in prion disease and many others neurodegenerative diseases remains an open question and a research priority. Indeed, in none of these diseases are the mechanisms of toxicity completely clear. While a large body of evidence indicates that the misfolded protein aggregates are the cause of the neurodegeneration, many studies link this toxicity to the existence of various intermediate structures, likely in the oligomeric state, prior to the fiber formation and/or their specific interaction with membranes [215, 216]. Indeed, in prion diseases it is well established that, in the absence of GPI-linked PrPC, PrPSc is innocuous, suggesting that PrP oligomers and fibrils are not toxic per se [217], and that PrPC may act as mediator of the toxic signal. Furthermore, the importance of certain physicochemical properties of the protein fragments forming the aggregate, such as size and glycosylation state, has also been highlighted by studies in prion disease, which uniquely comprise a wide range of disease phenotypes allowing for extensive molecular and clinicopathological correlations [125].
4.1. Insights from Studies on Naturally Occurring and Experimentally Transmitted Prion Diseases
From the study of affected brains we have learned that the events that are triggered by prion neuroinvasion and that result in neurodegeneration may vary significantly both in terms of resulting histopathology and speed of the neurodegenerative process. In humans the clinical course of a prion disease may range from a few weeks to at least one decade, and evidence from experimental transmissions and acquired prion diseases indicate that a similar heterogeneity likely characterize also the preclinical phase.
Histopathologically, while most prion diseases, including CJD, BSE, CWD, and most of scrapie cases (i.e., the classic transmissible spongiform encephalopathies or TSEs), are characterized by the triad of spongiform change, gliosis, and neuronal loss, some rare but very informative variants such as FI, GSS, or PrP-CAA may show very subtle or even absent spongiform change or be characterized by prominent extracellular amyloid plaques accumulating either in the neuropil or around blood vessels. Most significantly, in contrast to CJD, in which the abnormal PrPSc aggregates mainly consist of full-length protein together with GPI-anchored, N-terminal fragments truncated between residue 82 and residue 104 [129], in GSS or PrP-CAA affected patients the abnormal PrP plaque amyloid that accumulates is composed primarily of truncated internal PrP fragments (e.g., residues 82–153) that lack the GPI anchor and the glycosylated moiety [102, 146, 152]. In this respect, GSS patients carrying the P102L mutation can be considered a “quasinatural” experimental model. Indeed, while in some of these patients pure GSS histopathological features correlate with the presence of the GPI-anchorless PrP fragment, in others mixed CJD/GSS features (e.g., widespread spongiform changes cooccurring with amyloid plaques) correlate with the deposition of both types of PrPSc forms (e.g., GPI-anchored and glycosylated N-terminal PrPSc fragment + truncated internal PrP fragments lacking the GPI anchor). These observations strongly support the idea that PrP fragments have different neurotoxicities and cause distinct lesions as a consequence of their different properties, such as aggregation propensity [102]. In particular, the longer duration of illness in GSS patients can be explained postulating that the short GPI-anchorless PrPres fragments have a higher tendency toward aggregation and plaque formation and thus provide a relative protection with less neuronal dysfunction than the 21- or 19-kDa PrPres glycosylated fragments or full length PrPSc associated with CJD that form more diffuse and smaller deposits. Consistent with this hypothesis is also the observation that, among the GSS P102L patients, those showing the mixed CJD/GSS phenotype have, on average, a significantly shorter course [102].
More recently, the evidence obtained from studies on CJD and GSS patients has received strong support from a transgenic mouse model expressing anchorless PrP [218]. In these Tg44 mice scrapie infection results in an unusual type of slow fatal prion brain disease distinguished by widespread deposition of PrPSc amyloid in the CNS [219] and in extraneural sites such as heart, brown fat, white fat, and colon [220, 221]. In the CNS of infected Tg44 mice the gray matter vacuolation typical of prion diseases is minimal, and PrPSc is primarily deposited as perivascular amyloid [219]. In this model, most of the typical clinical and neuropathological characteristics of scrapie are either absent or greatly reduced, despite the accumulation of brain PrPSc to levels comparable to those in scrapie-infected wild-type mice. This reduced brain damage could be due either to a need for anchored PrPC on brain cells for toxicity induced by PrPSc and/or to a lower pathogenicity of PrPSc amyloid plaques compared to the more dispersed, amorphous, and membrane-associated PrPSc deposits seen in most other prion diseases. These findings highlight the role of GPI anchor in TSE pathogenesis [222]. It is likely that the anchoring of PrPSc aggregates to membranes by the GPIs could distort its local structure, composition, flexibility, fluidity, dynamics, integrity, and, hence, functionality. The results of several elegant EM studies corroborate these observations by showing that in all the naturally occurring TSEs of animals, as well as in experimental scrapie models of mice, there are a number of distinctive membrane changes, including membrane microfolding, membrane clefts, and abnormal endocytosis of dendrites, which are both directly linked to PrPSc and appear to be unique to prion diseases [223, 224]. These changes, however, were absent from Tg mice expressing only anchorless PrP and other Tg mice developing large amyloid plaques composed of abnormal prion protein [225].
While a definite progress has been made in understanding the divergent molecular pathology between classic TSEs and the “anchorless” PrP-amyloidosis, much less is known about the molecular basis of the different “neurotoxicity” associated with the various prion strains. Indeed, differences in the molecular and cellular pathology that correlate with the severity of the clinical phenotype have also been observed among classic TSEs such as sCJD. Subjects affected by the most common sCJD variant (e.g., the MM1 subtype), for example, do not accumulate higher amounts of PrPSc or develop more severe histopathological changes than the other sCJD variants despite their very rapid clinical course, sometimes lasting less than a month [115, 226]. Similarly, in a recent study in which we have correlated the amount of PrPSc deposition with the extent of microglial activation across the whole spectrum of sCJD subtypes, including the MM 2T or FI, we found that the degree of microglial activation differs significantly between disease subtypes and, above all, it does not correlate with the overall amount of PrPSc accumulation (Strammiello R and Parchi P, unpublished). Intriguingly, the most significant difference in the ratio between PrPSc amount and HLA-DR load was seen between two subtypes, the MM 2C and the MM 2T, sharing the average disease duration, codon 129 MM genotype, and PrPSc type 2. Overall, these data add to previous observations indicating that many critical properties of prions, including neurotoxicity, appear unrelated to the overall amount of PrPSc deposition. Furthermore, they indicate that there are strain-related differences in the apparent “neurotoxicity” associated with PrPSc deposition that must be addressed.
Another intriguing and largely unexplained issue of prion pathology concerns the regional specificity. In this respect, the study of FI, which is by far the most peculiar disease phenotype among those characterized by a “classic” PrPSc 27–30 deposition, has been very informative. The histopathological hallmark of FI, especially of the familial form linked to the D178N-129M PRNP haplotype, is a severe neuronal loss in the medial thalamic and inferior olivary nuclei [126]. These changes develop early since they are found in all affected subjects, irrespectively of the disease duration; furthermore they are found associated with amounts of PrPSc, which are at least tenfold lower than those detected in other sCJD subtypes where the neuronal loss in the thalamus is rarely so severe. In contrast, in the neocortex and, to a lesser extent, in the limbic cortex and the striatum of FFI patients, the amount of PrPSc increases with the duration of symptoms and eventually accumulates in significantly higher amounts than in the thalamus [227, 228]. Furthermore, the higher extent of PrPSc deposition correlates with the appearance of spongiform changes rather than with the degree of neuronal loss which remains milder than in the thalamus.
In conclusion, significant differences in the “neurotoxicity” associated with PrPSc deposition are also seen among classic TSE subtypes. However, in contrast with GSS, no significant data have been collected to explain how PrPSc may mediate these heterogeneous effects.
4.2. Insights from Studies on Animal Models
The first studies documenting the progression of neurodegeneration in prion disease dates back half a century and preceded the discovery of the prion protein. At that time experimental transmissions in primate and murine models already established that the appearance of spongiform change precedes neuronal loss and reactive astrogliosis [229]. It was later found that PrPSc deposition almost invariably represents the earliest event of the pathological cascade, which is immediately followed by microglial activation and the appearance of spongiform change. It was also found that the conversion of PrPC into PrPSc is critical to the neurotoxicity associated with prion diseases since neither loss of PrPC function nor deposition of PrPSc in absence of PrPC expression is sufficient to cause the prion-associated pathology [230, 231]. Having established the central role of both PrPSc and PrPC in prion pathogenesis, the critical issue has progressively become the search for a link between PrPSc, neurotoxicity, and infectivity. Although the temporal and anatomical correlation between PrPSc formation and the development of infectivity and neuropathological changes is often obvious in prion disease, the overall correlation between PrPSc levels, infectivity, and neurotoxicity can be weak or even absent. For example, transgenic mice expressing some mutant forms of PrPC that lack certain domains spontaneously develop neurological disorders, but no infectivity and bona-fide PrPSc are associated with prion protein aggregates accumulated in brain tissue of these animals [27, 232]. On the other hand, mice expressing GPI-anchorless prion protein show high levels of infectious PrP aggregate deposits, but reduced neurodegeneration compared to prion-infected wild-type mice [218]. Finally, there are subclinical infections in which there is abundant PrPSc but little symptomatology, for example, after inoculation of hamster prions into mice [233, 234]. Thus, it appears that infectious and neurotoxic forms of PrP could represent distinct molecular species, a view which is also supported by a recent study showing that prion propagation in brain proceeds via two distinct phases. More specifically, it has been shown that a clinically silent exponential phase, which rapidly reaches a maximal prion titre and is independent by PrPC expression, is followed by a plateau phase, which determines time to clinical onset in a manner inversely proportional to prion protein concentration [235]. Notably, however, the same data would also fit the model of PrPC-mediated PrPSc toxicity (see below), without requiring the existence of a toxic PrP as a distinct entity [236].
