

Abstract
AIM: To evaluate the risk associated with variants of the UNC5C gene recently suspected to predispose to familial colorectal cancer (CRC).
METHODS: We screened patients with familial CRC forms as well as patients with sporadic CRCs. In a first time, we analyzed exon 11 of the UNC5C gene in 120 unrelated patients with suspected hereditary CRC, 58 patients with suspected Lynch-associated cancer or polyposis, and 132 index cases of Lynch syndrome families with a characterized mutation in a DNA mismatch repair (MMR). Next, 1023 patients with sporadic CRC and 1121 healthy individuals were screened for the variants identified in patients with familial cancer.
RESULTS: Of 120 patients with familial CRC of unknown etiology, one carried the previously reported mis-sense mutation p.Arg603Cys (R603C) and another exhibited the unreported variant of unknown significance p.Thr617Ile (T617I). The p.Ala628Lys (A628K) mutation previously described as the main UNC5C risk variant for familial CRC was not detected in any cases of familial CRC of unknown etiology, but was present in a patient with familial gastric cancer and in two Lynch syndrome patients in co-occurrence with MMR mutations. A statistically non-significant increase in cancer risk was identified in familial CRC and/or other Lynch-associated cancers (1/178 patients vs 2/1121 healthy controls, OR = 3.2, 95%CI: 0.29-35.05, P = 0.348) and in sporadic CRCs (4/1023 patients vs 2/1121 healthy controls, OR = 2.2, 95%CI: 0.40-12.02, P = 0.364).
CONCLUSION: We confirm that UNC5C mutations are very rare in familial and sporadic CRCs, but further investigations are needed to justify routine UNC5C testing for diagnostic purposes.
Keywords: Colorectal cancer, UNC5C, Genetic predisposition, Familial study, Association study, Low risk
Core tip: UNC5C was recently described as a new gene potentially predisposing to familial forms of colorectal cancer (CRC). In order to evaluate the risk of CRC associated with the variants of UNC5C exon 11 suggested to underlie this predisposition, we screened both patients with familial CRC forms of hitherto unknown etiology, and patients with sporadic CRCs. Overall, we confirm that UNC5C mutations are very rare in familial and sporadic forms of CRC. Further identifications of families with UNC5C mutations are needed to justify routine UNC5C testing in patients with CRC or gastric cancer for potential genetic counseling purposes.
INTRODUCTION
Among the common cancerous diseases, colorectal cancer (CRC) is the second most frequently diagnosed and the second leading cause of death in developed countries, after breast cancer[1]. Approximately 70% of colorectal cancers are sporadic cases caused by additive/multiplicative effects of numerous genetic and environmental low-risk factors. The remaining 30% of cases are inherited forms of the disease caused by highly penetrant germline mutations in single genes[2]. So far, very few clinically well-characterized familial forms of CRC have been associated with highly penetrant germline mutations, accounting for less than 6% of overall CRC cases such as Lynch syndrome with mutations in MMR genes (MLH1, MSH2, MSH6 and PMS2), Familial adenomatous polyposis (FAP) and attenuated FAP associated with mutations in APC, the MUTYH-associated polyposis with biallelic mutations in MUTYH, Peutz-Jegher syndrome with mutations in STK11, and juvenile polyposis syndrome with mutations in SMAD4 or BMPR1A[3,4]. Thus, most inherited CRCs, i.e., about 20%-30% of overall CRCs, are of unknown etiology, suggesting that multiple cancer loci with high penetrance variants may not yet have been discovered.
The finding reported recently by Coissieux et al[5] fits well with this scheme of missing heritability[6]. In their article, the authors highlighted UNC5C as a new gene potentially predisposing to familial forms of CRC[5]. UNC5C is a member of the UNC5H family of transmembrane receptors and functions as a dependence receptor. UNC5C induces apoptosis when unbound to its specific ligand, the laminin-related secreted protein netrin-1, whereas its pro-apoptotic properties are conversely inhibited when it is bound to netrin-1, in which case UNC5C transmits positive signals of proliferation and migration[7]. This activity is the hallmark of all known netrin-1 receptors (UNC5A, UNC5B, UNC5C, UNC5D and DCC) and is thought to be a safeguard mechanism against tumor development[8]. Like the prototypical netrin-1 receptor DCC (deleted in colorectal cancer), the expression of UNC5C is lost or markedly reduced in a large proportion of cancers[9-11], especially in colorectal cancer, because of a loss of heterozygosity or promoter methylation occurring within the UNC5C gene[12-16]. Inactivation of UNC5C would not only contribute to tumor progression, but also to the initiation of CRC formation. This suggests that UNC5C is likely to be a tumor suppressor[13,14], and consequently, that its inactivation could be considered as a selective advantage for tumor cells.
Using a candidate gene strategy, Coissieux et al[5] identified putative high-penetrance risk alleles that almost completely co-segregated with disease phenotype in several families with forms of colorectal cancers of hitherto unknown etiology. Moreover, by screening individuals with a form of CRC distinct from Lynch syndrome, familial polyposis or hamartomatous syndromes, Mehlen’s team identified four putative pathogenic substitutions in UNC5C (NCBI accession number NM_003728), including one in exon 7 c.1057G>A/p.Asp353Asn (D353N) and three in exon 11 [c.1807C>T / p.Arg603Cys (R603C), c.1882_1883GC>AA/p.Ala628Lys (A628K), and c.1888C>G/p.Gln630Glu (Q630E)][5]. No statistically significant differences in frequencies were observed between probands and controls for D353N, R603C, and Q630E, and the effects on CRC risk were even discordant for exon 7 variant D353N between the French and American sub-populations of the study[5]. For this reason, the authors focused the majority of their study on variant A628K, which is located in the region coding for an intracellular ZU-5 domain that is essential for the pro-apoptotic activity of UNC5C[17,18]. They based their demonstration of a genetic predisposition essentially on two points: (1) the co-segregation of the variant with cancerous phenotypes in families with CRC, thus postulating an autosomal dominant mode of inheritance; and (2) the in vitro alteration of the pro-apoptotic function of UNC5C in the absence of netrin-1. In addition, the authors noted a higher frequency of the A628K variant in the patient population than in a control population. From these observations, Coisseux et al[5] inferred a major role of the UNC5C gene in the predisposition to familial forms of CRC. Given the small number of patients concerned by this potentially novel clinical parameter, which resulted in a lack of statistical significance, the authors recommended that other teams replicate their study in samples from independent patient populations.
