

Abstract
The poor overall prognosis of Gallbladder carcinoma (GBC) patients and the limited therapeutic regimens for these patients demonstrates the need for better therapeutic modalities, while the growing evidences have indicated that those genes contributed to epigenetic regulation may serve as therapeutic targets. The function of histone acetylation on growth and survival of GBC cells remains unknown. In present study, an RNAi screening of 16 genes involving histone acetyltransferases (HATs) was applied to GBC-SD cells and we found that KAT5 knockdown specifically inhibits the proliferation of GBC-SD cells by casp9-mediated apoptosis. Microarray data analysis showed that KAT5 RNAi may result in cleaved casp9 upregulation through p38MAPK activation in GBC-SD cells. The mRNA expression level of KAT5 was significantly upregulated in GBC tissues than in the adjacent normal tissues. In consistence with the mRNA level, the protein expression of KAT5 was markedly increased in tissues from patients with poor prognosis than those with good prognosis. These findings strongly indicated that KAT5 was implicated in GBC tumorigenesis and that its expression level was associated with the prognosis. Our work may also provide a potential therapeutic target for treatment of GBC patients.
Keywords: Gallbladder carcinoma, histone acetyltransferases, RNAi screening, KAT5, p38MAPK
Introduction
Gallbladder carcinoma (GBC), the most common and aggressive type of biliary tract cancer (BTC), is challenging to treat due to both its inability to be detected at an early stage and its poor sensitivity to conventional therapies [1,2]. Owing to its nonspecific symptoms, the majority of patients already have locally advanced or metastatic disease at the time of diagnosis [1]. According to the report from American Joint Committee on Cancer in 2010, the 5-year survival rate for advanced or metastatic (stage IIIA-IVB) GBC patients is below 10%. So far, complete surgical resection confers the only chance for cure; however, only 10% of patients with early stage disease are considered surgical candidates. Recurrence rates are high among the patients undergone “curative” resection and no adjuvant treatments have been established in this setting [2]. For patients with unresectable or metastatic GBC, the current chemotherapy is limited and gemcitabine-based therapy only presents 20-30% of the response rate and less than 1 year of the median overall survival [3-6]. The poor overall prognosis of GBC patients demonstrates the need for better therapeutic modalities.
The growing evidences suggest that epigenetic modifications as well as genetic alterations are implicated in cancer initiation and progression. It is recently proposed that epigenetic aberration may catalyze the occurrence of genetic alterations in somatic cells and the crosstalk between epigenetic and genetic alterations persistently plays critical role in the entire course of cancer initiation and progression [7]. Therefore, genes involved in epigenetic modifications are another type of potential therapeutic targets other than genetic mutation.
Histone modifications are post-translational chemical modifications on histone, including acetylation, methylation, phosphorylation, ubiquitination and sumoylation [8]. Histone modifications are important in transcriptional regulation and are often associated with DNA methylation [9], another type of epigenetic modifications. Disruption of normal patterns of covalent histone modifications is a hallmark of cancer [10]. Regarding histone acetylation, it is regulated by two types of enzymes, histone acetyltransferases (HATs) and histone deacetyltransferases (HDACs), which introduce and remove acetyl groups, respectively. The acetylation of lysine residues on histones is generally associated with active gene transcription. Several HAT aberrations have been reported in various cancers, such as mutations of E1A-binding protein p300 (EP300) in breast, colorectal and pancreatic cancer [11], mutations, translocations and deletions of cAMP response element-binding protein (CREBBP) in colon, stomach, endometrium, lung cancer and leukemia [8], mutation of KAT2B (PCAF) in epithelial cancer [11], translocations of KAT6A (MOZ, MYST3) and KAT6B (MORF, MYST4) in hematological malignancies [12]. These indicate the possibility of HATs genetic alterations in GBC. Moreover, several HATs genes, such as KAT2B (PCAF) [13], KAT2A (GCN5) [14], EP300 [15] and KAT8 [16], have been implicated to cancer cell growth and survival. Furthermore, 3 naturally occurring small molecules have been described as HAT inhibitors: curcumin, garcinol and anacardic acid. Curcumin is a specific inhibitor for EP300 and CREBBP and it has antitumor activities in a wide variety of cancers [17,18]. Garcinol and anacardic acid are both EP300 and KAT2B HAT inhibitors, and they both may improve cancer therapy [19]. Taken together, HATs are new potential targets for the design of therapeutics.
HDAC inhibitors, such as trichostatin A (TSA) [20], SAHA [21] and PCI-24781 [22], have been reported to inhibit the proliferation and growth of GBC cells in vitro and in vivo. However, the roles of HATs on GBC remain unknown and need to be defined.
In present study, an RNAi screening of 16 genes involving HATs was applied to GBC-SD cells and we found that KAT5 knockdown specifically inhibits the proliferation of GBC-SD cells by casp9-mediated apoptosis. Microarray data analysis showed that KAT5 inhibition might result in cleaved casp9 upregulation through p38MAPK activation. The mRNA expression of KAT5 was significantly higher in GBC tissues than in the adjacent normal tissues and the protein expression of KAT5 was also markedly higher in tissues from patients with poor prognosis than that with good prognosis, examined by real-time quantitative PCR and IHC, respectively. These proposed that KAT5 was implicated in GBC tumorigenesis and that its protein expression was associated with the prognosis.