Despite this largely unsolved complexity, as for other protein aggregation diseases, PrPSc oligomers currently attract most of attention and appear to be the preferred researcher's candidate to explain both prion toxicity and infectivity. However, while there appears to be little doubt that infectious prion particles consist of small PrP oligomers, it is much less clear whether oligomers, and if so which oligomers, are involved in prion toxicity. As far as the mechanism of mediated toxicity is concerned, current evidence supports the view that small oligomers formed on membrane-bound GPI-PrPC may act by compromising the integrity of cellular membranes or, more likely, by mediating a neurotoxic signal triggered from the extracellular milieu by PrPSc. Alternatively, PrPC may disrupt the endosomal compartment after being internalized [237]. Lines of evidence suggesting that PrPSc neurotoxicity may involve impairment of the normal physiological activity of PrPC have also been gathered, especially from the study of mutant forms of PrP that produce spontaneous neurodegeneration in transgenic mice without the formation of infectious PrPSc (reviewed in [238]). For example, Tg (PrPΔ32–134) mice, which express an N-terminally truncated form of PrP, spontaneously develop a neurodegenerative phenotype that is stoichiometrically reversed by coexpression of wild-type PrP, but only partially rescued by coexpression of a PrPC isoform carrying an insert mutation. The rescuing effect of wild-type PrP would implicate a molecular target for PrP, which is presumably a receptor or another cell-surface complex capable of transducing a signal to the interior of the cell. Based on these evidences, Harris and collaborators [239] have proposed that PrPSc (or other toxic forms of PrP), by interacting with the same putative membrane target, may subvert a normal function of PrPC to generate a neurotoxic signal. Although of significant interest, the proposed mechanism is in apparent contrast with the dominant mode of inheritance of familial prion diseases. Furthermore, the connection between the neurotoxic mechanisms activated by artificial mutants and those operative in “natural” prion diseases of humans and animals remain to be demonstrated. Whatever the nature and the mechanism of action of the toxic molecular species, there is a growing body of data to show that it is the synapses that are the first or most susceptible component of the neuron to succumb in the disease process rather than the death of the cell soma. Compromised synaptic function is currently thought to underlie the earliest symptoms in several neurodegenerative diseases, and loss of synapses, spines, and dendrites is thought to precede the loss of neuronal cell bodies [240–243]. Using an engineered mouse model Mallucci and collaborators [244] have shown that the block of PrPSc formation by knocking out PrPC in prion-infected mice during the course of disease prevented neuronal loss and progression to clinical disease. PrP knockout produced both long-term survival and neuroprotection and the disappearance of early spongiform change, thus indicating that spongiosis is a predegenerative change occurring in neurons which may represent an early morphological marker of functional impairment [244].
Using the same model, this group of researchers has recently demonstrated that the decline of synapse number and transmission is associated with an abrupt loss of synaptic proteins [245]. PrP replication and the consequent rise of PrP levels during disease would cause a sustained induction of the cellular unfolded protein response (UPR). Rising levels of unfolded proteins in the ER would cause the phosphorylation of PERK-P, followed by that of eIF2a, which ultimately causes a reduction of new protein synthesis. The resulting chronic blockade of protein synthesis would lead to synaptic failure, spongiform changes, and, ultimately, neuronal loss. Based on these findings, it has been proposed that the key trigger to prion neurodegeneration is the continued, unchecked activation of the UPR due to the rising levels of PrP during disease, with fatal repression of translation rates.
4.3. The Role of Microglia in PrPSc Clearing and Prion Disease-Associated Neurodegeneration
A major theme in studies of the role of microglia in neuropathology is the dichotomy between their contributions to neurodegeneration versus neuroprotection. Prion diseases are not an exception to this theme. Lines of evidence indicate that PrPSc can be efficiently cleared from the brain and that phagocytosis by microglia represents a prominent clearing mechanism [246, 247]. On the other hand, it has also been shown that activated microglia may assume an aggressive phenotype and release inflammatory cytokine fostering neuronal apoptosis and neurodegeneration [248].
Recent studies have contributed to shed some light into the molecular events regulating microglial activation during prion infection. In murine prion disease, the microglia was shown to activate early in the disease process, even in the absence of widespread histologically detectable PrPSc deposits [249]. This activated phenotype, which has been referred to as anti-inflammatory or benign, shows low levels of inflammatory cytokines and readily detectable levels of TGF-β and PGE2 [241, 250]. While there is no evidence that the enhanced levels of PGE2 are detrimental, nor that TGF-β is injurious, this situation may significantly worsen in the presence of systemic inflammation. Indeed, when mice were challenged systemically with endotoxin to mimic an intercurrent infection, this maneuver led to a dramatic switch in the microglial phenotype with an aggressive inflammatory cytokine profile and increased neuronal apoptosis [248]. This concept of rapid switching of the microglia phenotype is of course entirely in keeping with what is known about the degree of plasticity of the cells of the macrophage lineage. Systemic inflammation has a profound impact on a number of other animal models of neurological disease [251] and accelerates cognitive decline in Alzheimer's patients [252].
Acknowledgments
This research is supported by the Ministry of Health (Grant RF-2009-1474624), the University of Bologna (Grants RFO 2010), and the Gino Galletti Foundation.
References
- 1.Caughey B, Raymond GJ, Bessen RA. Strain-dependent differences in β-sheet conformations of abnormal prion protein. Journal of Biological Chemistry. 1999;273(48):32230–32235. doi: 10.1074/jbc.273.48.32230. [DOI] [PubMed] [Google Scholar]
- 2.Caughey BW. Secondary structure analysis of the scrapie-associated protein PrP 27–30 in water by infrared spectroscopy. Biochemistry. 1991;30(31):7672–7680. doi: 10.1021/bi00245a003. [DOI] [PubMed] [Google Scholar]
- 3.Govaerts C, Wille H, Prusiner SB, Cohen FE. Evidence for assembly of prions with left-handed β-helices into trimers. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(22):8342–8347. doi: 10.1073/pnas.0402254101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKinley MP, Meyer k. R, Kenaga L, et al. Scrapie prion rod formation in vitro requires both detergent extraction and limited proteolysis. Journal of Virology. 1991;65(3):1340–1351. doi: 10.1128/jvi.65.3.1340-1351.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nguyen JT, Inouye H, Baldwin MA, et al. X-ray diffraction of scrapie prion rods and PrP peptides. Journal of Molecular Biology. 1995;252(4):412–422. doi: 10.1006/jmbi.1995.0507. [DOI] [PubMed] [Google Scholar]
- 6.Peretz D, Williamson RA, Matsunaga Y, et al. A conformational transition at the N terminus of the prion protein features in formation of the scrapie isoform. Journal of Molecular Biology. 1997;273(3):614–622. doi: 10.1006/jmbi.1997.1328. [DOI] [PubMed] [Google Scholar]
- 7.Wille H, Bian W, McDonald M, et al. Natural and synthetic prion structure from X-ray fiber diffraction. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(40):16990–16995. doi: 10.1073/pnas.0909006106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Williamson RA, Peretz D, Pinilla C, et al. Mapping the prion protein using recombinant antibodies. Journal of Virology. 1998;72(11):9413–9418. doi: 10.1128/jvi.72.11.9413-9418.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zou W, Colucci M, Gambetti P, Chen SG. Characterization of prion proteins. Methods in Molecular Biology. 2003;217:305–314. doi: 10.1385/1-59259-330-5:305. [DOI] [PubMed] [Google Scholar]
- 10.Riek R, Wider G, Billeter M, Hornemann S, Glockshuber R, Wüthrich K. Prion protein NMR structure and familial human spongiform encephalopathies. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(20):11667–11672. doi: 10.1073/pnas.95.20.11667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pan K-M, Baldwin M, Nguyen J, et al. Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(23):10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeMarco ML, Silveira J, Caughey B, Daggett V. Structural properties of prion protein protofibrils and fibrils: an experimental assessment of atomic models. Biochemistry. 2006;45(51):15573–15582. doi: 10.1021/bi0612723. [DOI] [PubMed] [Google Scholar]
- 13.Smirnovas V, Baron GS, Offerdahl DK, Raymond GJ, Caughey B, Surewicz WK. Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nature Structural and Molecular Biology. 2011;18(4):504–506. doi: 10.1038/nsmb.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie Z, O’Rourke KI, Dong Z, et al. Chronic wasting disease of elk and deer and Creutzfeldt-Jakob disease: comparative analysis of the scrapie prion protein. Journal of Biological Chemistry. 2006;281(7):4199–4206. doi: 10.1074/jbc.M509052200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Capellari S, Strammiello R, Saverioni D, Kretzschmar H, Parchi P. Genetic Creutzfeldt-Jakob disease and fatal familial insomnia: insights into phenotypic variability and disease pathogenesis. Acta Neuropathologica. 2011;121(1):21–37. doi: 10.1007/s00401-010-0760-4. [DOI] [PubMed] [Google Scholar]
