

Abstract
Exposure of the immature brain to general anesthesia is common. The safety of this practice has recently been challenged in view of evidence that general anesthetics can damage developing mammalian neurons. Initial reports on immature rats raised criticism regarding the possibly unique vulnerability of this species, short duration of their brain development and a lack of close monitoring of nutritional and cardiopulmonary homeostasis during anesthesia. Therefore, we studied the neurotoxic effects of anesthesia in guinea pigs, whose brain development is longer and is mostly a prenatal phenomenon, so that anesthesia‐induced neurotoxicity studies of the fetal brain can be performed by anesthetizing pregnant female pigs. Because of their large size, these animals made invasive monitoring of maternal and, indirectly, fetal well‐being technically feasible. Despite adequate maintenance of maternal homeostasis, a single short maternal exposure to isoflurane, whether alone or with nitrous oxide and/or midazolam at the peak of fetal synaptogenesis, induced severe neuroapoptosis in the fetal guinea pig brain. As detected early in post‐natal life, this resulted in the loss of many neurons from vulnerable brain regions, demonstrating that anesthesia‐induced neuroapoptosis can cause permanent brain damage.
Keywords: Synaptogenesis, isoflurane, caspase, midazolam, neuronal death, nitrous oxide
INTRODUCTION
General anesthetics are unique pharmacological agents widely used in modern anesthesiology because of their potent inhibition of neuronal activity, which is clinically manifested as loss of consciousness and/or insensitivity to painful stimuli. Over the last 60 years, great advances in anesthesiology have made complex surgical interventions possible even in premature infants and fetuses in utero. Although exposure of the immature central nervous system to general anesthetics is considered to be safe, recent reports of in vivo (21, 22, 24, 61, 62) and in vitro (57) studies indicate that common general anesthetics may be damaging to developing neurons. For instance, several classes of intravenous anesthetics, such as barbiturates (3, 22, 42), benzodiazepines (3, 22, 42) and ketamine (21, 50, 53), have been shown to cause severe and widespread neuronal degeneration in the immature mammalian brain. In addition, we recently showed that exposure of immature rats at the peak of their brain development (ie, synaptogenesis) to the volatile anesthetic isoflurane, either alone or in combination with midazolam, an intravenous benzodiazepine, and nitrous oxide (N2O), an inhalational anesthetic, for 6 h triggered widespread neurodegeneration that was apoptotic in nature (24, 61). Moreover, when rats exposed to these anesthetics in infancy were tested as juveniles and adults, they were found to have persistent learning and memory deficits (24).
Thus, it appears that the use of common general anesthetics in clinically relevant doses may not be as innocuous to the developing mammalian brain (reviewed by 37) as was previously thought. However, several issues regarding the technical approach used in initial studies, particularly the duration of animals' exposure to anesthesia and the maintenance of their nutritional and cardiopulmonary homeostasis while under anesthesia, have raised severe criticism. For example, the neurotoxic anesthesia protocols were regarded as being prolonged (average 6 h), taking proportionally substantial time during the 2 to 3 weeks of rat and mouse brain development. As the majority of exposures to pediatric anesthesia are relatively short, studies comparing the susceptibility of species with different durations of brain development to anesthesia protocols of similar or shorter duration are crucial for appropriate risk assessment.
As noted, another important criticism regards the nutritional and cardiovascular status of immature animals while under anesthesia. In clinical settings, all vital functions and metabolic parameters of pediatric patients under anesthesia are closely monitored and tightly controlled. This was technically not feasible when infant rats and mice were studied, giving rise to the possibility that inadequacy of tissue perfusion, ventilation or oxygenation, as well as severe hypoglycemia (2, 44) during anesthesia could be, at least in part, responsible for anesthesia‐induced neurotoxicity. This notion was reinforced by the recent finding that mouse pups anesthetized with isoflurane developed severe hypoglycemia (29), which is known to jeopardize brain development and may cause learning disabilities (8, 33, 34).
Thus, our initial work on rats and mice, as well as that done in other laboratories, could not address two important issues: (i) can short exposure to anesthesia trigger apoptotic neurodegeneration in species with longer brain development? (ii) does close control over cardiorespiratory parameters (eg, blood pressure, heart rate, tissue oxygenation, and ventilation) and metabolic homeostasis (eg, blood glucose, bicarbonate levels and arterial blood pH) patterned after the actual clinical anesthesia setting have any affect on the severity of anesthesia‐induced developmental neuroapoptosis? It is technically impossible to monitor all of these parameters in immature rats because of their small size. Therefore, to address these important questions, we designed a study with guinea pigs, in which brain development lasts about 10 weeks, or approximately five times longer than in rats (9) and is mostly a prenatal phenomenon which allows tight control over vital functions via continuous monitoring of the pregnant mother during anesthesia. As anesthetics readily cross the utero‐placental barrier, our experimental design is based on the fact that fetal well‐being is highly dependent on maternal well‐being (16, 28, 39, 40, 48, 58). Thus, maintenance of maternal vital functions during anesthesia is an attainable way to assure fetal homeostasis.
Another question that needed to be addressed was whether exposure to anesthesia at the peak of brain development leads to neuronal loss in the most vulnerable brain regions. Although it is a useful marker for detecting developmental apoptosis, caspase activation is a rapidly occurring and transient process having somewhat different timing in different neurons within the same brain region, making it impossible to cumulatively assess neuronal damage in all vulnerable neurons at one time point. Therefore, we designed this study to assess overall neuronal loss many days after anesthesia treatment (first post‐natal week), by which time dying neurons were phagocytized and the neuronal debris had been removed.
The main hypothesis that guided this study was that, despite the assurance of adequate maternal nutritional and cardiorespiratory stability during a short exposure to clinically relevant anesthesia, the fetal guinea pig brain would suffer significant and widespread anesthesia‐induced apoptotic neurodegeneration. This, we hypothesized, would ultimately lead to neuronal loss in vulnerable brain regions.
EXPERIMENTAL METHODS
Animals
Pregnant Hartley guinea pigs were used for all experiments. We chose to use guinea pigs rather than rats, which have been used in earlier studies, because their brain growth is five times longer than that in rats and occurs predominantly prenatally. Thus, anesthesia‐induced neurotoxicity studies of the fetal brain can be performed by anesthetizing pregnant mothers. Because of the relatively large size of guinea pigs, invasive monitoring of maternal and indirectly fetal vital signs are technically feasible.
The duration of the guinea pig gestation is 59–72 days (41). We selected three groups of guinea pigs based on the stage of the fetal brain growth spurt: 20–25 days of pregnancy (early stage) (average maternal body weight, 973 ± 46 g); 35–40 days (peak stage) (average body weight, 1108 ± 25 g), and over 50 days (late stage) (average body weight, 1324 ± 55 g) (9). These guinea pigs were randomly assigned to one of three groups: the experimental group, in which animals were given continuous anesthesia (total number of animals was 17, bearing a total of 55 fetuses), a sham control group that was given continuous fentanyl infusion (total number of animals was 6, bearing a total of 16 fetuses), or the “true” control group, given no anesthesia for 4 h (total number of animals was 4, bearing a total of 11 fetuses). All possible efforts were made to minimize both the number of pregnant animals used and any pain they might experience. All experiments were approved by The Animal Use and Care Committee of the University of Virginia Health System and were conducted in accordance with the US Public Health Service's Policy on Human Care and Use of Laboratory Animals.
Anesthesia
Nitrous oxide (N2O) and oxygen (GTS, Allentown, PA, USA) were delivered using a calibrated flowmeter. Isoflurane (Abbott Laboratories, Chicago, IL, USA) was administered using an agent‐specific vaporizer that delivered a set percentage of this anesthetic. Midazolam (Sigma‐Aldrich Chemical Co., St. Louis, MO, USA) was dissolved in 10% dimethyl sulfoxide (DMSO) immediately before administration. For control experiments, 10% DMSO was used as a vehicle. Fentanyl, an intravenous narcotic, and rocuronium, an intravenous muscle relaxant, were purchased, respectively, from Abbott Laboratories and Baxter Pharmaceutical Solutions, LLC, Bloomington, IN, and administered using a multi‐channel infusion pump (Harvard Apparatus, PHD 2000).