Materials and methods
Cell culture
The human gallbladder carcinoma cell line GBC-SD was purchased from China Center for Type Culture Collection and maintained in RPMI 1640 medium (Gibco) supplemented with 10% FBS (Hyclone), penicillin (100 IU/ml) and Streptomycin (100 μg/ml) (Life Technologies) in a humidified atmosphere containing 5% CO2 at 37°C. Cells in the exponential growth phase were used for all the experiments.
shRNA construction and lentivirus infection
16 genes involving HATs were subjected to shRNA primer design and eight distinct shRNA fragments for each gene were constructed into lentivirus vector (Invitrogen, BLOCK-iT™ Lentiviral RNAi Expression System, K4944-00), the eight shRNA plasmids for each gene were mixed with equal amount and the mixtures were applied to shRNA lentivirus package and the obtained lentivirus were titered in HEK293T cells according to the manufacturer’s protocol. The obtained shRNA pools for each gene were used for RNAi screening. After the positive genes influenced proliferation of GBC-SD cells were selected by primary RNAi screening, lentivirus of the eight shRNA fragments for positive genes were packaged respectively as mentioned above. GBC-SD cells were infected by shRNA lentivirus at MOI of 10 in the presence of polybrene (8 μg/ml).
RNAi screening
GBC-SD cells were incubated in RPMI 1640 (10% FBS) at 1000 cells each well in 96-well plates, and then infected respectively with 16 HAT lentiviral shRNA pools for 96 h. Then the cell viability was detected by MTS assay (Promega, G3580) according to the operation manual. RNAi screening was completed in triplicate. The genes that inhibit cell viability of GBC-SD by higher than 35% following RNAi treatment were defined as positive hits.
Eight distinct shRNA species targeting each positive gene were used to revalidate hits from the screen. A significance threshold of p<0.05 (Student’s t test) was used for each individual siRNA. Validation of RNAi gene silencing was measured by quantitative PCR as described below.
Quantitative real-time PCR (qPCR)
GBC-SD cells were incubated in RPMI 1640 (10% FBS) at 8 × 104 cells each well in 6-well plates, and then infected respectively with eight distinct shRNA species targeting each positive gene for 96 h. Total RNA was isolated and synthesized to cDNA using PrimeScript RT reagent kit with gDNA Eraser (Takara, RR074A) for RT-PCR with mixture of oligo-dT and Random Primer (9 mer). The primers used for qPCR validation were list in Table 1. Real-time qPCR was performed on CFX-96 (Bio-lab), with endogenous control hActb. Gene expression was calculated relative to expression of hActb endogenous control and adjusted relative to expression in GBC-SD cells treated with control shRNA lentivirus.
Table 1.
Primers for qPCR validation
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Actb | GCATCCCCCAAAGTTCACAA | GGACTTCCTGTAACAACGCATCT |
| KAT5 | GGGGAGATAATCGAGGGCTG | CCACTCATCTTCGTTGTCCTG |
| ATF2 | CTTGAATGTTTAACGATGCCTTCA | TCCAAGCCTGCACAATAATGAC |
| EPC1 | AAGCAGCTCATTCACATACAGC | ACTTCATCTTCAGAATCCAAATCA |
Approximately 100 mg of fresh GBC tissues and adjacent normal tissues from GBC patients (n=11) were ground in liquid nitrogen and homogenized with 1 ml Trizol (Invitrogen, Life Technologies), respectively. Total RNA was isolated and synthesized to cDNA as described above. Gene expression was calculated relative to expression of hActb endogenous control.
Flowcytometry
Cells were harvested with trypsin-EDTA, washed with PBS, and fixed with 70% ethanol at -20°C for a few days. The fixed cells were pelleted, resuspended in 100 μL of hypotonic citric buffer (192 mmol/L Na2HPO4 and 4 mmol/L citric acid), and incubated for 30 minutes at room temperature. The cells were pelleted and suspended in PI/RNase/PBS (100 μg/mL propidium iodide and 10 μg/mL RNase A) overnight at 4°C. Analysis of DNA content was done on a FACSCalibur system (BD Immunocytometry Systems, San Jose, CA).
Protein isolation and western blotting
Cell pellets were resuspended in 1 × SDS loading buffer (1 mmol/L Na3VO4, 10 mmol/L NaF, 1 mmol/L PMSF) containing protease inhibitors. Lysates (20 μg each lane) were applied to SDS-PAGE. Immunoblotting of Abs specific for GAPDH (Abmart, 080922), p53 (ABclonal Biotechnology, A2063), Bax (ABclonal Biotechnology, A0207), CCNB1 (Epitomics, 1495-1), cleaved Casp3 (cell signaling, #9664), cleaved Casp9 (cell signaling, #9501), AKT (Santa Cruz, sc-8312), p-AKT (Santa Cruz, SC-7985-R, pS473), ERK (Abclonal, A0228) and p-ERK (Cell signaling, #9106S, pT202/204) were detected using HRP-conjugated anti-mouse (Promega) or anti-rabbit (Promega) and visualized by chemiluminescence detection system (Millipore, WBKLS0500).
Microarray analysis
GBC-SD cells (8 × 104) were grown in 2 ml of RPMI 1640 medium containing serum per well in a 6-well plate and then left untreated or treated with KAT5 shRNA lentivirus for 96 h. Every treatment was duplicated in the same experiment. All the samples were homogenized with 1 ml Trizol (Invitrogen, Life Technologies) and total RNAs were extracted according to the manufacturer’s instruction.
500 ng total RNA was used to synthesize double-strand cDNA and in vitro transcribed to cRNA, purified 10 μg cRNA was used to synthesize 2nd-cycle cDNA and then hydrolyzed by RNase H and purified. Above steps were performed with Ambion WT Expression Kit. 5.5 μg 2nd-cycle cDNA was fragmented and the single-stranded cDNA was labeled with GeneChip2 WT Terminal Labeling Kit and Controls Kit (Affymetrix, PN 702880). About 700 ng fragmented and labeled single-stranded cDNA were hybridized to an Affymetrix GeneChip Human Gene 1.0 ST array, which was washed and stained with GeneChip2 Hybridization, Wash and Stain kit (Affymetrix).