In order to address the question of the diagnostic potential of UNC5C suggested by Coissieux et al[5], we implemented a two-stage study to evaluate the role of UNC5C in the predisposition to CRC. More specifically, we focused on exon 11 of the UNC5C gene, which codes for the ZU-5 domain and contains A628K, the only variant found to significantly increase CRC risk in the French population studied by Coissieux et al[5]. We started by examining familial forms of cancer; we sought to find UNC5C variants in patients who had undergone prior routine screening for Lynch syndrome predisposition. One third of the patients exclusively presented a familial CRC of unknown etiology, thereby meeting the clinical criteria used by Coissieux et al[5]. The remaining two thirds consisted of two subgroups of patients; one that presented with cancer that fell within the spectrum of Lynch syndrome, albeit with no mutation in MMR genes, and the other that presented with Lynch syndrome with a known germline mutation in an MMR gene. We decided to extend our investigation to these two sub-groups because we were intrigued by the number of different cases of cancer other than CRC displayed by the families reported by Coissieux et al[5], with several patients affected by at least two types of cancer. In the second stage of our study, we tested patients and healthy controls from the ASTERISK population[19,20] to evaluate the possible role of UNC5C in the susceptibility to sporadic CRC. Taken together, the results we report here tend to challenge the hypothesis put forward by Mehlen’s team concerning the pivotal role of UNC5C in the predisposition to familial forms of CRC.
MATERIALS AND METHODS
Patients with familial colorectal or Lynch-associated cancers
The 310 unrelated patients included in the first stage of our study had all been previously screened and/or tested for Lynch syndrome predisposition by the Oncogenetic Laboratory within the Medical Genetics Department of the Nantes University Hospital (CHU de Nantes). They had all been recruited upon suspicion of a familial form of cancer, as they belong to families that meet either the criteria of the revised Bethesda guidelines (for patients included after 2007) or the criteria of Amsterdam I or II (for a few families of patients included before 2007)[21-23]. All probands were of Caucasian origin and had given informed consent for the genetic investigation of their predisposition to cancer. They provided a peripheral blood sample that was used for genomic DNA extraction with either the Nucleon BACC2 GE Healthcare® kit (GE Healthcare Bio-Sciences AB, Uppsala, Sweden), or the QuickGene DNA Whole Blood kit L (FujiFilm Life Sciences®, Tokyo, Japan). Whenever the patient’s tumor was available, MMR deficiency was screened by immunohistochemistry (IHC) and replication error (RER) phenotype analyses of MLH1, MSH2 and MSH6. Every time an MMR deficiency was suspected, the exons and intron/exon boundaries of the MLH1, MSH2, MSH6 and/or PMS2 genes were sequenced by Sanger technology and checked for large rearrangements by Multiplex ligation-dependent probe amplification.
According to clinical features, IHC, RER and sequencing results, patients were classified into three sub-groups detailed in Table 1, from which familial polyposis and hamartomatous polyposis syndromes were excluded.
Table 1.
Patients with familial colorectal or Lynch-associated cancers included in the study
| Patients with CRC (fulfilling Amsterdam II and/or Bethesda criteria) | Patients with family history of CRC and personal history of colorectal polyposis or of cancer in the Lynch syndrome tumor spectrum | Patients with Lynch syndrome (with identified MMR mutation or variant of unknown significance) | |
| Total number of patients | 120 | 581 | 132 |
| MSI | 50 | 13 | 132 |
| MSS | 37 | 16 | 0 |
| Unknown | 33 | 29 | 0 |
| Revised Bethesda criteria | 97 | 17 | 132 |
| MSI | 38 | 4 | 132 |
| MSS | 31 | 3 | 0 |
| Unknown | 28 | 10 | 0 |
| Amsterdam II criteria | 41 | 15 | ND |
| MSI | 14 | 4 | |
| MSS | 14 | 3 | |
| Unknown | 13 | 8 | |
| First-degree relatives with CRC | 23 | 13 | ND |
| MSI | 12 | 3 | |
| MSS | 6 | 3 | |
| Unknown | 5 | 7 | |
| Polyps (> 10 polyps) | 0 | 0 | 0 |
| Polyps (≤ 10 polyps) at age < 50 +/- first-degree relatives with polyps, MUTYH-negative | 0 | 41 | ND |
| MSI | 9 | ||
| MSS | 13 | ||
| Unknown | 19 |
Including 35 patients with personal history of colorectal polyposis and 23 patients with personal history of cancers included in the Lynch syndrome tumor spectrum (peritoneal carcinomatosis, oligoastrocytoma, endometrial carcinoma, gastric adenocarcinoma, or duodenal adenocarcinoma). CRC: Colorectal cancer; MSI: Microsatellite instability; MSS: Microsatellite stable; ND: Not data; MMR: Mismatch repair.
