

Abstract
S-nitrosoglutathione reductase (GSNOR) is a key denitrosylase and critically important for protecting immune and other cells from nitrosative stress. Pharmacological inhibition of GSNOR is being actively pursued as a therapeutic approach to increase S-nitrosoglutathione levels for the treatment of asthma and cystic fibrosis. In the present study, we employed GSNOR-deficient (GSNOR−/−) mice to investigate whether inactivation of GSNOR may increase susceptibility to pulmonary infection by Klebsiella pneumoniae, a common cause of nosocomial pneumonia. We found that compared to wild-type mice, bacterial colony forming units 48 hours after intranasal infection with K. pneumoniae were increased over four folds in lung and spleen and strikingly, over a thousand folds in blood of GSNOR−/− mice. Lung injury was comparable between infected wild-type and GSNOR−/− mice, but inflammation and injury was significantly elevated in spleen of GSNOR−/− mice. Whereas all wild-type mice survived 48 hours after infection, 10 of 23 GSNOR−/− mice died. Thus, GSNOR appears to play a crucial role in controlling pulmonary and systemic infection by K. pneumoniae. Our results suggest that patients treated in clinical trials with inhibitors of GSNOR should be carefully monitored for signs of infection.
Keywords: Nitric oxide, S-nitrosylation, S-nitrosoglutathione reductase, Klebsiella pneumoniae
1. Introduction
Nitric oxide (NO) affects the functions of a wide range of proteins through S-nitrosylation, the covalent modification of cysteine thiols [1]. Protein S-nitrosylation is increased by NO synthases (NOSs) but down-regulated by S-nitrosoglutathione reductase (GSNOR), a ubiquitous and highly conserved denitrosylase [2–4]. By preventing excessive protein S-nitrosylation, GSNOR plays an evolutionarily conserved, critical role in protecting against nitrosative stress [2, 3]. Studies of GSNOR-deficient (GSNOR−/−) mice have shown that GSNOR deficiency results in protection from asthma and myocardial infarction but also leads to increased susceptibility to septic shock, liver cancer, and lymphopenia [3–8].
Accumulating evidence in humans and animals suggest important roles of S-nitrosothiols (SNOs) in lung. S-nitrosoglutathione (GSNO) may represent a major source of bronchodilatory NO bioactivity [9] and it is reportedly depleted from airway lining fluid in human asthmatics [10, 11]. Reduction in airway GSNO has been reported to be associated with increased GSNOR activity [12]. Single nucleotide polymorphisms in the human GSNOR gene have been linked to increased risk of asthma and decreased responsiveness to β-agonist therapy in asthmatics [13–15]. GSNOR deficiency protects mice from airway hyper responsiveness in experimental asthma [5]. In addition, GSNO may increase expression and maturation of wild-type and F508 mutant cystic fibrosis transmembrane conductance regulatory protein [16, 17].
Given the involvement of dysregulated S-nitrosylation in multiple pathological conditions, a number of approaches to regulate S-nitrosylation therapeutically are being tried in multiple indications [18]. In the lung, GSNO releasing compounds and GSNOR inhibitors are being considered as potential therapeutic approaches for asthma [19] and cystic fibrosis [17]. However, since GSNOR−/− mice appear to be more susceptible to experimental peritoneal sepsis [3] as well as lung inflammation [4], we hypothesize that GSNOR deficiency may increase sensitivity to lung infection. Klebsiella pneumoniae is a common cause of nosocomial pneumonia. Recent increase in nosocomial infection by carbapenem-resistant enterobacteriaceae, which is associated with high mortality, is mostly observed in K. pneumoniae [20]. K. pneumoniae infection induces NOS activity and NO production in mouse and human alveolar macrophages [21, 22]. NO or related reactive nitrogen species (RNS) produced by the macrophages is indispensible for effective phagocytosis and killing of K. pneumoniae [21, 22]. Therefore, we have investigated whether GSNOR−/− mice are more susceptible to pulmonary infection by K. pneumoniae.