Microarray data analysis was done using Significance Analysis of Microarrays (SAM) method, as described before [23]. Functional annotation was performed to the differential expression genes with Ingenuity Pathway Analysis (IPA) online software.
Immunohistochemical staining
Immunohistochemical staining was performed as previously described [24]. Briefly, 3- to 4-μm thick paraffin sections were cut and mounted on Superfrost Plus slides that were exposed in a pressure cooker to EDTA buffer, pH 8.0, for antigen retrieval. An automated immunohistochemistry procedure was performed using the I6000 immunostainer (Biogenics, San Ramos, CA). Endogenous peroxidase activity was blocked by 10 minutes of treatment with 3% hydrogen peroxide in methanol. The sections were incubated with mouse monoclonal antibodies against KAT5 (Santa Cruz, sc-25378) for 30 minutes at room temperature and immunoperoxidase staining was accomplished using the Supersensitve Detection Kit with AEC or DAB (Zymed Labs, San Francisco, CA) as substrates, then counterstained with hematoxylin before coverslipping and reading by light microscopy. Statistical comparisons between groups were performed using the Wilcoxon test. All statistical analyses were performed using the SPSS software program (version 21, SPSS Japan Inc., Tokyo, Japan), and differences were considered to be statistically significant for values of p<0.05.
Results
RNAi screening to identify genes affecting proliferation of GBC-SD cells
To examine the determinants of gallbladder cancer cells proliferation, we designed an RNA interference screening targeting 16 known and putative HAT genes. The screening involved infecting a gallbladder cancer cell line, GBC-SD, with a 96-well plate arrayed library of shRNA triplicate. 96 h later, cell viability was measured to assess the effect of each shRNA on cell growth (Figure 1). We used a library of shRNA arrayed as SMARTpools; each SMARTpool (contained within one well of a 96-well plate) was composed of eight distinct shRNA species targeting different sequences of the same target transcript. The screening results showed that knockdown of 7 HAT genes (ATF2, CREBBP, EP300, EPC1, HAT1, KAT2A and KAT5) dramatically inhibited (relative viability was lower than 0.65 and p<0.05) the proliferation of GBC-SD cells (Figure 1).
Figure 1.
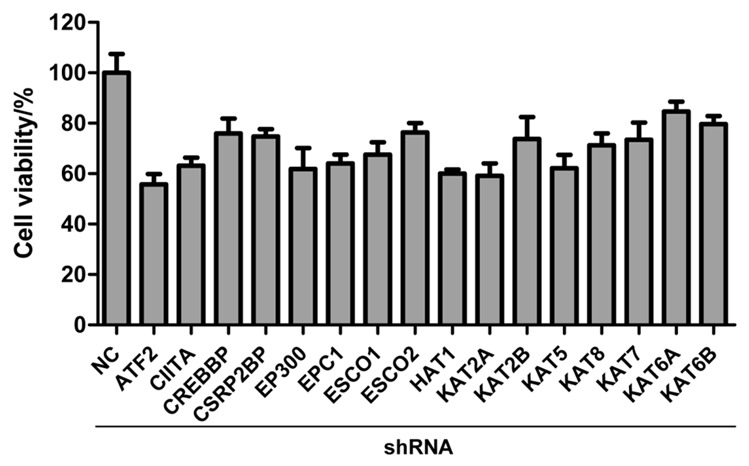
Cell viability in RNAi screening with shRNA library against 16 HAT genes. GBC-SD cells were treated with negative control (NC) shRNA or shRNA pools targeted to HAT genes. 96 h later, the cell viability was detected by MTS assay. RNAi screening was completed in triplicate. The error bar represents the standard deviation (SD).
We validated the performance of GBC-SD cells in the RNAi screen as follows: (1) infections of GBC-SD cells with shRNA targeting HATs genes previous reported to influence cellular viability, such as p300 [15] and KAT2B [13], were observed to cause a reduction in viability of 38% and 32%, respectively, compared to infection with a nontargeting shRNA, shControl (Figure 1), indicating that high-efficiency infection could be achieved; and (2) infection of GBC-SD with shControl did not reduce cellular viability more than 20%, compared to mock-infected cells, indicating that these cells could be infected without excessive nonspecific toxicity, which would reduce the sensitivity of the screen. The 7 shRNAs causing the most significantly reduction in cell viability (cell viability <0.65, p<0.05) were list in Table 2.
Table 2.
Seven positive hits in RNAi screening
| GENE | viability | P value | GENE | viability | P value |
|---|---|---|---|---|---|
| ATF2 | 0.558 | 0.001 | HAT1 | 0.6 | 0.001 |
| CREBBP | 0.635 | 0.005 | KAT2A | 0.592 | 0.001 |
| EP300 | 0.618 | 0.004 | KAT5 | 0.622 | 0.002 |
| EPC1 | 0.641 | 0.002 |
KAT5, ATF2 and EPC1 silencing specifically inhibits GBC-SD cell growth
To validate the specificity of the effects observed and rule out the possibility of off-target effects in RNAi screening, the most potent growth affecting hits were re-assayed using each of the eight different shRNA species that comprise the SMARTpools. For KAT5, ATF2 and EPC1, there are at least two shRNA fragments that markedly repressed GBC-SD cell growth following RNAi (Figure 2A), indicating that the growth inhibition by gene silencing is unlikely to be caused by off-targets effect. Moreover, the gene silencing efficacy of the two shRNA fragments caused the most outstanding inhibition on cell growth for these three genes were examined by quantitative PCR (Figure 2B). The results showed that the two shRNAs targeting to the 3 genes that caused the most significant effects on cell growth were also shown to cause the most significant gene silencing. Taken together, KAT5, ATF2 and EPC1 silencing specifically inhibits GBC-SD cell growth.
Figure 2.