Anesthesia treatments
Before the initiation of the assigned protocols, the pregnant guinea pigs were first put to sleep using isoflurane in oxygen (3‐L flow) delivered via mouth cone, after which atropine (0.05 mg/kg, s.c.) was administered. Once asleep, the animals were orotracheally intubated with a polyethylene tube (outside diameter, 2.5 mm, inside diameter 2.1 mm). Artificial ventilation was initiated with oxygen (40‐vol%), nitrogen (57–58‐vol%), and isoflurane as needed (2–3‐vol%), under conditions of 6–8 mL/kg tidal volume, and 23–40 strokes per minute, delivered by a SAR‐830 ventilator (CWE Inc., Ardmore, PA, USA). The exhaled gases (eg, oxygen, carbon dioxide, nitrous oxide and isoflurane) were continuously analyzed using an infrared analyzer (Datex Ultima, Helsinki, Finland). Postinduction of general anesthesia, one polyurethane catheter was inserted into the left carotid artery for continuous monitoring of blood pressure, heart rate and arterial blood gas; a second polyurethane catheter (PE50) was introduced into the right jugular vein for continuous infusion of intravenous fluid (Lactate Ringer solution at 4 mL/kg/h). To facilitate artificial ventilation, the pregnant animals were paralyzed by continuous intravenous infusion of the muscle relaxant rocuronium (15 nmol/kg/minute) (12), initiated after placement of an intravenous line and discontinued 30 minutes before termination of the anesthesia protocol.
After the placement of invasive lines, the pregnant animals were allowed 15–20 minutes to stabilize, at which point induction of an anesthesia protocol was initiated. The minute ventilation was adjusted to maintain stable physiological parameters, which were continually monitored. The experimental group of pregnant guinea pigs was divided into four subgroups; each subgroup was randomly assigned to receive one of the following clinically relevant anesthesia protocols: isoflurane alone (0.55‐vol%, ISO), isoflurane (0.55‐vol%) + N2O (75‐vol%), isoflurane (0.55‐vol%) + midazolam (1 mg/kg, i.m.), or isoflurane (0.55‐vol%) + nitrous oxide (75‐vol%) + midazolam (1 mg/kg, i.m.) (I + N + M) for 4 h. The inhaled oxygen concentration was approximately 24‐vol%. The use of nitrous oxide (75‐vol%) and midazolam (1 mg/kg, i.m.), either individually or in combination, was not possible due to ethical considerations. At the required doses, these agents do not provide adequate depth of anesthesia which is crucial to the comfort of intubated and mechanically ventilated guinea pigs. Sham control animals were exposed to the same procedures as were experimental animals, but fentanyl alone was used for the maintenance of anesthesia. Fentanyl was administered after placement of the intravenous line, first as a bolus (30 µg/kg, i.v. given slowly over 15 minutes), then by 4 h of continuous infusion (15 µg/kg/h) (46). “True” control animals were neither exposed to any anesthesia, nor to invasive monitoring so that the histopathological properties of the fetal control brains could be directly compared with those of age‐matched experimental and sham control fetal brains.
At the termination of any anesthesia protocol, the pregnant animals were kept normothermic and were closely inspected. As soon as they resumed a regular breathing pattern, airway reflexes and purposeful movements, the invasive lines were removed and the trachea was extubated. Soon after anesthesia, pregnant guinea pigs were capable of feeding well and did not require supplemental hydration.
Monitoring of maternal vital functions
The adequacy of ventilation and oxygenation was continuously monitored by analyzing the composition of maternal exhaled gases (end tidal oxygen and carbon dioxide, measured, respectively, in mmHg—EtO2 and EtCO2) collected from the exhalation limb of the anesthesia breathing circuit and by analyzing the composition of blood gases in the arterial blood obtained from the maternal left carotid artery (pH, partial pressure of carbon dioxide in mmHg—pCO2, partial pressure of oxygen in mmHg—pO2, bicarbonate concentration in mmol/L, oxygen saturation, SaO2 in %) (Nova Biomedical, Waltham, MA, USA). Maternal arterial blood pressure was continuously monitored using a blood pressure transducer (23Db transducer referred to the mid‐thoracic level) connected to a cannula (a polyethylene catheter‐PE‐50) inserted into the left carotid artery, which also allowed the sampling of arterial blood as needed. Systolic, diastolic and mean arterial pressures were obtained using custom software in Pascal computer language and an Intel digital computer. Maternal heart rate was derived from the pressure signal. Maternal animal core temperature was continuously monitored with a rectal temperature probe and maintained using heating pads. Maternal blood glucose levels were measured, using a glucometer (Accu‐check Advantage) at regular intervals during anesthesia (Nova Biomedical), hourly postanesthesia for 2 h, and immediately before the removal of fetuses.
Histopathological studies
The fetuses and infants were quickly weighed, deeply anesthetized and perfused with 4% paraformaldehyde in cacodylate buffer (0.1%), pH 7.4, for histopathology studies of the brain at different intervals after maternal anesthesia. The effects of anesthesia were tested at two time points, based on which histological analysis was performed. For caspase‐3 and ‐9 immunochemical staining (ICC), an excellent method for marking neurons that are in an early stage of apoptosis (17, 61), the interval was 2 h; for NeuN and Nissl staining, useful for assessing the overall neuronal density in any given brain region, the interval was 35–40 days after anesthesia (during the first week of post‐natal life). Whereas caspase‐3 ICC was used for all quantification analyses, caspase‐9 ICC was used for selected cases involving the most vulnerable fetal brains [35–40 days of gestational age (GA)] exposed to either “true” or sham control conditions or to a triple anesthesia combination.
For activated caspase‐9 and caspase‐3 staining, 50‐µm‐thick vibrotome sections of fetal brains were washed in 0.01 M phosphate‐buffered saline (PBS), quenched for 10 minutes in a solution of methanol containing 3% hydrogen peroxide, then incubated for 1 h in blocking solution (2% BSA/0.2% milk/0.1% triton X‐100 in PBS). This was followed by incubation overnight in caspase‐specific antibodies—either rabbit anti‐active caspase‐9, diluted 1:2000 or anti‐active caspase‐3, diluted 1:1000 in blocking solution (D175, Cell Signaling Technology, Beverly, MA, USA). After incubation with D175 primary antibody, the sections were incubated for 1 h in secondary antibody (goat anti‐rabbit 1:200 in blocking solution), then reacted in the dark with ABC reagents (standard Vectastain ABC Elite Kit, Vector Labs, Burlingame, CA, USA). The sections were then preincubated for 10 minutes in a filtered mixture containing 6 mL of 0.1 M Tris buffer, 2 mg 3, 3′‐diaminobenzidine (DAB), and 400 mg imidazole, then for 15 minutes in 6 mL of the same DAB/imidazole/Tris mixture containing 3‐µL H2O2.
To study the neuronal densities in selected brain regions at completion of brain development, infant brains (n = 21) were perfused and fixed with 4% paraformaldehyde in cacodylate buffer (0.1%), pH 7.4. 35–40 days after anesthesia treatment; that is, during the first week of post‐natal life. We used Nissl and NeuN staining, well‐established techniques for labeling neurons. The most susceptible brain regions, and therefore the ones of most interest, were, among the cortical areas, the retrosplenial, parietal, cingulate, occipital and piriform regions, and among the subcortical areas, the amygdala, subiculum, hippocampus and anterior thalamus (35). For Nissl staining, the fixed brains were blocked in paraffin, cut into 7‐µm sections, and stained. For NeuN staining, neurons were immunolabeled using a monoclonal antibody against neuron‐specific nuclear protein (NeuN; 1:100; Chemicon).
Quantitative histology
To determine the density of caspase‐3 and Nissl‐stained neurons in a given brain region, we used the optical disector and fractionator method. A counting frame (0.05 × 0.05 mm, disector height, 0.05 mm for caspase‐stained; 0.05 × 0.05 mm, disector height, 0.007 mm for Nissl‐stained) and a high numerical aperture objective lens were used to visualize neurons. Once a brain region of interest was identified, three random areas within that region were subsampled using Stereo Investigator 6.02.2 computer software, which enabled us to count all neuronal profiles of interest with equal probability of their being included in the sample and not counted repeatedly. Unbiased subsampling of each area in each brain region was performed by randomly selecting 10–12 viewing fields in each area. The counting frame was positioned for counting at different focal levels in three different coronal sections (Stereo Investigator System, MicroBright Field, Inc, Williston, VT, USA) (24, 59, 61, 62). The numerical density of caspase‐3 or Nissl‐stained neurons in any given region was expressed as the number of cells/mm3. By using the optical disector and fractionator method, we were able to estimate neuronal densities in each brain region of interest without determining their precise anatomical boundaries (59). The counting was done by an investigator who was blind to the experimental conditions.
Statistical analyses
Anesthesia‐induced pathomorphological changes (the stereological cell count data) were assessed using ANOVA models that included a between‐subject variable such as treatment group and fetal age (20–25, 35–40 and over 50 days in utero). The within‐subject variable was brain regions. When ANOVAs with repeated measures were conducted, the Bonferroni correction or Tukey's test was used to help maintain prescribed alpha levels (eg, 0.05).
