

Abstract
Background
Contractile discordance exacerbates cardiac dysfunction, aggravating heart failure outcome. Dissecting the genesis of mechanical dyssynchrony would enable an early diagnosis before advanced disease.
Methods and Results
High‐resolution speckle‐tracking echocardiography was applied in a knockout murine surrogate of adult‐onset human cardiomyopathy caused by mutations in cardioprotective ATP‐sensitive K+ (KATP) channels. Preceding the established criteria of cardiac dyssynchrony, multiparametric speckle‐based strain resolved nascent erosion of dysfunctional regions within cardiomyopathic ventricles of the KATP channel–null mutant exposed to hemodynamic stress. Not observed in wild‐type counterparts, intraventricular disparity in wall motion, validated by the degree, direction, and delay of myocardial speckle patterns, unmasked the disease substrate from asymptomatic to overt heart failure. Mechanical dyssynchrony preceded widening of the QRS complex and exercise intolerance and progressed into global myocardial discoordination and decompensated cardiac pump function, precipitating a low output syndrome.
Conclusions
The present study, with the use of high‐resolution imaging, prospectively resolved the origin and extent of intraventricular motion disparity in a KATP channel–knockout model of dilated cardiomyopathy. Mechanical dyssynchrony established as an early marker of cardiomyopathic disease offers novel insight into the pathodynamics of dyssynchronous heart failure.
Keywords: ATP‐sensitive K+ channel, heart failure, Kir6.2, QRS complex, speckle‐tracking
Introduction
Dilated cardiomyopathy is an intrinsic and progressive disorder of the myocardium and a major cause of end‐stage heart failure requiring device implantation or organ transplant.1 Accumulating evidence implicates genetic and environmental factors2; however, the natural course of disease pathophysiology is not fully understood. Dilated cardiomyopathy is typically diagnosed at an advanced stage, based on systemic symptoms and cardiac global evaluation.3 Recognizing early presentation, before overt symptomatology, would enable more timely management.
A case‐in‐point is the dilated cardiomyopathy CMD1O syndrome (Online Mendelian Inheritance in Man; OMIM 608569) caused by mutations in ATP‐sensitive K+ (KATP) channels.4 Dense in the ventricular sarcolemma, KATP channels are cardioprotective molecular rheostats implicated in the sustenance of wellness.5 These channels are integral in tissue energy conservation by coupling cellular energetics with membrane electrical activity. Mutations in KATP channel subunits have been associated with cardiomyopathic disorders.6 Linkage between KATP channel dysfunction and cardiac stress intolerance has been corroborated in knockout models,7–8 underscoring the vital role of intact channel function in ensuring protection against cardiac maladaptation and decompensation. In response to stress, KATP channel–deficient hearts demonstrate mild to severe cardiomyopathic phenotypes.9–10 Such a spectrum recapitulates the complexity of genotype–phenotype relationships in human heritable cardiomyopathies.11 Clinical manifestation of adult‐onset dilated cardiomyopathy includes an extended latent phase, with treatment delayed until detection of measurable functional and structural abnormalities.1–2 Therefore, strategies that would ensure subclinical disease diagnosis are warranted.
Based on the hypothesis that regional myocardial dysfunction precedes organ decompensation, high‐resolution cardiac imaging was used here in a KATP channel–null mutant to dissect myocardial pathodynamics before overt disease. This is the first study to prospectively resolve the origin and extent of intraventricular disparity of myocardial motion (ie, mechanical dyssynchrony) in a transgenic surrogate of a human cardiomyopathic syndrome. Detection of mechanical dyssynchrony, at a presymptomatic stage, anticipated organ failure in hearts genetically predisposed to disease.
Methods
Protocols were carried out in accordance with National Institutes of Health guidelines and with approval of the Mayo Clinic Institutional Animal Care and Use Committee.
Cardiomyopathy Model
Mice deficient in sarcolemmal KATP channels were generated by targeted disruption of the Kcnj11 gene encoding the Kir6.2 channel pore.12 Eight‐ to 12‐week‐old male wild‐type C57BL/6 (Harlan) and Kir6.2 knockout animals underwent, under 2% isoflurane anesthesia, transverse aortic constriction (TAC) to impose pressure overload.13 External constriction diameter was standardized using a 27‐gauge needle.14 Pain prophylaxis was implemented by an acetaminophen regimen (100 to 300 mg/kg in drinking water) 2 days before and 5 days after surgery. Prospective follow‐up was carried out in investigator‐blinded fashion for up to 100 days after TAC‐imposed stress load.
High‐Resolution Echocardiography
Cardiac function and structure of lightly anesthetized mice (0.5–1.5% of isoflurane) were quantified in vivo by transthoracic echocardiography with a 30‐MHz transducer (MS‐400; Vevo2100, VisualSonics) every 2 weeks and up to 100 days after TAC with average heart rates of 473±8 bpm and frame rates of 201±7/s. Left ventricular (LV) mass (mg) was derived as [(LVDd+IVST+PWT)3−LVDd3]×1.055, where LVDd is LV end‐diastolic dimension (mm), IVST is interventricular septum thickness (mm), and PWT is posterior wall thickness (mm). LV fractional shortening (%) was calculated as [(LVDd−LVDs)/LVDd]×100, where LVDs is LV end‐systolic dimension (mm). LV ejection fraction (%) was defined as [(LVVd−LVVs)/LVVd]×100, where LVVd is LV end‐diastolic volume, and LVVs is LV end‐systolic volume.15
Speckle Tracking
Consecutive cardiac cycles were acquired digitally from parasternal long‐axis and mid‐ventricular short‐axis views for assessment of radial, circumferential, and longitudinal strain and time‐to‐peak strain (VevoStrain, VisualSonics).16 The endocardium was traced with 48 sampling points within a 3.4‐ to 20‐mm length (70‐ to 400‐μm intervals), dividing the left ventricle into 6 segments. Strain was defined as change in length during myocardial contraction and relaxation. Peak strain (%) and time‐to‐peak strain (ms) were analyzed in each segment. Patterns of abnormal deformation were classified according to magnitude and timing of initiation and peak shortening.17 Specifically, dyssynchrony was defined as a pattern of early opposite deflection followed by reduced and delayed peak strain, akinesis as peak strain between −5% and 5%, and dyskinesis as motion opposite to contraction with peak systolic strain <−5% in radial and >5% in circumferential/longitudinal strain. The magnitude of dyssynchrony was further characterized by the absolute maximum delay and standard deviation of time‐to‐peak strain. Stretch in deformation imaging was defined as anomalous motion in the opposite direction to tissue shortening. Stretch‐to‐contraction ratio (%) was calculated as [(peak strain value of myocardial stretch)/(peak strain value of myocardial contraction)]×100.18 Preimposed inclusion criteria for prospective analysis of disease progression included survival >1 month post‐TAC and successful speckle‐based strain analysis in all 24 segments (6 segments in long‐ and short‐axis radial, circumferential, and longitudinal strain) throughout the 3‐month follow‐up. Data were collected and analyzed in a blinded fashion.