2. Materials and methods
2.1. Animals
Wild-type C57BL/6 and congenic GSNOR−/− mice were housed in ventilated filter-top cages with centralized water supplies and fed normal mouse chow (5058 PicoLab Mouse Diet 20) ad libitum in a specific pathogen-free facility at the University of California at San Francisco (UCSF). Six- to 12-week-old mice were used for all experiments. The experimental protocol was approved by the Institutional Animal Care and Use Committee of UCSF.
2.2. Pulmonary K. pneumoniae infection
K. pneumoniae (subsp. pneumoniae (Schroeter) Trevisan (ATCC® 43816™), serotype 2) from ATCC was grown in Luria Broth (LB) overnight at 37 °C in a shaking incubator. Fifty microliters of the overnight culture was used to inoculate 25 ml of LB and grown for 3–4 hours to achieve log phase. Bacterial colony forming units (CFUs) were estimated by OD600 measurements and confirmed by culturing on LB agar plates. This culture was diluted, and anesthetized mice were inoculated intranasally with 104 CFU K. pneumoniae in 30 μl volume. Inoculated mice were observed until fully recovered from anesthesia. Mice were checked twice daily for mortality until they were euthanized at 48 hours for bacterial load assessment.
2.3. Bacterial load quantification
Lungs and spleens were aseptically harvested from euthanized mice and homogenized in 500 μl of sterile phosphate buffered saline (PBS). The homogenates were serially diluted in PBS and 10 μl of each dilution were plated on LB agar plates and incubated overnight at 37°C, after which bacterial colonies were enumerated. The left lung was used for bacterial quantification. Spleen bacterial loads are normalized to the weight of the spleen that was homogenized. , Whole blood was serially diluted in PBS and 10 μl of each dilution were immediately plated on LB agar plates for quantification of blood bacterial titers.
2.4. Histopathology
Lung and spleen samples were fixed with formalin and embedded in paraffin. Tissue sections were stained with Hematoxylin and Eosin. The extent of lung injury induced by bacterial infection was quantified by histologically scoring neutrophils in the alveolar space, neutrophils in the interstitial space, hyaline membranes, proteinaceous debris in the airway, and alveolar septal thickening, according to the guidelines of the American Thoracic Society [23]. Histological analysis and scoring were performed in five random fields per section and in a blinded fashion. Spleen sections were scored for inflammation, necrosis/abscess formation, thrombus formation, and hyperemia essentially as outlined by Wiersinga et al. [24].
2.5. Statistical analysis
Survival was compared using the two-tailed Fisher’s exact test. Histological scores were compared with the Mann-Whitney U-test. Other data were analyzed with a two-tailed, unpaired Student’s t-test.
3. Results
3.1 K. pneumoniae is increased in lung of GSNOR−/− mice in a pneumonia model
To test whether GSNOR−/− mice are sensitive to lung infection, we employed a pulmonary infection model using K. pneumoniae [25]. We found that 48 hours after intranasal infection with K. pneumoniae, bacterial CFUs in the lungs of GSNOR−/− mice were three- to 19-fold greater than wild-type control in three independent experiments (Fig. 1A). Analyzed together as a group, the bacterial CFU in GSNOR−/− lung was significantly higher than that in wild-type mice (Fig. 1B; P = 0.003). Thus, GSNOR−/− mice are more susceptible to lung infection by K. pneumoniae.
Fig. 1.

Increased K. pneumoniae infection in the lungs of GSNOR−/− mice in a pneumonia model. (A) Bacterial loads in the left lung 48 hours after intranasal K. pneumoniae infection of wild-type (WT, open circles) and GSNOR−/− (closed diamonds) mice were quantified by colony forming units (CFU) for each of three independent experiments. (B) Aggregate lung data from the three experiments shown in (A). Each data point represents a single mouse, and horizontal lines represent the group mean. Bacterial loads in GSNOR−/− (n = 23) mice are significantly higher than wild-type controls (n = 17; P = 0.003). (C) Comparable levels of lung injury in K. pneumoniae-infected wild-type (open bars) and GSNOR−/− (closed bars) mice. PMN, polymorphonuclear leukocyte.