Patients with sporadic CRC and controls
A total of 1023 patients and 1121 healthy controls were tested. All patients had originally been recruited from the Pays de la Loire region in France between December 2002 and March 2006 as part of the French Association Study Evaluating RISK for sporadic colorectal cancer (ASTERISK)[19,20]. Ethics approval was granted for this study by the local (Comité Consultatif de Protection des Personnes dans la Recherche Biomédicale) and national (Commission Nationale de l’Informatique et des Libertés) ethics committees. After a clear explanation of the research protocol by a physician, a written informed consent was obtained from each participant, who thereafter provided a blood sample that was used for genomic DNA extraction with the Nucleon BACC2 GE Healthcare® kit (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). Eligibility criteria were: Caucasian origin, age at diagnosis ≥ 40 years and absence of family history of colorectal cancer or polyps. Cases were patients who had their first primary colorectal cancer diagnosed in one of the six public hospitals and five clinics located in the Pays de la Loire region that participated in the study. Cases were confirmed based on medical and pathology reports. Age- and sex-matched controls were recruited at two Health Examination Centers within the Pays de la Loire region, and the recruitment of controls ≥ 70 years of age was completed in the Departments of Internal Medicine and Hepatogastroenterology of the Nantes University Hospital, located in the same region. Controls were eligible to participate if they were Caucasian, ≥ 40 years of age, and had no family history of colorectal cancer or polyps. Each participant filled out a standardized questionnaire on family information, medical history, lifestyle and dietary intake.
UNC5C exon 11 genotyping
Genotyping was performed by automated Sanger sequencing in the 310 familial cases of cancer. The same strategy was also used in 300 healthy controls randomly chosen from the ASTERISK population. Genomic DNAs were amplified by Polymerase Chain Reaction (PCR with Platinum® Taq DNA Polymerase (Life Technologies, Saint Aubin, France), using forward primer 5’-GAAGCCATTCTCTGCTCTCC-3’ and reverse primer 5’-CATGGATTATTCTTGCATAGC-3’. PCR products were sequenced using ABIPRISM BigDye Terminator 1.1 Cycle Sequencing Ready Reaction (Applied Biosystems®, Courtaboeuf, France), and data were analyzed with the ABI PRISM 3130XL Genetic Analyzer and SeqScape® Software v2.5 (PE Applied Biosystems®, Foster City, CA). In the remaining individuals from the ASTERISK population, i.e., 1,023 patients and 821 healthy controls, genotyping of exon 11 was performed by High Resolution Melting (HRM), using the LightCycler® 480 High Resolution Melting Master (Roche Diagnostics, Meylan, France). The primers utilized (5’-GTCGTCCTCACTATGCATCAC-3’ and 5’-CTTTATCCCGACAGCAGCTC-3’) detected only the two variants of exon 11 found by sequencing (A628K and Q630E). In order to enable optimum variant detection, variant R603C was not tested, given its absence in the French patients studied by Coissieux et al[5]. Each DNA with a variant HRM profile was sequenced for confirmation following the above protocol. Detailed PCR conditions are available upon request.
Statistical analysis
The associations between UNC5C variants and risk for colorectal cancer were determined by calculating OR, 95%CI, and P values using the conditional maximum likelihood estimation (Fisher).
RESULTS
None of the 120 familial CRC patients included in our study who strictly matched Coissieux et al[5]’s clinical criteria were found to carry the main variant A628K, i.e., the variant with the most conclusive anti-apoptotic effect described by the authors (Figure 1). On the other hand, A628K was encountered in one of our patients who presented a gastric adenocarcinoma (Figure 1 and Family 1 in Figure 2). Oddly enough, this patient had three first-degree relatives with a gastric cancer, but no reported family member with colorectal polyps or adenocarcinoma. The only relative who could be tested here was the propositus’s sister, who was also found to be heterozygous for the variant. However, the gastric linitis plastica she had developed was a histologically different type of cancer compared to that of her brother, in whom CDH1 had been ruled out by molecular analysis.
Figure 1.
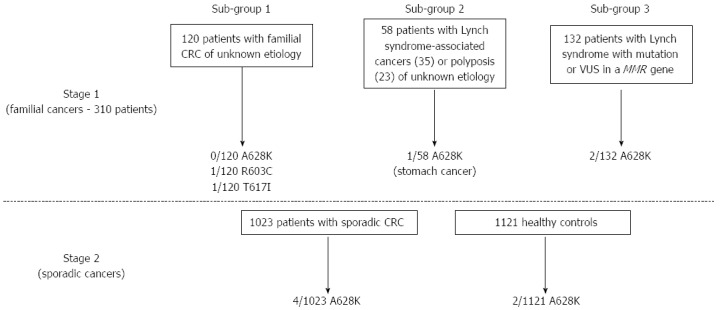
Flowchart of search strategy and results. The numbers of exon 11 UNC5C variants found are reported for each sub-group of individuals included in the study. CRC: Colorectal cancer; MMR: Mismatch repair; VUS: Variant of unknown significance.
Figure 2.