Electrocardiography
A 4‐limb lead electrocardiogram (LabChart 7, ADInstruments) was recorded to monitor heart rate, arrhythmic events, and QRS complex morphology.19 The QRS interval was measured from the beginning of a Q wave to termination of an S wave.
Systemic Effects
Maximum oxygen consumption (Oxymax, Columbus Instruments), exercise tolerance, and survivorship were collectively used to assess systemic effects of heart failure. Treadmill (Columbus Instruments) protocols included endurance capacity, using a stepwise increase in incline and velocity at 3‐minute intervals, and energy expenditure during exercise where a fixed speed of 10 m/minute for 35 minutes was carried out on a 20° inclination.5
Sarcomere Structure
Myocardial structure was evaluated by laser confocal microscopy (Zeiss LSM, Carl Zeiss) and transmission electron microscopy (JEOL 1200 EXII, Jeol Ltd) of LV tissues. For confocal microscopy, cardiac specific sarcomeric α‐actinin (1:200; Sigma‐Aldrich) along with 4′,6′‐diamidino‐2‐phenylindole (Molecular Probes) nuclear staining was used to visualize alignment of myocytes.20 For electron microscopy, tissue was fixed in phosphate‐buffered saline containing 1% glutaraldehyde and 4% formaldehyde (pH 7.2).21
Statistical Analysis
Data are presented as mean±SEM. Nonparametric Mann–Whitney U test was used to evaluate statistical significance (JMP 9, SAS Institute). Comparison between groups over time was performed by 2‐way repeated‐measures ANOVA. Kaplan–Meier analysis with log‐rank testing was applied for survival analysis. Linear regression analysis was performed, and the coefficient of correlation was determined. A P value <0.05 was predetermined as significant.
Results
KATP Channel–Dependent Cardiomyopathy Exhibits Mechanical Dyssynchrony
Pressure load imposed on the LV by TAC (Figure 1) unveiled cardiac stress intolerance in the setting of KATP channel deficiency (Figure 2). Compared with more‐resistant wild‐type counterparts, KATP channel–null mutants developed a significant cardiomyopathic phenotype characterized by hypertrophy (Figure 2A), reduced contractility (Figure 2B), and poor survivorship (2 weeks post‐TAC: wild‐type 86%, n=7; KATP channel knockout 34%, n=164; P<0.001), recapitulating clinical traits of the CMD1O syndrome.4,6,12 Consistently, the cardiomyopathic KATP channel–deficient hearts demonstrated post‐TAC end‐stage cardiomegaly, myocardial disarray, and fibrosis. In addition to evaluation of deterioration in global structure and function, conduction delay (ie, electrical dyssynchrony) and disparity in contraction (ie, mechanical dyssynchrony) were assessed by electrocardiography and speckle‐tracking echocardiography, respectively. The QRS interval, a parameter of ventricular conduction, did not differ between before and after TAC in either wild‐type or Kir6.2 knockout cohorts (Figure 2C). Within 2 weeks post‐TAC, parameters of discordant cardiac deformation were detected in KATP channel–deficient hearts, but not in the wild‐type (Figure 2D through 2F). Thus, KATP channel–dependent cardiomyopathy demonstrated early mechanical disparity.
Figure 1.
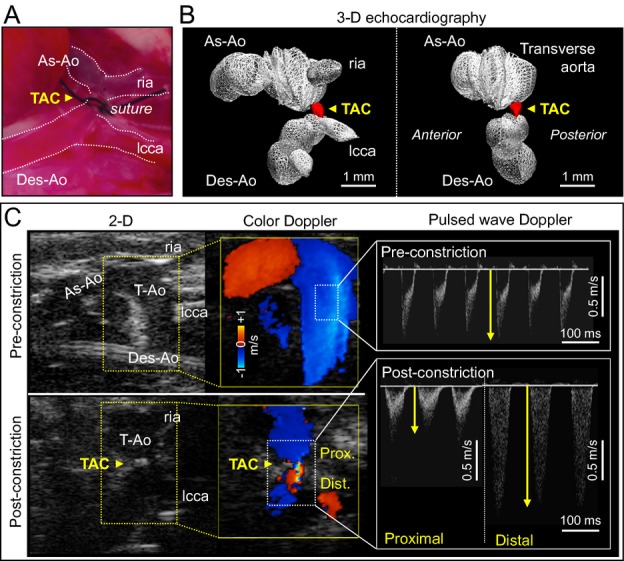
Hemodynamic stress induced by TAC. Constriction was surgically placed at the level of the transverse aorta (TAC, yellow arrowhead in A). The extent of TAC was determined by reduction in the diameter of the transverse aorta (red in B), which was originally ≈1 mm, and the increase in flow gradient between proximal and distal TAC sites (C). 2‐D/3‐D indicates 2‐/3‐dimensional; As‐Ao, ascending aorta; Des‐Ao, descending aorta; Dist., distal site of TAC; lcca, left common carotid artery; Prox., proximal site of TAC; ria, right innominate artery; TAC, transverse aortic constriction; T‐Ao, transverse aorta; yellow arrows, peak velocity.
Figure 2.
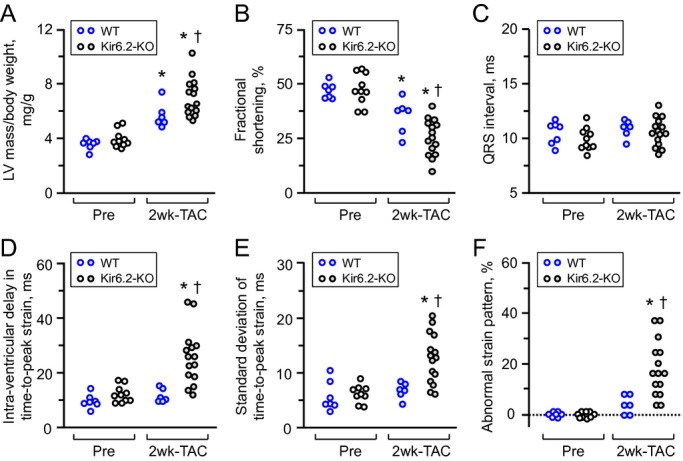
ATP‐sensitive K+ (KATP) channel knockout aggravates stress‐induced cardiomyopathy associated with mechanical dyssynchrony. Following transverse aortic constriction (TAC), KATP channel‐deficient hearts, due to Kir6.2 subunit knockout (Kir6.2‐KO, n=15), developed exaggerated left ventricular (LV) hypertrophy (A) with reduced contractility (B), compared with wild‐type hearts (WT, n=6). Within the initial 2 weeks (2 wk), mechanical dyssynchrony in the absence of QRS prolongation was present in the KATP channel knockout (C through F). In contrast, WT hearts did not demonstrate cardiac dyssynchrony post‐TAC (C through F). *P<0.05 vs Pre‐TAC; †P<0.05 vs WT.