- 16.van der Kamp MW, Daggett V. The consequences of pathogenic mutations to the human prion protein. Protein Engineering, Design and Selection. 2009;22(8):461–468. doi: 10.1093/protein/gzp039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y, Swietnicki W, Zagorski MG, Surewicz WK, Sönnichsen FD. Solution structure of the E200K variant of human prion protein. Implications for the mechanism of pathogenesis in familial prion diseases. Journal of Biological Chemistry. 2000;275(43):33650–33654. doi: 10.1074/jbc.C000483200. [DOI] [PubMed] [Google Scholar]
- 18.Swietnicki W, Petersen RB, Gambetti P, Surewicz WK. Familial mutations and the thermodynamic stability of the recombinant human prion protein. Journal of Biological Chemistry. 1998;273(47):31048–31052. doi: 10.1074/jbc.273.47.31048. [DOI] [PubMed] [Google Scholar]
- 19.Liemann S, Glockshuber R. Influence of amino acid substitutions related to inherited human prion diseases on the thermodynamic stability of the cellular prion protein. Biochemistry. 1999;38(11):3258–3267. doi: 10.1021/bi982714g. [DOI] [PubMed] [Google Scholar]
- 20.Apetri AC, Surewicz K, Surewicz WK. The effect of disease-associated mutations on the folding pathway of human prion protein. Journal of Biological Chemistry. 2004;279(17):18008–18014. doi: 10.1074/jbc.M313581200. [DOI] [PubMed] [Google Scholar]
- 21.Biljan I, Ilc G, Giachin G, et al. Toward the molecular basis of inherited prion diseases: NMR structure of the human prion protein with v210i mutation. Journal of Molecular Biology. 2011;412(4):660–673. doi: 10.1016/j.jmb.2011.07.067. [DOI] [PubMed] [Google Scholar]
- 22.Ilc G, Giachin G, Jaremko M, et al. NMR structure of the human prion protein with the pathological Q212P mutation reveals unique structural features. PLoS ONE. 2010;5(7) doi: 10.1371/journal.pone.0011715.e11715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee S, Antony L, Hartmann R, et al. Conformational diversity in prion protein variants influences intermolecular beta-sheet formation. EMBO Journal. 2010;29(1):251–262. doi: 10.1038/emboj.2009.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rossetti G, Giachin G, Legname G, Carloni P. Structural facets of disease-linked human prion protein mutants: a molecular dynamic study. Proteins. 2010;78(16):3270–3280. doi: 10.1002/prot.22834. [DOI] [PubMed] [Google Scholar]
- 25.Rossetti G, Cong X, Caliandro R, Legname G, Carloni P. Common structural traits across pathogenic mutants of the human prion protein and their implications for familial prion diseases. Journal of Molecular Biology. 2011;411(3):700–712. doi: 10.1016/j.jmb.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 26.Hsiao KK, Scott M, Foster D, Groth DF, DeArmond SJ, Prusiner SB. Spontaneous neurodegeneration in transgenic mice with mutant prion protein. Science. 1990;250(4987):1587–1590. doi: 10.1126/science.1980379. [DOI] [PubMed] [Google Scholar]
- 27.Nazor KE, Kuhn F, Seward T, et al. Immunodetection of disease-associated mutant PrP, which accelerates disease in GSS transgenic mice. EMBO Journal. 2005;24(13):2472–2480. doi: 10.1038/sj.emboj.7600717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manson JC, Jamieson E, Baybutt H, et al. A single amino acid alteration (101L) introduced into murine PrP dramatically alters incubation time of transmissible spongiform encephalopathy. EMBO Journal. 1999;18(23):6855–6864. doi: 10.1093/emboj/18.23.6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Asante EA, Gowland I, Grimshaw A, et al. Absence of spontaneous disease and comparative prion susceptibility of transgenic mice expressing mutant human prion proteins. Journal of General Virology. 2009;90(3):546–558. doi: 10.1099/vir.0.007930-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barron RM, Thomson V, Jamieson E, et al. Changing a single amino acid in the N-terminus of murine PrP alters TSE incubation time across three species barriers. EMBO Journal. 2001;20(18):5070–5078. doi: 10.1093/emboj/20.18.5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiesa R, Piccardo P, Ghetti B, Harris DA. Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron. 1998;21(6):1339–1351. doi: 10.1016/s0896-6273(00)80653-4. [DOI] [PubMed] [Google Scholar]
- 32.Dossena S, Imeri L, Mangieri M, et al. Mutant prion protein expression causes motor and memory deficits and abnormal sleep patterns in a transgenic mouse model. Neuron. 2008;60(4):598–609. doi: 10.1016/j.neuron.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 33.Hegde RS, Mastrianni JA, Scott MR, et al. A transmembrane form of the prion protein in neurodegenerative disease. Science. 1998;279(5352):827–834. doi: 10.1126/science.279.5352.827. [DOI] [PubMed] [Google Scholar]
- 34.Telling GC, Haga T, Torchia M, Tremblay P, DeArmond SJ, Prusiner SB. Interactions between wild-type and mutant prion proteins modulate neurodegeneration in transgenic mice. Genes and Development. 1996;10(14):1736–1750. doi: 10.1101/gad.10.14.1736. [DOI] [PubMed] [Google Scholar]
- 35.Sigurdson CJ, Nilsson KPR, Hornemann S, et al. De novo generation of a transmissible spongiform encephalopathy by mouse transgenesis. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(1):304–309. doi: 10.1073/pnas.0810680105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jackson WS, Borkowski AW, Faas H, et al. Spontaneous generation of prion infectivity in fatal familial insomnia knockin mice. Neuron. 2009;63(4):438–450. doi: 10.1016/j.neuron.2009.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jackson WS, Borkows AW, Watson NE, et al. Profoundly different prion diseases in knock-in mice carrying single PrP codon substitutions associated with human diseases. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(36):14759–14764. doi: 10.1073/pnas.1312006110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Friedman-Levi Y, Meiner Z, Canello T, et al. Fatal prion disease in a mouse model of genetic E200K Creutzfeldt-Jakob disease. PLoS Pathogens. 2011;7(11) doi: 10.1371/journal.ppat.1002350.e1002350 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39.Senatore A, Colleoni S, Verderio C, et al. Mutant PrP suppresses glutamatergic neurotransmission in cerebellar granule neurons by impairing membrane delivery of VGCC α2δ-1 subunit. Neuron. 2012;74(2):300–313. doi: 10.1016/j.neuron.2012.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Silva JL, Vieira TCRG, Gomes MPB, Rangel LP, Scapin SMN, Cordeiro Y. Experimental approaches to the interaction of the prion protein with nucleic acids and glycosaminoglycans: modulators of the pathogenic conversion. Methods. 2011;53(3):306–317. doi: 10.1016/j.ymeth.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 41.Ma J. The role of cofactors in prion propagation and infectivity. PLoS Pathogens. 2012;8(4) doi: 10.1371/journal.ppat.1002589.e1002589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kil SL, Caughey B. A simplified recipe for prions. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(23):9551–9552. doi: 10.1073/pnas.0703910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shaked GM, Meiner Z, Avraham I, Taraboulos A, Gabizon R. Reconstitution of prion infectivity from solubilized protease-resistant PrP and nonprotein components of prion rods. Journal of Biological Chemistry. 2001;276(17):14324–14328. doi: 10.1074/jbc.M007815200. [DOI] [PubMed] [Google Scholar]
- 44.Wong C, Xiong L-W, Horiuchi M, et al. Sulfated glycans and elevated temperature stimulate PrPSc-dependent cell-free formation of protease-resistant prion protein. EMBO Journal. 2001;20(3):377–386. doi: 10.1093/emboj/20.3.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yokoyama T, Takeuchi A, Yamamoto M, Kitamoto T, Ironside JW, Morita M. Heparin enhances the cell-protein misfolding cyclic amplification efficiency of variant Creutzfeldt-Jakob disease. Neuroscience Letters. 2011;498(2):119–123. doi: 10.1016/j.neulet.2011.04.072. [DOI] [PubMed] [Google Scholar]
- 46.Deleault NR, Geoghegan JC, Nishina K, Kascsak R, Williamson RA, Supattapone S. Protease-resistant prion protein amplification reconstituted with partially purified substrates and synthetic polyanions. Journal of Biological Chemistry. 2005;280(29):26873–26879. doi: 10.1074/jbc.M503973200. [DOI] [PubMed] [Google Scholar]
- 47.Deleault NR, Lucassen RW, Supattapone S. RNA molecules stimulate prion protein conversion. Nature. 2003;425(6959):717–720. doi: 10.1038/nature01979. [DOI] [PubMed] [Google Scholar]
- 48.Geoghegan JC, Valdes PA, Orem NR, et al. Selective incorporation of polyanionic molecules into hamster prions. Journal of Biological Chemistry. 2007;282(50):36341–36353. doi: 10.1074/jbc.M704447200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;411(6839):810–813. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 50.Wang F, Wang X, Yuan C-G, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science. 2010;327(5969):1132–1135. doi: 10.1126/science.1183748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deleault NR, Kascsak R, Geoghegan JC, Supattapone S. Species-dependent differences in cofactor utilization for formation of the protease-resistant prion protein in vitro. Biochemistry. 2010;49(18):3928–3934. doi: 10.1021/bi100370b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gonzalez-Montalban N, Makarava N, Savtchenko R, Baskakov IV. Relationship between conformational stability and amplification efficiency of prions. Biochemistry. 2011;50(37):7933–7940. doi: 10.1021/bi200950v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nandi PK, Leclerc E. Polymerization of murine recombinant prion protein in nucleic acid solution. Archives of Virology. 1999;144(9):1751–1763. doi: 10.1007/s007050050702. [DOI] [PubMed] [Google Scholar]