3.2. Comparable levels of lung injury in K. pneumoniae-infected wild-type and GSNOR−/− mice
To determine whether elevated bacterial loads in GSNOR−/− mice are associated with increased lung injury, lung sections from K. pneumoniae-infected wild-type (n = 13) and GSNOR−/− (n = 11) mice were analyzed and scored for the extent of neutrophils in the alveolar space and the interstitial space, hyaline membranes, proteinaceous debris in the airway, and alveolar septal thickening [23]. There was not a clear difference in the individual parameter scores or overall histological score between wild-type and GSNOR−/− mice (Fig. 1C). These data suggest that despite increased bacterial load in the lungs of GSNOR−/− mice, both wild-type and GSNOR−/− mice exhibit similar levels of lung injury.
3.3. Highly elevated bacterial loads in blood of K. pneumoniae-infected GSNOR−/− mice
When bacterial titers in the whole blood of K. pneumoniae-infected mice were analyzed, we found a striking 1,000- to 4,450-fold greater bacterial load in GSNOR−/− mice than in wild-type controls in three independent experiments (Fig. 2A). When analyzed together, blood bacterial titer was significantly higher in GSNOR−/− mice, increasing about 1,750 fold (P = 0.001; Fig. 2B). K. pneumoniae was not detected in two of 17 wild-type mice analyzed, whereas all of 13 GSNOR−/− mice had detectible levels of bacteria in their blood. Also, not included in the bacterial load analysis were several GSNOR−/− mice that died between 24 and 36 hours, presumably from high bacterial loads. Thus, pulmonary infection of K. pneumoniae leads to a much more severe systemic infection in GSNOR−/− mice.
Fig. 2.
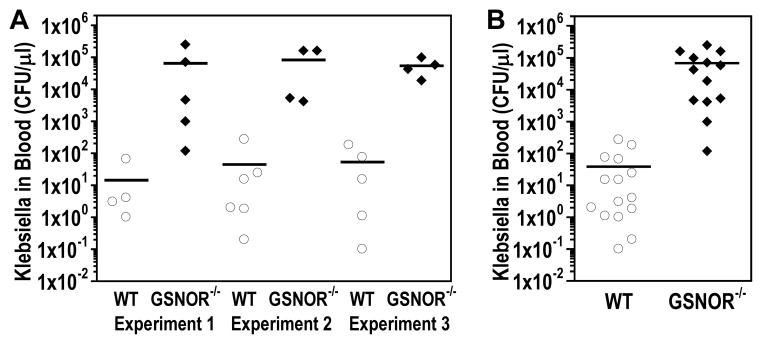
Elevated systemic dissemination of K. pneumoniae in GSNOR−/− mice. Bacterial titers in whole blood 48 hours after intranasal K. pneumoniae infection of wild-type (WT, open circles) and GSNOR−/− (closed diamonds) mice in three independent experiments individually (A) and aggregately (B). Each data point represents a single mouse, and horizontal lines represent the group mean. Blood bacterial titers are over 1,700 fold higher in GSNOR−/− mice (n = 13) compared to wild-type controls (n = 17, two mice without detectible titer not shown), P = 0.001.
3.4. Increased K. pneumoniae infection and injury in spleens of GSNOR−/− mice
We also assessed the bacterial load in the spleens of wild-type and GSNOR−/− mice after pulmonary K. pneumoniae infection. Bacterial loads in the spleen were similarly matched to that in the lung for each animal in both wild-type and GSNOR−/− strains (Fig. 3A). Histopathological examination revealed a substantial increase in pathology in spleen of K. pneumoniae-infected mice (Fig 3B and C). Notably, GSNOR−/− spleens exhibited elevated pathological scores in inflammation, thrombus formation, and hyperemia when compared with those of wild-type mice (Fig 3C). The levels of necrosis and abscess formation were not different between wild-type and GSNOR−/− mice. Thus, pulmonary infection of K. pneumoniae results in a significant increase in splenic infection and injury in GSNOR−/− mice.
Fig. 3.

Increased K. pneumoniae infection and injury in spleens of GSNOR−/− mice. (A) Ratios of bacterial loads in the spleen (CFU/g) versus those in the lung (CFU/left lung) in wild-type (open circles, n = 5) and GSNOR−/− (closed diamonds, n = 4) mice. The ratios vary little within and between groups. (B) Representative spleen histology in K. pneumoniae-infected wild-type and GSNOR−/− mice. Bottom panels represent an expanded portion of the above sections outlined. Bar = 50 μm. (C) Histological scoring of infection-associated spleen injury revealed significant differences in wild-type (open; n = 11) and GSNOR−/− (closed; n = 10) mice. *, P < 0.03.