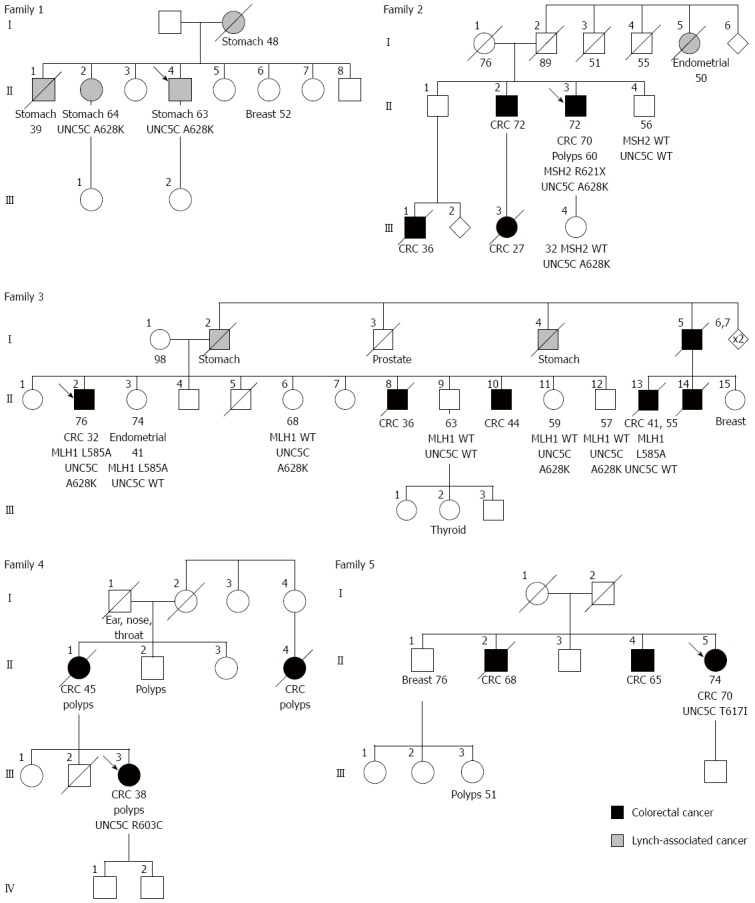
Pedigrees of the French families the proband of which was found to carry exon 11 UNC5C variant A628K (families 1-3), R603C (family 4) or T617I (family 5). A black filled-in circle or square shows that the individual developed a colorectal cancer, whereas a grey filled-in symbol indicates that the individual presented a cancer encompassed by the Lynch syndrome tumor spectrum. The type of cancer is precised below the affected individuals; the age at diagnosis is noted by figures next to the cancer type. Other figures below symbols indicate either the current age or the age of death of the individual. The variants carried by the individuals are noted below their respective symbols. The absence of any genotype means that no DNA sample was available for testing. CRC: Colorectal cancer; WT: Wild-type genotype.
The two cases of familial CRC from our study that presented the A628K variant were Lynch syndrome with characterized variants in MMR genes (Figure 1; Families 2 and 3 in Figure 2). In patient II.3 from family 2, UNC5C A628K co-occurred with a deleterious non-sense mutation of MSH2 c.1861C>T (p.Arg621*), which has been reported several times in the national UMD-MSH2 mutations database (http://www.umd.be/MSH2/) and in the International Society for Gastrointestinal Hereditary Tumours database (http://www.insight-group.org/mutations/). In patient II.2 from family 3, UNC5C A628K was found in co-occurrence with the variant of unknown significance (VUS) MLH1 c.1754T>G (p.Leu585Arg) that has also been reported in specific MMR databases. Despite its almost perfect co-segregation with the cancer phenotype in this particular family, this MLH1 variant could not be classified as likely pathogenic or even definitely pathogenic because the endometrial tumor in patient II.2’s sister was found to be microsatellite stable (MSS).
While the segregation study was not very informative in family 2, an absence of a co-segregation of the UNC5C A628K variant with cancer phenotype was observed in family 3 (Figure 2). On one hand, the propositus’s three siblings II.12, II.11 and II.6, who were 57, 59 and 68 years of age, were heterozygous for variant A628K and were all asymptomatic. On the other hand, the two siblings who were non-carriers of the UNC5C variant, II.13 and II.3, developed respectively two colorectal cancers at 41 and 55 years of age, and an endometrial cancer at 41 years of age.
Since no patients with a hitherto molecularly unexplained CRC were found to carry the A628K variant alone, risk calculation for CRC only was not possible. Nonetheless, by comparing A628K distribution between ASTERISK healthy controls and patients with familial colorectal or other Lynch-associated forms of cancer, we observed a non-significant increase in risk for cancer of the Lynch syndrome spectrum (OR = 3.2, 95%CI: 0.29-35.05, P = 0.348; Table 2). Even when considering molecularly unexplained cancer cases in combination with familial CRC cases with a known MMR mutation, the association of A628K with cancer risk did not achieve statistical significance (OR = 5.5, 95%CI: 0.91-32.87, P = 0.063). Further investigations in sporadic CRC cases from ASTERISK indicated a non-significant increase in risk of A628K for CRC (OR = 2.2, 95%CI: 0.40-12.02, P = 0.364; Table 2), which is very similar to the risk calculated in the US cohort included in Coissieux et al[5]’s study, with the difference that patients from this cohort had a familial form of CRC.
Table 2.