RESULTS
The adequacy of maternal oxygenation or ventilation, tissue perfusion and nutritional stability (Figure 1), as well as the stability of maternal vital signs (Figure 2), were regularly monitored and recorded every 30 and 15 minutes, respectively. We noted no significant differences between the experimental and sham pregnant guinea pigs in any physiological parameters at any point during the 4 h of anesthesia. The tight control is confirmed by the fact that recorded values were not significantly different from normal physiological ranges for guinea pigs (indicated with dotted lines in each graph) (5). Although blood glucose levels declined slightly in the experimental animals during the second half of exposure to anesthesia, neither the difference compared with sham controls nor variations in glucose stability were significant when compared with the normal physiological range (Figure 1). This finding suggests that anesthesia per se did not result in either severe hypo‐ or hyperglycemia. 1, 2 illustrate the results obtained from the experimental group that was exposed to a triple anesthesia combination (isoflurane, nitrous oxide and midazolam‐I + N + M, which was the most neurotoxic). Similar findings were obtained when isoflurane, alone or in combination with midazolam or nitrous oxide, was administered (data not shown).
Figure 1.
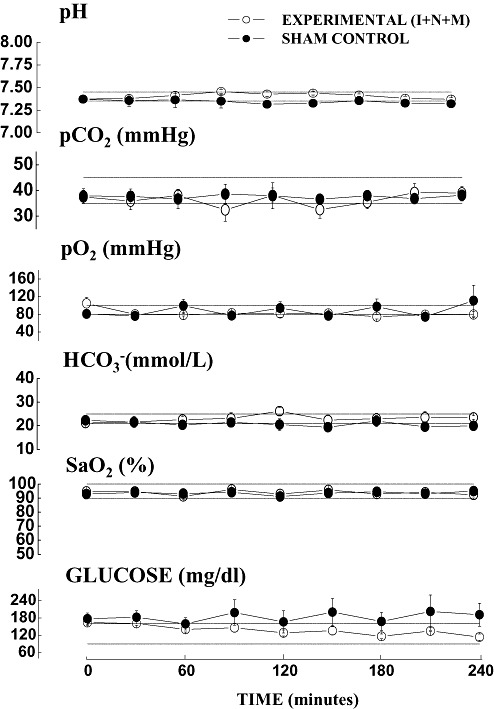
Anesthesia does not cause significant cardiorespiratory or metabolic disturbances in pregnant guinea pigs as determined by analysis of arterial blood gases. Conducting these analyses every 30 minutes. during exposure to anesthesia revealed no significant differences in any of the measured parameters in the experimental animals [exposed to isoflurane (0.55‐vol%) + N2O (75‐vol%) + midazolam (1 mg/kg, i.m.)—(I + N + M)] (n = 3 pregnant guinea pigs) compared with the sham animals (exposed to a continuous fentanyl infusion, 15 µg/kg/h) (n = 3 pregnant guinea pigs). The physiological range for guinea pigs for each parameter is indicated by a dotted line.
Figure 2.
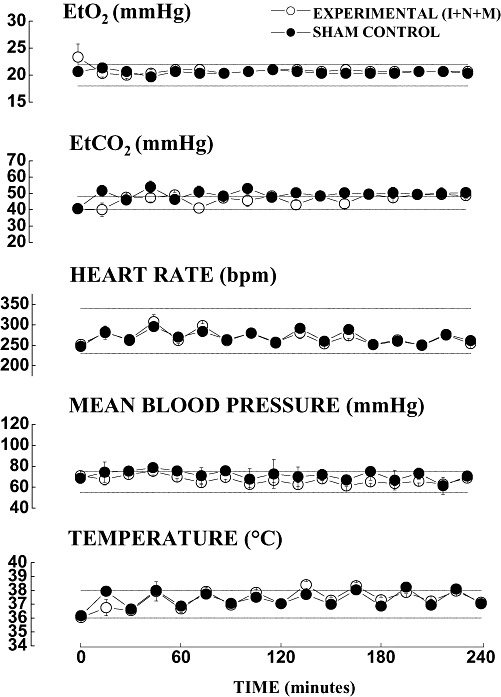
Anesthesia does not cause significant disturbances in the vital signs of pregnant guinea pigs. Close monitoring of vital signs was performed continuously and recorded every 15 minutes during exposure to anesthesia. No significant differences in any of the measured parameters in the experimental animals [exposed to isoflurane (0.55‐vol%) + N2O (75‐vol%) + midazolam (1 mg/kg, i.m.)––(I + N + M)] (n = 3 pregnant guinea pigs) compared with the sham animals (n = 3 pregnant guinea pigs). Physiological range for guinea pigs for each parameter is indicated by a dotted line.
To study the regional distribution and to compare the severity of anesthesia‐induced fetal neuroapoptosis caused by a variety of anesthesia protocols administered to the pregnant female pigs, we examined an extensive list of brain regions stained with caspase‐3 in fetal guinea pigs at the peak of their brain development (35–40 days GA) (3, 4, 1, 2).
Figure 3.
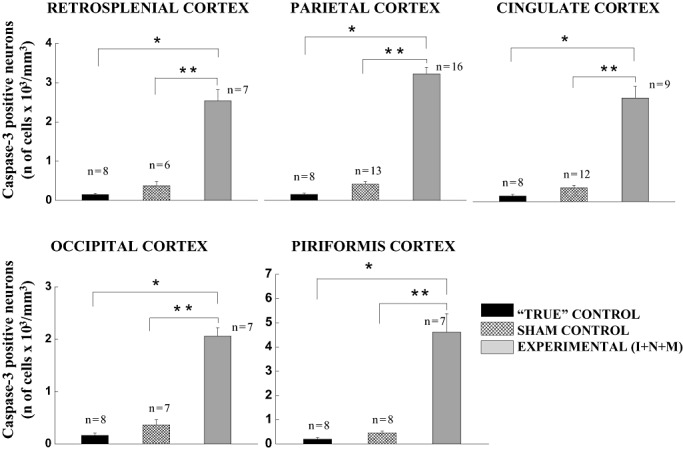
Anesthesia induces significant caspase‐3 activation in fetal guinea pig brains in several cortical brain regions. The sensitivity of cerebral cortices in fetal guinea pig brains (35–40 days gestational age) to general anesthesia is quantified as the number of caspase‐3‐positive cells/mm3 in the mid‐zone of the cortex (layers IV and V). The density of caspase‐3‐positive neurons in the “true” controls was minimal in all cortical regions (eg, retrosplenial, parietal, cingulate, occipital and piriform cortices). Although a somewhat higher degree of caspase‐3 labeling was detected in the sham controls (exposed to a continuous fentanyl infusion, 15 µg/kg/h), the difference was not statistically significant when compared with the “true” controls. Triple anesthesia combination containing isoflurane (0.55‐vol%), N2O (75 vol‐%), and midazolam [1 mg/kg, i.m. (I + N + M)] induced a significant increase in caspase‐3 labeling compared with the “true” and sham controls (*P < 0.001, and **P < 0.01, respectively). (The number of fetuses per each data point is indicated in the graph.)
Figure 4.
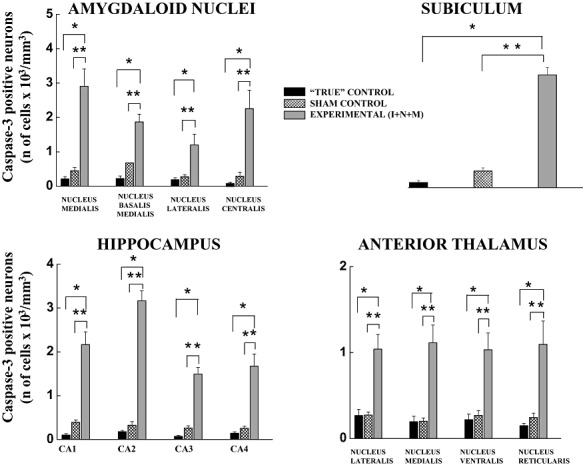
Anesthesia induces significant caspase‐3 activation in fetal guinea pig brains in several subcortical brain regions. The sensitivity of amygdala, subiculum, hippocampus and anterior thalamus in fetal guinea pig brains (35–40 days gestational age) to general anesthesia is quantified as the number of caspase‐3‐positive cells/mm3 in specific nuclei or regions, as indicated in each panel. Both control groups exhibit a low degree of developmental neuroapoptosis, with somewhat higher, although not significant, caspase‐3 staining in the sham controls (exposed to a continuous infusion of fentanyl, 15 µg/kg/h) compared with the “true” controls. The triple anesthesia combination containing isoflurane (0.55‐vol%), N2O (75‐vol%) and midazolam (1 mg/kg, i.m.) (I + N + M) induced a significant increase in caspase‐3 labeling compared with the “true” and sham controls (*P < 0.001, and **P < 0.01, respectively) (number of fetuses per each data point: 5–7 for amygdaloid nuclei, 5–10 for subiculum, 5–12 for hippocampus and 5–11 for anterior thalamus).