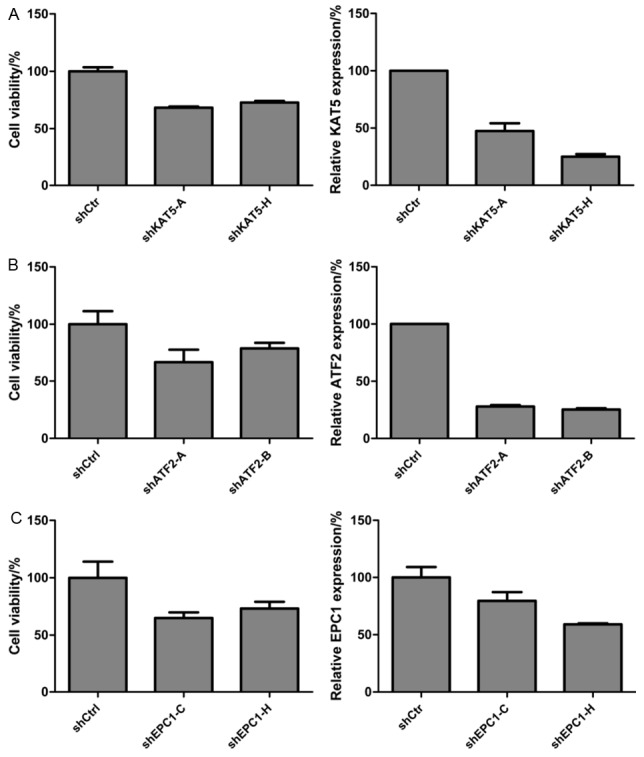
Silencing of KAT5, ATF2 and EPC1 specifically inhibits proliferation of GBC-SD cells. The cell viability (left panel) and the relative expression of target genes (right panel) were investigated by MTS assay and qPCR, respectively, following treatment with the most effective two shRNA fragments of KAT5 (A), ATF2 (B) and EPC1 (C). shKAT5-A means A fragment shRNA for KAT5, and so on. Every experiment was triplicate and the error bar represents the SD.
KAT5 is highly expressed in GBC tissues
To investigate the roles of these 3 genes in GBC, the mRNA expression of the 3 genes was compared between the GBC tissues and the adjacent normal tissues from 11 GBC patients (Figure 3). The data showed that KAT5 was significantly overexpressed in GBC tissues (p=0.01) compared to the adjacent normal tissues whereas there were no significant differences in ATF2 and EPC1 expression in the two tissues sets, suggesting KAT5 potentially plays roles on GBC initiation or progression and KAT5 may be a more valuable therapeutic target for GBC treatment than the other two genes. Therefore, the subsequent research will be focused on the mechanism by which KAT5 influences the growth of GBC-SD cells.
Figure 3.
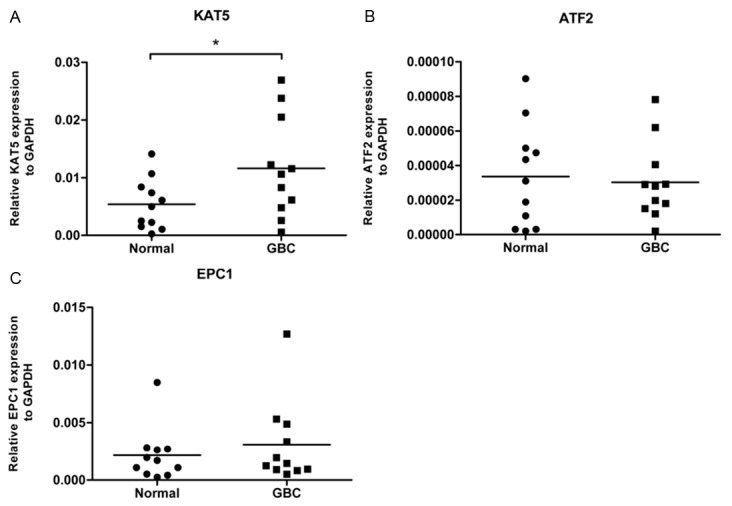
KAT5 mRNA was highly expressed in tumor tissues from GBC patients. Transcriptional levels of KAT5 (A), ATF2 (B) and EPC1 (C) were examined in 11 tumor tissues and 11 adjacent normal tissues from GBC patients by qPCR. The expression of these 3 genes was calculated relative to the endogenous GAPDH of each sample. The “*” in Figure 3A means that p<0.05.
KAT5 silencing results in apoptosis of GBC-SD cells
Several HATs, including p300, CREBBP (CBP), KAT2B (PCAF) and KAT2A (GCN5) have been implicated in the transcription activation of the cyclin-dependent kinase (cdk) inhibitor p21 as well as E2F1, Cyclin D1, and Cyclin E1 [13,14] and promoting cell growth and the G1/S phase transition. We explored the possibility that KAT5 knockdown leads to deregulation of cell cycle by flowcytometry. Interestingly, KAT5 silencing resulted in an unexpected significant increase in sub-G1 phase and a concomitant decrease in G1/S phase (Figure 4A, 4B), suggesting apoptosis is induced by KAT5 RNAi in GBC-SD cells. And then, expression of apoptosis- and cell cycle-related proteins, such as p53, Bax and CCNB1, was observed by immunoblotting to figure out the underlying signaling in this phenomenon. The results showed that there were no dramatic expression-changes in these 3 proteins (Figure 4B). Furthermore, cell survival related signaling, AKT and ERK were found to be not significantly changed. However, the cleaved Caspase-9 was markedly unregulated following KAT5 knockdown. Collectively, KAT5 knockdown induces apoptosis of GBC-SD cells by a special way.
Figure 4.