- 54.Cordeiro Y, Machado F, Juliano L, et al. DNA converts cellular prion protein into the β-sheet conformation and inhibits prion peptide aggregation. Journal of Biological Chemistry. 2001;276(52):49400–49409. doi: 10.1074/jbc.M106707200. [DOI] [PubMed] [Google Scholar]
- 55.Deleault NR, Piro JR, Walsh DJ, et al. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(22):8546–8551. doi: 10.1073/pnas.1204498109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deleault NR, Walsh DJ, Piro JR, et al. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(28):E1938–E1946. doi: 10.1073/pnas.1206999109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saá P, Sferrazza GF, Ottenberg G, Oelschlegel AM, Dorsey K, Lasmézas CI. Strain-specific role of RNAs in prion replication. Journal of Virology. 2012;86(19):10494–10504. doi: 10.1128/JVI.01286-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Taylor DR, Hooper NM. The prion protein and lipid rafts (review) Molecular Membrane Biology. 2006;23(1):89–99. doi: 10.1080/09687860500449994. [DOI] [PubMed] [Google Scholar]
- 59.Santuccione A, Sytnyk V, Leshchyns’ka I, Schachner M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. Journal of Cell Biology. 2005;169(2):341–354. doi: 10.1083/jcb.200409127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Roffé M, Beraldo FH, Bester R, et al. Prion protein interaction with stress-inducible protein 1 enhances neuronal protein synthesis via mTOR. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(29):13147–13152. doi: 10.1073/pnas.1000784107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Santos TG, Beraldo FH, Hajj GNM, et al. Laminin-gamma1 chain and stress inducible protein 1 synergistically mediate PrPC-dependent axonal growth via Ca2+ mobilization in dorsal root ganglia neurons. Journal of Neurochemistry. 2013;124(2):210–223. doi: 10.1111/jnc.12091. [DOI] [PubMed] [Google Scholar]
- 62.Santos TG, Silva IR, Costa-Silva B, Lepique AP, Martins VR, Lopes MH. Enhanced neural progenitor/stem cells self-renewal via the interaction of stress-inducible protein 1 with the prion protein. Stem Cells. 2011;29(7):1126–1136. doi: 10.1002/stem.664. [DOI] [PubMed] [Google Scholar]
- 63.Jen A, Parkyn CJ, Mootoosamy RC, et al. Neuronal low-density lipoprotein receptor-related protein 1 binds and endocytoses prion fibrils via receptor cluster 4. Journal of Cell Science. 2010;123(2):246–255. doi: 10.1242/jcs.058099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rushworth JV, Griffiths HH, Watt NT, Hooper NM. Prion protein-mediated toxicity of amyloid-beta oligomers requires lipid rafts and the transmembrane LRP1. Journal of Biological Chemistry. 2013;288(13):8935–8951. doi: 10.1074/jbc.M112.400358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Devanathan V, Jakovcevski I, Santuccione A, et al. Cellular form of prion protein inhibits reelin-mediated shedding of Caspr from the neuronal cell surface to potentiate Caspr-mediated inhibition of neurite outgrowth. Journal of Neuroscience. 2010;30(27):9292–9305. doi: 10.1523/JNEUROSCI.5657-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Naslavsky N, Stein R, Yanai A, Friedlander G, Taraboulos A. Characterization of detergent-insoluble complexes containing the cellular prion protein and its scrapie isoform. Journal of Biological Chemistry. 1997;272(10):6324–6331. doi: 10.1074/jbc.272.10.6324. [DOI] [PubMed] [Google Scholar]
- 67.Naslavsky N, Shmeeda H, Friedlander G, et al. Sphingolipid depletion increases formation of the scrapie prion protein in neuroblastoma cells infected with prions. Journal of Biological Chemistry. 1999;274(30):20763–20771. doi: 10.1074/jbc.274.30.20763. [DOI] [PubMed] [Google Scholar]
- 68.Klein TR, Kirsch D, Kaufmann R, Riesner D. Prion rods contain small amounts of two host sphingolipids as revealed by thin-layer chromatography and mass spectrometry. Biological Chemistry. 1998;379(6):655–666. doi: 10.1515/bchm.1998.379.6.655. [DOI] [PubMed] [Google Scholar]
- 69.Campana V, Sarnataro D, Zurzolo C. The highways and byways of prion protein trafficking. Trends in Cell Biology. 2005;15(2):102–111. doi: 10.1016/j.tcb.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 70.Pinheiro TJT. The role of rafts in the fibrillization and aggregation of prions. Chemistry and Physics of Lipids. 2006;141(1-2):66–71. doi: 10.1016/j.chemphyslip.2006.02.022. [DOI] [PubMed] [Google Scholar]
- 71.Caughey B, Raymond GJ. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. Journal of Biological Chemistry. 1991;266(27):18217–18223. [PubMed] [Google Scholar]
- 72.Borchelt DR, Taraboulos A, Prusiner SB. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. Journal of Biological Chemistry. 1992;267(23):16188–16199. [PubMed] [Google Scholar]
- 73.Caughey B, Raymond GJ, Ernst D, Race RE. N-terminal truncation of the scrapie-associated form of PrP by lysosomal protease(s): implications regarding the site of conversion of PrP to the protease-resistant state. Journal of Virology. 1991;65(12):6597–6603. doi: 10.1128/jvi.65.12.6597-6603.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Taraboulos A, Raeber AJ, Borchelt DR, Serban D, Prusiner SB. Synthesis and trafficking of prion proteins in cultured cells. Molecular Biology of the Cell. 1992;3(8):851–863. doi: 10.1091/mbc.3.8.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Taraboulos A, Serban D, Prusiner SB. Scrapie prion proteins accumulate in the cytoplasm of persistently infected cultured cells. Journal of Cell Biology. 1990;110(6):2117–2132. doi: 10.1083/jcb.110.6.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Béranger F, Mangé A, Goud B, Lehmann S. Stimulation of PrPC retrograde transport toward the endoplasmic reticulum increases accumulation of PrPSc in prion-infected cells. Journal of Biological Chemistry. 2002;277(41):38972–38977. doi: 10.1074/jbc.M205110200. [DOI] [PubMed] [Google Scholar]
- 77.Godsave SF, Wille H, Kujala P, et al. Cryo-immunogold electron microscopy for prions: toward identification of a conversion site. Journal of Neuroscience. 2008;28(47):12489–12499. doi: 10.1523/JNEUROSCI.4474-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ivanova L, Barmada S, Kummer T, Harris DA. Mutant prion proteins are partially retained in the endoplasmic reticulum. Journal of Biological Chemistry. 2001;276(45):42409–42421. doi: 10.1074/jbc.M106928200. [DOI] [PubMed] [Google Scholar]
- 79.Marijanovic Z, Caputo A, Campana V, Zurzolo C. Identification of an intracellular site of prion conversion. PLoS Pathogens. 2009;5(5) doi: 10.1371/journal.ppat.1000426.e1000426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lewis V, Hooper NM. The role of lipid rafts in prion protein biology. Frontiers in Bioscience. 2011;16:151–168. doi: 10.2741/3681. [DOI] [PubMed] [Google Scholar]
- 81.Pimpinelli F, Lehmann S, Maridonneau-Parini I. The scrapie prion protein is present in flotillin-1-positive vesicles in central- but not peripheral-derived neuronal cell lines. European Journal of Neuroscience. 2005;21(8):2063–2072. doi: 10.1111/j.1460-9568.2005.04049.x. [DOI] [PubMed] [Google Scholar]
- 82.Pattison IH, Millson GC. Scrapie produced experimentally in goats with special reference to the clinical syndrome. Journal of Comparative Pathology and Therapeutics. 1961;71:101–109. doi: 10.1016/s0368-1742(61)80013-1. [DOI] [PubMed] [Google Scholar]
- 83.Bruce ME, McConnell I, Fraser H, Dickinson AG. The disease characteristics of different strains of scrapie in Sinc congenic mouse lines: implications for the nature of the agent and host control of pathogenesis. Journal of General Virology. 1991;72(3):595–603. doi: 10.1099/0022-1317-72-3-595. [DOI] [PubMed] [Google Scholar]
- 84.Fraser H. The pathology of a natural and experimental scrapie. Frontiers of Biology. 1976;44:267–305. [PubMed] [Google Scholar]
- 85.Hecker R, Taraboulos A, Scott M, et al. Replication of distinct scrapie prion isolates is region specific in brains of transgenic mice and hamsters. Genes and Development. 1992;6(7):1213–1228. doi: 10.1101/gad.6.7.1213. [DOI] [PubMed] [Google Scholar]
- 86.Chesebro B. BSE and prions: uncertainties about the agent. Science. 1998;279(5347):42–43. doi: 10.1126/science.279.5347.42. [DOI] [PubMed] [Google Scholar]
- 87.Farquhar CF, Somerville RA, Bruce ME. Straining the prion hypothesis. Nature. 1998;391(6665):345–346. doi: 10.1038/34818. [DOI] [PubMed] [Google Scholar]
- 88.Kascsak RJ, Rubenstein R, Merz PA. Immunological comparison of scrapie-associated fibrils isolated from animals infected with four different scrapie strains. Journal of Virology. 1986;59(3):676–683. doi: 10.1128/jvi.59.3.676-683.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kascsak RJ, Rubenstein R, Merz PA. Biochemical differences among scrapie-associated fibrils support the biological diversity of scrapie agents. Journal of General Virology. 1985;66(8):1715–1722. doi: 10.1099/0022-1317-66-8-1715. [DOI] [PubMed] [Google Scholar]
- 90.Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. Journal of Virology. 1994;68(12):7859–7868. doi: 10.1128/jvi.68.12.7859-7868.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bessen RA, Kocisko DA, Raymond GJ, Nandan S, Lansbury PT, Caughey B. Non-genetic propagation of strain-specific properties of scrapie prion protein. Nature. 1995;375(6533):698–700. doi: 10.1038/375698a0. [DOI] [PubMed] [Google Scholar]
- 92.Telling GC, Parchi P, DeArmond SJ, et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274(5295):2079–2082. doi: 10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- 93.Safar J, Wille H, Itri V, et al. Eight prion strains have PrPSc molecules with different conformations. Nature Medicine. 1998;4(10):1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- 94.Thomzig A, Spassov S, Friedrich M, Naumann D, Beekes M. Discriminating scrapie and bovine spongiform encephalopathy isolates by infrared spectroscopy of pathological prion protein. Journal of Biological Chemistry. 2004;279(32):33847–33854. doi: 10.1074/jbc.M403730200. [DOI] [PubMed] [Google Scholar]