Table 1.
Cortical brain regions.
| Caspase‐3 positive neurons number of cells/ mm3 (fold increase from “true” control) | ||||
|---|---|---|---|---|
| True (n = 8) | ISO (n = 4) | ISO + N2O (n = 6) | ISO + midazolam (n = 3) | |
| Retrosplenial cortex | 149 ± 32 | 899 ± 112* (6) | 1194 ± 150* (8) | 1577 ± 511* (10.6) |
| Parietal cortex | 144 ± 33 | 697 ± 99* (4.8) | 1656 ± 252* (11.5) | 1007 ± 59* (7) |
| Cingulate cortex | 138 ± 43 | 433 ± 61* (3.1) | 1352 ± 296* (9.8) | 1247 ± 134* (9) |
| Occipital cortex | 167 ± 45 | 590 ± 25* (3.5) | 1286 ± 257* (7.7) | 1258 ± 541* (7.5) |
| Piriformis cortex | 203 ± 59 | 1236 ± 148* (6.1) | 2117 ± 159* (10.4) | 3177 ± 233* (15.7) |
P < 0.001 compared to “true” controls (see text).
Table 2.
Subcortical brain regions.
| Caspase‐3 positive neurons number of cells/mm3 (fold increase from “true” control) | |||||
|---|---|---|---|---|---|
| True (n = 5–7) | ISO (n = 4) | ISO + N2O (n = 6) | ISO + midazolam (n = 3) | ||
| Amygdaloid nuclei | Nucleus medialis | 213 ± 69 | 1017 ± 151* (4.8) | 730 ± 124* (3.4) | 1806 ± 455* (8.5) |
| Nucleus basalis medialis | 221 ± 80 | 966 ± 108* (4.4) | 831 ± 72* | 3160 ± 858* (14.3) | |
| Nucleus lateralis | 187 ± 58 | 523 ± 169* (2.8) | 980 ± 341* (5.2) | 728 ± 169* (3.9) | |
| Nucleus centralis | 72 ± 48 | 285 ± 149 (4) | 726 ± 203 (10.1) | 1399 ± 28* (19.4) | |
| Subiculum | 158 ± 65 | 1162 ± 319* (7.4) | 1343 ± 132* (8.5) | 2502 ± 356* (15.9) | |
| Hippocampus | CA1 | 104 ± 33 | 388 ± 103 (3.7) | 529 ± 149 (5.1) | 1501 ± 172* (14.4) |
| CA2 | 179 ± 29 | 1267 ± 107* (7.1) | 2376 ± 205* (13.3) | 1874 ± 98* (10.5) | |
| CA3 | 72 ± 23 | 788 ± 105* (11) | 1829 ± 113* (25.4) | 1472 ± 39* (20.4) | |
| CA4 | 145 ± 33 | 590 ± 155* (4.1) | 789 ± 116* (5.4) | 645 ± 98* (4.4) | |
| Anterior thalamus | Nucleus lateralis | 269 ± 71 | 349 ± 78 (1.3) | 952 ± 233* (3.5) | 1434 ± 265* (5.3) |
| Nucleus medialis | 195 ± 63 | 515 ± 139 (2.6) | 787 ± 138* (4) | 868 ± 104* (4.5) | |
| Nucleus ventralis | 219 ± 66 | 369 ± 106 (1.7) | 869 ± 191 (4) | 1597 ± 461* (7.3) | |
| Nucleus reticularis | 151 ± 26 | 402 ± 116 (2.7) | 662 ± 165 (4) | 1090 ± 202 (7.2) | |
P < 0.001 compared to “true” controls (see text).
While studying the developing fetal cortex, we observed that the neurons in the mid‐third zone (cortical layers IV and V) were consistently labeled with activated caspase‐3, whereas other cortical layers were minimally affected (cingulate cortical layer II) or were not affected by anesthesia treatments. Therefore, we focused our density studies on the mid zone because it appeared to be most sensitive. As shown in Table 1, the density of caspase‐3–positive neurons in the “true” controls varied somewhat in different cortical regions (eg, retrosplenial, parietal, cingulate, occipital and piriform cortices), although the number of neurons undergoing apoptosis was small. A low concentration of ISO (0.55‐vol%) induced a statistically significant increase in neuroapoptosis, on the average, three‐ to sixfold, in all cortical regions when compared with “true” controls (*P < 0.001). When either N2O (75‐vol%) or midazolam (1 mg/kg, i.m.) was combined with isoflurane (0.55‐vol%) (ISO + N2O and ISO + midazolam, respectively), we found further worsening of apoptotic damage compared with that in response to isoflurane alone. When compared with “true” controls, the degree of neuroapoptosis with double anesthesia combinations was, on the average, 7‐ to 16‐fold higher (*P < 0.001).
As Figure 3 shows, the effect of the triple anesthesia combination containing isoflurane (0.55‐vol%), nitrous oxide (75‐vol%), and midazolam (1 mg/kg, i.m.) [Experimental (I + N + M)] was extremely severe. For example, compared with “true” and sham controls, the increases in apoptotic damage were, on the average, 17‐ to 23‐fold (*P < 0.001) and 6‐ to 10‐fold (**P < 0.01), respectively, in all cortical regions of interest. Although a somewhat higher degree of apoptosis was detected in sham controls than in “true” controls, the difference was not statistically significant.
The studies of subcortical brain regions focused on the amygdala, subiculum, hippocampus and anterior thalamus because they were most vulnerable (Table 2, Figure 4). As indicated in Table 2, the “true” controls exhibited a low degree of developmental neuroapoptosis as measured by a low density of caspase‐3–positive neurons. However, exposure to isoflurane alone resulted in a significant increase in neuroapoptosis in the fetal amygdala (except in the nucleus centralis), subiculum and hippocampus (except in the CA1 region) compared with the “true” controls (*P < 0.001) (approximately 3‐ to 11‐fold higher density of caspase‐3‐stained neurons in isoflurane‐treated fetuses). Despite an approximately twofold higher density of caspase‐3‐positive neurons in anterior thalamic nuclei (eg, nucleus lateralis, nucleus medialis, nucleus ventralis, and nucleus reticularis) the difference was not statistically significant when compared with the “true” controls. When either N2O (75‐vol%) or midazolam (1 mg/kg, i.m.) was combined with isoflurane (0.55‐vol%), we found an overall worsening of apoptotic damage compared with that in response to isoflurane alone (except in the amygdaloid nuclei medialis and basalis medialis). When compared with the “true” controls, the degree of neuroapoptosis with double anesthesia combinations was, on the average, 4‐ to 25‐fold higher (*P < 0.001).
As shown in Figure 4, we detected a somewhat higher degree of apoptosis in sham than in “true” controls, although the difference was not statistically significant. However, when the triple anesthesia combination containing isoflurane (0.55‐vol%), N2O (75‐vol%), and midazolam (1 mg/kg, i.m.) [Experimental (I + N + M)] was tested, the neuroapoptosis was extremely severe overall. For example, compared with “true” and sham controls, the increases in apoptotic damage were, on the average, 4‐ to 31‐fold (*P < 0.001) and 3‐ to 10‐fold (**P < 0.01), respectively, in all subcortical regions of interest. Interestingly, in certain subcortical regions (eg, nucleus lateralis, ventralis and reticularis thalami; amygdaloid nucleus basalis medialis and the CA3 region of the hippocampus) double anesthesia combinations (Table 2) were as damaging or somewhat more damaging than was the triple combination (Figure 4), although the differences were not statistically significant.
When the vulnerability of the same brain regions were examined in fetal guinea pigs in the early stages of brain development (20–25 days GA), we found that a triple anesthesia cocktail was, again, most damaging compared with the “true” controls (an approximately 6‐ to 20‐fold increase in apoptosis) (P < 0.05), whereas isoflurane alone was not significantly more neurotoxic than the “true” controls (data not shown). The studies of fetal guinea pig brains in the late stages of development (50+ days) revealed a complete lack of sensitivity to the triple anesthesia cocktail; that is, we were unable to detect any signs of caspase‐3 activation in any vulnerable brain regions (n = 5 fetuses) (data not shown).