Conditional maximum likelihood estimation of the associations between UNC5C variants and risk for colorectal cancer n (%)
| Type of cancer | Patients mut/total | Controls mut/total | OR (95%CI) | P value |
| Variant A628K1 | ||||
| Familial CRCs without MMR mutations | 0/120 (0.00) | 2/1121 (0.18) | NA | NA |
| Familial CRCs or Lynch-associated cancers without MMR mutations | 1/178 (0.56) | 2/1121 (0.18) | 3.2 (0.29-35.05) | 0.348 |
| Familial CRCs or Lynch-associated cancers with or without MMR mutations | 3/310 (0.97) | 2/1121 (0.18) | 5.5 (0.91-32.87) | 0.063 |
| Sporadic CRCs (ASTERISK) | 4/1023 (0.39) | 2/1121 (0.18) | 2.2 (0.40-12.02) | 0.364 |
| Familial and sporadic CRCs | 7/1333 (0.53) | 2/1121 (0.18) | 3.0 (0.61-14.25) | 0.177 |
| Variants R603C, T617I, A628K, or Q630E2 | ||||
| Familial CRCs without MMR mutations | 2/120 (1.67) | 37/4300 (0.86) | 2.0 (0.47-8.20) | 0.3613 |
| Familial CRCs or Lynch-associated cancers without MMR mutations | 3/178 (1.69) | 37/4300 (0.86) | 2.0 (0.60-6.47) | 0.2613 |
| Familial CRCs or Lynch-associated cancers with or without MMR mutations | 5/310 (1.61) | 37/4300 (0.86) | 1.9 (0.74-4.84) | 0.1853 |
Estimates of the risk associated with variant A628K inferred from genotyping data obtained in patients with familial colorectal or Lynch-associated forms of cancer and in patients with sporadic colorectal cancers (CRCs) and healthy controls from the ASTERISK population;
Estimates of the risk associated with heterozygosity for variants R603C, T617I, A628K, or Q630E by comparison of the present study familial cancer cases with Caucasian individuals from the NHLBI GO Exome Sequencing Project used as controls Exome Variant Server, NHLBI Exome Sequencing Project (ESP), Seattle, WA [http://evs.gs.washington.edu/EVS/(date 08/2013 accessed)];
Applying Bonferroni correction, significance threshold was lowered to 0.05/4 = 0.0125. NA: Not available; MMR: Mismatch repair.
Two UNC5C variants were found in patients with CRC of hitherto unknown etiology: D353N in patient III.3 of family 4 and c.1850C>T/p.Thr617Ile (T617I) in patient II.1 of family 5 (Figure 2). No segregation study could be done in their respective families, in the absence of available samples and/or compliant relatives. To date, variant T617I has never be reported in any of the known mutations, neither in the dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/) nor in the Exome Variant Server (http://evs.gs.washington.edu/EVS/). According to the bioinformatic programs embedded in Alamut® software (Interactive Biosoftware, Rouen, France), including Align GVGD, Polyphen-2, SIFT and MutationTaster, amino acid 617 of UNC5C is conserved across mammalian species and a change in the protein would be benign.
Because a study limited to the A628K variant might be too restrictive, we estimated the risk for cancer associated with heterozygosity for any of the four variants R603C, T617I, A628K, and Q630E, i.e., the UNC5C exon 11 variants described by Coissieux et al[5] plus the novel T617I variant identified in our study. Specifically, we compared distribution of these variants in the familial cases of our study with that of the individuals from the NHLBI Exome Sequencing Project (ESP), assuming that Caucasian ESP individuals are genetically similar to our ASTERISK healthy control individuals. Even though we observed an increase in the risk of familial CRC without MMR mutations for subjects with any of the four variants (OR = 2.0, 95%CI: 0.47-8.20, P = 0.361; Table 2), the association was not statistically significant.
DISCUSSION
The present study was conducted to replicate recent publication findings of a major role for the UNC5C gene in the predisposition to familial forms of colorectal cancer of hitherto unknown etiology[5]. In the first stage of our study, we attempted to determine if the high CRC risk conferred by UNC5C variants could be confirmed in familial forms of CRC in our study. We focused on exon 11 of the gene, which codes for the intracellular ZU-5 domain, because this exon contains three of the four variants shown by Coissieux et al[5] to significantly alter the pro-apoptotic activity of UNC5C.
At least four of our observations diverge from those made by Coissieux et al[5], which weakens the hypothesis of a familial CRC predisposition due to UNC5C: (1) A628K was found in a family with gastric cancer (family 1); (2) A628K co-occurred with mutations in MMR genes (families 2 and 3); (3) A628K did not co-segregate with cancer phenotypes (family 3); and (4) no exon 11 variant was significantly associated with CRC risk.
Of interest, we found A628K mutation in a family with history of gastric carcinoma. This possible association between the new tissue location and A628K concurs with Coissieux et al[5]’s observation of several forms of cancer in the index cases or their relatives in their study, which can be explained by the extended inclusion criteria used in our study. We indeed followed the Amsterdam I, II or Bethesda guidelines[21-23] for Lynch syndrome screening, which include tumor sites encompassed by the clinical spectrum of Lynch syndrome (stomach, endometrium, etc.). Moreover, a down-regulation of UNC5C expression has already been reported in multiple forms of cancer, e.g., breast, ovary, uterus, stomach, lung and kidney cancers[12] and UNC5C methylation has been reported in gastric carcinomas[24].
Surprisingly, A628K was identified only in two Lynch syndrome cases with mutations previously identified in MMR genes. This fortuitous finding in two different families further complicates the interpretation of a possible association between UNC5C variants and risk of CRC, i.e., the co-segregation study is informative in only one family and no genotype-phenotype correlation was observed. Of course, the lack of co-segregation between A628K and cancer phenotype is not sufficient to definitively invalidate the hypothesis of a genetic predisposition according to a dominant mode of inheritance, considering likely phenocopies and low penetrance. It is also possible that carriers of the variant may have not yet developed any cancer. In fact, a heterozygous mutation in a predisposition gene greatly increases the probability of developing a cancer at an early age, but it does not mean that carriers of this mutation will necessarily develop a malignancy before 50-60 years of age. In contrast, as regard to the already known high-penetrance CRC predisposition genes, the likelihood of finding three asymptomatic carriers as well as two non-carriers affected with early onset cancer in the same family is relatively small. For instance, in Lynch syndrome, the cumulative risk of developing a CRC is 30%-80% in MMR mutations carriers and the mean age at CRC onset is 45 years[25]. It is also worth noting that co-segregation between A628K and CRC was not perfect in the families studied by Coissieux et al[5]. For example, at least one non-carrier, albeit with CRC, was found in each family, whereas a 64-year old carrier was reported as asymptomatic, underlining the aforementioned problem of phenocopies and suggesting that the penetrance of the variant may not be as high as originally thought.