KAT5 silencing induced apoptosis of GBC-SD cells through upregulation of cleaved Casp9. A: Cell population of each phase in cell cycle after KAT5 silencing. The left panel is the graph of flowcytometry results. The ratio changes in each phase were summarized in the right histogram. Sub-G1 phase was dramatically increased following KAT5 silencing, suggesting KAT5 knockdown induces apoptosis of GBC-SD cells. B: Western blotting for apoptosis- and growth-related proteins. The cleaved Casp9 was significantly upregulated after KAT5 knockdown.
Microarray analysis reveals KAT5 silencing causes activation of p38MAPK signaling
And then, to find the signaling underlying the pro-apoptosis by KAT5 silencing, GBC-SD cells treated with shCtr or shKAT5 were applied to DNA microarray analysis. The microarray data was analyzed by SAM method and the most significantly expression-changed 200 genes were applied to Ingenuity Pathway Analysis (IPA). IPA results showed that these differentially expressed genes were mainly enriched in the ATM signaling, cell cycle signaling, iNOS signaling and TGF-β signaling. Interestingly, p38-MAPK was predicted to be activated in the latter two signaling (Figure 5A). This prediction was based on the fact that 7 of 8 genes (TYR, RHOB, NOX1, DUSP1, CYP19A1, CDKN1A, CCL3L1 and CCNF) regulated by p38MAPK had expression direction consistent with activation of p38MAPK. Furthermore, DNA microarray data were validated by qPCR of 12 genes (Figure 5B). Expression trends of 11 genes were consistent between the microarray data and qPCR data, suggesting that the microarray data were reliable for further analysis.
Figure 5.
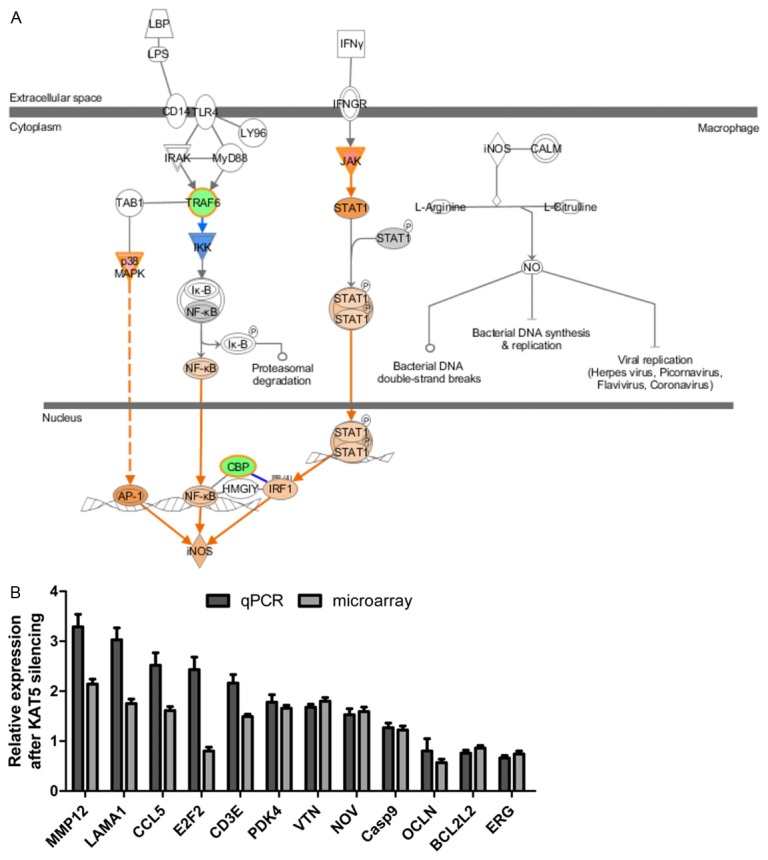
DNA microarray analysis. A: IPA results for the significantly expression-altered genes showed that i-NOS signaling was activated following KAT5 knockdown. The p38MAPK was one of the upstream of i-NOS signaling and it was predicted to be activated. B: qPCR validation of microarray data. Expression trends of 11 out of 12 genes were consistent between qPCR and microarray data.
High KAT5 protein expression is associated with poor prognosis
And then tissues from 46 patients with gallbladder cancer were used to detect the protein expression of KAT5 by immunochemistry (IHC). Among these 46 patients, 30 had good prognosis and 16 had poor prognosis. 3 sections from each tissue were selected to evaluate the protein expression. The relationship between the KAT5 protein expression and prognosis was examined through SPSS software. The results showed that KAT5 protein is highly expressed in patients with poor prognosis compared to those with good prognosis (0.066 vs 0.031, p=0.01), the typical photograph was shown in Figure 6, suggesting that KAT5 is not only in positive regulation of GBC cells growth but also in negatively correlated to prognosis.
Figure 6.
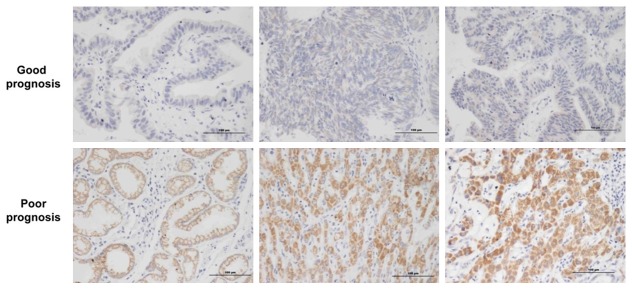
Immunohistochemistry (IHC) for KAT5 expression in GBC patients. KAT5 protein was highly expressed in tissues from patients with poor prognosis. The scale in each photograph is 100 μm.
Discussion
The poor overall prognosis of GBC patients and the limited therapeutic regimens for these patients demonstrates the need for better therapeutic modalities. The growing evidences suggest that genes involving in epigenetic regulation may serve as therapeutic targets with high potency in some types of cancer. But the function of histone acetylation on the growth and survival of GBC cells remains largely unknown.