3.5. Increased mortality in GSNOR−/− mice after K. pneumoniae infection
We monitored K. pneumoniae-infected wild-type and GSNOR−/− mice for survival twice daily for 48 hours following infection. All mice survived for the first 24 hours after infection with some GSNOR−/− mice exhibiting signs of illness. By 40 hours, wild-type mice mostly remained active while most GSNOR−/− mice became severely sick and four of 23 K. pneumoniae-infected GSNOR−/− mice died. Whereas all wild-type mice survived 48 hours after infection, 10 of 23 GSNOR−/− mice died (Fig. 4). These data suggest that GSNOR is important for protection against lethal infection by K. pneumoniae .
Fig. 4.
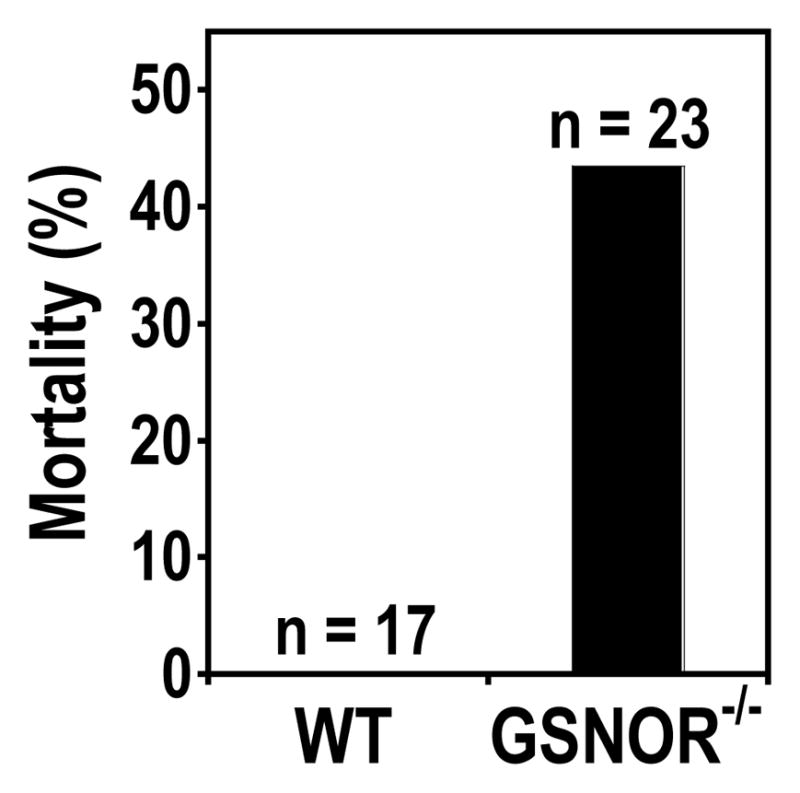
Increased mortality from K. pneumoniae infection in GSNOR−/− mice. Mortality of GSNOR−/− mice (Closed, n = 23) is significantly higher than WT control (n = 17; P = 0.002) 48 hours after intranasal infection with K. pneumoniae.
4. Discussion
In this study, we demonstrated that GSNOR−/− mice exhibited increased bacterial load and mortality in a pulmonary K. pneumoniae infection model. Bacterial loads 48 hours after infection were increased over four folds in lung and spleen and strikingly, over a thousand folds in blood of GSNOR−/− mice. Thus, GSNOR appears to play a crucial role in controlling pulmonary and systemic infection by K. pneumoniae.