In our study, we were not able to replicate the marginally significant association for an increase in CRC risk with UNC5C exon 11 variants reported by Coissieux et al[5] (OR = 8.8, 95%CI: 1.47-52.93, P = 0.0315). The divergence in the results observed between the two studies is in fact slighter than it may appear, as the groups of A628K carriers are very small in both cases. The power of association studies is indeed relatively limited for investigating rare variants, and achieving strong statistical significance would either require a very large sample size or necessitate more powerful methodologies such as burden tests. Thus, Genome-Wide Associations studies (GWAS) have failed to highlight loci already known in familial forms of cancer and other complex traits, due to their inability to detect highly-penetrant, rare variants[4,6]. On the contrary, by adopting a candidate gene strategy, Coissieux et al[5] were able to detect associations for some of these rare variants. However, the associations between the identified variants with CRC risk are somewhat fragile. This illustrates the difficulties to identify new high penetrance genetic variants involved in predisposition to CRC because of the high locus heterogeneity. The estimated 20%-30% of missing heritability for CRC can therefore be explained by a possible mosaic of rare familial variants, with an accumulation of private mutations. UNC5C could be one of the genes involved, but the identification of additional families is needed to confirm this. This could be facilitated by a more precise clinical definition of UNC5C-associated CRC cases.
It is quite tempting to draw parallels between UNC5C and DCC. In fact, a possible tumor suppressor role was postulated for both netrin receptors for several years[7,13,18,26]. However, no conclusive proof was produced until recently. With the development of murine models, it has been shown that, beside their role in tumor progression, UNC5C and DCC could also participate in the early events of colorectal carcinogenesis[1,2,13,27]. The hypothesis of tumor suppressor gene function is also supported by the fact that the UNC5C and DCC genes are located in frequently rearranged chromosomal regions and also the frequent hypermethylation of their promoters in colorectal tumors[15]. Nevertheless, for DCC, this putative role is considered as very controversial, notably because of its much rarer somatic mutations in colorectal cancers reported in literature or in specific databases (e.g., COSMIC), as compared to the nearby SMAD4 gene for example[28-34]. In addition, DCC has never been related to a familial form of colorectal malignancy, in contrast to SMAD4 - the mutations of which can cause Juvenile Polyposis Syndrome[28-34]. A query in the COSMIC database[28] showed that somatic UNC5C mutations are as infrequent in colorectal adenocarcinomas as DCC. Moreover, no clearly deleterious germline mutations, such as nonsense or frameshift mutations, were reported in UNC5C in patients with CRC, whereas these types of mutations are quite frequent in familial forms of cancers caused by tumor suppressor genes.
This raises the issue of whether UNC5C is the right candidate gene to explain colorectal cancer in the five families described by Coissieux et al[5]. In fact, the good co-segregation of the A628K variant with cancer phenotype is puzzling. In Coissieux’s study, all carriers of this variant shared a 108 kilo-base-pair minimum critical region within 4q22.3 determined by 34 SNPs. Genotyping a subset of these SNPs (rs65322545, rs6846193, rs17023311, rs1434534, rs3775048, rs11737434 and rs2289043) in our study patients was sufficient to discriminate carriers of variant A628K from carriers of other UNC5C exon 11 variants (data not shown). However, the power displayed in linkage analysis by 34 SNPs is quite low, because of a limited heterozygosity. Consequently, the minimum critical region defined by Coissieux et al[5] could not be as specific and significant as expected, leading us to postulate that UNC5C may not be the right susceptibility gene. The rare variants identified by Mehlen’s team may instead be markers that are in linkage disequilibrium with a real deleterious variant located, for example, within any other gene in the neighborhood of UNC5C. When considering the DCC/SMAD4 example, that other gene could be the nearby BMPR1B gene, which is centromeric to UNC5C. The risk for CRC conferred by variants of BMP genes has already been highlighted by GWAS[32,33], and a recent study pointed to a significant increase in CRC risk associated with genetic variation in BMPR1A, BMPR1B, BMPR2, BMP2 and BMP4[34].
In conclusion, our study shows that UNC5C is not a major CRC-predisposing gene that could compare for instance with MMR genes or APC. UNC5C exon 11 allelic variants were rare, with an incomplete penetrance in the families of our study. However, we were able to confirm a tendency towards an increase in cancer risk, not only for familial but also for sporadic CRC, which means that UNC5C alleles can be classified at least as low-moderate penetrance alleles. Previous studies have highlighted UNC5C as a potential tumor suppressor gene, but the simple presence of UNC5C variants in the families included in our study does not seem sufficient to confer a high risk for CRC. One may hypothesize that, in the families included in Coissieux et al[5]’s study, either A628K is in complete linkage disequilibrium with a causative mutation located outside UNC5C, or UNC5C participates in an oligogenic mechanism of carcinogenesis. In any case, it is probably too preliminary to apply Coissieux et al[5]’s finding to genetic counseling and routine genetic CRC-predisposition testing. The presence of variant UNC5C A628K in patients with gastric cancer in family 2 (Figure 2) also suggests that further investigations should be performed to address the impact of UNC5C on digestive cancers.
ACKNOWLEDGMENTS
Sébastien Küry and Stéphane Bezieau are very grateful and indebted to Professor Jean-Paul MOISAN for having given them the opportunity to work in the field of molecular genetics. He will miss them.
COMMENTS
Background
Colorectal cancer (CRC) is the second leading cause of cancer death in developed countries. Approximately 30% of CRCs are inherited forms of the disease caused by highly penetrant germline mutations in single genes. Yet, so far, very few CRC-predisposing genes have been identified which explain only a small proportion of the familial CRC cases.