To understand the role of histone acetylation in GBC cells, an RNAi screening with shRNAs library against 16 HAT genes was applied to GBC-SD cells. The screening results showed that knockdown of 7 HAT genes (ATF2, CREBBP, EP300, EPC1, HAT1, KAT2A and KAT5) dramatically inhibited (relative viability was lower than 0.65 and p<0.05) the proliferation of GBC-SD cells. And then, the effects of 3 genes (ATF2, EPC1 and KAT5) on proliferation of GBC-SD cells were validated and the off-target effect of shRNA was excluded, silencing of these 3 genes specifically inhibits the cell viability of GBC-SD cells.
ATF2 protein is a transcription factor important for normal cellular development and survival. In addition to its role as a transcription factor, ATF2 functions in the DNA damage response and in control of HAT complex activity. ATF2 exerts oncogenic or tumor suppressor activities, depending on the tissue and cell context. In melanoma [25] and our GBC models, ATF2 elicits oncogenic activities, knockdown of ATF2 suppresses the growth of cancer cells; while in breast cancer [25,26] and head and neck carcinoma [27], ATF2 exhibits tumor suppressor activities. Enhancer of polycomb 1 (EPC1) protein is a component of the NuA4 histone acetyltransferase complex and can act as both a transcriptional activator and repressor [28]. EPC1 is found to interact with EZH2 in a proliferation-specific manner [29], the alteration of EPC1 at 10p11.2 is one of the genetic events leading to development of adult T-cell leukemia/lymphoma [30]. KAT5 has a role in DNA repair and apoptosis and is typically thought as a tumor suppressor gene in lymphoma, colorectal cancer and melanoma [31-33], however, it is recently reported that KAT5 promotes prostate cancer cell proliferation [34], which is in consistent with our data in GBC cells. Collectively, these HAT genes may serve as oncogenic or tumor suppressor gene, depending on the tissue and cell context. In GBC cells, ATF2, EPC1 and KAT5 seem likely to be oncogenic.
And then, the transcriptional levels of these 3 HAT genes in 11 tumor tissues or the 11 adjacent normal tissues from GBC patients were investigated by qPCR. The results showed that KAT5 but not ATF2 and EPC1 mRNA is significantly highly expressed in tumor tissues than that in normal tissues, suggesting that KAT5 may potentially play roles on GBC initiation or progression and KAT5 may be a more valuable therapeutic target for GBC treatment than the other two genes. KAT5 mRNA expression has been reported to be reduced in gastric cancer [35], which is contrary to our results in GBC tissues. Furthermore, our IHC data proposed that highly KAT5 protein level is negatively correlated to the prognosis of GBC patients, which is also inconsistent with previous studies in melanoma and bladder cancer [33,36]. Because the amount of the used GBC tissues in our qPCR and IHC data was relative little, it is necessary to investigate the influence of the mRNA and protein expression of KAT5 on GBC initiation and progression and prognosis of patients in more tissues from GBC patients. Moreover, it is reported that KAT5 is frequently mono-allelic lost in human lymphomas and head-and-neck and mammary carcinomas, with concomitant reduction in mRNA levels [31]. Therefore, it is of great interest to evaluate the genomic alteration of KAT5 in GBC tissues in future.
It is suggested that KAT5-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis [37,38]. Furthermore, KAT5 is proposed to connect chromatin to DNA damage signaling, such as ATM signaling [39,40]. Consistent with these, our microarray data showed that ATM and cell cycle signaling were significantly altered following KAT5 knockdown. Flowcytometry assay showed that GBC-SD cells were induced to apoptosis after KAT5 silencing, however, the subsequent immunoblotting showed that cleaved Casp9 other than p53, BAX, CCNB1, ERK and AKT was increased dramatically. IPA results predicted that p38MAPK was activated following KAT5 silencing. p38MAPK has been proposed to mediate cancer cell apoptosis [41,42], this warrants further validation in more GBC cell lines and tissues.
Taken together, our RNAi screening reveals that 7 HAT genes are in positive regulation of proliferation of GBC-SD cells, and 3 of 7 genes have been more evaluated. KAT5 mRNA was found to be highly expressed in tumor tissues from GBC patients, compared to the adjacent normal tissues from GBC patients. Moreover, KAT5 protein was highly expressed in tumor tissues from GBC patients with poor prognosis, compared to those from GBC patients with good prognosis. These data suggest that KAT5 is probably associated with GBC initiation or progression and prognosis. Moreover, we also found that KAT5 silencing induced GBC cells to apoptosis, which might be because that cleaved Casp9 was dramatically upregulated and p38MAPK was predicted to be activated in GBC-SD cells with KAT5 knockdown. Our work may provide potential therapeutic target and predictive biomarker for treatment or prognosis of GBC patients.
Acknowledgements
This work is granted by National Natural Sci-ence Foundation of China (NSFC, No. 81172019).
Disclosure of conflict of interest
None.