Figure 5 illustrates caspase‐3 and caspase‐9 stages of apoptotic neurodegeneration observed in the mid‐third zone (cortical layers IV and V) of the parietal cortex of fetal brains (35–40 GA) exposed to the triple anesthesia combination, the most damaging anesthesia protocol. Early stage caspase‐9 activation a precursor step in caspase‐3 activation (32, 61, 63), is shown in the upper portion of Figure 5. Practically undetectable caspase‐9 labeling was noted in “true” controls and only very minimal labeling was noted in sham controls, whereas there was substantial caspase‐9 activation detected in the experimental cortex. Subsequent‐stage caspase‐3 labeling, a final step leading to DNA fragmentation and cell death, is illustrated in the lower portion of Figure 5. A substantial increase in caspase‐3 staining was noted in the experimental cortex compared with “true” and sham controls. Note similar laminar distribution of staining in both stages of apoptotic neurodegeneration, suggesting that caspase‐3 activation is, at least in part, mediated via the activation of caspase‐9.
Figure 5.
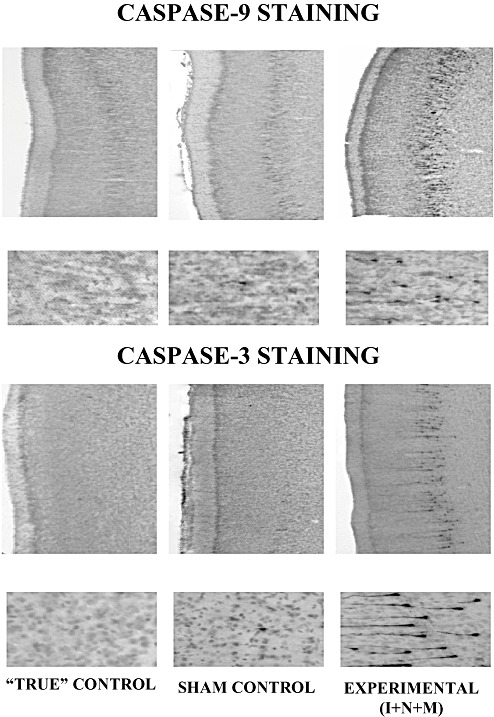
Various stages of anesthesia‐induced apoptotic neurodegeneration in the mid‐zone of parietal cortex (cortical layers IV and V). Exposure of fetal guinea pigs at the peak of their brain development (35–40 days gestational age) to the triple anesthesia combination (isoflurane, 0.55‐vol%, + N2O, 75‐vol%, + midazolam, 1 mg/kg, i.m.) (Experimental—I + N + M) induced significant caspase‐9 activation, a precursor step in caspase‐3 activation. Practically undetectable and only a very minimal caspase‐9 labeling was noted in the “true” (no anesthesia) and sham controls, (exposed to a continuous infusion of fentanyl, 15 µg/kg/h), respectively. Significant caspase‐3 labeling was noted in the experimental cortex compared with that of the two control groups. Note the similar laminar distribution of staining in both stages of apoptotic neurodegeneration (caspase‐9 microphotographs: magnification 10× in upper panels; 40× in lower panels) (caspase‐3 microphotographs: magnification 10× in upper panels; 60× lower panels).
Because caspase‐3 activation occurs when the neurons are committed to die, we set out to determine the severity of anesthesia‐induced overall neuronal loss in the most vulnerable brain regions once the dying neurons were phagocytized and the neuronal debris had been removed. Hence, we examined the most vulnerable regions in young guinea pig brain many days after anesthesia treatment (during the first week of post‐natal life) using Nissl staining. Because the triple combination of anesthetics given at the peak of the growth spurt of the guinea pig brain (35–40 days GA) was most damaging in the majority of vulnerable brain regions, we compared this experimental group with the “true” and sham controls treated at the same age. Figure 6 illustrates the neuronal densities in the mid‐zone (cortical layers IV and V) of the most vulnerable cortical regions (eg, retrosplenial, parietal, cingulate, occipital and piriform cortices) of the control (“true” and sham) and experimental animals. We found a significant difference in anesthesia‐induced neuronal loss in the experimental animals vs. the “true” and sham controls (*P < 0.01, and **P < 0.05, respectively). The overall anesthesia‐induced neuronal loss in the experimental animals varied from 40% to 50% compared with the controls. A comparison of the “true” and sham controls revealed similar neuronal densities in all cortical regions.
Figure 6.
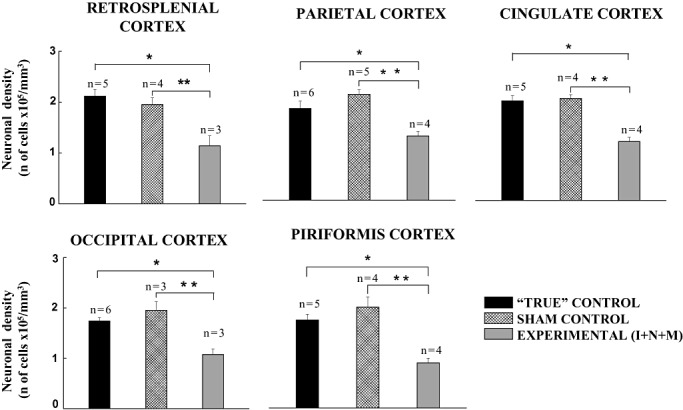
Anesthesia induces significant neuronal loss in vulnerable cortical brain regions. Neuronal density counts were obtained using Nissl‐stained slides. Fully developed guinea pig brains (first week of post‐natal life) of the “true” controls were compared with the experimental animals that had been exposed to the triple anesthesia (isoflurane, 0.55‐vol%, + N2O, 75‐vol%, + midazolam, 1 mg/kg, i.m.) or the sham procedure (continuous fentanyl infusion, 15 µg/kg/h) when they were at the peak of their development (35–40 days gestational age). The sham and “true” controls maintain similar neuronal densities, whereas the experimental animals show a significant decrease in neuronal densities in all vulnerable cortical brain regions compared with the “true” and sham control groups (*P < 0.01, and **P < 0.05, respectively). (The number of fetuses per each data point is indicated in the graph.)
The neuronal densities in vulnerable subcortical brain regions (eg, amygdala, subiculum, anterior thalamus and hippocampus) of the two control groups (“true” and sham) and the experimental (I + N + M) animals are evaluated in Figure 7. In general, we found a significant neuronal loss in the experimental animals when compared with the two control groups (*P < 0.01, and **P < 0.05, respectively); the overall anesthesia‐induced neuronal loss in the experimental group varied from 30% to 50% compared with the control groups. When the two control groups were compared, we found that the neuronal densities in all subcortical brain regions were similar. The representative microphotographs of hippocampal CA1 region (left lower panels) show a substantial decrease in neuronal densities in an experimental animal compared to a “true” control.
Figure 7.
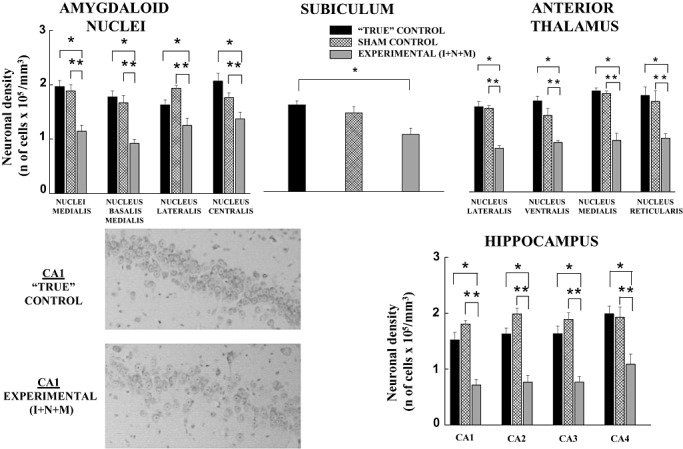
Anesthesia induces significant neuronal loss in vulnerable subcortical brain regions. Neuronal density counts were obtained using Nissl‐stained slides. Fully developed guinea pig brains (first week of post‐natal life) in the “true” control group were compared with the experimental animals that had been exposed to triple anesthesia (isoflurane, 0.55‐vol%, + N2O, 75‐vol%, + midazolam, 1 mg/kg, i.m.) or the sham procedure (continuous infusion of fentanyl, 15 µg/kg/h) when they were at the peak of their development (35–40 days gestational age). Note that the “true” and sham groups maintain similar neuronal densities (*P < 0.01, and **P < 0.05), whereas the experimental animals show a significant decrease in neuronal densities in all vulnerable subcortical regions. (Number of fetuses per each data point: 4–5 for amygdaloid nuclei, 3–5 for subiculum, 4–5 for anterior thalamus and 4–6 for hippocampus). The representative microphotographs of the hippocampal CA1 region indicate a lower density of Nissl stained cells in the “true” control animals compared with the experimental animals.