Research frontiers
In the area of cancer genetics, the finding of new CRC-predisposing genes represents a research hotspot, as it offers perspective on genetic testing and counseling for 20%-30% of overall CRC cases. In this context, variants of the UNC5C gene were recently reported to increase risk of familial CRC. If verified, this finding would have a significant impact for gastroenterologists, patients, genetic counselors, and for molecular diagnostic laboratories involved in the genetics of colorectal cancer. It would indeed make UNC5C a new gene to be tested in familial forms of cancer different from the ones routinely tested in diagnostic laboratories. In this study, the authors attempted to evaluate and clarify the risk of CRC associated with UNC5C candidate variants.
Innovations and breakthroughs
The possible role of UNC5C variants in the predisposition to CRC was assessed by a single study. Replication was therefore mandatory. Although they did not confirm the strong effect of UNC5C exon 11 variants on CRC risk, the authors showed however that UNC5C rare alleles could be considered at least as low-moderate penetrance alleles increasing overall CRC risk, regarding both familial and sporadic forms of CRC. Besides, the authors suggest a possible additional involvement of UNC5C in the predisposition to other digestive cancers, more especially gastric cancer.
Applications
The authors stressed that there is currently insufficient evidence to reliably propose routine UNC5C testing in patients with CRC of hitherto unknown etiology.
Terminology
The penetrance of a gene allele corresponds to the proportion of symptomatic individuals carrying the allele. A genetic predisposition to a cancer is the influence of a genetic factor on the occurrence of this cancer in an individual. In the study, the odds ratio is a statistical measure of the odds that a cancer will occur in presence of a genetic variant, compared to the odds of the cancer occurring in the absence of that variant. A genetic variant can be either a mutation or a polymorphism, according to its pathogenicity.
Peer review
The study presented interesting and seemingly controversial results, concerning role of A628K substitution in UNC5C receptor gene that has been studied before from Coissieux et al, 2012. These results imply medical and technical implication. The work is good and I recommend its publication.
Footnotes
P- Reviewers: Bujanda L, Pascale RM, Stanilova S, Vogt G S- Editor: Zhai HH L- Editor: A E- Editor: Ma S
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Jasperson KW, Tuohy TM, Neklason DW, Burt RW. Hereditary and familial colon cancer. Gastroenterology. 2010;138:2044–2058. doi: 10.1053/j.gastro.2010.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abulí A, Bessa X, González JR, Ruiz-Ponte C, Cáceres A, Muñoz J, Gonzalo V, Balaguer F, Fernández-Rozadilla C, González D, et al. Susceptibility genetic variants associated with colorectal cancer risk correlate with cancer phenotype. Gastroenterology. 2010;139:788–796, 796.e1-6. doi: 10.1053/j.gastro.2010.05.072. [DOI] [PubMed] [Google Scholar]
- 4.Bodmer W, Tomlinson I. Rare genetic variants and the risk of cancer. Curr Opin Genet Dev. 2010;20:262–267. doi: 10.1016/j.gde.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 5.Coissieux MM, Tomsic J, Castets M, Hampel H, Tuupanen S, Andrieu N, Comeras I, Drouet Y, Lasset C, Liyanarachchi S, et al. Variants in the netrin-1 receptor UNC5C prevent apoptosis and increase risk of familial colorectal cancer. Gastroenterology. 2011;141:2039–2046. doi: 10.1053/j.gastro.2011.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arakawa H. Netrin-1 and its receptors in tumorigenesis. Nat Rev Cancer. 2004;4:978–987. doi: 10.1038/nrc1504. [DOI] [PubMed] [Google Scholar]
- 8.Mehlen P, Delloye-Bourgeois C, Chédotal A. Novel roles for Slits and netrins: axon guidance cues as anticancer targets? Nat Rev Cancer. 2011;11:188–197. doi: 10.1038/nrc3005. [DOI] [PubMed] [Google Scholar]
- 9.Kaufmann S, Kuphal S, Schubert T, Bosserhoff AK. Functional implication of Netrin expression in malignant melanoma. Cell Oncol. 2009;31:415–422. doi: 10.3233/CLO-2009-0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lv D, Zhao W, Dong D, Qian XP, Zhang Y, Tian XJ, Zhang J. Genetic and epigenetic control of UNC5C expression in human renal cell carcinoma. Eur J Cancer. 2011;47:2068–2076. doi: 10.1016/j.ejca.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 11.Mao X, Boyd LK, Yáñez-Muñoz RJ, Chaplin T, Xue L, Lin D, Shan L, Berney DM, Young BD, Lu YJ. Chromosome rearrangement associated inactivation of tumour suppressor genes in prostate cancer. Am J Cancer Res. 2011;1:604–617. [PMC free article] [PubMed] [Google Scholar]