- 95.Spassov S, Beekes M, Naumann D. Structural differences between TSEs strains investigated by FT-IR spectroscopy. Biochimica et Biophysica Acta. 2006;1760(7):1138–1149. doi: 10.1016/j.bbagen.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 96.Legname G, Nguyen H-OB, Peretz D, Cohen FE, DeArmond SJ, Prusiner SB. Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(50):19105–19110. doi: 10.1073/pnas.0608970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bett C, et al. Biochemical properties of highly neuroinvasive prion strains. PLoS Pathogens. 2012;8(2) doi: 10.1371/journal.ppat.1002522.e100252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ayers JI, Schutt CR, Shikiya RA, Aguzzi A, Kincaid AE, Bartz JC. The strain-encoded relationship between PrPSc replication, stability and processing in neurons is predictive of the incubation period of disease. PLoS Pathogens. 2011;7(3) doi: 10.1371/journal.ppat.1001317.e1001317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pastrana MA, Sajnani G, Onisko B, et al. Isolation and characterization of a proteinase K-sensitive PrPSc fraction. Biochemistry. 2006;45(51):15710–15717. doi: 10.1021/bi0615442. [DOI] [PubMed] [Google Scholar]
- 100.Safar JG, Geschwind MD, Deering C, et al. Diagnosis of human prion disease. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(9):3501–3506. doi: 10.1073/pnas.0409651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tzaban S, Friedlander G, Schonberger O, et al. Protease-sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry. 2002;41(42):12868–12875. doi: 10.1021/bi025958g. [DOI] [PubMed] [Google Scholar]
- 102.Parchi P, Chen SG, Brown P, et al. Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann-Sträussler-Scheinker disease. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(14):8322–8327. doi: 10.1073/pnas.95.14.8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Benestad SL, Arsac J-N, Goldmann W, Nöremark M. Atypical/Nor98 scrapie: properties of the agent, genetics, and epidemiology. Veterinary Research. 2008;39(4, article 19) doi: 10.1051/vetres:2007056. [DOI] [PubMed] [Google Scholar]
- 104.Zou WQ, Puoti G, Xiao X, et al. Variably protease-sensitive prionopathy: a new sporadic disease of the prion protein. Annals of Neurology. 2010;68(2):162–172. doi: 10.1002/ana.22094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Polymenidou M, Prokop S, Jung HH, et al. Atypical prion protein conformation in familial prion disease with PRNP P105T mutation. Brain Pathology. 2011;21(2):209–214. doi: 10.1111/j.1750-3639.2010.00439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Monaco S, Fiorini M, Farinazzo A, et al. Allelic origin of protease-sensitive and protease-resistant prion protein isoforms in Gerstmann-Sträussler-Scheinker disease with the p102l mutation. PLoS ONE. 2012;7(2) doi: 10.1371/journal.pone.0032382.e32382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xiao X, Cali I, Dong Z, et al. Protease-sensitive prions with 144-bp insertion mutations. Aging. 2013;5(3):155–173. doi: 10.18632/aging.100543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Thackray AM, Hopkins L, Bujdoso R. Proteinase K-sensitive disease-associated ovine prion protein revealed by conformation-dependent immunoassay. Biochemical Journal. 2007;401(2):475–483. doi: 10.1042/BJ20061264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kim C, Haldiman T, Cohen Y, et al. Protease-sensitive conformers in broad spectrum of distinct PrPSc structures in sporadic Creutzfeldt-Jakob disease are indicator of progression rate. PLoS Pathogens. 2011;7(9) doi: 10.1371/journal.ppat.1002242.e1002242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kim C, Haldiman T, Surewicz K, et al. Small protease sensitive oligomers of PrPSc in distinct human prions determine conversion rate of PrPC . PLoS Pathogens. 2012;8(8) doi: 10.1371/journal.ppat.1002835.e1002835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Saverioni D, Notari S, Capellari S, et al. Analyses of protease resistance and aggregation state of abnormal prion protein across the spectrum of human prions. Journal of Biological Chemistry. 2013;288(39):27972–27985. doi: 10.1074/jbc.M113.477547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tuzi NL, Cancellotti E, Baybutt H, et al. Host PrP glycosylation: a major factor determining the outcome of prion infection. PLoS Biology. 2008;6(4, article e100) doi: 10.1371/journal.pbio.0060100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Casalone C, Zanusso G, Acutis P, et al. Identification of a second bovine amyloidotic spongiform encephalopathy: molecular similarities with sporadic Creutzfeldt-Jakob disease. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(9):3065–3070. doi: 10.1073/pnas.0305777101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Parchi P, Capellari S, Chen SG, et al. Typing prion isoforms. Nature. 1997;386(6622):232–234. doi: 10.1038/386232a0. [DOI] [PubMed] [Google Scholar]
- 115.Parchi P, Castellani R, Capellari S, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Annals of Neurology. 1996;39(6):767–778. doi: 10.1002/ana.410390613. [DOI] [PubMed] [Google Scholar]
- 116.Collinge J, Sidle KCL, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of “new variant” CJD. Nature. 1996;383(6602):685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 117.Helenius A, Aebi M. Intracellular functions of N-linked glycans. Science. 2001;291(5512):2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 118.O’Connor SE, Imperiali B. Modulation of protein structure and function by asparagine-linked glycosylation. Chemistry and Biology. 1996;3(10):803–812. doi: 10.1016/s1074-5521(96)90064-2. [DOI] [PubMed] [Google Scholar]
- 119.Cobb NJ, Surewicz WK. Prion diseases and their biochemical mechanisms. Biochemistry. 2009;48(12):2574–2585. doi: 10.1021/bi900108v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cancellotti E, Mahal SP, Somerville R, et al. Post-translational changes to PrP alter transmissible spongiform encephalopathy strain properties. EMBO Journal. 2013;32(5):756–769. doi: 10.1038/emboj.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Eigen M. On the nature of virus quasispecies. Trends in Microbiology. 1996;4(6):216–218. doi: 10.1016/0966-842X(96)20011-3. [DOI] [PubMed] [Google Scholar]
- 122.Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318(5852):930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 123.Li J, Browning S, Mahal SP, Oelschlegel AM, Weissmann C. Darwinian evolution of prions in cell culture. Science. 2010;327(5967):869–872. doi: 10.1126/science.1183218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ghetti BT, Kovacs GG, Piccardo P. Gerstmann-Sträussler-Scheinker disease. In: Dickson DW, Weller RO, editors. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. 2011. pp. 364–377. [Google Scholar]
- 125.Parchi P, Strammiello R, Giese A, Kretzschmar H. Phenotypic variability of sporadic human prion disease and its molecular basis: past, present, and future. Acta Neuropathologica. 2011;121(1):91–112. doi: 10.1007/s00401-010-0779-6. [DOI] [PubMed] [Google Scholar]
- 126.Parchi P, Capellari S, Gambetti P. Fatal familial and sporadic insomnia. In: Dickson DW, Weller RO, editors. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. 2011. pp. 346–349. [Google Scholar]
- 127.Gambetti P, Puoti G, Kong Q, Zou W. A new prion diseases: protease-sensitive prionopathy. In: Dickson DW, Weller RO, editors. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. 2011. pp. 350–353. [Google Scholar]
- 128.Ironside JW, Head MW, Will RG. Variant Creutzfeldt-Jacob disease. In: Dickson DW, Weller RO, editors. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. 2011. pp. 354–363. [Google Scholar]
- 129.Parchi P, Zou W, Wang W, et al. Genetic influence on the structural variations of the abnormal prion protein. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(18):10168–10172. doi: 10.1073/pnas.97.18.10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Annals of Neurology. 1999;46(2):224–233. [PubMed] [Google Scholar]
- 131.Parchi P, Cescatti M, Notari S, et al. Agent strain variation in human prion disease: insights from a molecular and pathological review of the National Institutes of Health series of experimentally transmitted disease. Brain. 2010;133(10):3030–3042. doi: 10.1093/brain/awq234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cali I, Castellani R, Yuan J, et al. Classification of sporadic Creutzfeldt-Jakob disease revisited. Brain. 2006;129(9):2266–2277. doi: 10.1093/brain/awl224. [DOI] [PubMed] [Google Scholar]
- 133.Parchi P, Strammiello R, Notari S, et al. Incidence and spectrum of sporadic Creutzfeldt-Jakob disease variants with mixed phenotype and co-occurrence of PrPSc types: an updated classification. Acta Neuropathologica. 2009;118(5):659–671. doi: 10.1007/s00401-009-0585-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Puoti G, Giaccone G, Rossi G, Canciani B, Bugiani O, Tagliavini F. Sporadic Creutzfeldt-Jakob disease: co-occurrence of different types of PrPSc in the same brain. Neurology. 1999;53(9):2173–2176. doi: 10.1212/wnl.53.9.2173. [DOI] [PubMed] [Google Scholar]
- 135.Murayama Y, Yoshioka M, Masujin K, et al. Sulfated dextrans enhance in vitro amplification of bovine spongiform encephalopathy PrPSc and enable ultrasensitive detection of bovine PrPSc . PLoS ONE. 2010;5(10) doi: 10.1371/journal.pone.0013152.e13152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(23):9741–9746. doi: 10.1073/pnas.0702662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Notari S, Capellari S, Giese A, et al. Effects of different experimental conditions on the PrPSc core generated by protease digestion: implications for strain typing and molecular classification of CJD. Journal of Biological Chemistry. 2004;279(16):16797–16804. doi: 10.1074/jbc.M313220200. [DOI] [PubMed] [Google Scholar]