Deconvolution of Mechanical Dyssynchrony
To dissect wall motion pathodynamics, high‐resolution speckle‐tracking echocardiography was performed prospectively post‐TAC. Specifically, disease progression was tracked within KATP channel–knockout cohorts that survived >1 month post‐TAC and whose speckle‐based strain was successfully analyzed in all 24 segments of the cardiomyopathic ventricles throughout follow‐up (n=15 at 2 weeks, n=8 at 1.5 months, and n=5 at 3 months post‐TAC). Temporal and spatial disparity were 2‐dimensionally deconvoluted in strain/R‐R interval maps (6 segments in Figure 3, 48 points in Figure 4A), and peak strain/anatomical maps (Figure 4B through 4D), respectively. Additionally, 3‐dimensional plots provided a comprehensive dataset of all sampling points throughout cardiac cycles (Figure 4E through 4G). Before stress imposition, the strain/R‐R interval map revealed a singular isosceles triangle–shaped peak, representing synchronous wall motion (Figure 3A). Poststress, KATP channel–deficient cardiomyopathic hearts were characterized by reduced shortening and varying amplitude, timing, and direction of wall motion, resulting in anisotropy underlying irregular ventricular contraction (Figure 3B through 3D). Intraventricular delay (yellow arrows in Figure 4A) and conflicting motion (blues areas in Figure 4A) were progressively exaggerated during follow‐up. In mid‐ventricular short‐axis view, peak strain was >30% in all sampling points prestress and decreased to <15% in 3 of 6 segments at 3 months poststress (Figure 4B through 4G). Peak radial strains progressively decreased (Figures 3 and 4). Similarly, longitudinal systolic strain declined as cardiomyopathy advanced, demonstrating a correlation with established parameters of cardiac dilatation (ie, LV end‐diastolic dimension, r=0.79; Figure 5A) and reduced ejection fraction (r=0.89; Figure 5B). At 3 months poststress, peak systolic strain persistently declined in longitudinal (P<0.0001; Figure 5C), circumferential (P<0.0001; Figure 5C), and long‐axis or short‐axis radial (P<0.0001; Figure 5D) directions. Beyond overall changes (Figure 5), the strain map revealed that myocardial dysfunction occurred inhomogeneously within the KATP channel–deficient ventricle (Figure 4D and 4G), not manifested in 2‐dimensional or M‐mode echocardiography. The interventricular septal wall was resilient (Figure 6), maintaining prestress values in peak systole (pre‐TAC 30.3±2.7%, 3 months post‐TAC 22.1±2.2%; P=0.07; Figure 6A and 6B) and in time‐to‐peak strain (pre‐TAC 67.8±3.7 ms, 3 months post‐TAC 63.7±6.1 ms; P=0.58; Figure 6C and 6D). Conversely, the lateral wall developed severe hypokinesis with maximum delay, demonstrating vulnerability to mechanical malfunction and significant conduction abnormality (peak strain: pre‐TAC 45.8±4.8%, 3 months post‐TAC 14.0±1.4%; P<0.01; Figure 6A and 6B; time‐to‐peak strain: pre‐TAC 63.4±3.1 ms, 3 months post‐TAC 87.5±6.5 ms P<0.01; Figure 6C and 6D). Speckle‐based strain analysis thus unmasked vulnerable areas of contractile defect that generate mechanical dyssynchrony in KATP channel–deficient stress‐vulnerable hearts.
Figure 3.
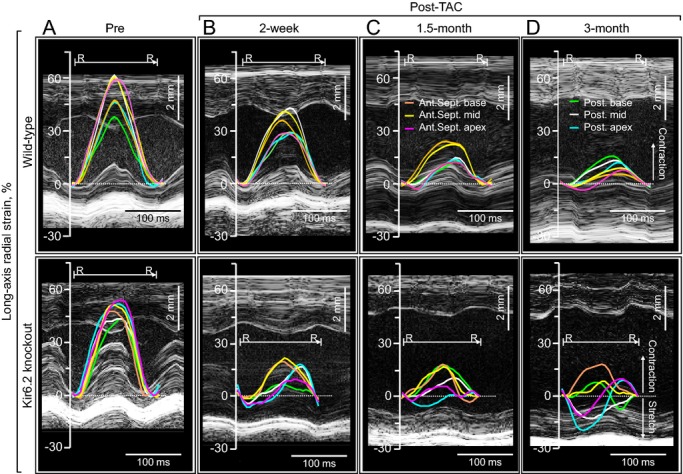
Progressive contractile discordance in KATP channel–deficient cardiomyopathy. Top, absence of mechanical dyssynchrony in the context of gradual reduction in force generation characterized the wild‐type postconstriction (Post‐TAC). Bottom, in contrast to an organized and timed ventricular contraction at baseline (Pre‐constriction, A), KATP channel‐deficient (due to Kir6.2 knockout) cardiomyopathic ventricular segments displayed a progressive decrease in peak contraction, early opposite deflection, and a disparity of time‐to‐peak strain (B and C), resulting in loss of regular systolic–diastolic cycles (D). Ant.Sept. apex indicates apical anterior septum; Ant.Sept. base, basal anterior septum; Ant.Sept. mid, mid anterior septum; KATP, ATP‐sensitive K+; Post. apex, posterior apex; Post. base, basal posterior wall; Post. mid, mid posterior wall; R‐R, R‐R interval; TAC, transverse aortic constriction.
Figure 4.
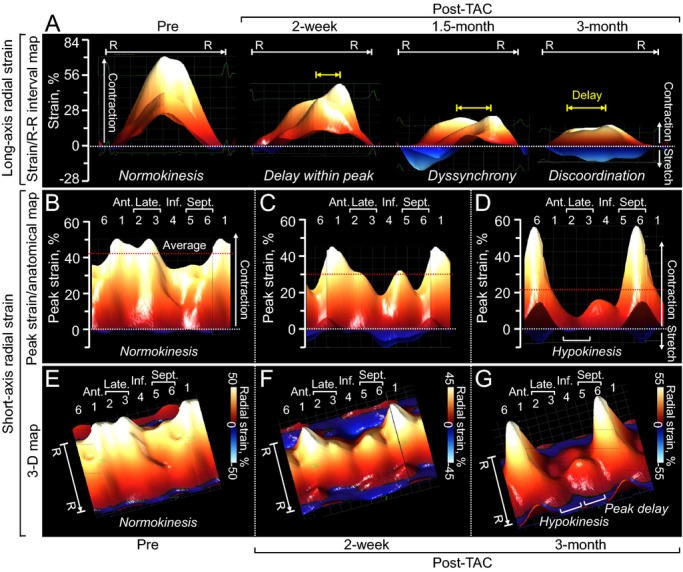
Two‐ and 3‐dimensional reconstruction of cardiomyopathic wall motion. Aberrant wall motion dynamics of the ATP‐sensitive K+ channel–deficient ventricle was documented in strain/R‐R interval (R‐R) map (A), peak strain/anatomical map (B through D), and 3‐dimensional (3‐D) map (E through G). 1 indicates anterior (Ant.); 2 plus 3, lateral (Late.); 2, anterolateral; 3, inferolateral; 4, inferior (Inf.); 5 plus 6, septum (Sept.); 5, inferior septum; 6, anterior septum; red dotted lines, average of peak strain at 48 sampling points; TAC, transverse aortic constriction; yellow arrows, delay within peak.