- 12.Thiebault K, Mazelin L, Pays L, Llambi F, Joly MO, Scoazec JY, Saurin JC, Romeo G, Mehlen P. The netrin-1 receptors UNC5H are putative tumor suppressors controlling cell death commitment. Proc Natl Acad Sci USA. 2003;100:4173–4178. doi: 10.1073/pnas.0738063100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bernet A, Mazelin L, Coissieux MM, Gadot N, Ackerman SL, Scoazec JY, Mehlen P. Inactivation of the UNC5C Netrin-1 receptor is associated with tumor progression in colorectal malignancies. Gastroenterology. 2007;133:1840–1848. doi: 10.1053/j.gastro.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shin SK, Nagasaka T, Jung BH, Matsubara N, Kim WH, Carethers JM, Boland CR, Goel A. Epigenetic and genetic alterations in Netrin-1 receptors UNC5C and DCC in human colon cancer. Gastroenterology. 2007;133:1849–1857. doi: 10.1053/j.gastro.2007.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hibi K, Mizukami H, Shirahata A, Goto T, Sakata M, Saito M, Ishibashi K, Kigawa G, Nemoto H, Sanada Y. Aberrant methylation of the UNC5C gene is frequently detected in advanced colorectal cancer. Anticancer Res. 2009;29:271–273. [PubMed] [Google Scholar]
- 16.Hibi K, Mizukami H, Shirahata A, Goto T, Sakata M, Sanada Y. Aberrant methylation of the netrin-1 receptor genes UNC5C and DCC detected in advanced colorectal cancer. World J Surg. 2009;33:1053–1057. doi: 10.1007/s00268-008-9909-x. [DOI] [PubMed] [Google Scholar]
- 17.Ackerman SL, Kozak LP, Przyborski SA, Rund LA, Boyer BB, Knowles BB. The mouse rostral cerebellar malformation gene encodes an UNC-5-like protein. Nature. 1997;386:838–842. doi: 10.1038/386838a0. [DOI] [PubMed] [Google Scholar]
- 18.Leonardo ED, Hinck L, Masu M, Keino-Masu K, Fazeli A, Stoeckli ET, Ackerman SL, Weinberg RA, Tessier-Lavigne M. Guidance of developing axons by netrin-1 and its receptors. Cold Spring Harb Symp Quant Biol. 1997;62:467–478. [PubMed] [Google Scholar]
- 19.Küry S, Buecher B, Robiou-du-Pont S, Scoul C, Sébille V, Colman H, Le Houérou C, Le Neel T, Bourdon J, Faroux R, et al. Combinations of cytochrome P450 gene polymorphisms enhancing the risk for sporadic colorectal cancer related to red meat consumption. Cancer Epidemiol Biomarkers Prev. 2007;16:1460–1467. doi: 10.1158/1055-9965.EPI-07-0236. [DOI] [PubMed] [Google Scholar]
- 20.Hutter CM, Chang-Claude J, Slattery ML, Pflugeisen BM, Lin Y, Duggan D, Nan H, Lemire M, Rangrej J, Figueiredo JC, et al. Characterization of gene-environment interactions for colorectal cancer susceptibility loci. Cancer Res. 2012;72:2036–2044. doi: 10.1158/0008-5472.CAN-11-4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Rüschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vasen HF, Mecklin JP, Khan PM, Lynch HT. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC) Dis Colon Rectum. 1991;34:424–425. doi: 10.1007/BF02053699. [DOI] [PubMed] [Google Scholar]
- 23.Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116:1453–1456. doi: 10.1016/s0016-5085(99)70510-x. [DOI] [PubMed] [Google Scholar]
- 24.Hibi K, Sakata M, Sakuraba K, Kitamura YH, Shirahata A, Goto T, Mizukami H, Saito M, Ishibashi K, Kigawa G, et al. Changes in UNC5C gene methylation during human gastric carcinogenesis. Anticancer Res. 2009;29:4397–4399. [PubMed] [Google Scholar]
- 25.Vasen HF, van der Meulen-de Jong AE, de Vos Tot Nederveen Cappel WH, Oliveira J. Familial colorectal cancer risk: ESMO clinical recommendations. Ann Oncol. 2009;20 Suppl 4:51–53. doi: 10.1093/annonc/mdp127. [DOI] [PubMed] [Google Scholar]
- 26.Grady WM. Making the case for DCC and UNC5C as tumor-suppressor genes in the colon. Gastroenterology. 2007;133:2045–2049. doi: 10.1053/j.gastro.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 27.Castets M, Coissieux MM, Delloye-Bourgeois C, Bernard L, Delcros JG, Bernet A, Laudet V, Mehlen P. Inhibition of endothelial cell apoptosis by netrin-1 during angiogenesis. Dev Cell. 2009;16:614–620. doi: 10.1016/j.devcel.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 28.Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Woodford-Richens KL, Rowan AJ, Gorman P, Halford S, Bicknell DC, Wasan HS, Roylance RR, Bodmer WF, Tomlinson IP. SMAD4 mutations in colorectal cancer probably occur before chromosomal instability, but after divergence of the microsatellite instability pathway. Proc Natl Acad Sci USA. 2001;98:9719–9723. doi: 10.1073/pnas.171321498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Riggins GJ, Thiagalingam S, Rozenblum E, Weinstein CL, Kern SE, Hamilton SR, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B. Mad-related genes in the human. Nat Genet. 1996;13:347–349. doi: 10.1038/ng0796-347. [DOI] [PubMed] [Google Scholar]
- 31.Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 32.Peters U, Hutter CM, Hsu L, Schumacher FR, Conti DV, Carlson CS, Edlund CK, Haile RW, Gallinger S, Zanke BW, et al. Meta-analysis of new genome-wide association studies of colorectal cancer risk. Hum Genet. 2012;131:217–234. doi: 10.1007/s00439-011-1055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Houlston RS, Webb E, Broderick P, Pittman AM, Di Bernardo MC, Lubbe S, Chandler I, Vijayakrishnan J, Sullivan K, Penegar S, et al. Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat Genet. 2008;40:1426–1435. doi: 10.1038/ng.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slattery ML, Lundgreen A, Herrick JS, Kadlubar S, Caan BJ, Potter JD, Wolff RK. Genetic variation in bone morphogenetic protein and colon and rectal cancer. Int J Cancer. 2012;130:653–664. doi: 10.1002/ijc.26047. [DOI] [PMC free article] [PubMed] [Google Scholar]