References
- 1.Misra S, Chaturvedi A, Misra NC, Sharma ID. Carcinoma of the gallbladder. Lancet Oncol. 2003;4:167–176. doi: 10.1016/s1470-2045(03)01021-0. [DOI] [PubMed] [Google Scholar]
- 2.Zhu AX, Hong TS, Hezel AF, Kooby DA. Current management of gallbladder carcinoma. Oncologist. 2010;15:168–181. doi: 10.1634/theoncologist.2009-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Knox JJ, Hedley D, Oza A, Feld R, Siu LL, Chen E, Nematollahi M, Pond GR, Zhang J, Moore MJ. Combining gemcitabine and capecitabine in patients with advanced biliary cancer: a phase II trial. J. Clin. Oncol. 2005;23:2332–2338. doi: 10.1200/JCO.2005.51.008. [DOI] [PubMed] [Google Scholar]
- 4.Kim ST, Park JO, Lee J, Lee KT, Lee JK, Choi SH, Heo JS, Park YS, Kang WK, Park K. A Phase II study of gemcitabine and cisplatin in advanced biliary tract cancer. Cancer. 2006;106:1339–1346. doi: 10.1002/cncr.21741. [DOI] [PubMed] [Google Scholar]
- 5.Andre T, Tournigand C, Rosmorduc O, Provent S, Maindrault-Goebel F, Avenin D, Selle F, Paye F, Hannoun L, Houry S, Gayet B, Lotz JP, de Gramont A, Louvet C GERCOR Group. Gemcitabine combined with oxaliplatin (GEMOX) in advanced biliary tract adenocarcinoma: a GERCOR study. Ann Oncol. 2004;15:1339–1343. doi: 10.1093/annonc/mdh351. [DOI] [PubMed] [Google Scholar]
- 6.Valle J, Wasan H, Palmer DH, Cunningham D, Anthoney A, Maraveyas A, Madhusudan S, Iveson T, Hughes S, Pereira SP, Roughton M, Bridgewater J ABC-02 Trial Investigators. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;362:1273–1281. doi: 10.1056/NEJMoa0908721. [DOI] [PubMed] [Google Scholar]
- 7.You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. 2012;22:9–20. doi: 10.1016/j.ccr.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8:286–298. doi: 10.1038/nrg2005. [DOI] [PubMed] [Google Scholar]
- 9.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 10.Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, Iyer NG, Perez-Rosado A, Calvo E, Lopez JA, Cano A, Calasanz MJ, Colomer D, Piris MA, Ahn N, Imhof A, Caldas C, Jenuwein T, Esteller M. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 11.Miremadi A, Oestergaard MZ, Pharoah PD, Caldas C. Cancer genetics of epigenetic genes. Hum Mol Genet. 2007;16 Spec No 1:R28–49. doi: 10.1093/hmg/ddm021. [DOI] [PubMed] [Google Scholar]
- 12.Yang XJ. The diverse superfamily of lysine acetyltransferases and their roles in leukemia and other diseases. Nucleic Acids Res. 2004;32:959–976. doi: 10.1093/nar/gkh252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Love IM, Sekaric P, Shi D, Grossman SR, Androphy EJ. The histone acetyltransferase PCAF regulates p21 transcription through stress-induced acetylation of histone H3. Cell Cycle. 2012;11:2458–2466. doi: 10.4161/cc.20864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen L, Wei T, Si X, Wang Q, Li Y, Leng Y, Deng A, Chen J, Wang G, Zhu S, Kang J. Lysine Acetyltransferase GCN5 Potentiates the Growth of Non-small Cell Lung Cancer via Promotion of E2F1, Cyclin D1, and Cyclin E1 Expression. J Biol Chem. 2013;288:14510–14521. doi: 10.1074/jbc.M113.458737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang H, Pinello CE, Luo J, Li D, Wang Y, Zhao LY, Jahn SC, Saldanha SA, Planck J, Geary KR, Ma H, Law BK, Roush WR, Hodder P, Liao D. Small-Molecule Inhibitors of Acetyltransferase p300 Identified by High-Throughput Screening Are Potent Anticancer Agents. Mol Cancer Ther. 2013;12:610–620. doi: 10.1158/1535-7163.MCT-12-0930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang S, Liu X, Zhang Y, Cheng Y, Li Y. RNAi screening identifies KAT8 as a key molecule important for cancer cell survival. Int J Clin Exp Pathol. 2013;6:870–877. [PMC free article] [PubMed] [Google Scholar]
- 17.Balasubramanyam K, Varier RA, Altaf M, Swaminathan V, Siddappa NB, Ranga U, Kundu TK. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J Biol Chem. 2004;279:51163–51171. doi: 10.1074/jbc.M409024200. [DOI] [PubMed] [Google Scholar]
- 18.Mukhopadhyay A, Banerjee S, Stafford LJ, Xia C, Liu M, Aggarwal BB. Curcumin-induced suppression of cell proliferation correlates with down-regulation of cyclin D1 expression and CDK4-mediated retinoblastoma protein phosphorylation. Oncogene. 2002;21:8852–8861. doi: 10.1038/sj.onc.1206048. [DOI] [PubMed] [Google Scholar]
- 19.Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17:330–339. doi: 10.1038/nm.2305. [DOI] [PubMed] [Google Scholar]
- 20.Xu LN, Wang X, Zou SQ. Effect of histone deacetylase inhibitor on proliferation of biliary tract cancer cell lines. World J Gastroenterol. 2008;14:2578–2581. doi: 10.3748/wjg.14.2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamaguchi J, Sasaki M, Sato Y, Itatsu K, Harada K, Zen Y, Ikeda H, Nimura Y, Nagino M, Nakanuma Y. Histone deacetylase inhibitor (SAHA) and repression of EZH2 synergistically inhibit proliferation of gallbladder carcinoma. Cancer Sci. 2010;101:355–362. doi: 10.1111/j.1349-7006.2009.01387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kitamura T, Connolly K, Ruffino L, Ajiki T, Lueckgen A, DiGiovanni J, Kiguchi K. The therapeutic effect of histone deacetylase inhibitor PCI-24781 on gallbladder carcinoma in BK5. erbB2 mice. J Hepatol. 2012;57:84–91. doi: 10.1016/j.jhep.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fogel M, Gutwein P, Mechtersheimer S, Riedle S, Stoeck A, Smirnov A, Edler L, Ben-Arie A, Huszar M, Altevogt P. L1 expression as a predictor of progression and survival in patients with uterine and ovarian carcinomas. Lancet. 2003;362:869–875. doi: 10.1016/S0140-6736(03)14342-5. [DOI] [PubMed] [Google Scholar]