- 138.Notari S, Strammiello R, Capellari S, et al. Characterization of truncated forms of abnormal prion protein in Creutzfeldt-Jakob disease. Journal of Biological Chemistry. 2008;283(45):30557–30565. doi: 10.1074/jbc.M801877200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Monari L, Chen SG, Brown P, et al. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: different prion proteins determined by a DNA polymorphism. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(7):2839–2842. doi: 10.1073/pnas.91.7.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pan T, Colucci M, Wong B-S, et al. Novel differences between two human prion strains revealed by two-dimensional gel electrophoresis. Journal of Biological Chemistry. 2001;276(40):37284–37288. doi: 10.1074/jbc.M107358200. [DOI] [PubMed] [Google Scholar]
- 141.Aucouturier P, Kascsak RJ, Frangione B, Wisniewski T. Biochemical and conformational variability of human prion strains in sporadic Creutzfeldt-Jakob disease. Neuroscience Letters. 1999;274(1):33–36. doi: 10.1016/s0304-3940(99)00659-x. [DOI] [PubMed] [Google Scholar]
- 142.Kobayashi A, Satoh S, Ironside JW, Mohri S, Kitamoto T. Type 1 and type 2 human PrPSc have different aggregation sizes in methionine homozygotes with sporadic, iatrogenic and variant Creutzfeldt-Jakob disease. Journal of General Virology. 2005;86(1):237–240. doi: 10.1099/vir.0.80389-0. [DOI] [PubMed] [Google Scholar]
- 143.Cali I, Castellani R, Alshekhlee A, et al. Co-existence of scrapie prion protein types 1 and 2 in sporadic Creutzfeldt-Jakob disease: its effect on the phenotype and prion-type characteristics. Brain. 2009;132(10):2643–2658. doi: 10.1093/brain/awp196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pirisinu L, di Bari M, Marcon S, et al. A new method for the characterization of strain-specific conformational stability of protease-sensitive and protease-resistant PrPSc . PLoS ONE. 2010;5(9) doi: 10.1371/journal.pone.0012723.e12723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Choi YP, Peden AH, Gröner A, Ironside JW, Head MW. Distinct stability states of disease-associated human prion protein identified by conformation-dependent immunoassay. Journal of Virology. 2010;84(22):12030–12038. doi: 10.1128/JVI.01057-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Piccardo P, Dlouhy SR, Lievens PMJ, et al. Phenotypic variability of Gerstmann-Sträussler-Scheinker disease is associated with prion protein heterogeneity. Journal of Neuropathology and Experimental Neurology. 1998;57(10):979–988. doi: 10.1097/00005072-199810000-00010. [DOI] [PubMed] [Google Scholar]
- 147.Piccardo P, Liepnieks JJ, William A, et al. Prion proteins with different conformations accumulate in Gerstmann-Sträussler-Scheinker disease caused by A117V and F198S mutations. American Journal of Pathology. 2001;158(6):2201–2207. doi: 10.1016/S0002-9440(10)64692-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Piccardo P, Seiler C, Dlouhy SR, et al. Proteinase-K-resistant prion protein isoforms in Gerstmann-Sträussler-Scheinker disease (Indiana kindred) Journal of Neuropathology and Experimental Neurology. 1996;55(11):1157–1163. doi: 10.1097/00005072-199611000-00007. [DOI] [PubMed] [Google Scholar]
- 149.Tagliavini F, Lievens PM-J, Tranchant C, et al. A 7-kDa prion protein (PrP) fragment, an integral component of the PrP region required for infectivity, is the major amyloid protein in Gerstmann-Sträussler-Scheinker disease A117V. Journal of Biological Chemistry. 2001;276(8):6009–6015. doi: 10.1074/jbc.M007062200. [DOI] [PubMed] [Google Scholar]
- 150.Tagliavini F, Prelli F, Ghiso J, et al. Amyloid protein of Gerstmann-Sträussler-Scheinker disease (Indiana kindred) is an 11 kd fragment of prion protein with an N-terminal glycine at codon 58. EMBO Journal. 1991;10(3):513–519. doi: 10.1002/j.1460-2075.1991.tb07977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tagliavini F, Prelli F, Porro M, et al. Amyloid fibrils in Gerstmann-Sträussler-Scheinker disease (Indiana and Swedish kindreds) express only PrP peptides encoded by the mutant allele. Cell. 1994;79(4):695–703. doi: 10.1016/0092-8674(94)90554-1. [DOI] [PubMed] [Google Scholar]
- 152.Jansen C, Parchi P, Capellari S, et al. A second case of Gerstmann-Sträussler-Scheinker disease linked to the G131V mutation in the prion protein gene in a Dutch patient. Journal of Neuropathology and Experimental Neurology. 2011;70(8):698–702. doi: 10.1097/NEN.0b013e3182270c54. [DOI] [PubMed] [Google Scholar]
- 153.Gambetti P, Dong Z, Yuan J, et al. A novel human disease with abnormal prion protein sensitive to protease. Annals of Neurology. 2008;63(6):697–708. doi: 10.1002/ana.21420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Head MW, Lowrie S, Chohan G, Knight R, Scoones DJ, Ironside JW. Variably protease-sensitive prionopathy in a PRNP codon 129 heterozygous UK patient with co-existing tau, α synuclein and Aβ pathology. Acta Neuropathologica. 2010;120(6):821–823. doi: 10.1007/s00401-010-0766-y. [DOI] [PubMed] [Google Scholar]
- 155.Head MW, Yull HM, Ritchie DL, et al. Variably protease-sensitive prionopathy in the UK: a retrospective review 1991–2008. Brain. 2013;136(4):1102–1115. doi: 10.1093/brain/aws366. [DOI] [PubMed] [Google Scholar]
- 156.Jansen C, Head MW, van Gool WA, et al. The first case of protease-sensitive prionopathy (PSPr) in the Netherlands: a patient with an unusual GSS-like clinical phenotype. Journal of Neurology, Neurosurgery and Psychiatry. 2010;81(9):1052–1055. doi: 10.1136/jnnp.2009.175646. [DOI] [PubMed] [Google Scholar]
- 157.Xiao X, Yuan J, Haïk S, et al. Glycoform-selective prion formation in sporadic and familial forms of prion disease. PLoS ONE. 2013;8(3) doi: 10.1371/journal.pone.0058786.e58786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mastrianni JA, Nixon R, Layzer R, et al. Prion protein conformation in a patient with sporadic fatal insomnia. The New England Journal of Medicine. 1999;340(21):1630–1638. doi: 10.1056/NEJM199905273402104. [DOI] [PubMed] [Google Scholar]
- 159.Korth C, Kaneko K, Groth D, et al. Abbreviated incubation times for human prions in mice expressing a chimeric mouse-human prion protein transgene. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(8):4784–4789. doi: 10.1073/pnas.2627989100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Nonno R, di Bari MA, Cardone F, et al. Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathogens. 2006;2(2, article e12) doi: 10.1371/journal.ppat.0020012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Brown P, Gibbs CJ, Jr., Rodgers-Johnson P, et al. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Annals of Neurology. 1994;35(5):513–529. doi: 10.1002/ana.410350504. [DOI] [PubMed] [Google Scholar]
- 162.Bishop MT, Will RG, Manson JC. Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(26):12005–12010. doi: 10.1073/pnas.1004688107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Moda F, Suardi S, di Fede G, et al. MM2-thalamic Creutzfeldt-Jakob disease: neuropathological, biochemical and transmission studies identify a distinctive prion strain. Brain Pathology. 2012;22(5):662–669. doi: 10.1111/j.1750-3639.2012.00572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Bruce ME, Will RG, Ironside JW, et al. Transmissions to mice indicate that “new variant” CJD is caused by the BSE agent. Nature. 1997;389(6650):498–501. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- 165.Hill AF, Desbruslais M, Joiner S, et al. The same prion strain causes vCJD and BSE. Nature. 1997;389(6650):448–450. doi: 10.1038/38925. [DOI] [PubMed] [Google Scholar]
- 166.Wadsworth JDF, Joiner S, Linehan JM, et al. Kuru prions and sporadic Creutzfeldt-Jakob disease prions have equivalent transmission properties in transgenic and wild-type mice. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(10):3885–3890. doi: 10.1073/pnas.0800190105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wadsworth JDF, Asante EA, Desbruslais M, et al. Human prion protein with valine 129 prevents expression of variant CJD phenotype. Science. 2004;306(5702):1793–1796. doi: 10.1126/science.1103932. [DOI] [PubMed] [Google Scholar]
- 168.Asante EA, Linehan JM, Gowland I, et al. Dissociation of pathological and molecular phenotype of variant Creutzfeldt-Jakob disease in transgenic human prion protein 129 heterozygous mice. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(28):10759–10764. doi: 10.1073/pnas.0604292103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bishop M, Hart P, Aitchison L, et al. Predicting susceptibility and incubation time of human-to-human transmission of vCJD. The Lancet Neurology. 2006;5(5):393–398. doi: 10.1016/S1474-4422(06)70413-6. [DOI] [PubMed] [Google Scholar]
- 170.Béringue V, Le Dur A, Tixador P, et al. Prominent and persistent extraneural infection in human PrP transgenic mice infected with variant CJD. PLoS ONE. 2008;3(1, article e1419) doi: 10.1371/journal.pone.0001419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Takeuchi A, Kobayashi A, Ironside JW, Mohri S, Kitamoto T. Characterization of variant Creutzfeldt-Jakob disease prions in prion protein-humanized mice carrying distinct codon 129 genotypes. Journal of Biological Chemistry. 2013;288(30):21659–21666. doi: 10.1074/jbc.M113.470328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Tateishi J, Kitamoto T, Hoque MZ, Furukawa H. Experimental transmission of Creutzfeldt-Jakob disease and related diseases to rodents. Neurology. 1996;46(2):532–537. doi: 10.1212/wnl.46.2.532. [DOI] [PubMed] [Google Scholar]