The important contribution to host defense against K. pneumoniae by GSNOR may result from its protection of immune cells from nitrosative stress. K. pneumoniae infection induces NOS activity and NO production in macrophages [21, 22], which is indispensible for effective phagocytosis and killing of K. pneumoniae [21, 22]. NO production by immune cells can cause nitrosative stress, which in absence of GSNOR can cause extensive cell death [2, 4]. Thus during immune response to K. pneumoniae infection, GSNOR may be essential for protection against NO-induced damage and death of immune cells in lung and spleen. Splenic macrophages, responsible for recycling of hemoglobin-derived iron [26], might sustain increased formation of S-nitrosothiols from NO [27] and thus critically depend on disposal of S-nitrosothiols by GSNOR. Lack of this protection in GSNOR−/− mice may account for increased damage and inflammation in spleen. Injury of the spleens, including particularly splenic macrophages, may disrupt their important function in the trapping and clearance of blood-borne pathogens [26], resulting in the marked failure of systemic control of the bacterial infection in GSNOR−/− mice. In addition, GSNOR−/− mice suffer increased apoptosis from nitrosative stress in thymic development that results in decreased CD4 T cells. While mice lacking αβ-T cells showed no increased susceptibility to K. pneumoniae in a pneumonia model, mice lacking γδ-T cells displayed unimpaired clearance of pulmonary bacterial but mildly increased peripheral blood dissemination and mortality [28]. It remains to be determined whether GSNOR−/− mice exhibit a loss or deficiency of γδ-T cells.
GSNOR may contribute to the control of K. pneumoniae infection by preventing S-nitrosylation and inactivation of surfactant protein D (SP-D) or other proteins important for host defense. SP-D is secreted by lung epithelial cells and binds pathogen-associated molecular patterns as part of innate immune response [29]. SP-D can bind K. pneumoniae LPS [30] and improve phagocytosis and killing [31]. The binding to K. pneumoniae LPS requires higher-order multimerization of SP-D [30], which is inhibited by S-nitrosylation [32]. Whether GSNOR deficiency increases S-nitrosylation of SP-D or other antibacterial proteins during K. pneumoniae infection remains to be determined.
Pharmacologic approaches of inhibiting GSNOR activity have reached the point of clinical development with the recent announcement of the first cystic fibrosis patients treated with the first-in-class GSNOR inhibitor, N6022 (N30 Pharmaceuticals). Clinical development is ongoing for other indications including asthma. Our results suggest that GSNOR deficiency can increase sensitivity to K. pneumoniae, a common cause of nosocomial infection. Thus, clinical trials using S-notrosylating agents or GSNOR inhibitors should closely monitor patients for signs of infection.
Highlights.
GSNOR−/− mice are hypersensitive to pulmonary infection by Klebsiella pneumoniae.
Bacterial loads were increased over 4-fold in GSNOR−/− lung and 1000-fold in blood.
GSNOR−/− mice suffered significantly more inflammation and injury in spleen.
All wild-type mice survived the infection, but 40% of GSNOR−/− mice died.
Acknowledgments
Grant Support
This work was supported by the National Institutes of Health (R01CA55578 and R01CA122359 to L.L.).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 2.Liu L, Hausladen A, Zeng M, Que L, Heitman J, Stamler JS. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature. 2001;410:490–494. doi: 10.1038/35068596. [DOI] [PubMed] [Google Scholar]
- 3.Liu L, Yan Y, Zeng M, Zhang J, Hanes MA, Ahearn G, McMahon TJ, Dickfeld T, Marshall HE, Que LG, Stamler JS. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116:617–628. doi: 10.1016/s0092-8674(04)00131-x. [DOI] [PubMed] [Google Scholar]