Figure 5.
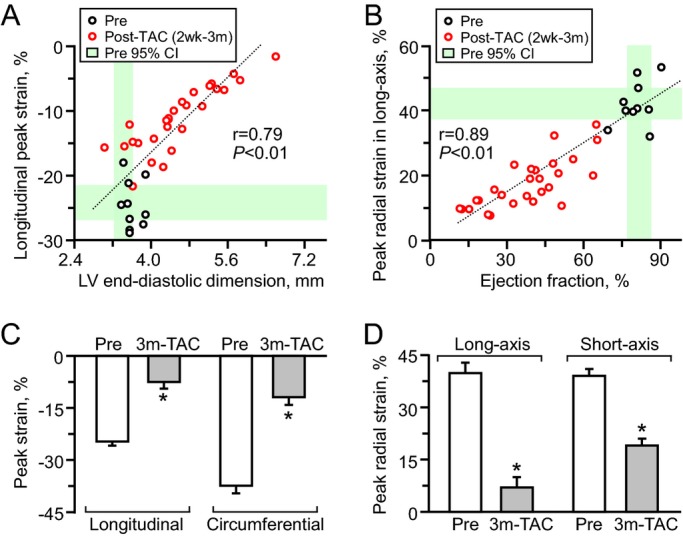
Global cardiac contractile dysfunction. Peak strain values in speckle‐tracking echocardiography had a linear correlation with cardiac dilatation (n=38; A) and reduced contractility (n=38; B) measured in M‐mode/2‐dimensional echocardiography. Peak strains significantly decreased following transverse aortic constriction (TAC) in ATP‐sensitive K+ channel knockout (Pre n=10, Post n=5; C and D). *P<0.05 vs Pre. 2 wk indicates 2 weeks; 3 m‐TAC, 3 months post‐TAC; CI, confidence interval; LV, left ventricular.
Figure 6.
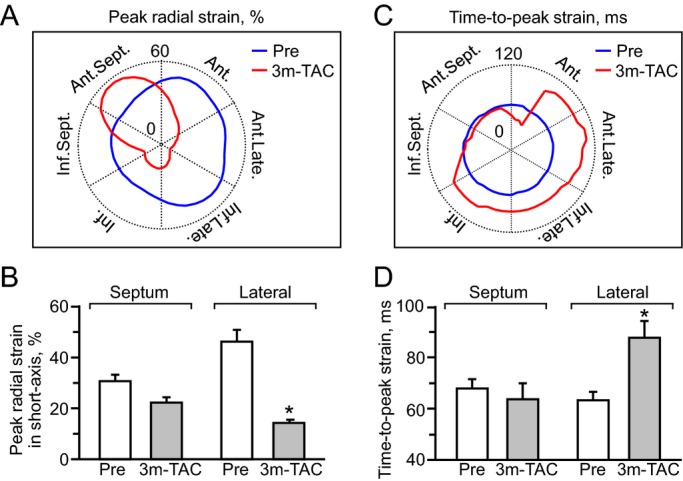
Hypokinesis with conduction delay. At 3‐month follow‐up, the septal wall maintained myocardial contractility (A and B), while the lateral wall developed hypokinesis with conduction delay (C and D). 3 m‐TAC indicates 3 months post transverse aortic constriction (TAC) in ATP‐sensitive K+ channel knockout (n=5). (A and C): Ant. indicates anterior; Ant.Late., anterolateral; Ant.Sept., anterior septum; Inf., inferior; Inf.Late., inferolateral; Inf.Sept., inferior septum; Late., lateral; blue and red lines indicate peak strain value pre‐ and post‐TAC, respectively. B and D: *P<0.05 vs Pre (n=10).
Contractile Discordance Precedes QRS Widening
QRS prolongation was significant only after 3 months of follow‐up without a noticeable change in the axis of the QRS complex (Figure 7A through 7D, top), heart rate (pre‐TAC 479±16 bpm, 3 months post‐TAC 531±49 bpm, P=0.14), or PQ interval (pre‐TAC 42.1±1.5 ms, 3 months post‐TAC 38.2±2.1 ms, P=0.30). In contrast to the late onset of QRS prolongation, evidence of nascent LV mechanical discordance was present as early as 2 weeks poststress (Figure 7A through 7D, bottom). Intraventricular delay in time‐to‐peak strain increased at early stage, from 12.3±1.1 ms prestress to 25.8±2.5 ms at 2 weeks poststress (P<0.001) and 38.6±4.2 ms at 3 months (P<0.01; Figure 7E), which preceded the time course of QRS prolongation (pre‐TAC 10.0±0.3 ms, 2 weeks post‐TAC 10.7±0.3 ms; P=0.19, 3 months post‐TAC 18.3±4.3 ms; P<0.05; Figure 7E). Likewise, the standard deviation of time‐to‐peak strain doubled from 6.4±0.5 ms prestress to 13.0±1.1 ms at 2 weeks poststress (P<0.001) and tripled at 3 months (18.8±2.7 ms; P<0.01 versus prestress; Figure 7F). Compared with the early onset of intraventricular delay in peak shortening, mechanical discoordination (ie, opposite deformation within the LV) developed later. At 2 weeks to 1.5 months poststress, conflicting wall motion was limited in early systole (blue areas in Figures 4A, 7B and 7C) and did not significantly affect myocardial contractility (stretch‐to‐contraction ratio: prestress 1.2±0.3%, 2 weeks post‐TAC 3.5±1.1%; P=0.12). The extension of discoordination over late‐systolic and diastolic phases compromised cardiac performance, resulting in loss of regular systolic–diastolic cycle (stretch‐to‐contraction ratio in strain: 3 months post‐TAC 26.3±14.3%; P<0.05 versus prestress; Figure 7D, inset). Areas of myocardial stretch in strain/R‐R interval maps amplified from 7.8±1.3% at prestress to 39.8±4.4% at 3 months in long‐axis view (P<0.01; blue areas in Figure 4A). In addition to scalar markers (Figure 7D through 7F), abnormal strain patterns also became apparent during the 3‐month follow‐up (percentage of abnormal segments: pre‐TAC 0%, 2 weeks post‐TAC 18.3±2.8%; P<0.001, 3 months post‐TAC 39.2±9.0%; P<0.001; Figure 7G). Thus, disparities of contractile timing were prominent in KATP channel–deficient cardiomyopathic hearts despite a normal to near‐normal QRS complex and preceded destructive conflicting wall motion.
Figure 7.