- 173.Piccardo P, Manson JC, King D, Ghetti B, Barron RM. Accumulation of prion protein in the brain that is not associated with transmissible disease. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(11):4712–4717. doi: 10.1073/pnas.0609241104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Asante EA, Linehan JM, Smidak M, et al. Inherited prion disease A117V is not simply a proteinopathy but produces prions transmissible to transgenic mice expressing homologous prion protein. PLoS Pathogens. 2013;9(9) doi: 10.1371/journal.ppat.1003643.e100364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kimberlin RH, Walker CA. Characteristics of a short incubation model of scrapie in the golden hamster. Journal of General Virology. 1977;34(2):295–304. doi: 10.1099/0022-1317-34-2-295. [DOI] [PubMed] [Google Scholar]
- 176.Kimberlin RH, Walker CA. Pathogenesis of scrapie (strain 263k) in hamsters infected intracerebrally, intraperitoneally or intraocularly. Journal of General Virology. 1986;67(2):255–263. doi: 10.1099/0022-1317-67-2-255. [DOI] [PubMed] [Google Scholar]
- 177.Kuczius T, Haist I, Groschup MH. Molecular analysis of bovine spongiform encephalopathy and scrapie strain variation. Journal of Infectious Diseases. 1998;178(3):693–699. doi: 10.1086/515337. [DOI] [PubMed] [Google Scholar]
- 178.Hope J, Wood SCER, Birkett CR, et al. Molecular analysis of ovine prion protein identifies similarities between BSE and an experimental isolate of natural scrapie, CH1641. Journal of General Virology. 1999;80(1):1–4. doi: 10.1099/0022-1317-80-1-1. [DOI] [PubMed] [Google Scholar]
- 179.Horiuchi M, Nemoto T, Ishiguro N, Furuoka H, Mohri S, Shinagawa M. Biological and biochemical characterization of sheep scrapie in Japan. Journal of Clinical Microbiology. 2002;40(9):3421–3426. doi: 10.1128/JCM.40.9.3421-3426.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Stack MJ, Chaplin MJ, Clark J. Differentiation of prion protein glycoforms from naturally occurring sheep scrapie, sheep-passaged scrapie strains (CH1641 and SSBP1), Bovine Spongiform Encephalopathy (BSE) cases and Romney and Cheviot breed sheep experimentally inoculated with BSE using two monoclonal antibodies. Acta Neuropathologica. 2002;104(3):279–286. doi: 10.1007/s00401-002-0556-2. [DOI] [PubMed] [Google Scholar]
- 181.Baron T, Crozet C, Biacabe A-G, et al. Molecular analysis of the protease-resistant prion protein in scrapie and bovine spongiform encephalopathy transmitted to ovine transgenic and wild-type mice. Journal of Virology. 2004;78(12):6243–6251. doi: 10.1128/JVI.78.12.6243-6251.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Vulin J, Biacabe A-G, Cazeau G, Calavas D, Baron T. Molecular typing of protease-resistant prion protein in transmissible spongiform encephalopathies of small ruminants, France, 2002–2009. Emerging Infectious Diseases. 2011;17(1):55–63. doi: 10.3201/eid1701.100891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Baron TGM, Madec J-Y, Calavas D. Similar signature of the prion protein in natural sheep scrapie and bovine spongiform encephalopathy-linked diseases. Journal of Clinical Microbiology. 1999;37(11):3701–3704. doi: 10.1128/jcm.37.11.3701-3704.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Groschup MH, Kuczius T, Junghans F, Sweeney T, Bodemer W, Buschmann A. Characterization of BSE and scrapie strains/isolates. Archives of Virology. 2000;(16):217–226. doi: 10.1007/978-3-7091-6308-5_21. [DOI] [PubMed] [Google Scholar]
- 185.Sweeney T, Kuczius T, McElroy M, Gomerez Parada M, Groschup MH. Molecular analysis of Irish sheep scrapie cases. Journal of General Virology. 2000;81(6):1621–1627. doi: 10.1099/0022-1317-81-6-1621. [DOI] [PubMed] [Google Scholar]
- 186.Thackray AM, Hopkins L, Spiropoulos J, Bujdoso R. Molecular and transmission characteristics of primary-passaged ovine scrapie isolates in conventional and ovine PrP transgenic mice. Journal of Virology. 2008;82(22):11197–11207. doi: 10.1128/JVI.01454-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Baron T, Biacabe A-G. Molecular behaviors of “CH1641-Like” sheep scrapie isolates in ovine transgenic mice (TgOvPrP4) Journal of Virology. 2007;81(13):7230–7237. doi: 10.1128/JVI.02475-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Nicot S, Baron TGM. Strain-specific proteolytic processing of the prion protein in prion diseases of ruminants transmitted in ovine transgenic mice. Journal of General Virology. 2010;91(2):570–574. doi: 10.1099/vir.0.014464-0. [DOI] [PubMed] [Google Scholar]
- 189.Baron T, Bencsik A, Vulin J, et al. A C-terminal protease-resistant prion fragment distinguishes ovine “CH1641-like” scrapie from bovine classical and L-type BSE in ovine transgenic mice. PLoS Pathogens. 2008;4(8) doi: 10.1371/journal.ppat.1000137.e1000137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Foster JD, Dickinson AG. The unusual properties of CH1641, a sheep-passaged isolate of scrapie. Veterinary Record. 1988;123(1):5–8. doi: 10.1136/vr.123.1.5. [DOI] [PubMed] [Google Scholar]
- 191.Kimberlin RH, Walker CA. Evidence that the transmission of one source of scrapie agent to hamsters involves separation of agent strains from a mixture. Journal of General Virology. 1978;39(3):487–496. doi: 10.1099/0022-1317-39-3-487. [DOI] [PubMed] [Google Scholar]
- 192.Bruce ME, Boyle A, Cousens S, et al. Strain characterization of natural sheep scrapie and comparison with BSE. Journal of General Virology. 2002;83(3):695–704. doi: 10.1099/0022-1317-83-3-695. [DOI] [PubMed] [Google Scholar]
- 193.Benestad SL, Sarradin P, Thu B, Schönheit J, Tranulis MA, Bratberg B. Cases of scrapie with unusual features in Norway and designation of a new type, Nor98. Veterinary Record. 2003;153(7):202–208. doi: 10.1136/vr.153.7.202. [DOI] [PubMed] [Google Scholar]
- 194.de Bosschere H, Roels S, Benestad SL, Vanopdenbosch E. Scrapie case similar to Nor98 diagnosed in Belgium via active surveillance. Veterinary Record. 2004;155(22):707–708. doi: 10.1136/vr.155.22.707. [DOI] [PubMed] [Google Scholar]
- 195.Gavier-Widén D, Nöremark M, Benestad S, et al. Recognition of the Nor98 variant of scrapie in the Swedish sheep population. Journal of Veterinary Diagnostic Investigation. 2004;16(6):562–567. doi: 10.1177/104063870401600611. [DOI] [PubMed] [Google Scholar]
- 196.Onnasch H, Gunn HM, Bradshaw BJ, Benestad SL, Bassett HF. Two Irish cases of scrapie resembling Nor98. Veterinary Record. 2004;155(20):636–637. doi: 10.1136/vr.155.20.636. [DOI] [PubMed] [Google Scholar]
- 197.Loiacono CM, Thomsen BV, Hall SM, et al. Nor98 scrapie identified in the United States. Journal of Veterinary Diagnostic Investigation. 2009;21(4):454–463. doi: 10.1177/104063870902100406. [DOI] [PubMed] [Google Scholar]
- 198.Klingeborn M, Wik L, Simonsson M, Renström LHM, Ottinger T, Linné T. Characterization of proteinase K-resistant N- and C-terminally truncated PrP in Nor98 atypical scrapie. Journal of General Virology. 2006;87(6):1751–1760. doi: 10.1099/vir.0.81618-0. [DOI] [PubMed] [Google Scholar]
- 199.Götte DR, Benestad SL, Laude H, Zurbriggen A, Oevermann A, Seuberlich T. Atypical scrapie isolates involve a uniform prion species with a complex molecular signature. PLoS ONE. 2011;6(11) doi: 10.1371/journal.pone.0027510.e27510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Pirisinu L, Nonno R, Esposito E, et al. Small ruminant nor98 prions share biochemical features with human Gerstmann-Sträussler-Scheinker disease and variably protease-sensitive prionopathy. PLoS ONE. 2013;8(6) doi: 10.1371/journal.pone.0066405.e66405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Biacabe A-G, Laplanche J-L, Ryder S, Baron T. Distinct molecular phenotypes in bovine prion diseases. EMBO Reports. 2004;5(1):110–114. doi: 10.1038/sj.embor.7400054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Jacobs JG, Langeveld JPM, Biacabe A-G, et al. Molecular discrimination of atypical bovine spongiform encephalopathy strains from a geographical region spanning a wide area in Europe. Journal of Clinical Microbiology. 2007;45(6):1821–1829. doi: 10.1128/JCM.00160-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Bruce M, Chree A, McConnell I, Foster J, Pearson G, Fraser H. Transmission of bovine spongiform encephalopathy and scrapie to mice: strain variation and the species barrier. Philosophical Transactions of the Royal Society of London B. 1994;343(1306):405–411. doi: 10.1098/rstb.1994.0036. [DOI] [PubMed] [Google Scholar]
- 204.Buschmann A, Gretzschel A, Biacabe A-G, et al. Atypical BSE in Germany—proof of transmissibility and biochemical characterization. Veterinary Microbiology. 2006;117(2–4):103–116. doi: 10.1016/j.vetmic.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 205.Capobianco R, Casalone C, Suardi S, et al. Conversion of the BASE prion strain into the BSE strain: the origin of BSE? PLoS Pathogens. 2007;3(3, article e31) doi: 10.1371/journal.ppat.0030031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Baron T, Vulin J, Biacabe A-G, et al. Emergence of classical BSE strain properties during serial passages of H-BSE in wild-type mice. PLoS ONE. 2011;6(1) doi: 10.1371/journal.pone.0015839.e15839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Biacabe A-G, Jacobs JG, Bencsik A, Langeveld JPM, Baron TGM. H-type bovine spongiform encephalopathy: complex molecular features and similarities with human prion diseases. Prion. 2007;1(1):61–68. doi: 10.4161/pri.1.1.3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Richt JA, Hall SM. BSE case associated with prion protein gene mutation. PLoS Pathogens. 2008;4(9) doi: 10.1371/journal.ppat.1000156.e1000156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Gough KC, Maddison BC. Prion transmission: prion excretion and occurrence in the environment. Prion. 2010;4(4):275–282. doi: 10.4161/pri.4.4.13678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Marsh RF, Kincaid AE, Bessen RA, Bartz JC. Interspecies transmission of chronic wasting disease prions to squirrel monkeys (Saimiri sciureus) Journal of Virology. 2005;79(21):13794–13796. doi: 10.1128/JVI.79.21.13794-13796.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Williams ES, Miller MW. Transmissible spongiform encephalopathies in non-domestic animals: origin, transmission and risk factors. Revue Scientifique et Technique. 2003;22(1):145–156. doi: 10.20506/rst.22.1.1385. [DOI] [PubMed] [Google Scholar]