DISCUSSION
In this study, we show that the commonly used general anesthetic isoflurane, alone or in combination with either nitrous oxide or midazolam or both, induces severe and widespread apoptotic neurodegeneration in fetal guinea pigs at the peak of the brain growth spurt. This damage ultimately leads to a loss of a significant number of neurons in vulnerable regions of the brain, suggesting that anesthesia‐induced neuroapoptosis can result in permanent brain damage. The intravenous narcotic fentanyl did not significantly increase apoptotic damage in vulnerable fetal brain regions and consequently did not induce a significant decrease in neuronal densities later on during development.
The quantification analyses of anesthesia‐induced developmental neuroapoptosis presented here are based on the activation of caspase‐3 because it represents the final and converging step in apoptotic activation, the point at which a neuron is committed to die. The fact that activated caspase‐9 labeling in fetal brains exposed to a severely neurotoxic triple anesthesia combination shows a similar pattern of distribution suggests that caspase‐3 activation is mediated, at least in part, via the activation of caspase‐9. In the future, detailed mechanistic studies should address the involvement and relative importance of different apoptotic cascades in anesthesia‐induced apoptotic neurodegeneration in fetal guinea pig brain.
The brain's growth spurt consists of several stages that are precisely timed and vary in duration among species. In humans, its duration is fairly long, spanning both the in utero (last trimester of pregnancy) and post‐natal (first couple of years) periods. In guinea pigs and rats, its duration is much shorter (10 and 2 weeks, respectively) and takes place either mostly in utero (in guinea pigs) or mostly after birth (in rats) (9). In guinea pigs, the species of interest for this study, synaptogenesis, an important stage of the brain's growth spurt, includes the period of intense formation of synapses, which starts midway during in utero life and culminates by day 50 (25, 27, 31). Studies of not only the number of synaptic junctions but synaptic maturity (eg, the number of dense projections per junction, the length of the junction and the narrowing of the synaptic cleft) have determined that at the end of the gestational period (59–72 days) (41) the guinea pig brain is almost fully developed (25); at birth, the guinea pig is essentially fully functional and its eyes are open (31).
To appropriately evaluate the risks posed by commonly used anesthetics in human anesthesia, it is important to understand whether a species with a longer brain growth spurt is less vulnerable to brief exposure to anesthesia. Our study indicates that, at the peak of brain growth, only 4 h of anesthesia, which represents a brief time during the guinea pig brain development (on the average, 70 days), caused significant and widespread neuroapoptotic damage in the fetal brain. This damage is comparable in intensity to the damage to the immature rat brain after 6 h of anesthesia (24, 61, 62) despite the fact that the duration of brain development in guinea pigs is approximately five times longer than it is in rats.
However, according to the present study and a previously published report (61), it appears that the timing of exposure to anesthesia is more important than its duration; that is, the same anesthesia protocol administered when development of the immature brain is at its peak causes significantly more damage than if the insult occurs later in development. The reason is that the neuronal tasks at the peak of brain development are multifaceted and include several important elements (eg, migration, formation of synapses, differentiation, maturation). These elements are precisely timed and require highly coordinated neuronal communications (see 63 for a review), factors that make the immature neurons especially vulnerable if the circumstances of their normal functioning have been altered by an external insult such as exposure to anesthesia.
In daily practice, the tight control of vital signs and functions during anesthesia is crucially important to assure adequate homeostasis. By anesthetizing pregnant guinea pigs, we were able to mimic the clinically relevant anesthesia setup and to monitor the adequacy of maternal ventilation, oxygenation, and tissue perfusion at any point in time, which was technically impossible with immature rats because of their small size. We found that despite strict control of all maternal vital parameters, the amount of fetal neuronal damage that occurred in the experimental animals was many‐fold greater than was the case in either the sham or “true” controls. It is important to note that in designing these experiments, because direct monitoring of fetal ventilation, oxygenation and perfusion in guinea pigs was not technically feasible, we relied on the common assumption that careful monitoring and tight control over maternal vital functions and well‐being during anesthesia assured adequate well‐being of a fetus. This assumption has been tested in the practice of obstetric anesthesia and in several animal studies. The results have shown that any abnormalities in maternal cardiopulmonary functions could have a negative impact on the fetus (1, 4, 54) and that therapeutic interventions aimed at correcting maternal vital functioning could be beneficial for the fetus (11, 16). Because all anesthetics, including the ones used in our study, easily cross the utero‐placental barrier (10, 18, 30, 36, 60) without significantly affecting in utero placental blood flow (6, 11, 43), we believe that pregnant guinea pigs are a good model for assessing fetal anesthesia‐induced neurotoxicity. Because the apoptotic neurodegeneration observed in the experimental fetal brains was more severe than that observed in the sham and “true” control fetal brains, despite adequate maintenance of maternal vital functions, we conclude that general anesthesia per se is the main culprit, not a hypoxia/hypercarbia or metabolic imbalance (7).
Although fentanyl's potential to induce neuroapoptosis during the brain growth spurt has not been studied, several reports suggest that chronic administration of other narcotics (eg, morphine, buprenorphine) very early during development (in neurogenesis) as well as later (in adulthood) can modulate a variety of apoptotic markers (eg, c‐jun, caspase‐3) (13, 19). This results in obvious changes in neuronal morphology, such as reduced dendritic arborization and density of dendritic spines (49, 52) that, in turn, may lead to abnormalities in the formation of neuronal circuitries. Our findings with continuous infusion of fentanyl suggest that brief exposure to this intravenous narcotic, unlike brief exposure to volatile anesthetic isoflurane, does not induce significant neuroapoptosis or significant neuronal deletion when compared with the controls. However, establishing whether prolonged exposure to fentanyl (eg, for days and weeks) during critical stages of the brain growth spurt could induce severe apoptotic neurodegeneration would be important, especially because continuous administration of fentanyl is a part of the standard sedation protocol in many neonatal and pediatric intensive care units.
The development of neuronal circuitries in the mammalian brain is complex, and although certain stages are activity‐independent (56), formation of the majority of neuronal synapses is highly dependent on constant neuronal signaling. Thus, refinement and fine‐tuning of synaptic connections is practically impossible when this signaling is severely inhibited (26). Based on our findings, it seems that combining different general anesthetics to achieve the desired depth of anesthesia increases the vulnerability of the developing brain to neuroapoptotic damage. For instance, although a low, clinically relevant, concentration of isoflurane alone (at 0.55‐vol%, which is only 0.5 MAC—the minimum alveolar anesthetic concentration that prevents purposeful movement to supramaximal noxious stimulation in 50% guinea pigs) (51) was pro‐apoptotic at the peak of brain growth spurt, the severity of neuroapoptosis was significantly augmented by the addition of very low concentrations of nitrous oxide (at 75‐vol% which is only 0.375–0.5 MAC for guinea pigs) (55) and a low dose midazolam (commonly used in sedation protocols) (45). It remains to be determined whether the higher toxicity potential of anesthesia cocktails compared with a single anesthetic is caused by a more profound generalized inhibition of neuronal activity caused by an anesthetic combination or caused by, at least in part, a combined and synchronized modulatory effect(s) on two of the most important neuro‐transmitter systems during the development—glutamate (nitrous oxide‐induced inhibition of N‐methyl‐D‐aspartate system) (23) and γ‐amino‐butyric acid‐GABA (isoflurane/midazolam‐induced over‐stimulation of GABAergic system) (14).
The development and function of NMDA and GABAA receptor systems in fetal guinea pigs could be modulated by maternal exposure to environmental challenges. For example, maternal hypoxia has been shown to cause selective and differential modification of recognition, co‐activation and modulation the NMDA receptor ion channel complex in fetal guinea pig cortices. This resulted in increased affinity and basal activation, ultimately leading to hyperstimulation of the NMDA receptor ion channel complex and a possible increase in fetal susceptibility to hypoxia (38). Maternal exposure to ethanol resulted in alterations in fetal hippocampal GABAA receptor expression, and pharmacological properties, thus contributing to hippocampal‐related behavioral abnormalities and cognitive deficits in guinea pig offspring (20). Although detailed studies of the function and subunit composition of these two receptor systems in all brain regions of fetal guinea pigs are not available, it is reasonable to propose that the brain‐region‐specific differences in vulnerability of the fetal guinea pig brain to a variety of anesthetic protocols could be caused by unique receptor/ion channel composition and interplay at each point during fetal brain development.