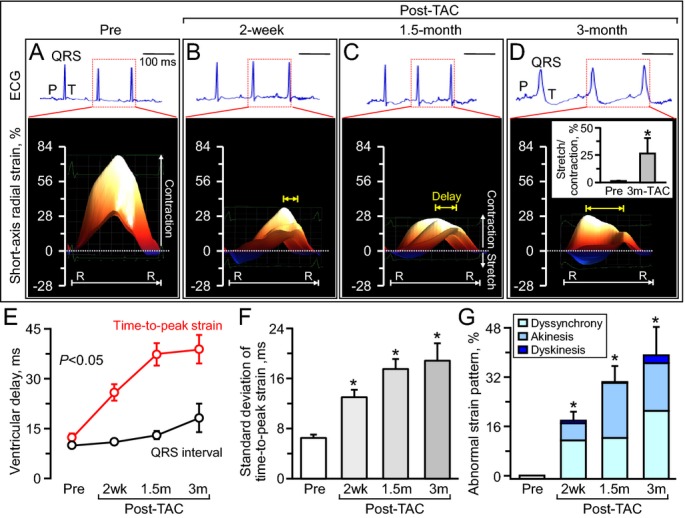
Mechanical dyssynchrony before QRS widening. Serial monitoring of electrocardiography (ECG: top panels in A through D) and speckle‐tracking echocardiography (bottom panels in A through D) demonstrated that mechanical dyssynchrony, validated by delay (E, yellow arrows in B through D), disparity (F) and abnormal strain patterns (B through D, and G), preceded the time course of initiation of electrocardiographic manifestation (E). R‐R indicates R‐R interval; TAC, transverse aortic constriction in ATP‐sensitive K+ channel knockout; 2 wk, 2 weeks (n=15); 1.5 m, 1.5 months (n=8); 3 m, 3 months (n=5); scale, 100 ms; *P<0.05 vs Pre (n=10).
Speckle‐Based Strain Offers a Refined Diagnosis of Disease Onset
At 3‐month follow‐up under stress, KATP channel–null mutants developed overt symptoms of heart failure, including poor exercise capacity, severe cachexia, and pronounced mortality (life span, 69.6±1.0 days poststress; 62% mortality between 2 weeks and 3 months post‐TAC; Figure 8). Of note, generalized seizures, skeletal muscle weakness, or differences in serum glucose levels post‐TAC were nondemonstrable in either wild‐type or KATP channel–knockout cohorts. Using current clinical guidelines requiring a low ejection fraction plus a wide QRS complex to meet indications for resynchronization, cardiomyopathic KATP channel–knockout hearts were diagnosed only late in disease as dyssynchronous (ie, at 3 months poststress) (Figure 8B). In contrast, speckle‐based myocardial motion mapping detected significant mechanical disturbance as early as 2 weeks poststress (Figure 8A). In fact, speckle‐based strain unmasked progression of dyssynchrony at all stages of follow‐up. Notably, a hypokinesis pattern with intraventricular delay in peak strain was detectable within 2 weeks, advanced hypokinesis and delay at 1.5 months, and loss of coordinated myocardial motion within 3 months (Figure 7). Intraventricular delay in time‐to‐peak strain was a sensitive marker throughout the time course of disease progression (relative change to pre‐TAC: 110.5±20.7% at 2 weeks, P<0.001, 214.5±34.3% at 3 months; P<0.01; Figure 8A). Standard deviation of time‐to‐peak strain progressively increased compared with pre‐TAC value, to 100.6±18.4% at 2 weeks, P<0.001, and 190.4±41.3% at 3 months, P<0.01 (Figure 8A). Thus, dyssynchrony and discoordination of tissue deformation provides a sensitive readout of disease onset and progression based on the deviation of regional and global mechanical dynamics within KATP channel‐deficient hearts.
Figure 8.
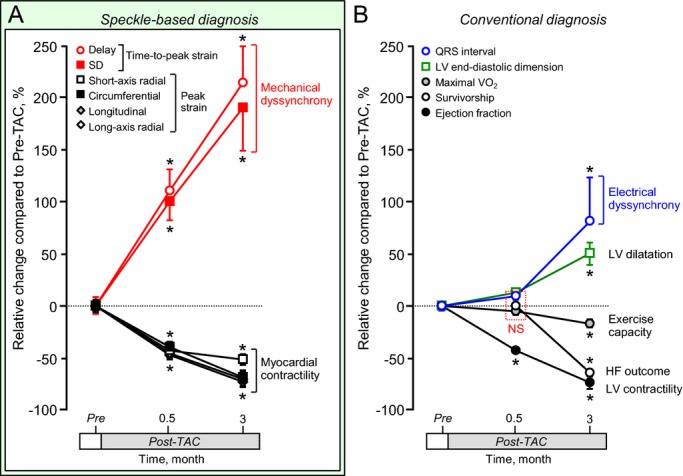
Imaging of myocardial motion reveals mechanical deformation within normal to near‐normal QRS hearts before systemic heart failure. Speckle‐tracking echocardiography detected myocardial mechanical disturbance as early as 2 weeks poststress in ATP‐sensitive K+ channel–deficient cardiomyopathy (A), at time point when electrocardiographic and systemic abnormalities were not yet prominent (B). Sample sizes were 10 before stress imposition (Pre), 15 at 2 weeks, and 5 in 3 months poststress. *P<0.05 vs Pre. Delay indicates intraventricular delay; HF, heart failure; LV, left ventricle; maximal Vo2, maximal oxygen consumption; TAC, transverse aortic constriction.
Discussion
Dyssynchrony – the disparity of electrical and mechanical activity – is increasingly recognized as a participating mechanism in defining poor heart failure outcome.22–26 Detrimental effects of dyssynchronous wall motion on pump function and remodeling have been associated with structural disease or conduction abnormalities.27–28 Early diagnosis is essential for adequate management of chronic disease; still, the origin and progression of cardiac dyssynchrony have yet to be fully characterized.29 Accordingly, the present study used speckle‐tracking imaging to unmask initial mechanical disturbances revealing a sensitive dyssynchrony readout in the setting of KATP channel–deficient cardiomyopathy.
Multimodal imaging enabled prospective noninvasive measurement of quantifiable parameters defining myocardial dynamics. Multiparametric analysis of speckle‐tracking signals30 was optimized and validated in a transgenic surrogate of the KATP channel–dependent CMD1O cardiomyopathic syndrome.4,12 KATP channel–deficient hearts demonstrated unique processes of cardiac dyssynchrony characterized by a shift from intraventricular delay of peak contraction (ie, mechanical dyssynchrony) to deleterious conflicting wall motion (ie, mechanical discoordination). Indeed, in this model, mechanical dyssynchrony was recognized early, before QRS widening and heart failure symptomatology. Lack of changes in the QRS axis and atrioventricular conduction indicated that the late widening of the QRS complex in the setting of KATP channel deficiency is due to a ventricular conduction delay rather than to onset of proximal bundle‐branch block. Existing preclinical models of cardiac dyssynchrony induce mechanical discordance as a consequence of preimposed electrical abnormality, via either bundle‐branch block or pacing.28,31 Right ventricular pacing and left bundle‐branch block models share prominent QRS widening and, accordingly, represent an advanced stage of electrical and mechanical dyssynchrony in the absence of a primary myocardial pathology.32 The present study establishes transgenic KATP channel–deficient cardiomyopathy with gradual QRS widening as a unique model of disease progression enabling examination of mechanical before electrical disparity.