- 212.Miller MW, Williams ES. Horizontal prion transmission in mule deer. Nature. 2003;425(6953):35–36. doi: 10.1038/425035a. [DOI] [PubMed] [Google Scholar]
- 213.Angers RC, Kang H-E, Napier D, et al. Prion strain mutation determined by prion protein conformational compatibility and primary structure. Science. 2010;328(5982):1154–1158. doi: 10.1126/science.1187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Johnson CJ, Herbst A, Duque-Velasquez C, et al. Prion protein polymorphisms affect chronic wasting disease progression. PLoS ONE. 2011;6(3) doi: 10.1371/journal.pone.0017450.e17450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Caughey B, Lansbury PT., Jr. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annual Review of Neuroscience. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 216.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nature Reviews Molecular Cell Biology. 2007;8(2):101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 217.Brandner S, Isenmann S, Raeber A, et al. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature. 1996;379(6563):339–343. doi: 10.1038/379339a0. [DOI] [PubMed] [Google Scholar]
- 218.Chesebro B, Trifilo M, Race R, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005;308(5727):1435–1439. doi: 10.1126/science.1110837. [DOI] [PubMed] [Google Scholar]
- 219.Chesebro B, Race B, Meade-White K, et al. Fatal transmissible amyloid encephalopathy: a new type of prion disease associated with lack of prion protein membrane anchoring. PLoS Pathogens. 2010;6(3) doi: 10.1371/journal.ppat.1000800.e1000800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Race B, Meade-White K, Oldstone MBA, Race R, Chesebro B. Detection of prion infectivity in fat tissues of scrapie-infected mice. PLoS Pathogens. 2008;4(12) doi: 10.1371/journal.ppat.1000232.e1000232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Trifilo MJ, Yajima T, Gu Y, et al. Prion-induced amyloid heart disease with high blood infectivity in transgenic mice. Science. 2006;313(5783):94–97. doi: 10.1126/science.1128635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Caughey B, Baron GS, Chesebro B, Jeffrey M. Getting a grip on prions: oligomers, amyloids, and pathological membrane interactions. Annual Review of Biochemistry. 2009;78:177–204. doi: 10.1146/annurev.biochem.78.082907.145410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Jeffrey M, McGovern G, Sisó S, González L. Cellular and sub-cellular pathology of animal prion diseases: relationship between morphological changes, accumulation of abnormal prion protein and clinical disease. Acta Neuropathologica. 2011;121(1):113–134. doi: 10.1007/s00401-010-0700-3. [DOI] [PubMed] [Google Scholar]
- 224.Jeffrey M. Review: membrane-associated misfolded protein propagation in natural Transmissible Spongiform Encephalopathies (TSEs), synthetic prion diseases and Alzheimer's disease. Neuropathology and Applied Neurobiology. 2013;39(3):196–216. doi: 10.1111/nan.12004. [DOI] [PubMed] [Google Scholar]
- 225.Jeffrey M, McGovern G, Chambers EV, et al. Mechanism of PrP-amyloid formation in mice without transmissible spongiform encephalopathy. Brain Pathology. 2012;22(1):58–66. doi: 10.1111/j.1750-3639.2011.00508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Parchi P, Capellari S, Gambetti P. Intracerebral distribution of the abnormal isoform of the prion protein in sporadic Creutzfeldt-Jakob disease and fatal insomnia. Microscopy Research and Technique. 2000;50(1):16–25. doi: 10.1002/1097-0029(20000701)50:1<16::AID-JEMT4>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 227.Parchi P, Petersen RB, Chen SG, et al. Molecular pathology of fatal familial insomnia. Brain Pathology. 1998;8(3):539–548. doi: 10.1111/j.1750-3639.1998.tb00176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Parchi P, Castellani R, Cortelli P, et al. Regional distribution of protease-resistant prion protein in fatal familial insomnia. Annals of Neurology. 1995;38(1):21–29. doi: 10.1002/ana.410380107. [DOI] [PubMed] [Google Scholar]
- 229.Masters CL, Richardson EP., Jr. Subacute spongiform encephalopathy (Creutzfeldt-Jakob disease). The nature and progression of spongiform change. Brain. 1978;101(2):333–344. doi: 10.1093/brain/101.2.333. [DOI] [PubMed] [Google Scholar]
- 230.Bueler H, Fischer M, Lang Y, et al. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356(6370):577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- 231.Mallucci GR, Ratté S, Asante EA, et al. Post-natal knockout of prion protein alters hippocampal CA1 properties, but does not result in neurodegeneration. EMBO Journal. 2002;21(3):202–210. doi: 10.1093/emboj/21.3.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Chiesa R, Piccardo P, Quaglio E, et al. Molecular distinction between pathogenic and infectious properties of the prion protein. Journal of Virology. 2003;77(13):7611–7622. doi: 10.1128/JVI.77.13.7611-7622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Hill AF, Joiner S, Linehan J, Desbruslais M, Lantos PL, Collinge J. Species-barrier-independent prion replication in apparently resistant species. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(18):10248–10253. doi: 10.1073/pnas.97.18.10248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Race R, Raines A, Raymond GJ, Caughey B, Chesebro B. Long-term subclinical carrier state precedes scrapie replication and adaptation in a resistant species: analogies to bovine spongiform encephalopathy and variant Creutzfeldt-Jakob disease in humans. Journal of Virology. 2001;75(21):10106–10112. doi: 10.1128/JVI.75.21.10106-10112.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Sandberg MK, Al-Doujaily H, Sharps B, Clarke AR, Collinge J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature. 2011;470(7335):540–542. doi: 10.1038/nature09768. [DOI] [PubMed] [Google Scholar]
- 236.Aguzzi A, Falsig J. Prion propagation, toxicity and degradation. Nature Neuroscience. 2012;15(7):936–939. doi: 10.1038/nn.3120. [DOI] [PubMed] [Google Scholar]
- 237.Caughey B, Baron GS. Prions and their partners in crime. Nature. 2006;443(7113):803–810. doi: 10.1038/nature05294. [DOI] [PubMed] [Google Scholar]
- 238.Solomon IH, Schepker JA, Harris DA. Prion neurotoxicity: insights from prion protein mutants. Current Issues in Molecular Biology. 2010;12(2):51–62. [PMC free article] [PubMed] [Google Scholar]
- 239.Solomon IH, Huettner JE, Harris DA. Neurotoxic mutants of the prion protein induce spontaneous ionic currents in cultured cells. Journal of Biological Chemistry. 2010;285(34):26719–26726. doi: 10.1074/jbc.M110.134619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Jeffrey M, McGovern G, Goodsir CM, Brown KL, Bruce ME. Sites of prion protein accumulation in scrapie-infected mouse spleen revealed by immuno-electron microscopy. Journal of Pathology. 2000;191(3):323–332. doi: 10.1002/1096-9896(200007)191:3<323::AID-PATH629>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 241.Cunningham C, Deacon R, Wells H, et al. Synaptic changes characterize early behavioural signs in the ME7 model of murine prion disease. European Journal of Neuroscience. 2003;17(10):2147–2155. doi: 10.1046/j.1460-9568.2003.02662.x. [DOI] [PubMed] [Google Scholar]
- 242.Chiesa R, Piccardo P, Dossena S, et al. Bax deletion prevents neuronal loss but not neurological symptoms in a transgenic model of inherited prion disease. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(1):238–243. doi: 10.1073/pnas.0406173102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Scheff SW, Price DA, Schmitt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiology of Aging. 2006;27(10):1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 244.Mallucci G, Dickinson A, Linehan J, Klöhn P-C, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302(5646):871–874. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- 245.Moreno JA, Radford H, Peretti D, et al. Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature. 2012;485(7399):507–511. doi: 10.1038/nature11058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Beringue V, Demoy M, Lasmézas CI, et al. Role of spleen macrophages in the clearance of scrapie agent early in pathogenesis. Journal of Pathology. 2000;190(4):495–502. doi: 10.1002/(SICI)1096-9896(200003)190:4<495::AID-PATH535>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 247.Luhr KM, Nordström EK, Löw P, Ljunggren H-G, Taraboulos A, Kristensson K. Scrapie protein degradation by cysteine proteases in CD11c+ dendritic cells and GT1-1 neuronal cells. Journal of Virology. 2004;78(9):4776–4782. doi: 10.1128/JVI.78.9.4776-4782.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. Journal of Neuroscience. 2005;25(40):9275–9284. doi: 10.1523/JNEUROSCI.2614-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Williams AE, Lawson LJ, Perry VH, Fraser H. Characterization of the microglial response in murine scrapie. Neuropathology and Applied Neurobiology. 1994;20(1):47–55. doi: 10.1111/j.1365-2990.1994.tb00956.x. [DOI] [PubMed] [Google Scholar]
- 250.Perry VH, Cunningham C, Boche D. Atypical inflammation in the central nervous system in prion disease. Current Opinion in Neurology. 2002;15(3):349–354. doi: 10.1097/00019052-200206000-00020. [DOI] [PubMed] [Google Scholar]
- 251.Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nature Reviews Immunology. 2007;7(2):161–167. doi: 10.1038/nri2015. [DOI] [PubMed] [Google Scholar]
- 252.Holmes C, El-Okl M, Williams AL, Cunningham C, Wilcockson D, Perry VH. Systemic infection, interleukin 1β, and cognitive decline in Alzheimer’s disease. Journal of Neurology Neurosurgery and Psychiatry. 2003;74(6):788–789. doi: 10.1136/jnnp.74.6.788. [DOI] [PMC free article] [PubMed] [Google Scholar]