Several pathways of anesthesia‐induced apoptotic cell death have been described, including mitochondria‐dependent and ‐independent (death receptor‐dependent) (61, 62), as well as neurotrophic factor‐dependent (32), suggesting that anesthesia‐induced developmental neuroapoptosis is a complex phenomenon. However, it has been established that caspase‐3 activation is the final step at which a developing neuron is committed to die, demonstrating that immunocytochemical analysis using activated caspase‐3 is a reliable method for mapping neurons dying by apoptosis (21, 22, 24, 42, 61, 62).
Physiological “pruning” of the redundant neurons is commonly observed in the developing mammalian brain. It is estimated that less than 1% of the total neuronal population (with some regional variations) succumb to apoptosis (21, 22). Thus, only a small percentage of the neuronal population is expected not to survive during the normal brain growth spurt. Our findings with “true” control animals support this notion. However, based on our evaluation of the experimental animals, it appears that general anesthesia increases neuronal apoptosis many times over, leading to an alarming increase in neuronal “pruning” (as much as 50% in most vulnerable brain regions). Similar severity of damage has been described in the hippocampus (approximately 30% neuronal loss in the CA1 region) of young post‐natal guinea pigs chronically exposed to ethanol in utero (15) which also resulted in impaired cognitive functioning (47). Although the functional importance of anesthesia‐induced neuronal loss in guinea pigs remains to be determined, our studies of rats suggest that early exposure to general anesthesia during the brain growth spurt causes permanent impairment of learning and memory capabilities (24).
In conclusion, our study demonstrates that a single short maternal exposure to commonly used general anesthesia induces significant neuroapoptotic degeneration in developing fetal guinea pig brains. This leads to a loss of developing neurons in the most vulnerable brain regions.
ACKNOWLEDGMENTS
Our research is supported by NIH/NICHD HD 44517 (V.J‐T.) and Dr. Harold Carron endowment (V.J‐T.). V.J‐T. is an Established Investigator of the American Heart Association.
This work was presented in part at the Neuroscience Meeting, Washington, DC, 2005 and Anesthetic and Life support Drug Advisory Committee Meeting, FDA, Rockville, MD, 2007.
REFERENCES
- 1. Adamsons K, Mueller‐Heubach E, Myers RE (1971) Production of fetal asphyxia in the rhesus monkey by administration of catecholamine to the mother. Am J Obstet Gynecol 109:248–262. [DOI] [PubMed] [Google Scholar]
- 2. Anand KJ, Soriano SG (2004) Anesthetic agents and the immature brain: are these toxic or therapeutic? Anesthesiology 101:527–530. [DOI] [PubMed] [Google Scholar]
- 3. Bittigau P, Sifringer M, Genz K, Reith E, Pospischil D, Govindarajalu S et al (2002) Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc Natl Acad Sci USA 99:15089–15094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brann AW, Myers RE (1975) Central nervous system findings in the newborn monkey following severe in utero partial asphyxia. Neurology 25:327–338. [DOI] [PubMed] [Google Scholar]
- 5. Brown JN, Thorne PR, Nuttall AL (1989) Blood pressure and other physiological responses in awake and anesthetized guinea pigs. Lab Anim Sci 39:142–148. [PubMed] [Google Scholar]
- 6. Conklin KA, Graham CW, Murad S, Randall FM, Katz RL, Cabalum T et al (1980) Midazolam and diazepam: maternal and fetal effects in pregnant ewe. Obstet Gynecol 56:471–474. [PubMed] [Google Scholar]
- 7. Daval JL, Vert P (2004) Apoptosis and neurogenesis after transient hypoxia in the developing rat brain. Semin Perinatol 28:257–263. [DOI] [PubMed] [Google Scholar]
- 8. Dobbing J (1985) Infant nutrition and later achievement. Am J Clin Nutr 41:477–484. [DOI] [PubMed] [Google Scholar]
- 9. Dobbing J, Sands J (1979) Comparative aspects of the brain growth spurt. Early Hum Dev 3:79–83. [DOI] [PubMed] [Google Scholar]
- 10. Dwyer R, Fee JP, Moore J (1995) Uptake of halothane and isoflurane by mother and baby during caesarean section. Br J Anaesthesiol 74:379–383. [DOI] [PubMed] [Google Scholar]
- 11. Eisenach JC (1999) Uteroplacental blood flow. In: Obstetric Anesthesia, 2nd edn. Chestnut DH (ed.), pp. 43–56. Mosby: St Louis. [Google Scholar]
- 12. Epemolu O, Bom A, Hope F, Mason R (2003) Reversal of neuromuscular blockade and simultaneous increase in plasma rocuronium concentration after the intravenous infusion of the novel reversal agent Org 25969. Anesthesiology 99:632–637. [DOI] [PubMed] [Google Scholar]
- 13. Fan XL, Zhang JS, Zhang XQ, Ma L (2003) Chronic morphine treatment and withdrawal induce up‐regulation of c‐Jun N‐terminal kinase 3 gene expression in rat brain. Neuroscience 122: 997–1002. [DOI] [PubMed] [Google Scholar]
- 14. Franks NP, Lieb WR (1994) Molecular and cellular mechanisms of general anaesthesia. Nature 367:607–614. [DOI] [PubMed] [Google Scholar]
- 15. Gibson MA, Butters NS, Reynolds JN, Brien JF (2000) Effects of chronic prenatal ethanol exposure on locomotor activity, and hippocampal weight, neurons, and nitric oxide synthase activity of the young postnatal guinea pig. Neurotoxicol Teratol 22:183–192. [DOI] [PubMed] [Google Scholar]
- 16. Haydon ML, Gorenberg DM, Nageotte MP, Ghamsary M, Rumney PJ, Patillo C, Garite TJ (2006) The effect of maternal oxygen administration on fetal pulse oximetry during labor in fetuses with nonreassuring fetal heart rate patterns. Am J Obstet Gynecol 195:735–738. [DOI] [PubMed] [Google Scholar]
- 17. Hengartner MO (2000) The biochemistry of apoptosis. Nature 407:770–776. [DOI] [PubMed] [Google Scholar]
- 18. Herman NL (1999) The placenta: anatomy, physiology and transfer of drugs. In: Obstetric Anesthesia, 2nd edn. Chestnut DH (ed.), pp. 57–74. Mosby: St Louis. [Google Scholar]
- 19. Hu S, Sheng WS, Lokensgard JR, Peterson PK (2002) Morphine induces apoptosis of human microglia and neurons. Neuropharmacology 42:829–836. [DOI] [PubMed] [Google Scholar]
- 20. Igbal U, Dringenberg HC, Brien JF, Reynolds JN (2004) Chronic prenatal ethanol exposure alters hippocampal GABA(A) receptors and impairs spatial learning in the guinea pig. Behav Brain Res 150:117–125. [DOI] [PubMed] [Google Scholar]
- 21. Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vöckler J, Dikranian K et al (1999) Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 283:70–74. [DOI] [PubMed] [Google Scholar]
- 22. Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K et al (2000) Ethanol‐induced apoptotic neurodegeneration and fetal alcohol syndrome. Science 287:1056–1060. [DOI] [PubMed] [Google Scholar]
- 23. Jevtovic‐Todorovic V, Todorović SM, Mennerick S, Powell S, Dikranian K, Benshoff N, Zorumski CF, Olney JW (1998) Nitrous oxide (laughing gas) is an NMDA antagonist, neuroprotectant and neurotoxin. Nat Med 4:460–463. [DOI] [PubMed] [Google Scholar]
- 24. Jevtovic‐Todorovic V, Hartman RE, Izumi Y, Benshoff ND, Dikranian K, Zorumski CF, Olney JW, Wozniak DF (2003) Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci 23:876–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jones DG, Dittmer MM, Reading LC (1974) Synaptogenesis in guinea‐pig cerebral cortex: a glutaraldehyde‐PTA study. Brain Res 70:245–259. [DOI] [PubMed] [Google Scholar]
- 26. Katz LC, Shatz CJ (1996) Synaptic activity and the construction of cortical circuits. Science 274:1133–1138. [DOI] [PubMed] [Google Scholar]