Mechanical disturbance in the setting of a normal or near‐normal QRS complex has been controversial, in particular with regard to the need for therapeutic intervention.33–36 Introduction of cardiac resynchronization therapy, based on pacemaker implantation to harmonize ventricular contractions, has addressed severe heart failure where both electrical and mechanical abnormalities coexist.37 Clinical guidelines consider cardiac resynchronization therapy indications based on global parameters, including heart failure symptoms, reduced LV ejection fraction, and a wide QRS complex.38 Indeed, current guidelines do not incorporate assessment of myocardial mechanical discordance into criteria for cardiac resynchronization therapy candidate selection.39 This limitation is largely due to lack of standardized methodology capable of evaluating isolated mechanical dyssynchrony, independent of confounding electrical interaction.40–41 By combining a clinically relevant transgenic model and a high‐resolution imaging platform, this study longitudinally traced the evolution of regional mechanical dyssynchrony from an asymptomatic and normal QRS to an advanced stage associated with intraventricular conduction delay ultimately leading to terminal dyssynchronous heart failure.42 Collectively, the present study demonstrates in the setting of KATP channel–deficient cardiomyopathy that mechanical dyssynchrony precedes current criteria of cardiac dyssynchrony and thus could serve as an early marker of disease.
The size and hyperkinetic status of the murine heart43–44 had so far excluded transgenic models from cardiac dyssynchrony research. This study now establishes such a prospect, demonstrating comprehensive quantification of mechanical dyssynchrony based on the degree, timing, and direction of abnormal myocardial motions. By using advanced echocardiography equipped with a 30‐MHz probe at an average frame rate of 200 per second, regional myocardial deformations were reliably mapped within a diameter of <5 mm and at a 2‐ to 10‐mm depth in murine hearts beating on average at >470 bpm. In conjunction with recent studies in wild‐type murine models of ischemic and nonischemic heart failure,16,18,45–46 the present study expands the application of speckle‐tracking echocardiography to unmask progressive mechanical dyssynchrony underlying genetic cardiomyopathy.
Properties of ventricular conduction are regulated by membrane action potential, along with cell‐to‐cell connectivity and structure. KATP channel–deficient cardiomyopathic hearts display myocardial hypertrophy, tissue disarray, interstitial fibrosis, and a paucity of tight junctions,14 all of which could contribute to the observed pathophenotype. The underlying pathobiology of the CMD1O cardiomyopathy includes KATP channel‐dependent calcium dysregulation precipitating nuclear up‐regulation of calcium‐dependent proremodeling myocyte enhancer factor‐2 and nuclear factor of activated T cells pathways.13 Proteomics dissection has identified that deletion of the KATP channel pore subunit significantly alters the ventricular proteome leading to overrepresentation of “cardiovascular disease” pathology revealed by systems network analysis.47–48 Although the KATP channel Kir6.2 subunit is not exclusive to the heart,12 exclusion of noncardiac signs in the CMD1O model, as well as in separate experiments in denervated and isolated kir6.2‐null heart preparations,8,49 underscored cardiac‐dependent consequences of the KATP channel–knockout phenotype. While the present study uses the KATP channel–dependent cardiomyopathy model, it is conceivable that distinct cardiomyopathies may display mechanical dyssynchrony in the absence of QRS prolongation.50–52
In conclusion, here, high‐resolution cardiac imaging detected early signs of progressive cardiomyopathy. By mapping the origin and extent of aberrant myocardial motion, speckle‐tracking enabled presymptomatic diagnosis of dyssynchronous heart failure associated with KATP channel dysfunction.
Clinical Perspective Summary
Contractile discordance compromises cardiac pump efficiency, aggravating heart failure outcome. A strategy that would evaluate mechanical dyssynchrony independently from confounding electrophysiological manifestations would advance the deconvolution of disease pathobiology. High‐resolution speckle‐tracking imaging was here prospectively applied in a transgenic dilated cardiomyopathy model caused by ablation of cardioprotective ATP‐sensitive K+ channel function. Serial monitoring of speckle‐based strain pinpointed the nascent spatial and temporal disparity of myocardial motion before, and independent from, electrocardiographic deterioration. The degree, direction, and delay in myocardial speckle strains unmasked progressive mechanical dyssynchrony. This study thus demonstrates that mechanical dyssynchrony is a prodrome of dyssynchronous heart failure in genetic cardiomyopathy, identifying an early marker of disease.
Sources of Funding
Dr Yamada is a recipient of American Heart Association National Scientist Development Grant (11SDG7210074) and a Mayo Clinic Center for Regenerative Medicine Career Development Award. Dr Purushothaman is supported by T32GM065841 Medical Scientist Training Program at Mayo Clinic. This work was funded by the National Institutes of Health, Marriott Heart Disease Research Program, and Mayo Clinic. A.T. holds the Marriott Family Professorship in Cardiovascular Research, and is the Michael S. and Mary Sue Shannon Director, Mayo Clinic Center for Regenerative Medicine.
Disclosures
Dr Prinzen receives research support from Medtronic and EBR. Dr Auricchio reports consulting for Medtronic, St. Jude Medical, Sorin Group, Biotronik GmBH, Cordis Biological Delivery Systems, EBR, and Abbott and receives speaker fees from Medtronic, St. Jude Medical, Sorin Group, Biotronik GmBH, Cordis Biological Delivery Systems, EBR, and Abbott. The other authors report no conflicts of interest relevant to this work.
Acknowledgments
We thank Diane M. Jech, RDCS, for echocardiography analysis, Jonathan J. Nesbitt for animal surgery, Lois A. Rowe for histology, Courtney Rust for strain mapping, and Nicolas L. Carlblom and Sarah Burrington for oxygen consumption tests. We are grateful to Drs. Takashi Miki (Chiba University) and Susumu Seino (Kobe University) for the initial derivation of the KATP channel–knockout mice.