- 25.Bhoumik A, Ronai Z. ATF2: a transcription factor that elicits oncogenic or tumor suppressor activities. Cell Cycle. 2008;7:2341–2345. doi: 10.4161/cc.6388. [DOI] [PubMed] [Google Scholar]
- 26.Lau E, Ronai ZA. ATF2 - at the crossroad of nuclear and cytosolic functions. J Cell Sci. 2012;125:2815–2824. doi: 10.1242/jcs.095000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duffey D, Dolgilevich S, Razzouk S, Li L, Green R, Gorti GK. Activating transcription factor-2 in survival mechanisms in head and neck carcinoma cells. Head Neck. 2011;33:1586–1599. doi: 10.1002/hed.21648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doyon Y, Selleck W, Lane WS, Tan S, Cote J. Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol Cell Biol. 2004;24:1884–1896. doi: 10.1128/MCB.24.5.1884-1896.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Attwooll C, Oddi S, Cartwright P, Prosperini E, Agger K, Steensgaard P, Wagener C, Sardet C, Moroni MC, Helin K. A novel repressive E2F6 complex containing the polycomb group protein, EPC1, that interacts with EZH2 in a proliferation-specific manner. J Biol Chem. 2005;280:1199–1208. doi: 10.1074/jbc.M412509200. [DOI] [PubMed] [Google Scholar]
- 30.Nakahata S, Saito Y, Hamasaki M, Hidaka T, Arai Y, Taki T, Taniwaki M, Morishita K. Alteration of enhancer of polycomb 1 at 10p11.2 is one of the genetic events leading to development of adult T-cell leukemia/lymphoma. Genes Chromosomes Cancer. 2009;48:768–776. doi: 10.1002/gcc.20681. [DOI] [PubMed] [Google Scholar]
- 31.Gorrini C, Squatrito M, Luise C, Syed N, Perna D, Wark L, Martinato F, Sardella D, Verrecchia A, Bennett S, Confalonieri S, Cesaroni M, Marchesi F, Gasco M, Scanziani E, Capra M, Mai S, Nuciforo P, Crook T, Lough J, Amati B. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature. 2007;448:1063–1067. doi: 10.1038/nature06055. [DOI] [PubMed] [Google Scholar]
- 32.Mattera L, Escaffit F, Pillaire MJ, Selves J, Tyteca S, Hoffmann JS, Gourraud PA, Chevillard-Briet M, Cazaux C, Trouche D. The p400/Tip60 ratio is critical for colorectal cancer cell proliferation through DNA damage response pathways. Oncogene. 2009;28:1506–1517. doi: 10.1038/onc.2008.499. [DOI] [PubMed] [Google Scholar]
- 33.Chen G, Cheng Y, Tang Y, Martinka M, Li G. Role of Tip60 in human melanoma cell migration, metastasis, and patient survival. J Invest Dermatol. 2012;132:2632–2641. doi: 10.1038/jid.2012.193. [DOI] [PubMed] [Google Scholar]
- 34.Shiota M, Yokomizo A, Masubuchi D, Tada Y, Inokuchi J, Eto M, Uchiumi T, Fujimoto N, Naito S. Tip60 promotes prostate cancer cell proliferation by translocation of androgen receptor into the nucleus. Prostate. 2010;70:540–554. doi: 10.1002/pros.21088. [DOI] [PubMed] [Google Scholar]
- 35.Sakuraba K, Yokomizo K, Shirahata A, Goto T, Saito M, Ishibashi K, Kigawa G, Nemoto H, Hibi K. TIP60 as a potential marker for the malignancy of gastric cancer. Anticancer Res. 2011;31:77–79. [PubMed] [Google Scholar]
- 36.Laurberg JR, Brems-Eskildsen AS, Nordentoft I, Fristrup N, Schepeler T, Ulhoi BP, Agerbaek M, Hartmann A, Bertz S, Wittlinger M, Fietkau R, Rodel C, Borre M, Jensen JB, Orntoft T, Dyrskjot L. Expression of TIP60 (tat-interactive protein) and MRE11 (meiotic recombination 11 homolog) predict treatment-specific outcome of localised invasive bladder cancer. BJU Int. 2012;110:E1228–1236. doi: 10.1111/j.1464-410X.2012.11564.x. [DOI] [PubMed] [Google Scholar]
- 37.Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006;24:827–839. doi: 10.1016/j.molcel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 38.Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24:841–851. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun Y, Jiang X, Price BD. Tip60: connecting chromatin to DNA damage signaling. Cell Cycle. 2010;9:930–936. doi: 10.4161/cc.9.5.10931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaidi A, Jackson SP. KAT5 tyrosine phosphorylation couples chromatin sensing to ATM signalling. Nature. 2013;498:70–74. doi: 10.1038/nature12201. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 41.Dong J, Huang S, Caikovski M, Ji S, McGrath A, Custorio MG, Creighton CJ, Maliakkal P, Bogoslovskaia E, Du Z, Zhang X, Lewis MT, Sablitzky F, Brisken C, Li Y. ID4 regulates mammary gland development by suppressing p38MAPK activity. Development. 2011;138:5247–5256. doi: 10.1242/dev.069203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de la Cruz-Morcillo MA, Valero ML, Callejas-Valera JL, Arias-Gonzalez L, Melgar-Rojas P, Galan-Moya EM, Garcia-Gil E, Garcia-Cano J, Sanchez-Prieto R. P38MAPK is a major determinant of the balance between apoptosis and autophagy triggered by 5-fluorouracil: implication in resistance. Oncogene. 2012;31:1073–1085. doi: 10.1038/onc.2011.321. [DOI] [PubMed] [Google Scholar]