- 4.Yang Z, Wang ZE, Doulias PT, Wei W, Ischiropoulos H, Locksley RM, Liu L. Lymphocyte Development Requires S-nitrosoglutathione Reductase. J Immunol. 2010;185:6664–6669. doi: 10.4049/jimmunol.1000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Que LG, Liu L, Yan Y, Whitehead GS, Gavett SH, Schwartz DA, Stamler JS. Protection from Experimental Asthma by an Endogenous Bronchodilator. Science. 2005;308:1618–1621. doi: 10.1126/science.1108228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lima B, Lam GK, Xie L, Diesen DL, Villamizar N, Nienaber J, Messina E, Bowles D, Kontos CD, Hare JM, Stamler JS, Rockman HA. Endogenous S-nitrosothiols protect against myocardial injury. Proc Natl Acad Sci U S A. 2009;106:6297–6302. doi: 10.1073/pnas.0901043106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tang CH, Wei W, Hanes MA, Liu L. Hepatocarcinogenesis Driven by GSNOR Deficiency Is Prevented by iNOS Inhibition. Cancer Res. 2013;73:2897–2904. doi: 10.1158/0008-5472.CAN-12-3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei W, Li B, Hanes MA, Kakar S, Chen X, Liu L. S-nitrosylation from GSNOR deficiency impairs DNA repair and promotes hepatocarcinogenesis. Sci Transl Med. 2010;2:19ra13. doi: 10.1126/scitranslmed.3000328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaston B, Reilly J, Drazen JM, Fackler J, Ramdev P, Arnelle D, Mullins ME, Sugarbaker DJ, Chee C, Singel DJ, et al. Endogenous nitrogen oxides and bronchodilator S-nitrosothiols in human airways. Proc Natl Acad Sci USA. 1993;90:10957–10961. doi: 10.1073/pnas.90.23.10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dweik RA, Comhair SA, Gaston B, Thunnissen FB, Farver C, Thomassen MJ, Kavuru M, Hammel J, Abu-Soud HM, Erzurum SC. NO chemical events in the human airway during the immediate and late antigen-induced asthmatic response. Proc Natl Acad Sci U S A. 2001;98:2622–2627. doi: 10.1073/pnas.051629498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaston B, Sears S, Woods J, Hunt J, Ponaman M, McMahon T, Stamler JS. Bronchodilator S-nitrosothiol deficiency in asthmatic respiratory failure. Lancet. 1998;351:1317–1319. doi: 10.1016/S0140-6736(97)07485-0. [DOI] [PubMed] [Google Scholar]
- 12.Que LG, Yang Z, Stamler JS, Lugogo NL, Kraft M. S-nitrosoglutathione reductase: an important regulator in human asthma. Am J Respir Crit Care Med. 2009;180:226–231. doi: 10.1164/rccm.200901-0158OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choudhry S, Que LG, Yang Z, Liu L, Eng C, Kim SO, Kumar G, Thyne S, Chapela R, Rodriguez-Santana JR, Rodriguez-Cintron W, Avila PC, Stamler JS, Burchard EG. GSNO reductase and beta2-adrenergic receptor gene-gene interaction: bronchodilator responsiveness to albuterol. Pharmacogenet Genomics. 2010;20:351–358. doi: 10.1097/FPC.0b013e328337f992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu H, Romieu I, Sienra-Monge JJ, Estela Del Rio-Navarro B, Anderson DM, Jenchura CA, Li H, Ramirez-Aguilar M, Del Carmen Lara-Sanchez I, London SJ. Genetic variation in S-nitrosoglutathione reductase (GSNOR) and childhood asthma. J Allergy Clin Immunol. 2007;120:322–328. doi: 10.1016/j.jaci.2007.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moore PE, Ryckman KK, Williams SM, Patel N, Summar ML, Sheller JR. Genetic variants of GSNOR and ADRB2 influence response to albuterol in African-American children with severe asthma. Pediatr Pulmonol. 2009;44:649–654. doi: 10.1002/ppul.21033. [DOI] [PubMed] [Google Scholar]
- 16.Zaman K, McPherson M, Vaughan J, Hunt J, Mendes F, Gaston B, Palmer LA. S-nitrosoglutathione increases cystic fibrosis transmembrane regulator maturation. Biochem Biophys Res Commun. 2001;284:65–70. doi: 10.1006/bbrc.2001.4935. [DOI] [PubMed] [Google Scholar]
- 17.Zaman K, Carraro S, Doherty J, Henderson EM, Lendermon E, Liu L, Verghese G, Zigler M, Ross M, Park E, Palmer LA, Doctor A, Stamler JS, Gaston B. S-nitrosylating agents: a novel class of compounds that increase cystic fibrosis transmembrane conductance regulator expression and maturation in epithelial cells. Mol Pharmacol. 2006;70:1435–1442. doi: 10.1124/mol.106.023242. [DOI] [PubMed] [Google Scholar]