References
- 1.Watkins H, Ashrafian H, Redwood C. Inherited cardiomyopathies. N Engl J Med. 2011; 364:1643-1656 [DOI] [PubMed] [Google Scholar]
- 2.Piran S, Liu P, Morales A, Hershberger RE. Where genome meets phenome: rationale for integrating genetic and protein biomarkers in the diagnosis and management of dilated cardiomyopathy and heart failure. J Am Coll Cardiol. 2012; 60:283-289 [DOI] [PubMed] [Google Scholar]
- 3. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012. Eur Heart J. 2012; 33:1787-1847 [DOI] [PubMed] [Google Scholar]
- 4.Terzic A, Alekseev AE, Yamada S, Reyes S, Olson TM. Advances in cardiac ATP‐sensitive K+ channelopathies from molecules to populations. Circ Arrhythm Electrophysiol. 2011; 4:577-585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alekseev AE, Reyes S, Yamada S, Hodgson‐Zingman DM, Sattiraju S, Zhu Z, Gerbin M, Coetzee WA, Goldhamer DJ, Terzic A, Zingman LV. Sarcolemmal ATP‐sensitive K+ channels control energy expenditure determining body weight. Cell Metab. 2010; 11:58-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bienengraeber M, Olson TM, Selivanov VA, Kathmann EC, O'Cochlain F, Gao F, Karger AB, Ballew JD, Hodgson DM, Zingman LV, Pang YP, Alekseev AE, Terzic A. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat Genet. 2004; 36:382-387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kane GC, Behfar A, Yamada S, Perez‐Terzic C, O'Cochlain F, Reyes S, Dzeja PP, Miki T, Seino S, Terzic A. KATP channel knockout compromises the metabolic benefit of exercise training resulting in cardiac deficits. Diabetes. 2004; 53:S169-S715 [DOI] [PubMed] [Google Scholar]
- 8.Zingman LV, Hodgson DM, Bast PH, Kane GC, Perez‐Terzic C, Gumina RJ, Pucar D, Bienengraeber M, Dzeja PP, Miki T, Seino S, Alekseev AE, Terzic A. Kir6.2 is required for adaptation to stress. Proc Natl Acad Sci USA. 2002; 99:13278-13283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arrell DK, Zlatkovic‐Lindor J, Yamada S, Terzic A. KATP channel‐dependent metaboproteome decoded: systems approaches to heart failure prediction, diagnosis, and therapy. Cardiovasc Res. 2011; 90:258-266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kane GC, Behfar A, Dyer RB, O'Cochlain DF, Liu XK, Hodgson DM, Reyes S, Miki T, Seino S, Terzic A. KCNJ11 gene knockout of the Kir6.2 KATP channel causes maladaptive remodeling and heart failure in hypertension. Hum Mol Genet. 2006; 15:2285-2297 [DOI] [PubMed] [Google Scholar]
- 11.Lakdawala NK, Thune JJ, Colan SD, Cirino AL, Farrohi F, Rivero J, McDonough B, Sparks E, Orav EJ, Seidman JG, Seidman CE, Ho CY. Subtle abnormalities in contractile function are an early manifestation of sarcomere mutations in dilated cardiomyopathy. Circ Cardiovasc Genet. 2012; 5:503-510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olson TM, Terzic A. Human KATP channelopathies: diseases of metabolic homeostasis. Pflugers Arch. 2010; 460:295-306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamada S, Kane GC, Behfar A, Liu XK, Dyer RB, Faustino RS, Miki T, Seino S, Terzic A. Protection conferred by myocardial ATP‐sensitive K+ channels in pressure overload‐induced congestive heart failure revealed in KCNJ11 Kir6.2‐null mutant. J Physiol. 2006; 577:1053-1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamada S, Nelson TJ, Crespo‐Diaz RJ, Perez‐Terzic C, Liu XK, Miki T, Seino S, Behfar A, Terzic A. Embryonic stem cell therapy of heart failure in genetic cardiomyopathy. Stem Cells. 2008; 26:2644-2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zlatkovic‐Lindor J, Arrell DK, Yamada S, Nelson TJ, Terzic A. ATP‐sensitive K+ channel‐deficient dilated cardiomyopathy proteome remodeled by embryonic stem cell therapy. Stem Cells. 2010; 28:1355-1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bauer M, Cheng S, Jain M, Ngoy S, Theodoropoulos C, Trujillo A, Lin FC, Liao R. Echocardiographic speckle‐tracking based strain imaging for rapid cardiovascular phenotyping in mice. Circ Res. 2011; 108:908-916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carasso S, Rakowski H, Witte KK, Smith P, Carasso D, Garceau P, Sasson Z, Parker JD. Left ventricular strain patterns in dilated cardiomyopathy predict response to cardiac resynchronization therapy: timing is not everything. J Am Soc Echocardiogr. 2009; 22:242-250 [DOI] [PubMed] [Google Scholar]
- 18.Yamada S, Nelson T, Kane G, Martinez‐Fernandez A, Crespo‐Diaz RJ, Ikeda Y, Terzic C, Terzic A. iPS cell intervention rescues ventricular wall motion disparity achieving biological cardiac resynchronization post‐infarction. J Physiol. 2013; 591:4335-4349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamada S, Nelson TJ, Behfar A, Crespo‐Diaz RJ, Fraidenraich D, Terzic A. Stem cell transplant into preimplantation embryo yields myocardial infarction‐resistant adult phenotype. Stem Cells. 2009; 27:1697-1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Behfar A, Yamada S, Crespo‐Diaz R, Nesbitt JJ, Rowe LA, Perez‐Terzic C, Gaussin V, Homsy C, Bartunek J, Terzic A. Guided cardiopoiesis enhances therapeutic benefit of bone marrow human mesenchymal stem cells in chronic myocardial infarction. J Am Coll Cardiol. 2010; 56:721-734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hodgson DM, Zingman LV, Kane GC, Perez‐Terzic C, Bienengraeber M, Ozcan C, Gumina RJ, Pucar D, O'Coclain F, Mann DL, Alekseev AE, Terzic A. Cellular remodeling in heart failure disrupts KATP channel‐dependent stress tolerance. EMBO J. 2003; 22:1732-1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Auricchio A, Prinzen FW. Update on the pathophysiological basics of cardiac resynchronization therapy. Europace. 2008; 10:797-800 [DOI] [PubMed] [Google Scholar]
- 23.Kass DA. Pathobiology of cardiac dyssynchrony and resynchronization. Heart Rhythm. 2009; 6:1660-1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bertini M, Sengupta PP, Nucifora G, Delgado V, Ng AC, Marsan NA, Shanks M, van Bommel RJ, Schalij MJ, Narula J, Bax JJ. Role of left ventricular twist mechanics in the assessment of cardiac dyssynchrony in heart failure. JACC Cardiovasc Imaging. 2009; 2:1425-1435 [DOI] [PubMed] [Google Scholar]
- 25.Onishi T, Onishi T, Marek JJ, Ahmed M, Haberman SC, Oyenuga O, Adelstein E, Schwartzman D, Saba S, Gorcsan J., III Mechanistic features associated with improvement in mitral regurgitation after cardiac resynchronization therapy and their relation to long‐term patient outcome. Circ Heart Fail. 2013; 6:685-693 [DOI] [PubMed] [Google Scholar]
- 26.Linde C, Gold MR, Abraham WT, St John Sutton M, Ghio S, Cerkvenik J, Daubert CREVERSE Study Group Long‐term impact of cardiac resynchronization therapy in mild heart failure: 5‐year results from the REsynchronization reVErses Remodeling in Systolic left vEntricular dysfunction (REVERSE) study. Eur Heart J. 2013; 34:2592-2599 [DOI] [PubMed] [Google Scholar]