- 27. Lennon AM, Francon J, Fellous A, Nunez J (1980) Rat, mouse, and guinea pig brain development and microtubule assembly. J Neurochem 35:804–813. [DOI] [PubMed] [Google Scholar]
- 28. Levison G, Shnider SM, De Lorimier AA, Steffenson JL (1974) Effects of maternal hyperventilation on uterine blood flow and fetal oxygenation and acid‐base status. Anesthesiology 40:340–347. [DOI] [PubMed] [Google Scholar]
- 29. Loepke AW, McCann JC, Kurth CD, McAuliffe JJ (2006) The physiologic effects of isoflurane anesthesia in neonatal mice. Anesth Analg 102:75–80. [DOI] [PubMed] [Google Scholar]
- 30. Loftus JR, Hill H, Cohen SE (1995) Placental transfer and neonatal effects of epidural sufentanil and fentanyl administered with bupivacaine during labor. Anesthesiology 83:300–308. [DOI] [PubMed] [Google Scholar]
- 31. Lohmann SM, Ueda T, Greengard P (1978) Ontogeny of synaptic phosphoproteins in brain. Proc Natl Acad Sci USA 75:4037–4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lu LX, Yon JH, Carter LB, Jevtovic‐Todorovic V (2006) General anesthesia activates BDNF‐dependent neuroapoptosis in the developing rat brain. Apoptosis 11:1603–1615. [DOI] [PubMed] [Google Scholar]
- 33. Lucas A, Morley R, Cole TJ, Gore SM, Lucas PJ, Crowle P et al (1990) Early diet in preterm babies and developmental status at 18 months. Lancet 335:1477–1481. [DOI] [PubMed] [Google Scholar]
- 34. Lucas A, Morley R, Cole TJ (1998) Randomized trial of early diet in preterm babies and later intelligence quotient. BMJ 317:1481–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Luparello TJ (1967) Stereotaxic Atlas of the Forebrain of the Guinea Pig. S. Karger, Basel, Switzerland. [Google Scholar]
- 36. Marx GF, Joshi CW, Orkin LR (1970) Placental transmission of nitrous oxide. Anesthesiology 32:429–432. [DOI] [PubMed] [Google Scholar]
- 37. Mellon RD, Simone AF, Rappaport BA (2007) Use of anesthetic agents in neonates and young children. Anesth Analg 104:509–520. [DOI] [PubMed] [Google Scholar]
- 38. Mishra OP, Delivoria‐Papadopoulos M (1992) NMDA receptor modification in the fetal guinea pig brain during hypoxia. Neurochem Res 17:1211–1216. [DOI] [PubMed] [Google Scholar]
- 39. Motoyama EK, Rivard G, Acheson F, Cook CD (1967) The effect of changes in maternal pH and PCO2 on the PO2 of fetal lambs. Anesthesiology 28:891–903. [DOI] [PubMed] [Google Scholar]
- 40. Newman W, McKinnon L, Phillips L, Paterson P, Wood C (1967) Oxygen transfer from mother to fetus during labor. Am J Obstet Gynecol 99:61–70. [DOI] [PubMed] [Google Scholar]
- 41. Noonan D (1994) The guinea pig (Cavia porcellus). ANZCCART News 7:1–8. [Google Scholar]
- 42. Olney JW, Wozniak DF, Jevtovic‐Todorovic V, Farber NB, Bittigau P, Ikonomidou C et al (2002) Drug‐induced apoptotic neurodegeneration in the developing brain. Brain Pathol 12:488–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Palahniuk RJ, Shnider SM (1974) Maternal and fetal cardiovascular and acid‐base changes during halothane and isoflurane anesthesia in pregnant ewe. Anesthesiology 41:462–472. [DOI] [PubMed] [Google Scholar]
- 44. Perouansky M (2007) Liaisons dangereuses? General anaesthetics and long‐term toxicity in the CNS. Eur J Anaesthesiol 24:107–115. [DOI] [PubMed] [Google Scholar]
- 45. Quesenberry KE, Hillyer EV (1997) Ferrets, Rabbits and Rodents: Clinical Medicine and Surgery, pp. 243–258. WB Saunders, Philadelphia. [Google Scholar]
- 46. Rajan V, Beharry KDA, Williams P, Modanlou HD (1998) Pharmacodynamic effects and pharmacokinetic profile of continuous infusion fentanyl in newborn piglets. Biol Neonate 74:39–47. [DOI] [PubMed] [Google Scholar]
- 47. Richardson DP, Byrnes ML, Brien JF, Reynolds JN, Dringenberg HC (2002) Impaired acquisition in the water maze and hippocampal long‐term potentiation after chronic prenatal ethanol exposure in the guinea‐pig. Eur J Neurosci 16:1593–1598. [DOI] [PubMed] [Google Scholar]
- 48. Rivard G, Motoyama EK, Acheson F, Cook CD, Reynolds EO (1967) The relation between maternal and fetal oxygen tensions in sheep. Am J Obstet Gynecol 97:925–930. [DOI] [PubMed] [Google Scholar]
- 49. Robinson TE, Kolb B (1999) Morphine alters the structure of neurons in the nucleus accumbens and neocortex of rats. Synapse 33:160–162. [DOI] [PubMed] [Google Scholar]
- 50. Scallet AC, Schmued LC, Slikker W Jr, Grunberg N, Faustino PJ, Davis H et al (2004) Developmental neurotoxicity of ketamine: morphometric confirmation, exposure parameters, and multiple fluorescent labeling of apoptotic neurons. Toxicol Sci 81:364–370. [DOI] [PubMed] [Google Scholar]
- 51. Seifen AB, Kennedy RH, Bray JP, Seifen E (1989) Estimation of minimum alveolar concentration (MAC) for halothane, enflurane and isoflurane in spontaneously breathing guinea pigs. Lab Anim Sci 39:579–581. [PubMed] [Google Scholar]
- 52. Sklair‐Tavron L, Shi WX, Lane SB, Harris HW, Bunney BS, Nestler EJ (1996) Chronic morphine induces visible changes in the morphology of mesolimbic dopamine neurons. Proc Natl Acad Sci U S A 93:11202–11207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Slikker W Jr, Zou X, Hotchkiss CE, Divine RL, Sadovova N, Twaddle NC et al (2007) Ketamine‐induced neuronal cell death in the perinatal rhesus monkey. Toxicol Sci 98:145–158. [DOI] [PubMed] [Google Scholar]
- 54. Stek AM, Fisher BK, Baker RS, Lang U, Tseng CY, Clark KE (1993) Maternal and fetal cardiovascular responses to methamphetamine in the pregnant sheep. Am J Obstet Gynecol 169:888–897. [DOI] [PubMed] [Google Scholar]
- 55. Stevens WC, Eger EI II, White A, Biava CG, Gibbons RD, Shargel R (1977) Comparative toxicities of enflurane, fluroxene and nitrous oxide at subanaesthetic concentrations in laboratory animals. Can Anaesth Soc J 24:479–490. [DOI] [PubMed] [Google Scholar]
- 56. Tessier‐Lavigne M, Goodman CS (1996) The molecular biology of axon guidance. Science 274:1123–1133. [DOI] [PubMed] [Google Scholar]
- 57. Vutskits L, Gascon E, Tassonyi E, Kiss JZ (2005) Clinically relevant concentrations of propofol but not midazolam alter in vitro dendritic development of isolated gamma‐aminobutyric acid‐positive interneurons. Anesthesiology 102:970–976. [DOI] [PubMed] [Google Scholar]
- 58. Walker A, Maddern L, Day E, Renou P, Talbot J, Wood C (1971) Fetal scalp tissue oxygen tension measurements in relation to maternal dermal oxygen tension and fetal heart rate. J Obstet Gynaecol Br Commonw 78:1–12. [DOI] [PubMed] [Google Scholar]
- 59. West MJ (1999) Stereological methods for estimating the total number of neurons and synapses: issues of precision and bias. Trends Neurosci 22:51–61. [DOI] [PubMed] [Google Scholar]
- 60. Wilson CM, Dundee JW, Moore J, Howard PJ, Collier PS (1987) A comparison of the early pharmacokinetics of midazolam in pregnant and nonpregnant women. Anaesthesia 42:1057–1062. [DOI] [PubMed] [Google Scholar]
- 61. Yon J‐H, Daniel‐Johnson J, Carter LB, Jevtovic‐Todorovic V (2005) Anesthesia induces neuronal cell death in the developing rat brain via the intrinsic and extrinsic apoptotic pathways. Neuroscience 35:815–827. [DOI] [PubMed] [Google Scholar]
- 62. Yon J‐H, Carter LB, Reiter RJ, Jevtovic‐Todorovic V (2006) Melatonin reduces the severity of anesthesia‐induced apoptotic neurodegeneration in the developing rat brain. Neurobiol Dis 21:522–530. [DOI] [PubMed] [Google Scholar]
- 63. Yuan J, Yankner BA (2000) Apoptosis in the nervous system. Nature 407:802–808. [DOI] [PubMed] [Google Scholar]