- 18.Foster MW, Hess DT, Stamler JS. Protein S-nitrosylation in health and disease: a current perspective. Trends Mol Med. 2009;15:391–404. doi: 10.1016/j.molmed.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Colagiovanni DB, Drolet DW, Langlois-Forget E, Piche MP, Looker D, Rosenthal GJ. A nonclinical safety and pharmacokinetic evaluation of N6022: a first-in-class S-nitrosoglutathione reductase inhibitor for the treatment of asthma. Regul Toxicol Pharmacol. 2012;62:115–124. doi: 10.1016/j.yrtph.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 20.Vital signs: carbapenem-resistant Enterobacteriaceae. MMWR Morb Mortal Wkly Rep. 2013;62:165–170. [PMC free article] [PubMed] [Google Scholar]
- 21.Hickman-Davis JM, O'Reilly P, Davis IC, Peti-Peterdi J, Davis G, Young KR, Devlin RB, Matalon S. Killing of Klebsiella pneumoniae by human alveolar macrophages. Am J Physiol Lung Cell Mol Physiol. 2002;282:L944–956. doi: 10.1152/ajplung.00216.2001. [DOI] [PubMed] [Google Scholar]
- 22.Tsai WC, Strieter RM, Zisman DA, Wilkowski JM, Bucknell KA, Chen GH, Standiford TJ. Nitric oxide is required for effective innate immunity against Klebsiella pneumoniae. Infect Immun. 1997;65:1870–1875. doi: 10.1128/iai.65.5.1870-1875.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, Kuebler WM. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. 2011;44:725–738. doi: 10.1165/rcmb.2009-0210ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wiersinga WJ, Wieland CW, Dessing MC, Chantratita N, Cheng AC, Limmathurotsakul D, Chierakul W, Leendertse M, Florquin S, de Vos AF, White N, Dondorp AM, Day NP, Peacock SJ, van der Poll T. Toll-like receptor 2 impairs host defense in gram-negative sepsis caused by Burkholderia pseudomallei (Melioidosis) PLoS Med. 2007;4:e248. doi: 10.1371/journal.pmed.0040248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sutherland RE, Olsen JS, McKinstry A, Villalta SA, Wolters PJ. Mast cell IL-6 improves survival from Klebsiella pneumonia and sepsis by enhancing neutrophil killing. J Immunol. 2008;181:5598–5605. doi: 10.4049/jimmunol.181.8.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mebius RE, Kraal G. Structure and function of the spleen. Nat Rev Immunol. 2005;5:606–616. doi: 10.1038/nri1669. [DOI] [PubMed] [Google Scholar]
- 27.Stamler JS, Singel DJ, Loscalzo J. Biochemistry of nitric oxide and its redox-activated forms. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- 28.Moore TA, Moore BB, Newstead MW, Standiford TJ. Gamma delta-T cells are critical for survival and early proinflammatory cytokine gene expression during murine Klebsiella pneumonia. J Immunol. 2000;165:2643–2650. doi: 10.4049/jimmunol.165.5.2643. [DOI] [PubMed] [Google Scholar]
- 29.Chroneos ZC, Sever-Chroneos Z, Shepherd VL. Pulmonary surfactant: an immunological perspective. Cell Physiol Biochem. 2010;25:13–26. doi: 10.1159/000272047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sahly H, Ofek I, Podschun R, Brade H, He Y, Ullmann U, Crouch E. Surfactant protein D binds selectively to Klebsiella pneumoniae lipopolysaccharides containing mannose-rich O-antigens. J Immunol. 2002;169:3267–3274. doi: 10.4049/jimmunol.169.6.3267. [DOI] [PubMed] [Google Scholar]
- 31.Ofek I, Mesika A, Kalina M, Keisari Y, Podschun R, Sahly H, Chang D, McGregor D, Crouch E. Surfactant protein D enhances phagocytosis and killing of unencapsulated phase variants of Klebsiella pneumoniae. Infect Immun. 2001;69:24–33. doi: 10.1128/IAI.69.1.24-33.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo CJ, Atochina-Vasserman EN, Abramova E, Foley JP, Zaman A, Crouch E, Beers MF, Savani RC, Gow AJ. S-nitrosylation of surfactant protein-D controls inflammatory function. PLoS Biol. 2008;6:e266. doi: 10.1371/journal.pbio.0060266. [DOI] [PMC free article] [PubMed] [Google Scholar]