- 27. HRS/ACCF expert consensus statement on pacemaker device and mode selection. J Am Coll Cardiol. 2012; 60:682-703 [DOI] [PubMed] [Google Scholar]
- 28.Verbeek XA, Vernooy K, Peschar M, van der Nagel T, van Hunnik A, Prinzen FW. Quantification of interventricular asynchrony during LBBB and ventricular pacing. Am J Physiol. 2001; 283:H1370-H1378 [DOI] [PubMed] [Google Scholar]
- 29.Liu L, Tockman B, Girouard S, Pastore J, Walcott G, KenKnight B, Spinelli J. Left ventricular resynchronization therapy in a canine model of left bundle branch block. Am J Physiol. 2002; 282:H2238-H2244 [DOI] [PubMed] [Google Scholar]
- 30. 2012 EHRA/HRS expert consensus statement on cardiac resynchronization therapy in heart failure: implant and follow‐up recommendations and management. Heart Rhythm. 2012; 9:1524-1576 [DOI] [PubMed] [Google Scholar]
- 31.Russell K, Opdahl A, Remme EW, Gjesdal O, Skulstad H, Kongsgaard E, Edvardsen T, Smiseth OA. Evaluation of left ventricular dyssynchrony by onset of active myocardial force generation: a novel method that differentiates between electrical and mechanical etiologies. Circ Cardiovasc Imaging. 2010; 3:405-414 [DOI] [PubMed] [Google Scholar]
- 32.Strik M, van Middendorp LB, Vernooy K. Animal models of dyssynchrony. J Cardiovasc Transl Res. 2012; 5:135-145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Auricchio A, Prinzen FW. Non‐responders to cardiac resynchronization therapy: the magnitude of the problem and the issues. Circ J. 2011; 75:521-527 [DOI] [PubMed] [Google Scholar]
- 34.Van Bommel RJ, Delgado V, Schalij MJ, Bax JJ. Critical appraisal of the use of cardiac resynchronization therapy beyond current guidelines. J Am Coll Cardiol. 2010; 56:754-762 [DOI] [PubMed] [Google Scholar]
- 35.Thibault B, Harel F, Ducharme A, White M, Ellenbogen KA, Frasure‐Smith N, Roy D, Philippon F, Dorian P, Talajic M, Dubuc M, Guerra PG, Macle L, Rivard L, Andrade J, Khairy PLESSER‐EARTH Investigators Cardiac resynchronization therapy in patients with heart failure and a QRS complex <120 milliseconds: the evaluation of resynchronization therapy for heart failure (LESSER‐EARTH) trial. Circulation. 2013; 127:873-881 [DOI] [PubMed] [Google Scholar]
- 36.Ruschitzka F, Abraham WT, Singh JP, Bax JJ, Borer JS, Brugada J, Dickstein K, Ford I, Gorcsan J, III, Gras D, Krum H, Sogaard P, Holzmeister JEchoCRT Study Group Cardiac‐resynchronization therapy in heart failure with a narrow QRS complex. N Engl J Med. 2013; 369:1395-1405 [DOI] [PubMed] [Google Scholar]
- 37.Al‐Majed NS, McAlister FA, Bakal JA, Ezekowitz JA. Meta‐analysis: cardiac resynchronization therapy for patients with less symptomatic heart failure. Ann Intern Med. 2011; 154:401-412 [DOI] [PubMed] [Google Scholar]
- 38. 2013 ESC guidelines on cardiac pacing and cardiac resynchronization therapy: the task force on cardiac pacing and resynchronization therapy of the European Society of Cardiology (ESC). Developed in collaboration with the European Heart Rhythm Association (EHRA). Europace. 2013; 15:1070-1118 [DOI] [PubMed] [Google Scholar]
- 39. ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2013; 128:1810-1852 [DOI] [PubMed] [Google Scholar]
- 40.Kass DA. An epidemic of dyssynchrony: but what does it mean? J Am Coll Cardiol. 2008; 51:12-17 [DOI] [PubMed] [Google Scholar]
- 41.Geyer H, Caracciolo G, Abe H, Wilansky S, Carerj S, Gentile F, Nesser HJ, Khandheria B, Narula J, Sengupta PP. Assessment of myocardial mechanics using speckle tracking echocardiography: fundamentals and clinical applications. J Am Soc Echocardiogr. 2010; 23:351-369 [DOI] [PubMed] [Google Scholar]
- 42.Kandala J, Altman RK, Park MY, Singh JP. Clinical, laboratory, and pacing predictors of CRT response. J Cardiovasc Transl Res. 2012; 5:196-212 [DOI] [PubMed] [Google Scholar]
- 43.Scherrer‐Crosbie M, Thibault HB. Echocardiography in translational research: of mice and men. J Am Soc Echocardiogr. 2008; 21:1083-1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.American Heart Association Council on Basic Cardiovascular Sciences, Council on Clinical Cardiology, and Council on Functional Genomics and Translational Biology Animal models of heart failure: a scientific statement from the American Heart Association. Circ Res. 2012; 111:131-150 [DOI] [PubMed] [Google Scholar]
- 45.Peng Y, Popovic ZB, Sopko N, Drinko J, Zhang Z, Thomas JD, Penn MS. Speckle tracking echocardiography in the assessment of mouse models of cardiac dysfunction. Am J Physiol Heart Circ Physiol. 2009; 297:H811-H820 [DOI] [PubMed] [Google Scholar]
- 46.Bauer M, Cheng S, Unno K, Lin FC, Liao R. Regional cardiac dysfunction and dyssynchrony in a murine model of afterload stress. PLoS One. 2013; 8:e59915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arrell DK, Zlatkovic J, Kane GC, Yamada S, Terzic A. ATP‐sensitive K+ channel knockout induces cardiac proteome remodeling predictive of heart disease susceptibility. J Proteome Res. 2009; 8:4823-4834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arrell DK, Terzic A. Systems proteomics for translational network medicine. Circ Cardiovasc Genet. 2012; 5:478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu XK, Yamada S, Kane GC, Alekseev AE, Hodgson DM, O'Cochlain F, Jahangir A, Miki T, Seino S, Terzic A. Genetic disruption of Kir6.2, the pore‐forming subunit of ATP‐sensitive K+ channel, predisposes to catecholamine‐induced ventricular dysrhythmia. Diabetes. 2004; 53:S165-S168 [DOI] [PubMed] [Google Scholar]
- 50.Hor KN, Wansapura JP, Al‐Khalidi HR, Gottliebson WM, Taylor MD, Czosek RJ, Nagueh SF, Akula N, Chung ES, Benson WD, Mazur W. Presence of mechanical dyssynchrony in Duchenne muscular dystrophy. J Cardiovasc Magn Reson. 2011; 13:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Basaran Y, Tigen K, Karaahmet T, Isiklar I, Cevik C, Gurel E, Dundar C, Pala S, Mahmutyazicioglu K, Basaran O. Fragmented QRS complexes are associated with cardiac fibrosis and significant intraventricular systolic dyssynchrony in nonischemic dilated cardiomyopathy patients with a narrow QRS interval. Echocardiography. 2011; 28:62-68 [DOI] [PubMed] [Google Scholar]
- 52.D'Andrea A, Mele D, Nistri S, Riegler L, Galderisi M, Agricola E, Losi MA, Ballo P, Mondillo S, Badano LPWorking Group Nucleus on Echocardiography of Italian Society of Cardiology The prognostic impact of dynamic ventricular dyssynchrony in patients with idiopathic dilated cardiomyopathy and narrow QRS. Eur Heart J Cardiovasc Imaging. 2013; 14:183-189 [DOI] [PubMed] [Google Scholar]